
Abstract
Introduction
A
Low-power laser irradiation (LPLI) has been used by many health specialists and general practitioners to treat a broad range of illnesses. Currently, we developed high-fluence, low-power laser irradiation (HF-LPLI) as a novel cancer treatment modality using a mitochondria-targeted laser (635 nm) and explored the mechanism involved in the interaction between the light and its photoacceptor. Our results clearly demonstrated that HF-LPLI initiated its effects via targeted cytochrome c oxidase photoinactivation and that the tumor-killing efficacy was dependent on the subsequent mitochondrial superoxide anion burst via electron transport chain. We conclude that the mitochondria-targeting HF-LPLI is feasible and effective, and may be of significant clinical importance in treating solid cancer.
The ideal treatment modality for cancer should achieve tumor destruction via a minimally invasive local intervention. As the gateway of the intrinsic pathway for apoptosis, mitochondrial destruction represents a point of no return in many models of apoptosis (22). As a result, mitochondria have been considered potential targets for cancer therapy (12). Previously, we found that HF-LPLI (633 nm, 120 J/cm2) could induce cancer cell apoptosis in vitro via an intrinsic mitochondrial pathway by triggering the generation of reactive oxygen species (ROS) (40, 41). We demonstrated the mitochondrial pathway by HF-LPLI, as evidenced by the inactivation of caspase-8 (41), the activation of caspase-9 (5), and the release of cytochrome c (40). We also found that HF-LPLI induced the mitochondrial pathway via the induction of ROS-mediated mitochondrial permeability transition (MPT) (40). Another pro-apoptotic signaling pathway comprising the inactivation of protein kinase B/glycogen synthase kinase 3 beta on HF-LPLI was also explored (16). Although the initial mechanism involved in HF-LPLI-induced ROS generation is still unknown, these reports suggest that LPLI at higher doses can be used for cancer therapy via laser focusing and mitochondrial targeting.
The photobiological reactions of LPLI involve the absorption of photons at a specific wavelength by functioning photoacceptor molecules (19, 33). Cytochrome c oxidase (COX) is the terminal enzyme (complex IV) of the electron transport chain (ETC) in eukaryotic cells and mediates the transfer of electrons from cytochrome c to molecular oxygen (O2) (34). COX has been increasingly shown to be the photoacceptor and photosignal transducer in the red-to-NIR region of light (7, 19, 28). It has long been known that electronic excitation by light stimulates redox processes in organic dyes to intensify electron transfer (24). Similarly, it is quite possible that LPLI makes more electrons available for the reduction of O2 in the catalytic center of COX (heme a3–CuB site). The increase in the availability of electrons can be the crucial result of LPLI in situations in which all the four electrons are unavailable for the reduction of O2. The COX absorption of LPLI at low fluence has been reported to increase its enzymatic activity, increase the mitochondrial transmembrane potential (ΔΨm), and increase the levels of ATP, cyclic adenosine monophosphate, and ROS, leading to increased energy availability and signal transduction (7, 10, 19, 39). However, the initial interaction between light and COX under HF-LPLI is still unknown.
Inducing local cellular damage via the modulation of ROS production is likely to cause less systematic side effect compared with other modalities, and its potential in the treatment of various health conditions has already been demonstrated clinically. Here, we develop a novel cancer treatment modality using HF-LPLI and investigate the initial mechanism of ROS generation by HF-LPLI. We found that HF-LPLI at the range of red light (635 nm) could selectively photoinactivate its endogenous photoacceptor COX to generate a mitochondrial superoxide anion (O2 −•) burst, resulting in oxidative damage to cancer cells. This mitochondria-targeting phototherapy reaches sufficient antitumor efficacy without the administration of exogenous chemicals.
Results
In vitro tumor killing efficacy of HF-LPLI via ROS generation
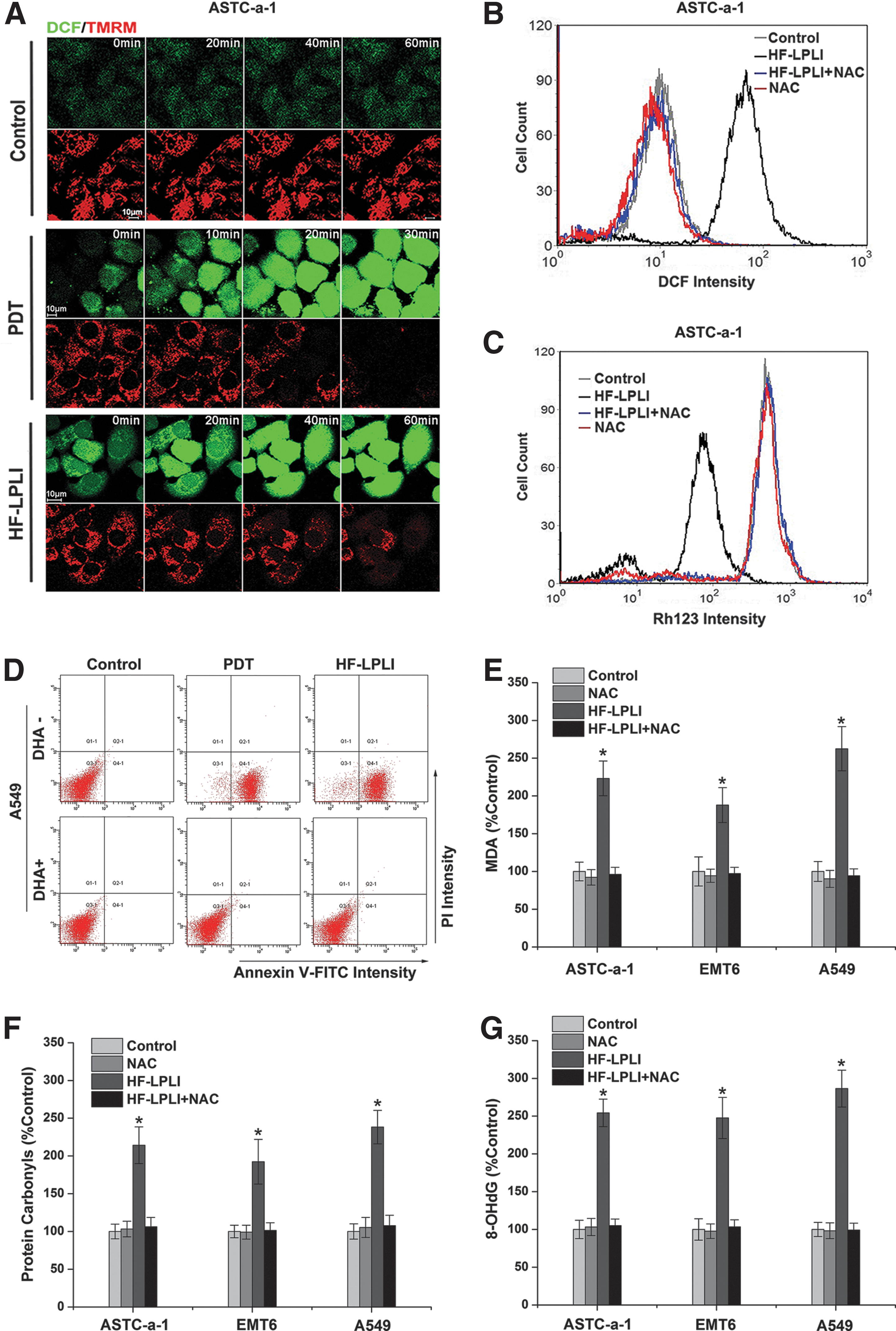
Our previous study showed that either HF-LPLI or photodynamic therapy (PDT) could induce cell apoptosis via the mitochondrial pathway by triggering the generation of ROS (41, 42). To confirm the damage effect of ROS generated by HF-LPLI, we studied the correlation between ROS generation and mitochondrial injury in human lung adenocarcinoma cell line (ASTC-a-1) cells (Fig. 1). The cells were stained with 2′,7′-dichlorodihydrofluorescein diacetate, succinimidyl ester (H2DCFDA, SE), and tetramethylrhodamine methyl ester (TMRM) dyes and imaged using confocal microscopy under various conditions. The representative sequential images show that the decrease in TMRM intensity corresponds closely to the increase in 2′,7′-dichlorofluorescein (DCF) intensity after both PDT and HF-LPLI treatment, indicating that HF-LPLI induces a decrease in ΔΨm along with ROS generation, similar to the results with PDT (Fig. 1A). Using flow cytometry (FACS) analysis, we then detected ROS generation (Fig. 1B) and ΔΨm collapse (Fig. 1C) in ASTC-a-1 cells, and cell apoptosis in human lung adenocarcinoma cell line (A549; Fig. 1D), ASTC-a-1 (Supplementary Figs. S1 and S2; Supplementary Data are available online at
Mitochondrial O2 −• burst by HF-LPLI
To explore the initial site of ROS generation, A549 cells stained with H2DCFDA, SE dye, and expressing DsRed-mit (labels mitochondria) were imaged using confocal microscopy immediately after HF-LPLI or PDT treatment. As shown in Figure 2A, the co-localization of the DCF and DsRed-mit emissions clearly shows that the initial site of ROS generation by HF-LPLI is primarily in mitochondria, similar to the results with PDT. Studies performed in ASTC-a-1 and A549 cells with dihydroethidium (DHE) and MitoTraker Deeper Red 633 (MTR, labels mitochondria) dyes revealed that the initial site of O2 −• generation by HF-LPLI was primarily in mitochondria (Fig. 2B). We used fluoresceinyl cypridina luciferin analog (FCLA)-based chemiluminescence (CL) to detect O2 −• and singlet oxygen (1O2) generation during treatment of HF-LPLI (Fig. 2C). The subcellular fractions of primary mouse liver cells incubated with FCLA were subjected to HF-LPLI with or without superoxidase dismutase (SOD) exposure. Positive CL signals, representing the generation of O2 −• and 1O2, were only detected in the mitochondrial fraction. SOD exposure significantly reduced the HF-LPLI-triggered CL signal in the mitochondrial fraction.
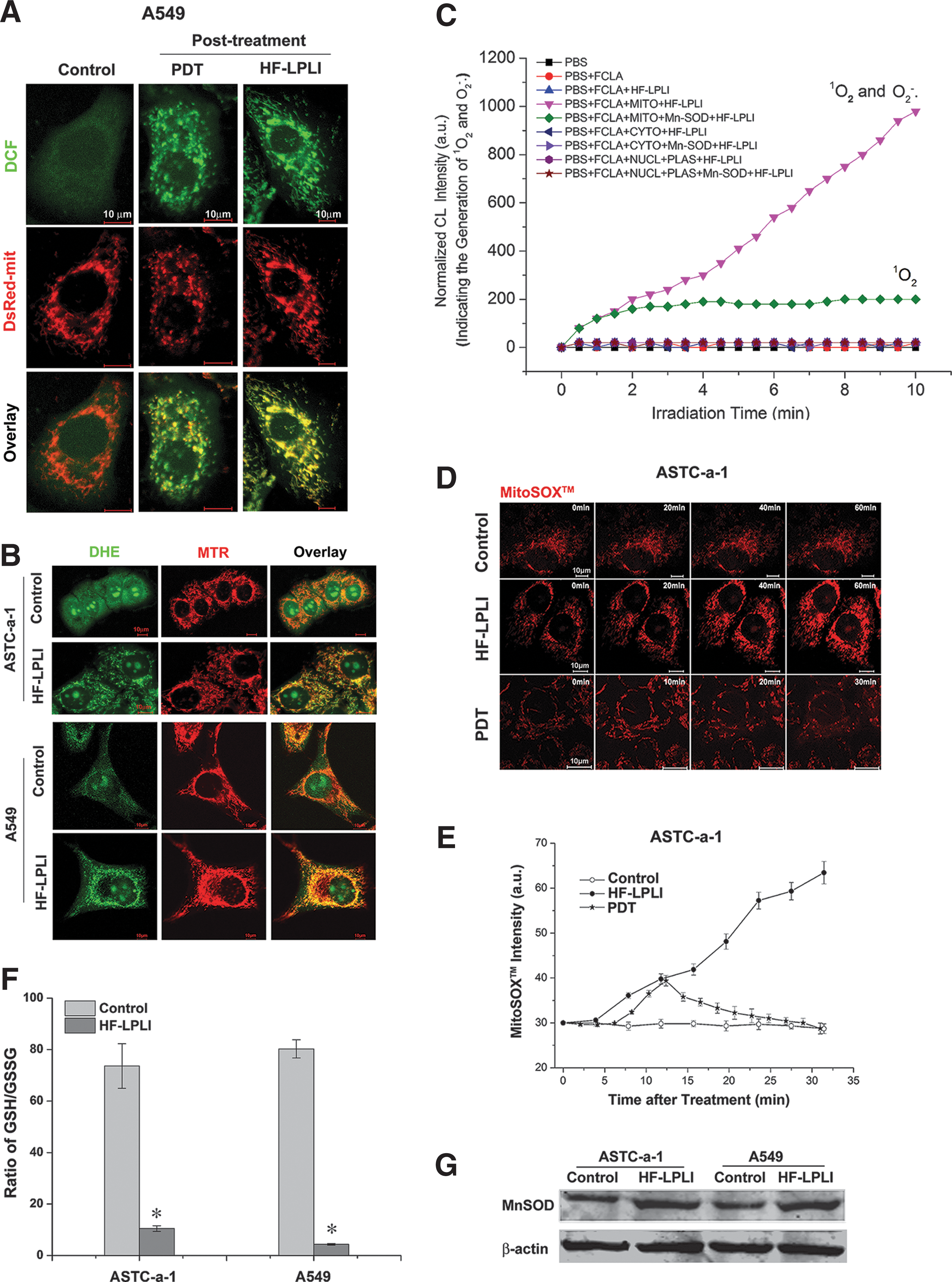
To confirm the generation of mitochondrial O2 −• after HF-LPLI, A549 cells stained with MitoSOX™ dye were subjected to FACS analysis (Supplementary Fig. S3). The MitoSOX™ intensity obviously increased after HF-LPLI, compared with the low increase observed in the control cells. SOD exposure largely inhibited HF-LPLI-induced mitochondrial O2 −• generation. We monitored the dynamic generation of mitochondrial O2 −• after HF-LPLI or PDT treatment in ASTC-a-1 cells stained with MitoSOX™ dye using confocal microscopy (Fig. 2D, E). The MitoSOX™ intensity increased quickly after HF-LPLI treatment, indicating a mitochondrial O2 −• burst. After PDT treatment, the MitoSOX™ intensity increased in the first 10 min and then decreased gradually, indicating the rupture of the mitochondrial membrane (48). These results suggest that HF-LPLI induces a selectively mitochondrial O2 −• burst with mitochondrial membrane integrity, while PDT ruptures the mitochondrial membrane after the initial oxidized reaction of the photosensitizer.
The expression of manganese superoxide dismutase (MnSOD) was induced by HF-LPLI in both ASTC-a-1 and A549 cells (Fig. 2G), suggesting a protective mechanism initiated by these cells against oxidative stress. Oxidative stress always causes an intracellular increase in oxidized glutathione (GSSG) levels concurrent with a decrease in glutathione (GSH) levels. Cellular GSSG/GSH ratio in both ASTC-a-1 and A549 cells increased 2 h after HF-LPLI treatment (Fig. 2F).
In vitro tumor cell-killing efficacy of HF-LPLI depends on the mitochondrial O2 −• burst
We generated two respiration-deficient cell lines ρ0ASTC-a-1 and ρ0A549 (Supplementary Figs. S4 and S5) to inhibit the mitochondrial O2 −• generation via ETC. To analyze the mitochondrial O2 −• generation between wild-type cells and ρ0 cells, we used FACS analysis with MitoSOX™ dye (Fig. 3A, B) and CL detection with FCLA probe (Fig. 3C). After HF-LPLI, no positive MitoSOX™ signal was detected in the ρ0 cells compared with a strong signal detected in wild-type cells (Fig. 3A, B), indicating that HF-LPLI fails to induce mitochondrial O2 −• burst in ETC-deficient cells. Similar results were obtained using CL detection (Fig. 3C).
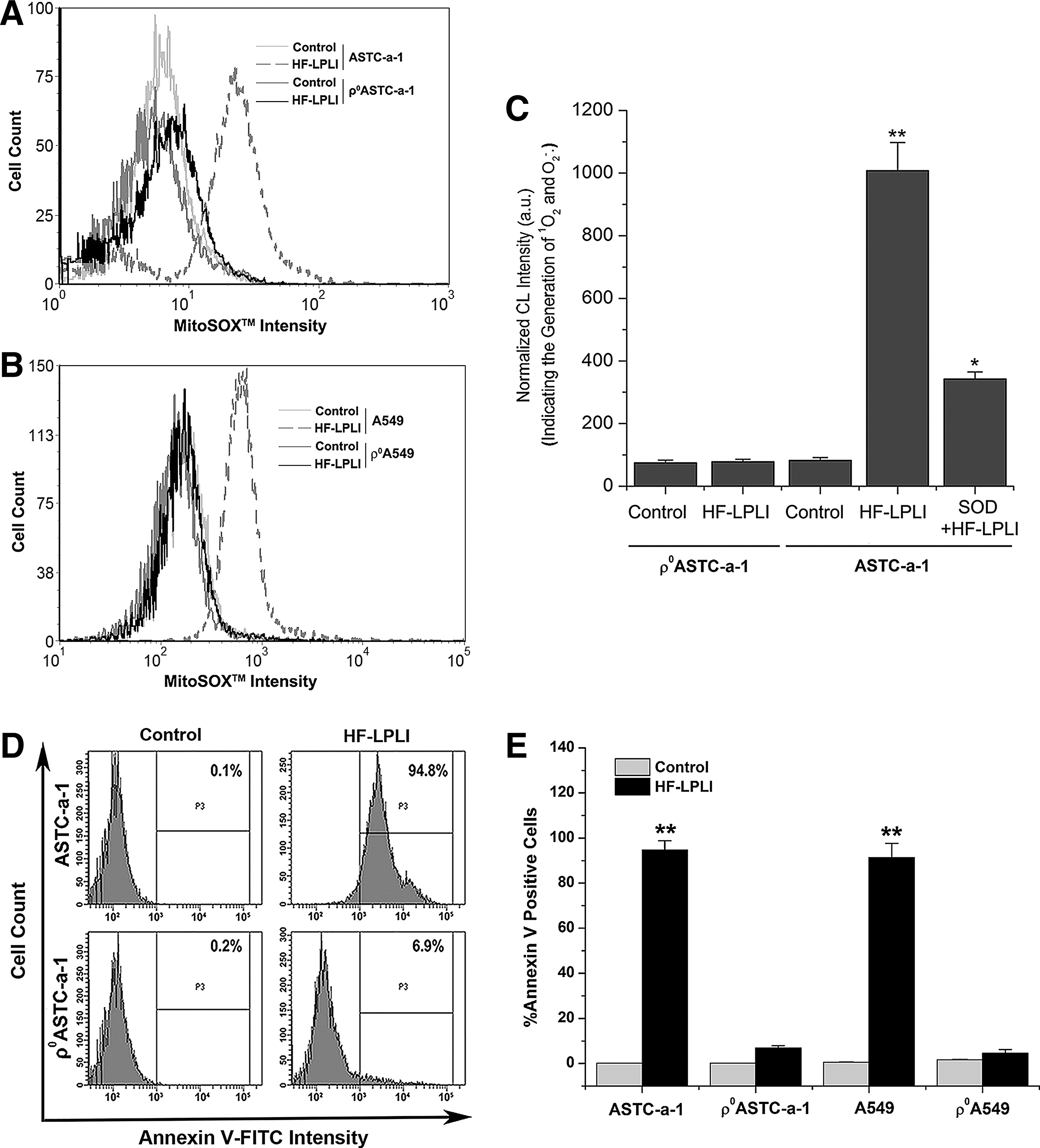
To determine the effect of mitochondrial O2 −• burst on cell apoptosis after HF-LPLI, wild-type cells and ρ0 cells were analyzed using FACS with annexin V-fluorescein isothiocyanate (FITC) staining. Ten hours after HF-LPLI, the wild-type cells showed a high level of apoptosis, while the ρ0ASTC-a-1 and ρ0A549 cells had only a low level of apoptosis, as shown in Figure 3D and E, and Supplementary Figure S6. These results indicate that the mitochondrial O2 −• burst via ETC is highly likely the determinant of HF-LPLI-induced cell apoptosis.
Photoinactivation of COX induced by HF-LPLI causes mitochondrial O2 −• burst
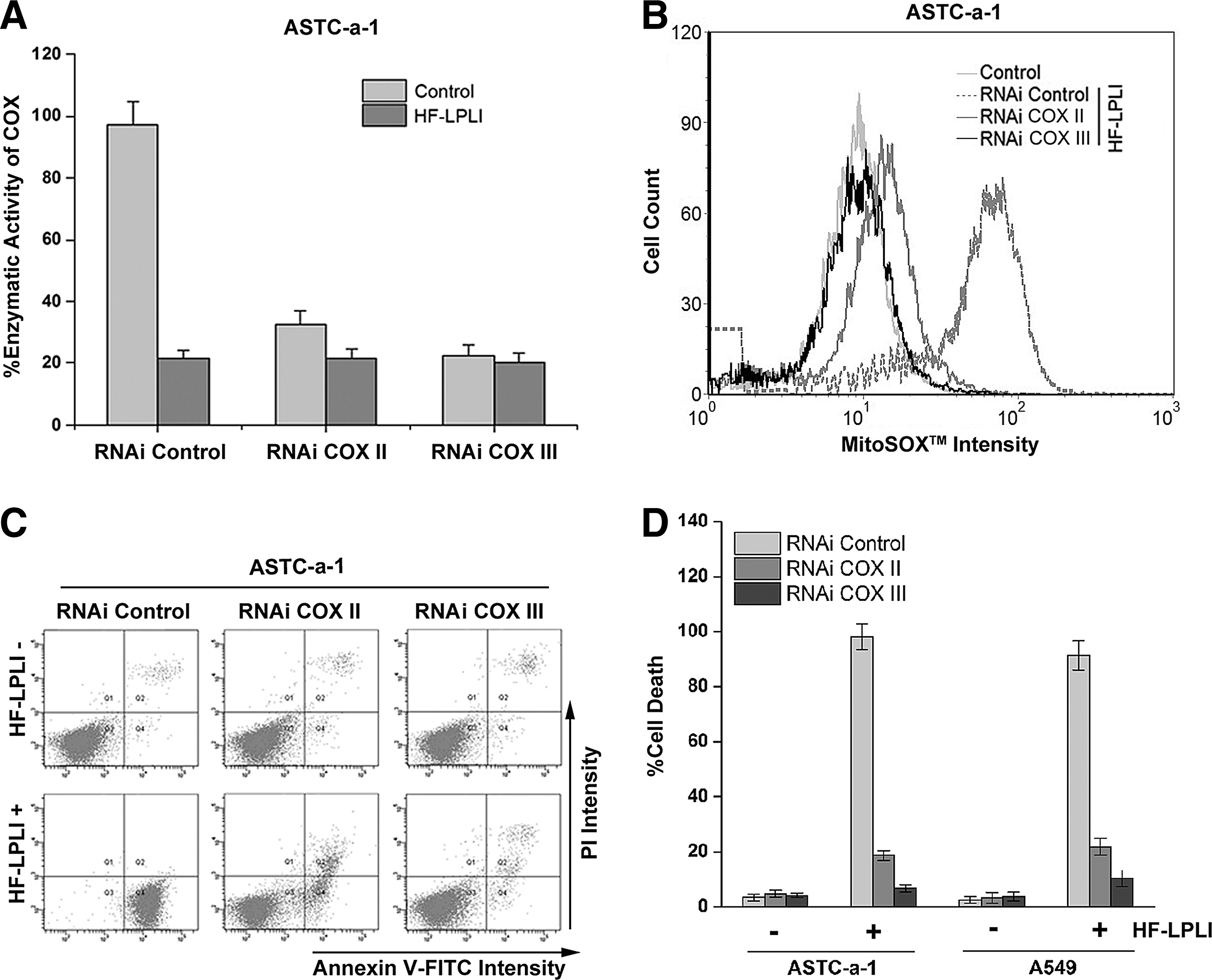
COX is believed to be the photoacceptor of LPLI in the red-to-NIR region (7, 19, 28). The effect of HF-LPLI on COX was investigated by evaluating the enzymatic activity of COX. The COX activity in ASTC-a-1 cells treated with 120 J/cm2 HF-LPLI was decreased to 0.25-fold of the control cells, demonstrating that HF-LPLI significantly inhibits the enzymatic activity (Fig. 4A). However, no enzymatic activity of COX could be detected in the ρ0ASTC-a-1 cells, and there was no change under HF-LPLI treatment (Fig. 4B).
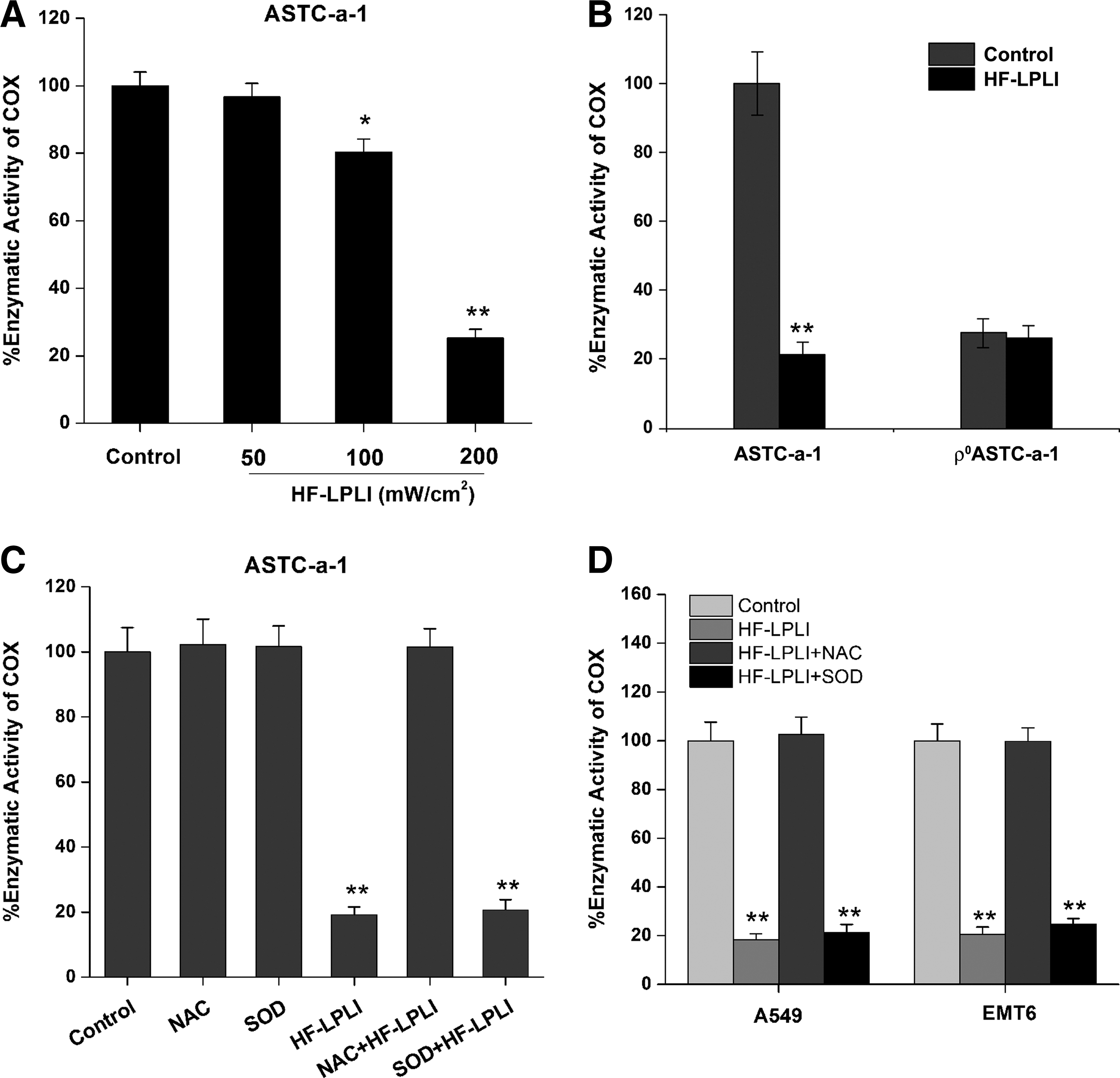
SOD exposure had almost no effect on the inactivation of COX in ASTC-a-1 cells after HF-LPLI (Fig. 4C), indicating that the mitochondrial O2 −• burst most likely results from COX photoinactivation. In contrast, NAC exposure completely reversed the inactivation of COX by HF-LPLI (Fig. 4C), indicating that in situ oxidative damage is most likely the cause of COX photoinactivation. Similar results were obtained in A549 and EMT6 cell lines (Fig. 4D). These results suggest that HF-LPLI causes COX inactivation via the initial photosensitized products and then triggers a mitochondrial O2 −• burst via ETC.
We used a gene silencing approach to explore whether the changes in the enzyme activity of COX contributed to the mitochondrial O2 −• burst and the oxidative damage of cells after HF-LPLI. The more efficient small interfering RNAs specifically targeting the subunits of COX were selected to suppress COX activity in ASTC-a-1 (Fig. 5A) and A549 (Supplementary Fig. S7) cells. The ROS levels in COX-deficient cells were assayed using H2DCFDA, SE dye. No obvious changes in the ROS level were detected in the COX II or COX III knockdown cells (Data not shown). As shown in Figure 5B, HF-LPLI-induced mitochondrial O2 −• generation depends on the activity of COX, because the MitoSOX™ intensity in COX III knockdown cells treated with HF-LPLI was nearly the same as that in control cells. The analysis of cell apoptosis based on annexin V-FITC/propidium iodide (PI) double staining showed that when the activity of COX was completely inhibited, the cell apoptosis by HF-LPLI was completely prevented (Fig. 5C, D and Supplementary Fig. S8). The results showed that photoinactivation of COX caused mitochondrial O2 −•·burst and cell apoptosis.
In vivo tumor-killing efficacy of HF-LPLI
The tumor-killing efficacy of HF-LPLI was evaluated with ASTC-a-1 xenografted tumor model. First, the power intensity and irradiation time of HF-LPLI at the fluence of 1200 J/cm2 was screened by evaluating the mean tumor volume. The tumor size was recorded for 36 days, and the results showed that HF-LPLI at the fluence of 500 mW/cm2 for 40 min was the most efficacious in tumor reduction, resulting in tumors which were significantly smaller than those of the control groups (Fig. 6A). For the survival studies, mice were monitored for 100 days after tumor inoculation. Kaplan–Meier survival curves showed that 95% of the HF-LPLI-treated mice and 100% of the Photofrin-PDT-treated mice were still alive on day 60, whereas no control mice survived beyond day 40 (Fig. 6B). A further study was performed to investigate the tumor-killing efficacy with hematoxylin and eosin (H&E) staining and annexin V-FITC staining 1 day after HF-LPLI treatment. A high scathe level was observed in the cells treated with HF-LPLI (Fig. 6C). These results demonstrated that HF-LPLI was an efficacious modality for cancer treatment.
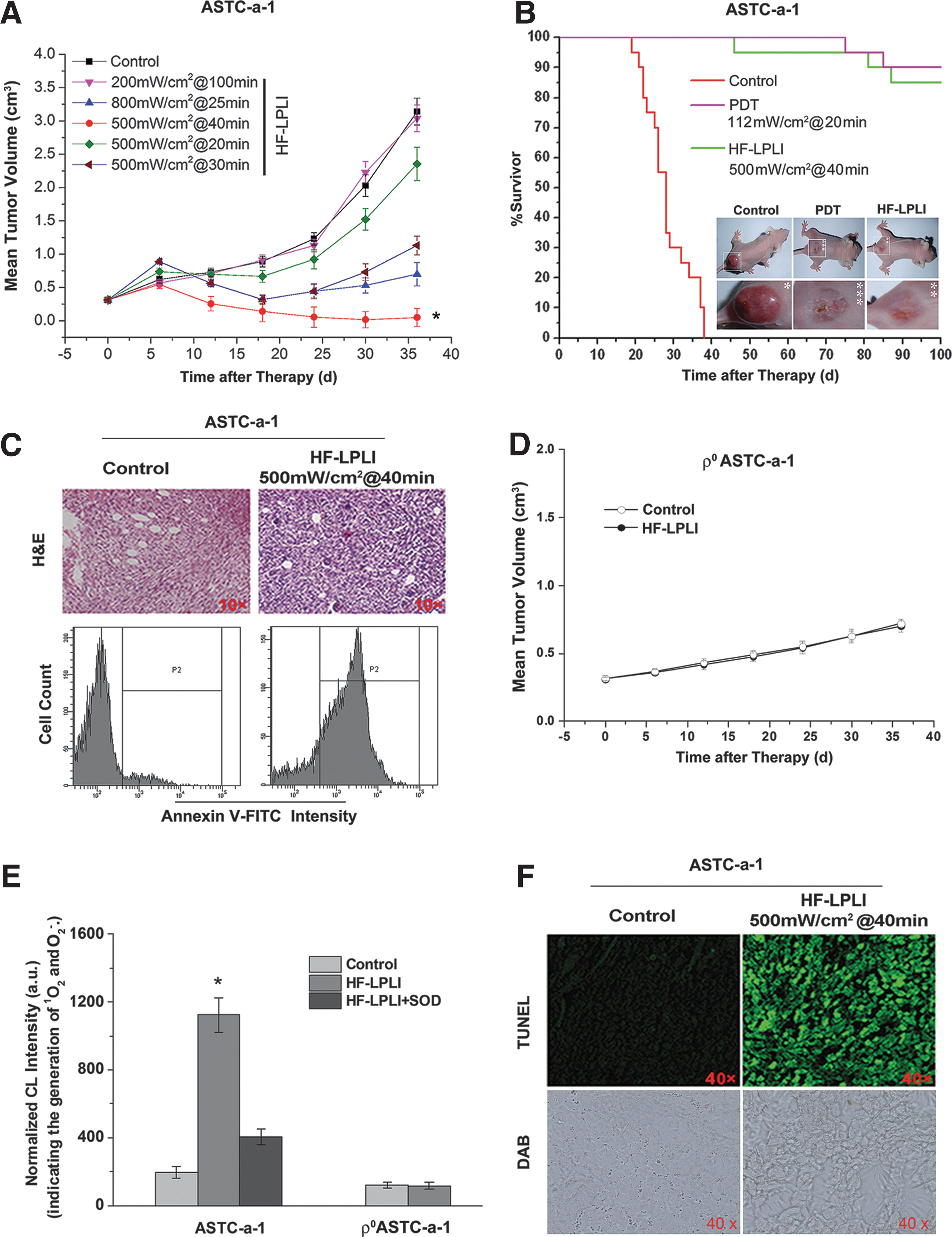
We investigated the tumor-killing efficacy of HF-LPLI with a ρ0ASTC-a-1 xenografted tumor model. Compared with the control group, there was no reduction of tumor size in the HF-LPLI-treated group (Fig. 6D). Next, we monitored ROS generation in vivo using FCLA-based CL during HF-LPLI treatment (Fig. 6E). The injection of SOD was used to scavenge the O2 −• generated. The results clearly demonstrate that HF-LPLI caused in vivo O2 −• generation, as evidenced by the strong CL signal detected during HF-LPLI treatment in ASTC-a-1 tumor model. However, there was no O2 −• generation detection in ρ0ASTC-a-1 tumor model treated by HF-LPLI. We detected the enzymatic activity of COX 1 h post HF-LPLI treatment in the ASTC-a-1 tumor model, and performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay 1 day after HF-LPLI treatment. Compared with the control group, the 3,3′-diaminobenzidine (DAB) level was significantly decreased in the HF-LPLI-treated group, demonstrating that HF-LPLI significantly inhibits the enzymatic activity of COX in this tumor model, according to high-level cell death by HF-LPLI (Fig. 6F). These results suggest that the in vivo tumor-killing mechanism of HF-LPLI is an oxidative damage effect with COX photoinactivation.
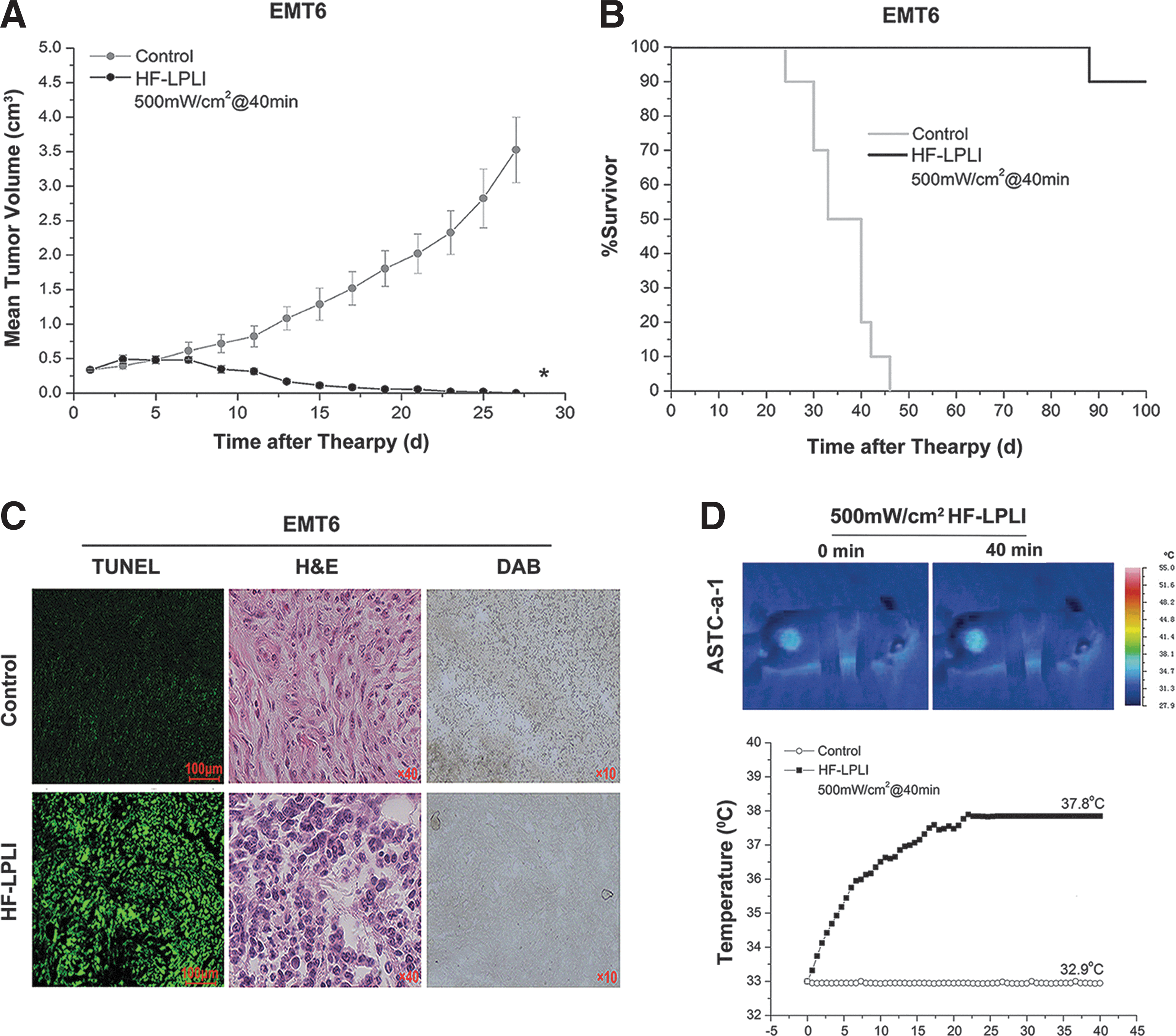
The EMT6 tumor model was also used to analyze the tumor-killing efficacy of HF-LPLI at 500 mW/cm2 for 40 min. In the HF-LPLI-treated group, the tumor burden was significantly smaller than that of the control groups (Fig. 7A). The survival rates were 90% up to day 100 after tumor inoculation (Fig. 7B). A high scathe level was observed in the tumor treated with HF-LPLI as assessed by either TUNEL assay or H&E staining, corresponded to the decreased enzymatic activity of COX (Fig. 7C). In addition, a thermal imaging camera was used to evaluate the effect of temperature changes on the surface of ASTC-a-1 tumor model during HF-LPLI treatment at the fluence of 500 mW/cm2 for 40 min. The results show that the temperature reached to 37.8°C after HF-LPLI (Fig. 7D). This indicates that thermal effect is an insignificant contributor to the antitumor effect of HF-LPLI.
Discussion
Here, we report a novel modality for cancer phototherapy using HF-LPLI in an experimental model, explored the mechanism of ROS generation, which is the crucial initial step of cell death induced by HF-LPLI, and evaluated the antitumor effects of HF-LPLI. Our study reveals that HF-LPLI selectively photoinactivates its endogenous photoacceptor COX, induces a mitochondrial O2 −• burst via ETC, and, ultimately, produces oxidative damage on cancer cells. We demonstrate that the first reaction after laser absorption is the photo-oxidative damage of COX, which causes an in situ inhibition of the enzymatic activity of COX, leading to a significant mitochondrial O2 −• burst via ETC. This modality utilizes the two properties of COX, photoacceptor of HF-LPLI and complex IV of ETC, to kill cancer cells. Our study shows that HF-LPLI initiates its effects via targeted COX photoinactivation and that the tumor-killing efficacy depends on the subsequent mitochondrial O2 −• burst.
HF-LPLI-induced oxidative damage can be clearly demonstrated by the oxidation of GSH (Fig. 2F), a metabolic signal or an early warning of apoptosis (8). Oxidative stress by HF-LPLI can also be evidenced by the damage to cellular macromolecules, including DNA, phospholipids, and proteins (Fig. 1E–G). The importance of a tight regulation of the mitochondrial redox balance is emphasized by the broad spectrum of mitochondrial antioxidant enzymes (27). Here, we discovered an elevated expression of MnSOD stimulated by HF-LPLI (Fig. 2G). Mainly located in mitochondrial matrix, MnSOD is well known as one of the major antioxidant enzymes against superoxide-free radicals (11). Overexpression of MnSOD has been reported to attenuate mitochondrial ROS generation, protect respiratory function, and even block apoptosis (11). Therefore, MnSOD up-regulation seems an important protective mechanism by which cancer cells counteract injuries caused by ROS.
It has been suggested that the mechanism of LPLI at the cellular level is based on the absorption of red-to-NIR light by COX (21). This photo-reactivity of COX is dependent on its four redox active metal centers: the binuclear CuA, CuB, heme a, and heme a3, all of which have absorbency in the red-to-NIR range (21). In ideal case, the action spectrum resembles the absorption spectrum of the molecule absorbing the light. Therefore, the bands in various action spectra in visible-to-NIR radiation in living cells were identified by analogy with the metal-ligand systems absorption spectra characteristic in this spectral range. This analysis allowed us to conclude that all bands in various action spectra with rather similar peak positions may be related to the COX (18, 20, 21). It is also evidenced that all bands present in the action spectra were present in the absorption spectra of the living cell monolayer as well (21). Dermatologists and photobiologists now consider COX as the photoacceptor in human skin when they received NIR radiation (31). However, the primary mechanism of light action after photon absorption has not yet been established, although several hypotheses exist. Currently, we found that COX activity was inhibited by HF-LPLI and that quenching ROS completely reversed this inhibition (Fig. 4). These observations suggest that irradiation may induce structural and functional changes to some of the COX components through an in situ photosensitized reaction. The photosensitized products are most likely 1O2 or O2 −•, or possibly even both (Fig. 2C). The mixed valence form of COX has been demonstrated to be required for its physiological function in the ETC (14, 34). Studies also suggest that the photoacceptor of red-to-NIR is one of the intermediate forms of COX redox cycle (18, 21). The photosensitized reaction triggered by HF-LPLI may contribute to the increased COX oxidation, making the enzyme fail to keep normal electron flow. On the other hand, it has been suggested that the changes in the degree of oxidation of the chromophores of COX caused by the irradiation are accompanied by conformational changes in their vicinity. Even small structural changes in the binuclear site of COX control both the rates of O2 reduction and the rates of the internal electron- and proton-transfer reactions (14, 34).
Chen and Pervaiz reported that Bcl-2-mediated increase in COX activity was via the enhanced presence of COX Va and Vb in the mitochondria, indicative of a more complete and stable COX enzyme (3, 4). A similar result was shown in our current study that A549 cells overexpressing Bcl-2 transiently had nearly a twofold increase in COX activity (Supplementary Figs. S9 and S10). It is known that an elevated COX activity can accelerate the rate of electron flux through the entire ETC (34). The inhibition of COX activity in these conditions will cause a quick and more leak of electrons from complexes I and/or III to trigger an ROS burst. Therefore, cancer cells with elevated COX activity are highly susceptive to events that lead to oxidative stress and dysfunction of the mitochondrial machinery (13). Our current findings support this view, because once the activity of COX was inhibited by RNA interfere (RNAi), cell death rate declined (Fig. 5A, C and Supplementary Figs. S7 and S8). However, no obvious effect of Bcl-2 overexpression on HF-LPLI-induced cell apoptosis was observed (Supplementary Fig. S11). This phenomenon may be explained by the canonical role of Bcl-2 in sequestrating pro-apoptotic proteins such as Bax and Bak, because Bax knockdown by RNAi had been reported to slightly delay HF-LPLI-induced cell apoptosis (40).
Although COX is not a source of ROS, it is known that the inhibition of the oxidase can facilitate ROS production from complex I or III (6). The HF-LPLI-triggered mitochondrial O2 −• burst was due to COX photoinhibition, as evidenced by two clues in our current study. One is that the addition of SOD had no effect on COX inhibition by HF-LPLI, indicating that the mitochondrial O2 −• burst is a result of COX photoinhibition (Fig. 4C, D). Another is when the activity of COX was inhibited by COX III gene silencing, and the mitochondrial O2 −• burst was fully prevented after HF-LPLI (Fig. 5B). A mitochondrial O2 −• burst can induce cellular signaling cascades (14, 34). It has been reported that a mitochondrial ROS burst triggers the intrinsic pathway through the permeabilization of the outer mitochondrial membrane via the opening of MPT pores (22). All of the key apoptotic events in the intrinsic pathway were observed in HF-LPLI-treated cells, including the opening of transition pores, the release of cytochrome c, and the activation of caspase-9 and caspase-3 (5, 40, 41, 43). The mechanism how MPT pore is formed is rather complex and involves transcriptional and other regulatory mechanisms, including the alteration of the homeostasis of the Bcl-2 family proteins. Using isolated mitochondria, Narita et al. reported that proapoptotic Bcl-2 family proteins, Bax or Bak, can release cytochrome c by interacting with MPT pore components, in particular, the voltage-dependent anion channel (26). However, in our earlier study, Bax was not activated until outer mitochondrial membrane permeabilization by HF-LPLI (40), indicating that Bax is not participated in the induction of MPT. It is reported that truncated Bid (tBid) added to mouse liver mitochondria stimulated cytochrome c release via mechanisms which did not require the BH3-domain of tBid or the presence of Bak (Bax), but were sensitive to cyclosporine (CsA) (32). The ability of CsA to block tBid-induced release of cytochrome c suggested that the MPT pore complex was involved in this remodeling process. However, our previous results showed that Bid was not cleaved during the cell apoptosis process by HF-LPLI (41). This result indicates that Bid is not required for HF-LPLI-induced MPT.
Our in vivo results demonstrated that, with a rational dosimetry, HF-LPLI could achieve high antitumor efficacy. Interestingly, HF-LPLI at 500 mW/cm2 for 40 min resulted in the highest tumor-killing efficacy (Fig. 6A), suggesting that there is a critical window of irradiation fluence. With excessively high laser power intensity (800 mW/cm2, Fig. 6A), rapid mitochondrial injury as a result of higher temperature and/or higher initial ROS production is likely to cause the cell to fail to generate physiological oxidative stress, resulting in a reduction in the subsequent cell death.
The reactivity of COX as the photoacceptor of red-to-NIR light depends on the absorption of light by its four redox active metal centers: the binuclear CuA, CuB, heme a, and heme a3 (21). COX consists of 13 subunits in mammalian cells with the three catalytic subunits, COX I, II, and III, coded by the mitochondrial DNA. Heme a, heme a3, and CuB are ligated to COX I, while CuA is ligated to COX II (30, 45). It is reported that increased levels of RNA transcripts of COX I and II have been observed in liver, breast, and prostate neoplastic cells compared with normal tissue (23, 29). Therefore, it is reasonable to speculate that the increased levels of COX subunits will enhance the selectivity of HF-LPLI to treat these cancers. On the other hand, the catalytic efficiency of the COX can be enhanced by the nuclear-encoded COX subunits, such as COX IV, Vb, VIa, and VIb (1, 36). Recent studies have demonstrated an up-regulation and an increased involvement of COX Va, Vb, and COX IV in a variety of cancers, such as colorectal cancer, squamous cell cancer of the larynx, intraductal carcinoma of the breast, and prostate cancer (2, 15, 38). More recently, it is directly demonstrated that epithelial cancer cells have higher levels of COX activity than normal adjacent epithelial cells, and even higher than stromal cells (37). It is known that an elevated COX activity can accelerate the rate of electron flux through the entire ETC. Hence, the inhibition of COX activity in these conditions will cause a quick and more leak of electrons, from complexes I and/or III of the ETC, to trigger ROS breakout. Therefore, cancer cells with elevated COX activity are highly vulnerable to events that lead to oxidative stress and dysfunction of the mitochondrial machinery. It is also reported that drug-resistant leukemic cell lines have more active mitochondrial function, which is associated with a greater susceptibility to tumor necrosis factor α-induced respiratory inhibition (17). In our current study, low cell death rate was obtained once the activity of COX was inhibited by RNAi (Fig. 5A, C and Supplementary Figs. S7 and S8). This indicates that HF-LPLI has potentially significant applications against cancer with higher COX activity.
With the dose of 500 mW/cm2 for 40 min, we investigated the antitumor efficacy of HF-LPLI using two different solid tumor models. The tumor histopathology staining revealed a significant number of dead cells in the mouse tumors that received HF-LPLI (Figs. 6C and 7C). After HF-LPLI, the animal survival rate was improved compared with that of the control groups (Figs. 6B and 7B). Within several weeks, the treated area was healed (Fig. 6B inset). The in vivo generation of ROS in tumors during HF-LPLI therapy was also monitored using FCLA probe-based CL detection (Fig. 6E). These phenomena were completely absent in ρ0ASTC-a-1 xenografted tumor model, without tumor burden decrease (Fig. 6D) and ROS generation (Fig. 6E). DAB staining analysis demonstrated that HF-LPLI inhibited the enzymatic activity of COX in the tumor model (Figs. 6F and 7C), according to high-level cell death (Figs. 6F and 7C). These results suggest that the in vivo HF-LPLI modality of tumors is an oxidative damage effect via the ETC, with COX photoinactivation.
Previously, we reported that low-dose Photofrin-PDT could induce endogenous ROS production via ETC, which contributed to increased cell apoptosis (47). The results of this study suggest that mitochondria are not only targets but also a source of ROS during low-dose PDT, which may provide a novel approach to improve PDT applications. In our present study, HF-LPLI was able to directly inhibit the enzymatic activity of COX to trigger a mitochondrial O2 −• burst without the involvement of any exogenous photosensitizer. Thus, the systematic toxicity, specifically the skin photosensitization, was completely eliminated. Finally, we demonstrated the mitochondria-targeting HF-LPLI as a novel modality of laser cancer therapy, which utilized COX as an endogenous photoacceptor. The enzymatic activity of COX was inhibited by the initial irradiation, and this gave rise to a burst of mitochondrial O2 −•, resulting in effective tumor cell death. This modality is characterized by minimal phototoxicity to normal tissue and has the advantage of being a minimally invasive treatment with promising potential antitumor activities.
Materials and Methods
Cell lines, tumor models, and reagents
The A549 and EMT6 were obtained from American Type Culture Collection (ATCC, Manassas, VA). The ASTC-a-1 were obtained from Jinan University (Guangzhou, China) and used within 2 months after resuscitation. These cell lines were authenticated based on viability, recovery, growth, morphology, and isoenzymology by their suppliers. The ASTC-a-1 and A549 cells were grown in Dulbecco's modified Eagle's medium (Life Technologies, Inc., Grand Island, NY), and the EMT6 cells were grown in RPMI 1640 (GIBCO, Grand Island, NY) supplemented with 15% fetal bovine serum (Gibco-BRL, Gaithersburg, MD), 50 U/ml penicillin, and 50 μg/ml streptomycin in 5% CO2 at 37°C in a humidified incubator. In all experiments, 70–85% confluent cultures were used.
ASTC-a-1 cells or ρ0ASTC-a-1 cells (1×106) in a 100 μl solution were injected into the flank region of female BALB/c nude mice aged 6–8 weeks. EMT6 cells (5×105) in a 50 μl solution were injected into the flank region of female BALB/c mice aged 6–8 weeks. The animals were used when the tumors reached a size of ≈300 mm3. The mice were randomly divided into different experimental groups and received various treatments.
H2DCFDA, SE (10 μM), DHE (10 μM), MitoSOX™ (5 μM), TMRM (100 nM), and MTR (100 nM) probes were purchased from Molecular Probes, Inc. (Eugene, OR). Rhodamine 123 (5 μM) probe, DHA (250 μM), NAC (250 μM), and MnSOD (50 U/ml) were purchased from Sigma-Aldrich (St. Louis, MO). pDsRed-mit was presented by Prof. Yukiko Gotoh (University of Tokyo).
Laser treatment
For in vitro HF-LPLI, the cells were irradiated with a fiberoptic light delivery system (635 nm, CW semiconductor laser, NL-FBA-2.0-635; nLight Photonics Corporation, Vancouver, WA) for 10 min at a fluence rate of 50–200 mW/cm2. For in vitro Photofrin-PDT, cells were incubated in the dark with Photofrin (2.5 μg/ml) in complete growth medium for 20 h and then rinsed with phosphate-buffered saline (PBS) before irradiation. The cells were irradiated with the 635-nm laser for 30 s.
For in vivo HF-LPLI, the tumor-bearing mice were directly irradiated with the 635-nm laser. The total fluence delivered to the tumors was 1200 J/cm2 with different fluence rate and irradiation time. During laser irradiation, the mice were anesthetized with an intraperitoneal injection (i.p.) of pentobarbital sodium (2%) and were restrained in a specially designed holder. For in vivo Photofrin-PDT, Photofrin was administered i.p. at a dose of 10 mg/kg 24 h before irradiation. The light was delivered to the tumors using the 635-nm laser. The power density at the irradiated area was 112 mW/cm2. The total laser fluence delivered to the tumors was 134 J/cm2. After treatment, the mice were observed daily, and the tumors were measured every other day for a period of 100 days. The control samples were maintained on the laser table for the same amount of time used in the irradiated groups, but the laser source was not activated (sham irradiation).
Live cell ROS detection using fluorescence probes
H2DCFDA, SE is a ROS-sensitive probe that can be used to detect ROS production in living cells. It passively diffuses into cells, where its acetate groups are cleaved by intracellular esterases, releasing the corresponding dichlorodihydrofluorescein derivatives H2DCF. H2DCF oxidation yields a fluorescent adduct, DCF that is trapped inside the cell. Cells were incubated at 37°C in the dark with 10 μM H2DCFDA, SE in serum-free medium for 30 min and then, the DCF fluorescence was recorded using laser scanning microscope (LSM). Intracellular accumulation of O2 −• was estimated using MitoSOX™, which selectively targets mitochondria and is oxidized by O2 −•, emitting red fluorescence on binding to nucleic acids. Cells were loaded at 37°C with 5 μM MitoSOX™ for 10 min in the dark, rinsed with PBS before fluorescence measurement by LSM. DHE is a cell-permeable blue fluorescent dye that on entering cells interacts with O2 −• to form oxyethidium, which intercalates with nucleic acids and emits a red fluorescence which is detectable qualitatively by fluorescent microscopy. To assess O2 −• production, cells were cultured in the dark with 10 μM DHE diluted in PBS for 30 min at 37°C followed by a wash in PBS before being visualized by LSM.
COX activity assay
The enzymatic activity of COX in living cells was assayed by measuring cytochrome c oxidation using a COX Activity Testing Kit (Genmed Scientifics, Inc., Arlington, MA). The COX activity was determined by measuring the decrease in absorbance at 550 nm using an Infinite 2000 plate reader (TECAN, Mönnedorf, Switzerland). To analyze the enzymatic activity of COX in situ, DAB staining (Genmed, Boston, MA) was performed 1 h after each indicated treatment, and the middle sections of each tumor were formalin fixed and paraffin embedded following a standard routine.
Cell death assays
FACS was performed on an FACScanto II flow cytometer (BD Bioscience, San Jose, CA). For cellular apoptosis analysis, the annexin V-FITC/PI Apoptosis Detection Kit (BD PharMingen, San Diego, CA) was used as a standard reagent with excitation at 488 nm. The fluorescent emission of FITC was measured at BP515–545 nm, and the emission of DNA-PI complexes was measured at BP564–606 nm. The number of events considered in FACS was 10,000 per each independent experiment. For in situ tumor cell death analysis, H&E and TUNEL staining (Genmed, Boston, MA) was performed 1 day after the indicated treatments. The middle sections of each tumor were formalin fixed and paraffin embedded following a standard routine.
Statistics
For the fluorescence emission intensity analysis, a background subtraction was performed for all data. Each experiment was performed at least thrice. Statistical analysis was applied using the two-tailed Student's t-test. Representative data are shown for all other cases.
Footnotes
Acknowledgments
The authors acknowledge the financial support rendered by the National Basic Research Program of China (2011CB910402; 2010CB732602), the Program for Changjiang Scholars and Innovative Research Team in University (IRT0829), the National Natural Science Foundation of China (81101741, 61308111), the Research Fund for the Doctoral Program of Higher Education of China (20114407120002), and the Natural Science Foundation of Guangdong Province, China (S2011040003254).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.