
Abstract
Introduction
T
Roles of hydrogen sulfide (H2S) on intracellular Ca2+ homeostasis and its underlying mechanism are ambiguous in exocrine pancreas. We demonstrated, for the first time, the Ca2+-releasing effect of H2S via the production of nitric oxide. We also demonstrated that this effect is induced via the pathway in which activation of each soluble guanylate cyclase, protein kinase G, Gq-protein, and phospholipase C is involved. This hypothesis may provide a useful key to clarify the physiological and/or pathological mechanisms of action of H2S and eventually may yield clues for potential therapeutic exploitation.
Recent studies have revealed several physiological and pathophysiological functions of H2S. It has been shown to relax vascular smooth muscle, induce vasodilation of isolated blood vessels, and reduce blood pressure (43), indicating that it is a cardinal regulator of blood pressure, whereas some contradictory result was reported (18). H2S has been identified as a potent anti-inflammatory (66) and antioxidant molecule (22). It regulates expression of chemokines, cytokines, and adhesion molecules and has a biphasic effect in acute pancreatitis and associated lung injury (50,53). The physiological functions of H2S in the brain have been suggested to include Ca2+ homeostasis, suppression of oxidative stress, modulation of neurotransmission (44), and enhancement of N-methyl-D-aspartate (NMDA) receptor-mediated responses and they facilitate the induction of hippocampal long-term potentiation (1). Among its presumptive molecular targets, H2S is known to act on a number of other ion channels such as those of Ca2+ and K+ (12,69,70). H2S activates KATP and transient receptor potential (TRP) channels (51,71), whereas it inhibits the big conductance Ca2+-sensitive K+ channels (BKCa) (57) and T- and L-type Ca2+ channels (31,52). Other targets may be active sites inside the cell such as proteins, enzymes, and transcription factors (27).
Ca2+ plays essential roles in various cellular functions, including muscle contraction, control of cell growth, activation of platelets, control of secretion, and apoptosis. In pancreatic acinar cells, Ca2+ has a central role in the secretory process. It is a trigger, promoter, and modulator in different events leading to digestive enzyme secretion (13,14). Most of the studies on H2S in the exocrine pancreas have aimed at interpreting mechanism(s) of H2S by which pancreatitis, nociception, and apoptosis are induced. However, the available data are controversial. H2S has been shown to reduce caerulein-induced inflammation in pancreatic acini through a phosphoinositide-3 kinase signaling pathway (54). In contrast, H2S has both pro- and anti-inflammatory properties (3). Excessive production may contribute to the pathogenesis of inflammatory diseases, and reduction of its production may be of potential therapeutic value in inflammatory diseases (67,68). Hui et al. (17) demonstrated that endogenous vascular H2S increased in rats with septic shock and endotoxic shock which suggested that endogenous H2S was involved in physiological and pathophysiological process during shock. As possible target molecules, sodium hydrosulfide (NaHS)/H2S most probably exerts its influence on T-type Ca2+ channels, leading to nociception and endogenous H2S produced by CSE, and T-type Ca2+ channels may be involved in pancreatitis-related pain (35).
To elucidate the pathophysiological roles of H2S, it is of utmost importance to investigate its possible effects on intracellular Ca2+ homeostasis in pancreatic acinar cells, as intracellular Ca2+ is a double-edged sword and its regulatory disturbance is thought to be related to some types of functional damage (14). In the present study, therefore, an attempt was made to evaluate the possible roles of H2S in the physiology and pathology of pancreatic exocrine tissue with special reference to its participation in regulating intracellular Ca2+ dynamics. Recently, we have reported the presence of cross-talk between Ca2+ and NO in pancreatic acini and this cascade may, at least partially, participate in physiological secretagogue-evoked Ca2+ dynamics in pancreatic acinar cells (32,33). Although both NO- and H2S-producing enzymes are known to be expressed in pancreatic acinar cells, and the fact that H2S can interact with NO has been indicated in smooth muscles, vascular tissue, and the brain (16,25), no studies have examined such possible interactions in pancreatic acinar cells. In the present study, therefore, we attempted to define whether cross-talk between H2S and NO was present and, if so, how intensively such cross-talk correlated with regulation of the intracellular Ca2+ concentration ([Ca2+]i) dynamics in pancreatic acinar cells.
Results
Effect of H2S on [Ca2+]i in pancreatic acinar cells
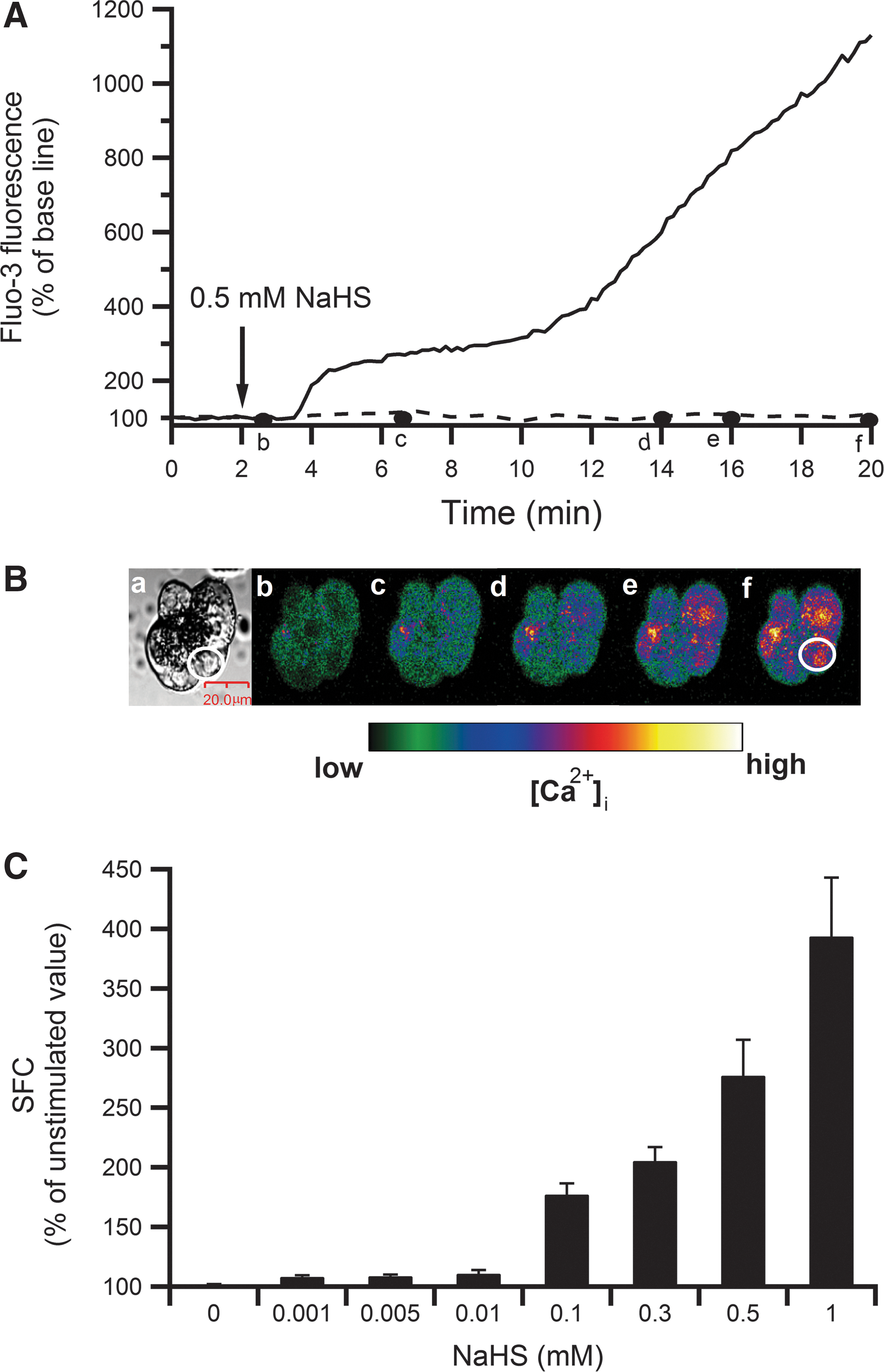
To investigate the effect of H2S on [Ca2+]i in pancreatic acinar cells, the H2S donor NaHS was used. Figure 1A shows a typical example of a [Ca2+]i increase induced by 0.5 mM NaHS in the cell indicated by the white oval region of interest (ROI) in translucent and pseudocolor images of the acinus shown in Figure 1B. The H2S-induced [Ca2+]i increase occurred rapidly after application of NaHS and the response seemed to be biphasic, with a transient initial increase reaching a plateau followed by a long-lasting gradual increase. Figure 1C shows that NaHS dose dependently (1–1000 μM) increased [Ca2+]i with an approximately fourfold increase at 1000 μM NaHS (p<0.001 by analysis of variance [ANOVA]). NaHS at a concentration of 1–10 μM caused faint but significant increases in [Ca2+]i.
Source of Ca2+ in H2S-induced [Ca2+]i increase
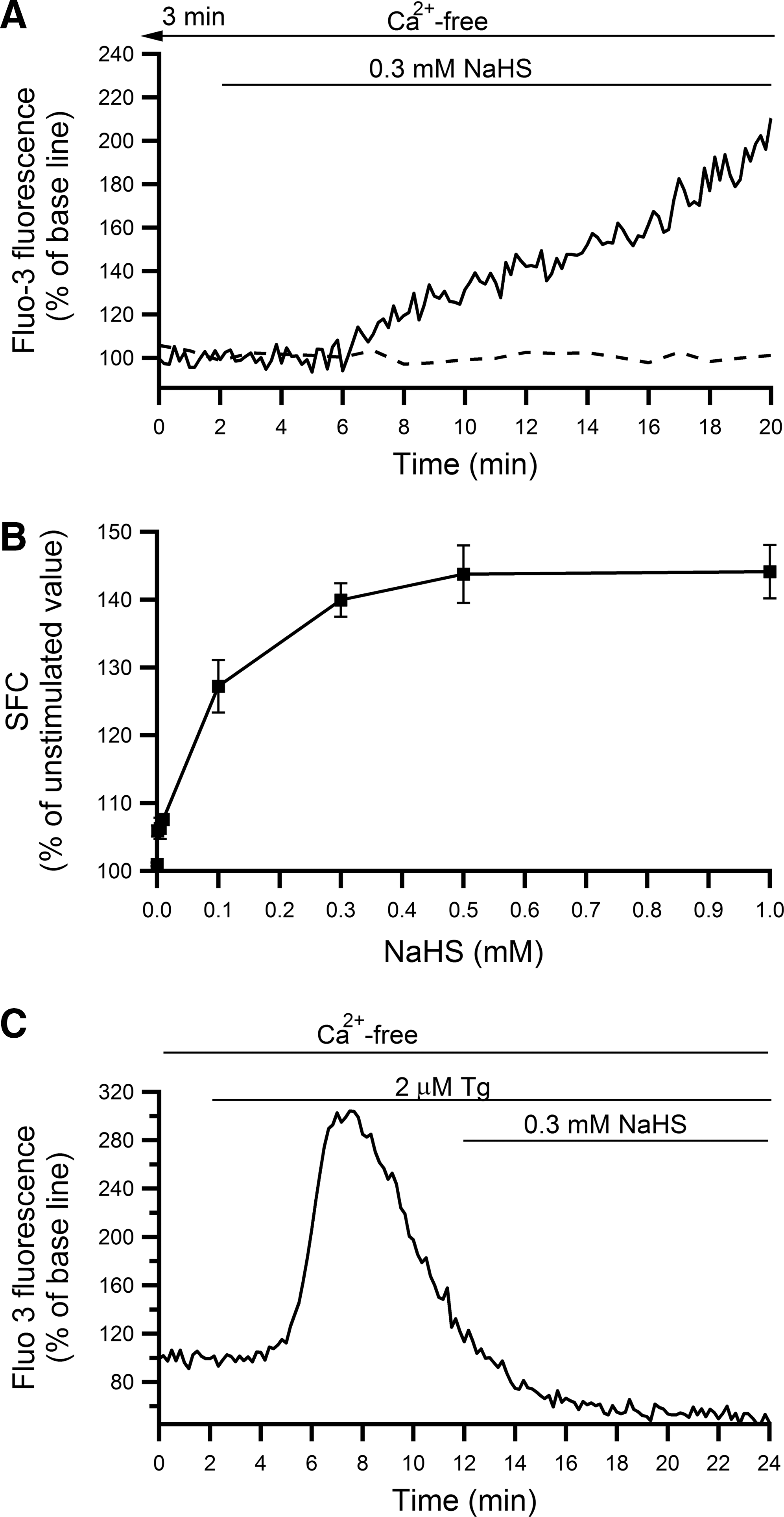
Elevation of intracellular [Ca2+]i can be due to extracellular Ca2+ influx or release of Ca2+ from intracellular stores or both. To examine the contribution of Ca2+ influx, [Ca2+]i changes were evaluated in acini perfused with ethylene glycol tetraacetic acid (EGTA)-containing, Ca2+-free buffer. Figure 2A shows a typical example of [Ca2+]i changes in an acinar cell perfused for 5 min with the Ca2+-free buffer, then treated with 0.3 mM NaHS in the absence of extracellular Ca2+. Different concentrations of NaHS (1–1000 μM) could still induce [Ca2+]i elevation in the absence of extracellular Ca2+ with an appreciable increase at 1000 μM NaHS with an EC50 of 73.3 μM (Fig. 2B). However, the NaHS-induced Ca2+ elevation was significantly attenuated in the absence of extracellular Ca2+, especially at higher concentrations of NaHS (500 and 1000 μM), indicating that both extracellular and intracellular Ca2+ were involved in H2S-induced Ca2+ increase (Fig. 2B). Endoplasmic reticulum (ER) is one of the intracellular Ca2+ storage sites and Ca2+-ATPase transports calcium from the cytosol to ER, so it restores low Ca2+ in cytosol and high Ca2+ in ER. To examine the involvement of Ca2+ ATPase, thapsigargin (Tg), a Ca2+-ATPase inhibitor was employed. Acini pretreated with 2 μM Tg for 10 min in the Ca2+-free buffer before the application of 0.3 mM NaHS showed complete inhibition of [Ca2+]i elevation (Fig. 2C). These data confirmed that intracellular Ca2+, in combination with extracellular Ca2+, was responsible for [Ca2+]i elevation in response to H2S.
Endogenous production of H2S
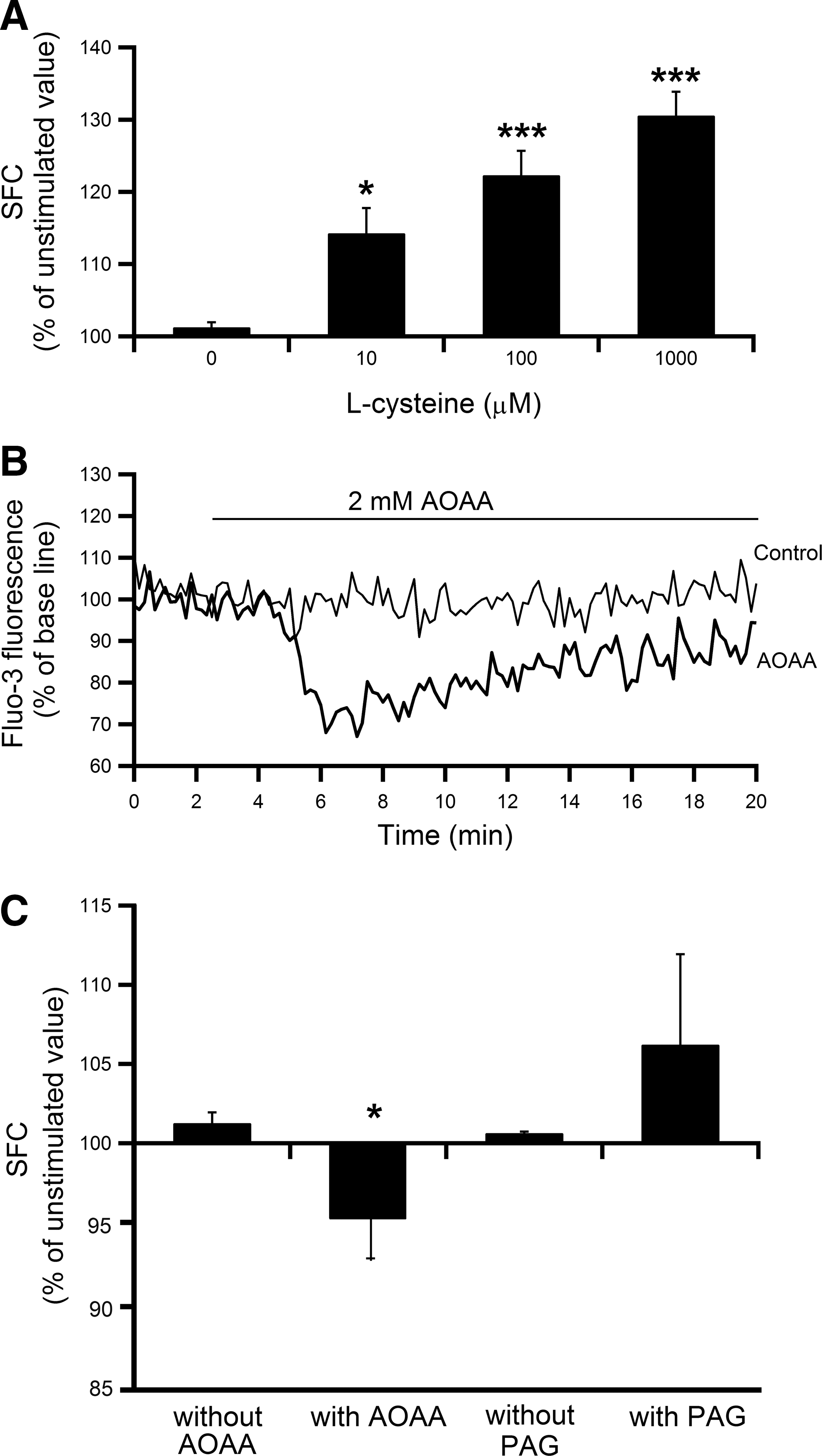
The relevance of endogenous H2S to [Ca2+]i homeostasis was examined using L-cysteine, the substrate of the H2S-producing enzymes CBS and CSE. Addition of L-cysteine caused a concentration-dependent (10–1000 μM) increase in [Ca2+]i (Fig. 3A). In addition, effects of 2 mM aminooxyacetic acid (AOAA), a CBS inhibitor, and 2 mM DL-propargylglycine (PAG), a CSE inhibitor, on basal [Ca2+]i were examined. Two millimolar AOAA significantly decreased basal [Ca2+]i (Fig. 3B, C), but PAG was without effect (Fig. 3C).
H2S-induced [Ca2+]i increase is largely dependent on NO production
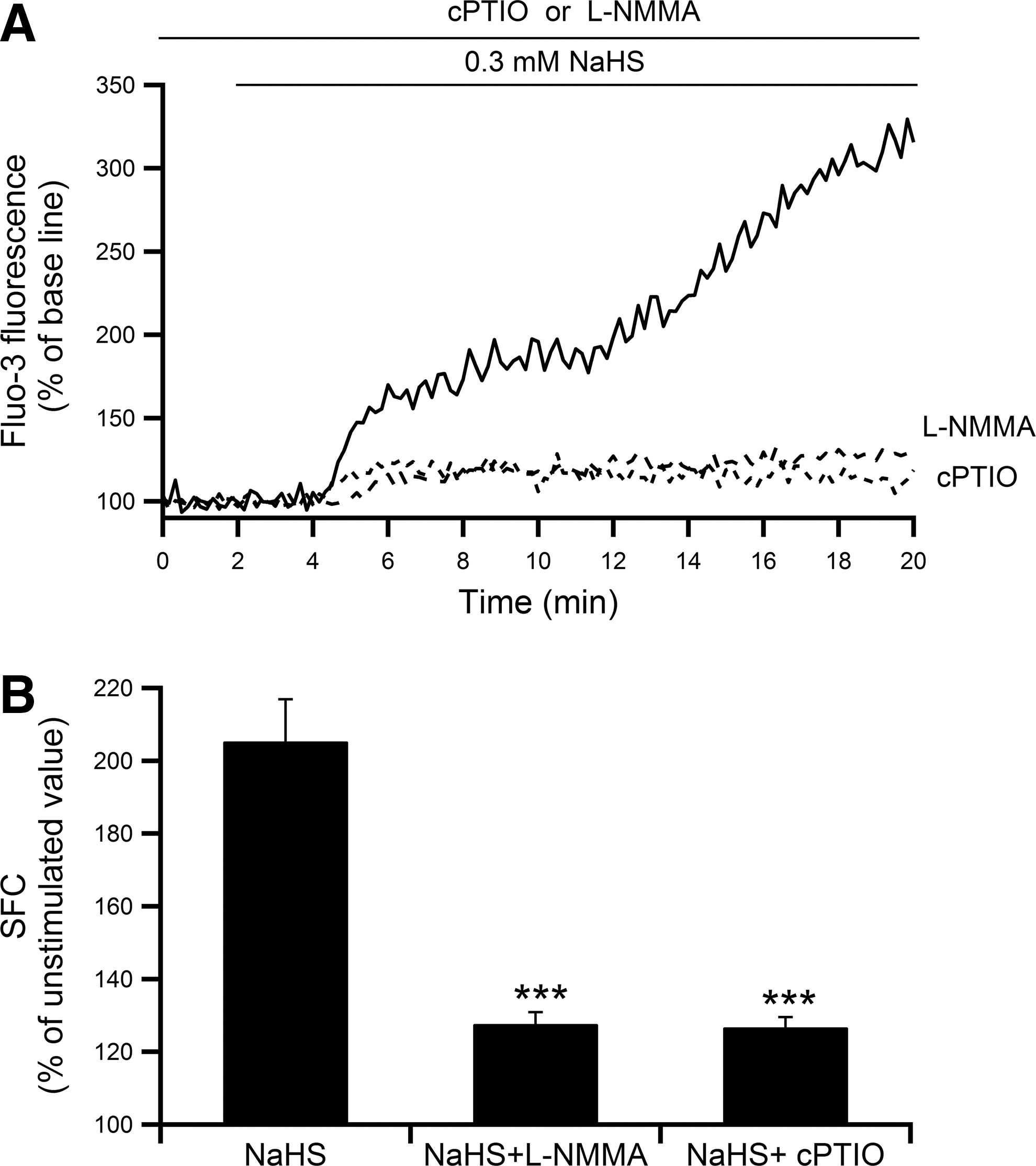
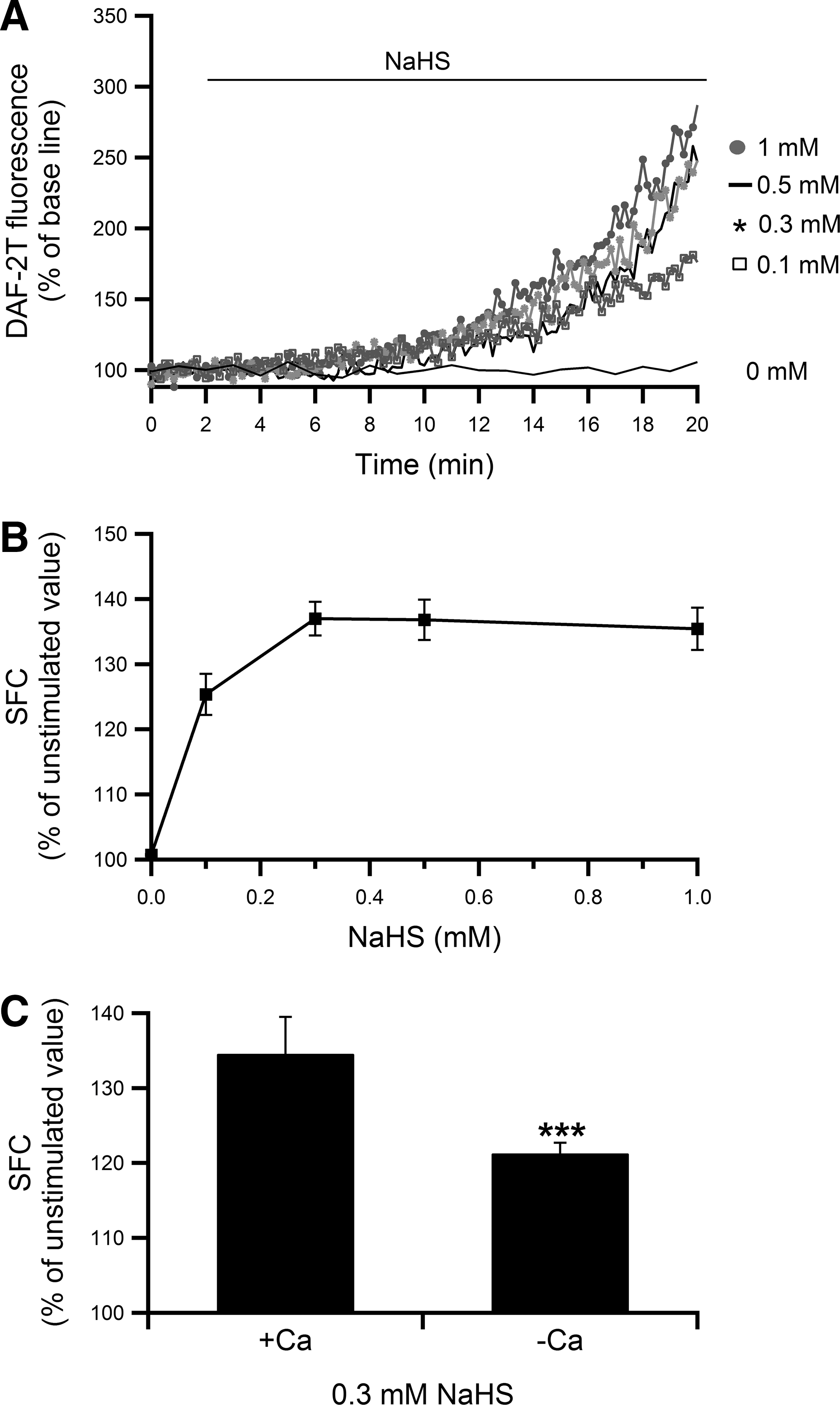
An additive effect has been demonstrated between H2S and NO in relaxation of smooth muscle, indicating a strong interaction between the two gases (16). To assess the possible involvement of NO in the NaHS-induced [Ca2+]i increase in pancreatic acinar cells as well, effects of 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO), an NO scavenger, and NG-monomethyl L-arginine (L-NMMA), a non-specific inhibitor of all nitric oxide synthase (NOS) isoforms, were examined. Pretreatment of acini with 100 μM L-NMMA or 0.5 mM cPTIO significantly attenuated the NaHS-induced [Ca2+]i increase (Fig. 4A, B). The summed area of fluorescence changes (SFC) was decreased by 37% in the presence of each chemical (the increment from the unstimulated value was decreased by 74%). We next examined whether H2S itself could stimulate NO production by using a fluorescent NO probe, diaminofluorescein-2/diaminofluorescein-2 triazole (DAF-2/DAF-2T). As shown in Figure 5A, different concentrations of NaHS (0.1–1 mM) induced gradual and continuous NO production in the presence of extracellular Ca2+, and NaHS dose dependently increased NO production (Fig. 5B) (p<0.001 by ANOVA). The EC50 was calculated to be 64.8 μM. The NaHS-induced NO production was partially abolished in the absence of extracellular Ca2+ (Fig. 5C). The SFC was decreased by 11% (the increment from the unstimulated value was decreased to 41%). These results indicated that NaHS induced a [Ca2+]i increase via NOS activation and subsequent NO production. Moreover, pretreatment with 100 μM L-NMMA significantly reduced the extent of NaHS-induced DAF-2/DAF-2T fluorescence increase by 40% compared with that without L-NMMA (data not shown).
Effect of phospholipase C inhibitor U73122 on H2S-induced [Ca2+]i increase
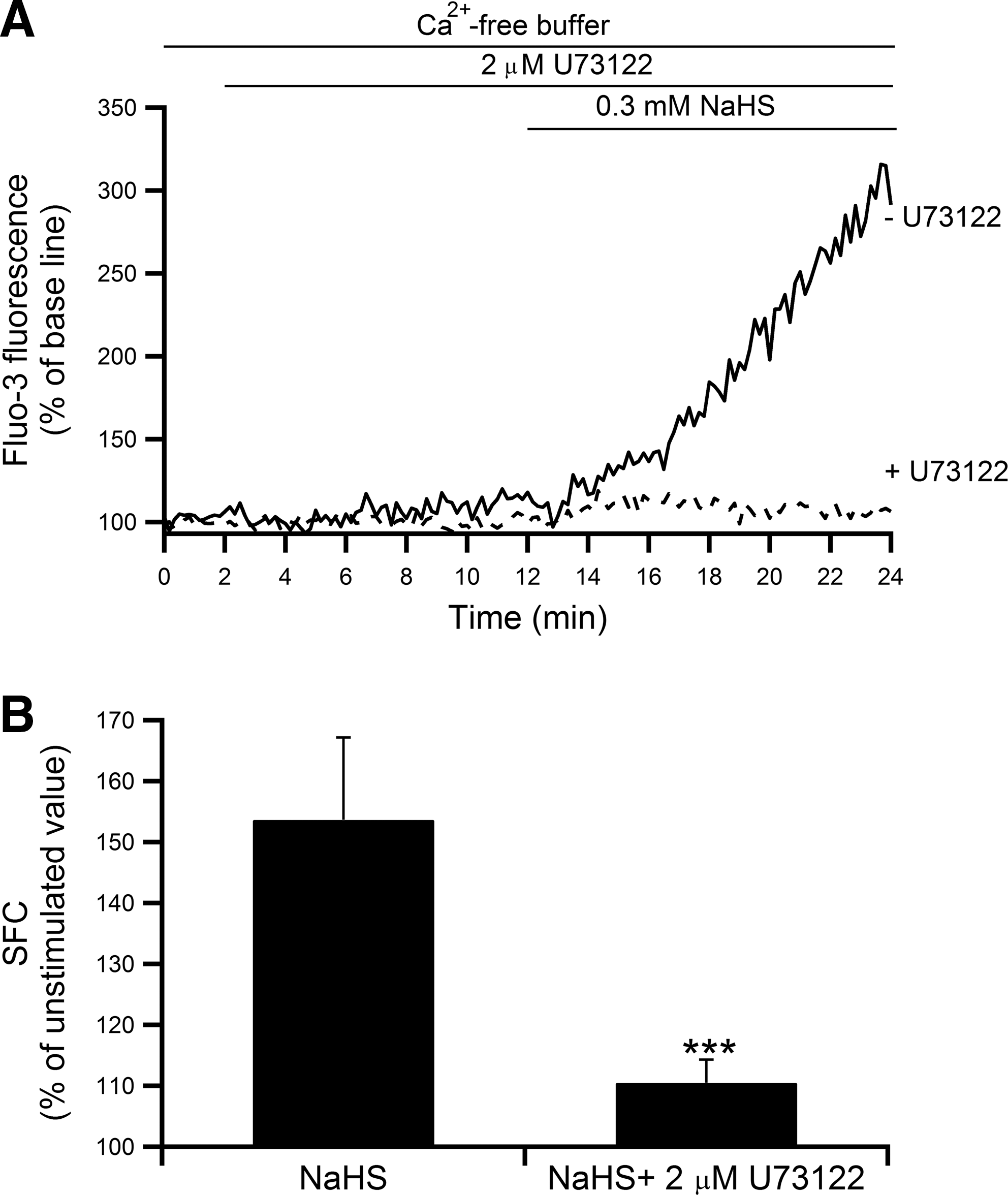
We next conducted a series of experiments to investigate the intracellular signaling mechanism(s) involved in H2S-induced intracellular Ca2+ release. First, we examined whether the phospholipase C (PLC) pathway was involved in the [Ca2+]i increase in response to H2S treatment. A specific inhibitor of phospholipase C, U73122, was utilized in the absence of extracellular Ca2+. Pretreatment with 2 μM U73122 for 10 min significantly suppressed the 0.3 mM NaHS-induced [Ca2+]i increase (Fig. 6A) and caused a significant 28% reduction of the SFC (the increment from the unstimulated value was decreased by 81%) (Fig. 6B), suggesting that H2S activated PLC, which eventually caused [Ca2+]i elevation.
Effect of H2S involves G-protein-inositol 1,4,5-trisphosphate and cyclic guanosine monophosphate/protein kinase G pathway
Second, we examined the involvement of inositol 1,4,5-trisphosphate (IP3) in the NaHS-induced [Ca2+]i increase by using xestospongin C, a potent, cell-permeable inhibitor of the IP3 receptor (IP3R). Figure 7A and B shows that 3 μM xestospongin C almost completely inhibited the [Ca2+]i increase induced by 0.3 mM NaHS. The SFC was reduced by 23% (the increment from the unstimulated value was decreased by 75%), which indicated that the H2S-induced [Ca2+]i increase was partly triggered via the PLC/IP3 signaling pathway.
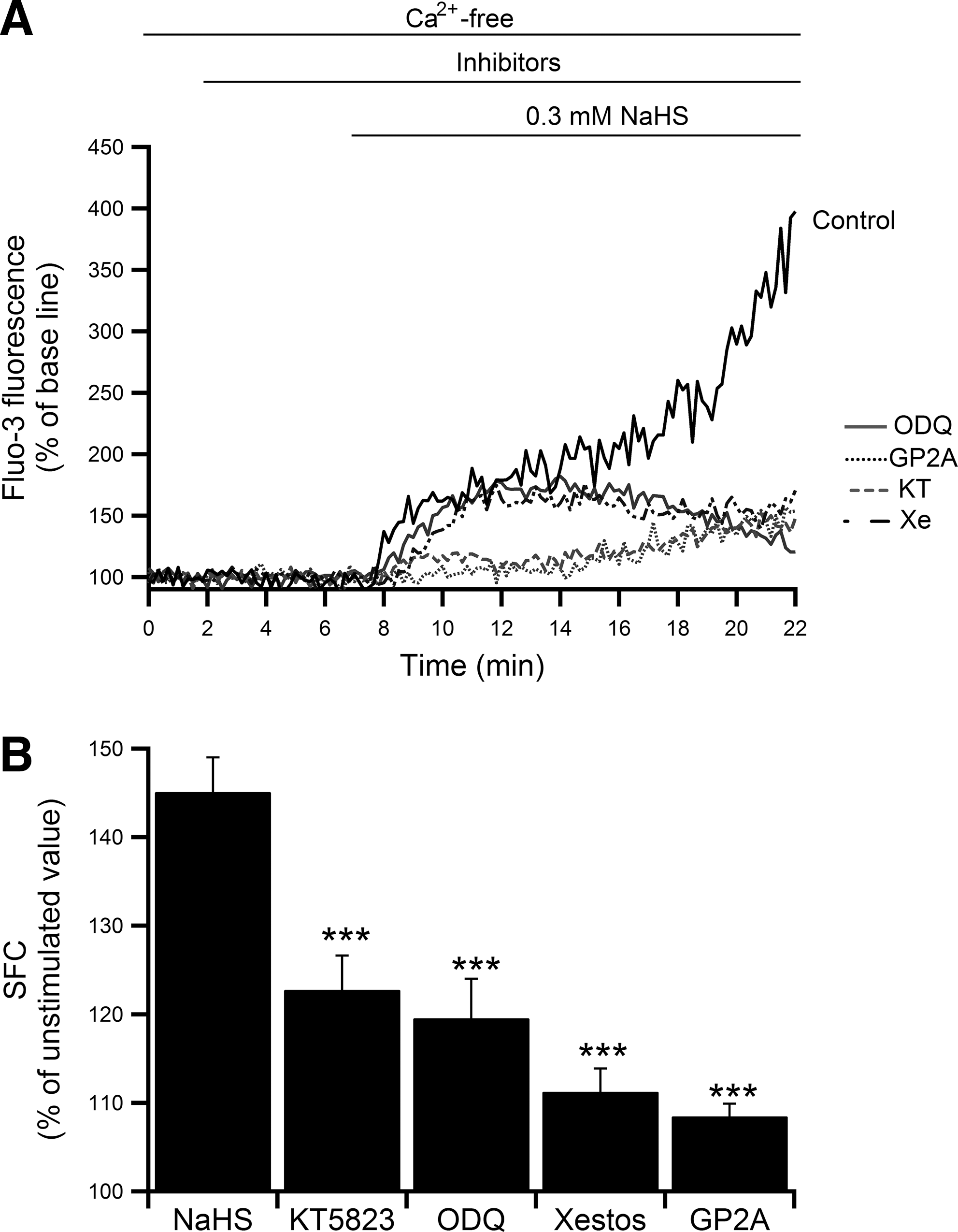
To further investigate the intracellular signals involved in the H2S-induced [Ca2+]i increase in pancreatic acinar cells, we investigated the contribution of soluble guanylate cyclase (sGC) by using 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a potent and selective inhibitor of sGC. Pretreatment with ODQ at 100 μM significantly abolished the [Ca2+]i increase elicited by 0.3 mM NaHS, and the SFC decreased by 17% (the increment from the unstimulated value was decreased by 56%) (Fig. 7). Since the downstream product of sGC is cyclic guanosine monophosphate (cGMP), these data suggested that the effect of H2S on [Ca2+]i involved production of cGMP. It has been reported that the intracellular action of cGMP is primarily mediated by cGMP-dependent protein kinase G (PKG). Whether PKG mediated the NaHS-induced [Ca2+]i increase was examined by using a highly cell-permeable and selective inhibitor of PKG, KT5823. Pretreatment with 2 μM KT5823 significantly suppressed the NaHS-induced [Ca2+]i increase and the SFC decreased by 15% (the increment from the unstimulated value was decreased by 49%) (Fig. 7). These results indicated that the H2S-induced [Ca2+]i increase was caused via a cGMP/PKG-dependent pathway.
Several sites of action have been proposed to account for cGMP-dependent regulation of cytosolic Ca2+, including inhibition of receptor/G-protein coupling in smooth muscle (28), PKG activation of the BK channel either by acting as the direct catalyst for phosphorylation (10) or by activating a protein phosphatase that dephosphorylates the BK channel (72), and cGMP/PKG regulation of IP3R (15). With the data mentioned earlier, the question now is how cGMP activates PLC. We focused on G-protein, as PLC is activated by an α-subunit cleaved from trimeric G-protein. Acini pretreated with the Gq-protein antagonist peptide GP2A (10 μM) showed marked inhibition of the NaHS-induced [Ca2+]i increase compared with the control and the SFC decreased by 25% (the increment from the unstimulated value was decreased by 81%) (Fig. 7). These results indicated that H2S-induced [Ca2+]i elevation occurred through a pathway including G-protein.
Discussion
The current study showed that the superfusion of isolated pancreatic acini with NaHS increased [Ca2+]i in a dose-dependent manner. The increase was biphasic with an initial rapid rise followed by a long-lasting elevation (Fig. 1). L-cysteine-induced dose-dependent increase in [Ca2+]i and the decrease in the basal [Ca2+]i level by the addition of a CBS inhibitor indicates that low concentrations of endogenous H2S act to keep the basal [Ca2+]i level within a narrow range. These findings suggest that H2S regulates [Ca2+]i homeostasis in pancreatic acinar cells. The NaHS/H2S-induced [Ca2+]i rise has been widely reported in various tissues such as astrocytes, microglial cells, neuronal cells, and distal colonic epithelium (24,34,42,63). In contrast, inhibition of the [Ca2+]i increase has been demonstrated in vascular, gastrointestinal, urogenital, atrial, and ventricular smooth muscle, and pancreatic islet cells, causing vasodilation, cardiac muscle relaxation, and inhibition of insulin release (20,27,40,56,62). A protective effect against ischemia-reperfusion injury is also reported (64).
In the exocrine pancreas, H2S was initially demonstrated to be a pro-inflammatory factor involved in caerulein-induced pancreatitis (5,53). Later, however, anti-inflammatory effects were also reported (50,54). H2S has also been suggested to be related to pancreatitis-originated nociception (11,35), probably via T-type Ca2+ channels and TRPV1, leading to extracellular signal-regulated kinase (ERK) phosphorylation that causes pain. Since Ca2+ plays crucial roles in both physiology and pathophysiology in a variety of tissues, it is conceivable that, depending on the magnitude of the increase in [Ca2+]i, the final results induced by H2S may be divergent. In the exocrine pancreas, since Ca2+ would function as a double-edged sword (14), it is conceivable that excessive production of H2S may be related to the pathogenesis of inflammatory diseases and reduction of its formation could be of potential therapeutic value in inflammation (67,68).
The sources of Ca2+ utilized for [Ca2+]i elevation were both extracellular and intracellular, as the H2S-induced [Ca2+]i increase was attenuated but persisted even in the absence of extracellular Ca2+. The extent of the dependence on extracellular Ca2+ appeared to be large when the acini were treated with higher NaHS concentrations, indicating that Ca2+ entry could be a major component causing [Ca2+]i elevation at these concentrations. The H2S-induced [Ca2+]i increase was completely abolished by pretreatment with Tg (Fig. 2C), indicating that intracellular Ca2+ sequestering organella which possessed Ca2+ ATPase were involved in Ca2+ mobilization. This is consistent with previous studies showing that the H2S-induced Ca2+ waves in astrocytes and [Ca2+]i increase in microglial and neuronal cells were also brought about by both Ca2+ influx and the release of intracellularly stored Ca2+ (24,34,63).
H2S-induced NOS activation and NO production
A previous study demonstrated that an NO donor, sodium nitroprusside (SNP), up-regulated H2S production and another NO donor, S-nitroso-N-acetyl-DL-penicillamine (SNAP), increased the transcriptional level of CSE in cultured aortic smooth muscle cells (70). Subsequent studies conducted by other groups revealed the functioning of a retrograde pathway, that is, H2S exerts its effect, at least in part, via the activation of endothelial nitric oxide synthase (eNOS) by phosphorylation (31) via serine/threonine kinase/protein kinase B (Akt/PKB) activation (7,64). NO production by H2S mediated by an increase in [Ca2+]i was also indicated (39). Moreover, H2S reacts with S-nitrosothiols to form thionitrous acid (HSNO). At the cellular level, HSNO can be metabolized to afford NO+, NO, and NO− species (9). It has been proposed that H2S functions as a paracrine signaling molecule (8). Conversely, an inhibitory effect by H2S on lipopolysaccaride-induced NO production was reported in macrophages, in which up-regulation of heme oxygenase-1 was involved (36). Further, inhibition by NaHS of all three NOS isoforms was demonstrated (23). These findings suggested that H2S and NO could be symbiotic or interdependent in eliciting some physiological and/or pathological effects (26) and suggested the existence of cross-talk between NO and H2S (6).
Based on the idea mentioned earlier, we scrutinized potential increases of [NO]i by H2S and found that NOS activation and NO production were indeed involved in the H2S-induced [Ca2+]i increase in pancreatic acini, as the NaHS-evoked [Ca2+]i rise was largely decreased by the NOS inhibitor L-NMMA and NO scavenger cPTIO (Fig. 4). More directly, [NO]i measurement with DAF-2/DAF-2T clearly showed that NO was actually formed by the treatment of acini with NaHS, whose effect was gradual and long lasting (Fig. 5A, B). What we would like to especially stress is the temporal change in [NO]i; the [NO]i started increasing with some delay after NaHS addition and this time course appeared to be synchronized with the second phase of the [Ca2+]i increase (Fig. 5A vs. Fig. 4A solid line). This most likely indicated that the second phase of the NaHS-induced [Ca2+]i could be largely attributed to the NaHS-induced [NO]i increase. A possible underlying mechanism will be discussed next. In support of this hypothesis, pharmacokinetic analyses of the dose-response relationship for the NaHS-induced NO production (Fig. 5B) and the [Ca2+]i increase in the absence of extracellular Ca2+ (Fig. 2B) demonstrated that these EC50 values were in close proximity, being 73.3 and 64.8 μM, respectively. All these lines of evidence strongly suggested that NOS activation and the resultant NO production were largely involved in the downstream cascade of H2S action in pancreatic acinar cells, indicative of the multifaceted and complicated nature of the biological actions of H2S. Altogether, it is likely that the first H2S-induced increase in [Ca2+]i was due to a solo effect by H2S and the secondary long-lasting increase was caused by synergism between H2S and H2S-produced NO.
Intracellular mechanism of H2S-induced [Ca2+]i elevation
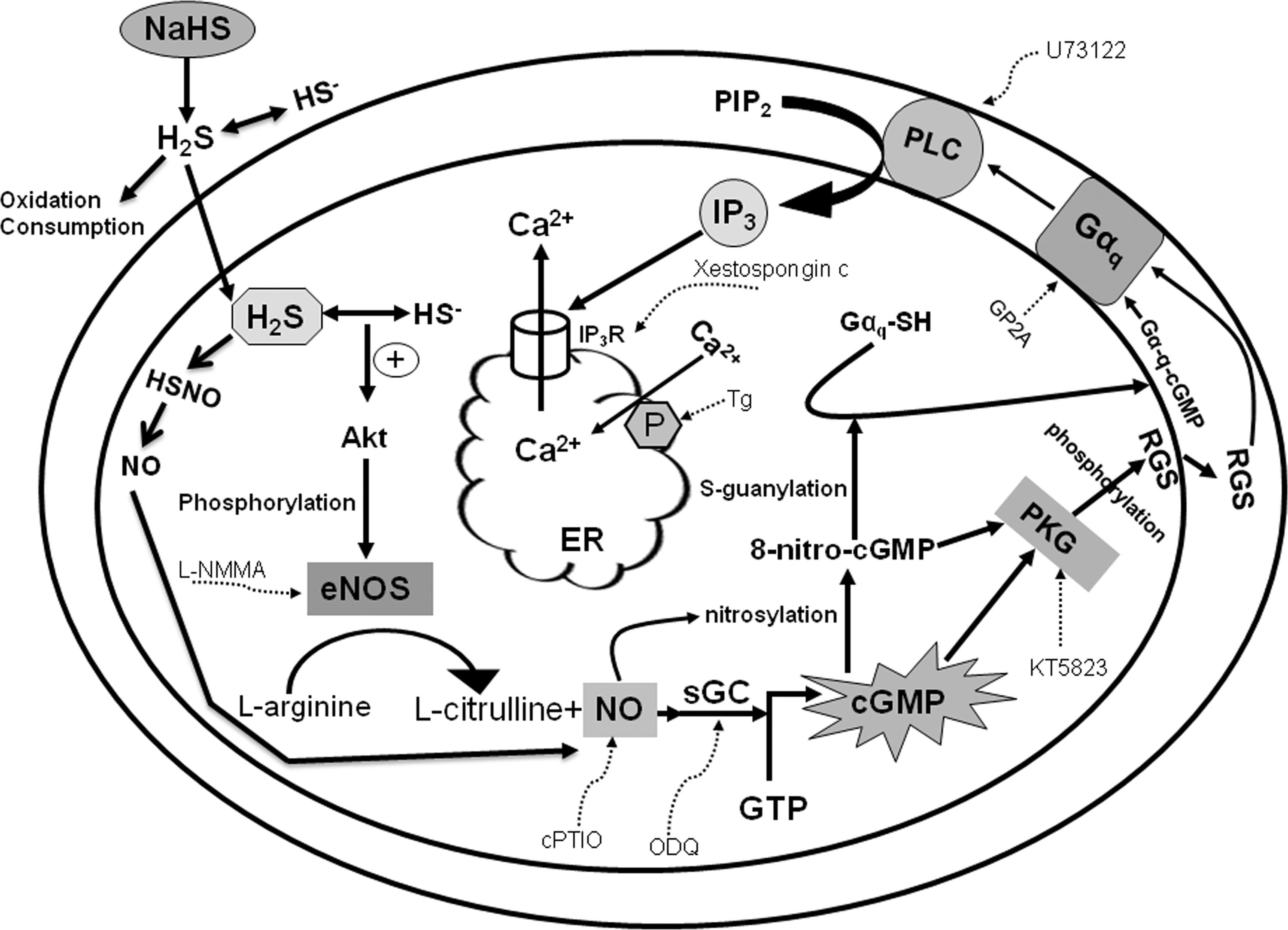
Presumptive targets of H2S proposed so far include the KATP channel and myosin-light-chain phosphatase, which cause vasodilation and atrial and ventricular relaxation. Other sites of action may be BKCa, L-type and T-type Ca2+ channels, Cl− channels and TRPV channels, and some kinases such as mitogen-activated protein kinase, ERK, and PKC are also molecular targets of H2S (27). In the endocrine pancreas, H2S has been reported to open the KATP channel and reduce exocytosis of insulin (20,40,62). It is well understood that hampering intracellular Ca2+ homeostasis results in distortion of normal cellular functions. In a recent report, we proposed the presence of a novel pathway underlying regulation of NO-induced intracellular Ca2+ dynamics in rat pancreatic acinar cells (32,33), and here we show evidence that such a pathway synergistically operates in the H2S-triggered [Ca2+]i increase. In the present study, similar characteristics were found in the NaHS-induced [Ca2+]i elevation. First, the NaHS-induced [Ca2+]i was significantly attenuated by the PLC inhibitor U73122 (Fig. 6). This provided evidence that H2S might directly or indirectly activate PLC and the resultant Ca2+ response. This is contradictory to the result reported in microglial cells, in which U73122 failed to affect the action of H2S on the intracellular Ca2+ store (24), but a result similar to the present one has been reported for neuronal SH-SYS5Y cells, in which U73122 significantly suppressed the H2S-induced [Ca2+]i elevation (63). Second, the NaHS-induced [Ca2+]i elevation was found to be greatly reduced in the presence of the IP3R blocker xestospongin C (Fig. 7), which indicated that IP3-IP3R interaction was present in the downstream of PLC activation to cause Ca2+ release from the IP3-sensitive intracellular Ca2+ store. Third, the increase of [Ca2+]i by NaHS was markedly depressed by pretreatment with the G-protein antagonist GP2A (Fig. 7). This provided evidence that G-protein, possibly Gq-protein, was involved in PLC-IP3-IP3R pathway. Fourth, the NaHS-induced [Ca2+]i increase was inhibited by the presence of the sGC inhibitor ODQ and the PKG inhibitor KT5823 (Fig. 7). This suggested that the sGC-cGMP-PKG cascade was functioning even in the NaHS-caused [Ca2+]i increase in pancreatic acinar cells. These four findings are analogous to what we previously observed in the SNP-induced [Ca2+]i increase in pancreatic acinar cells (32,33), strongly indicating that the NO-triggered process was also included in the NaHS-induced increase in [Ca2+]i. Additional evidence that supports this hypothesis would be the significant production of NO by NaHS as mentioned earlier. Some contradictory reports have indicated that H2S inhibits NO-induced intracellular cGMP accumulation in the salamander retina (41) and that it does not affect sGC or the level of cGMP in the course of vascular relaxation (69). Moreover, H2S does not affect the sGC or the cGMP concentration, but it increases the intracellular adenosine 3′, 5′-cyclic monophosphate (cAMP) level, especially in neurons (21). On the other hand, cGMP was reported to be involved in the combined effect of NO and H2S on pacemaker currents generated by interstitial cells of Cajal (65). A clear-cut interpretation for these controversial findings is not apparent, but they may partly be due to the types of cells utilized. In any case, our present findings implied that, in pancreatic acini, the cGMP-PKG cascade was involved in the H2S-provoked [Ca2+]i increase and the putative overall cascade seems identical to our recently proposed model (32,33).
However, certain missing links in our model remain to be identified. The first missing link is how cGMP activates membranous Gq-protein and the second one is how H2S activates NOS. To obtain an overall model, the following reports appear to be beneficial, providing us with significant hints. According to Akaike et al. (2) and Saito et al. (46), NO, in addition to activating sGC, nitrosylates various intracellular substances as post-translational modifications, producing 8-nitro-cGMP in the case of cGMP. The nitrosylated cGMP then binds with Gαq in the cell membrane forming Gαq-cGMP, which can activate PLC. Tang et al. (55) documented that cGMP-activated PKG could phosphorylate a regulator of G-protein signaling-2 and its translocation from cytosol to the cellular membrane, which can also activate the Gαq subunit. Thus, it is highly possible that cGMP can activate Gq-protein, leading to PLC activation. Yong et al. (64) have demonstrated that endogenous production of H2S after ischemia-reperfusion phosphorylates Akt, a serine/threonine kinase, followed by eNOS activation, inducing a protective effect of NO against cellular injury. Minamishima et al. (31) also reported that H2S phosphorylates Akt and NOS3. Coletta et al. (7) proposed, in their recent report, a model in which H2S and NO synergistically affect angiogenesis and vasodilation.
In conclusion, the present study suggests the hypothesis that H2S accelerates NO production via NOS activation, which, in turn, activates sGC and elevates the intracellular cGMP level. The latter can activate G-protein followed by PLC activation and mobilize intracellularly stored Ca2+ that is triggered by IP3 bound to IP3R (Fig. 8). This pathway is identical to that proposed for the NO-induced [Ca2+]i increase in pancreatic acinar cells. To best of our knowledge, this is the first report that highlights a novel pathway by which H2S modulates intracellular Ca2+ dynamics and the resultant physiology and/or pathology of pancreatic acinar cells.
Materials and Methods
Chemicals
Chromatographically purified collagenase (CLSPA) was obtained from Worthington Biochemical (Lakewood, NJ). A Gq-protein antagonist peptide, GP2A, and a PKG inhibitor, KT5823, were purchased from Calbiochem (La Jolla, CA). U-73122, bovine serum albumin (BSA), AOAA, and soybean trypsin inhibitor (type1-S) were purchased from Sigma (St. Louis, MO). Fluo-3/AM, EGTA, L-NMMA, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and cPTIO were from Dojindo (Kumamoto, Japan). Eagle's minimum essential medium (MEM) was purchased from Invitrogen (Carlsbad, CA). DAF-2/DA was from Daiichikagaku (Tokyo, Japan). Cell-Tak was from BD Biosciences (San Jose, CA). DL-propargylglycine (PAG) was purchased from Cayman Chem. (Ann Arbor, MI). Other chemicals, including the H2S donor NaHS and L-cysteine hydrochloride, were obtained from Wako Pure Chemicals (Osaka, Japan).
Recently, it is claimed that circulating sulfide (HS−, H2S, S2−) level is very low, achieving nM level at highest (37,38). Tissue sulfide concentration is not easy to measure or estimate, as multiple factors are related to determining actual concentration. H2S can easily diffuse from the site of production (8) and readily permeate the cell (29) but, on the other hand, H2S is rapidly consumed by tissues in the presence of oxygen. Exogenously applied H2S is quickly and efficiently consumed by tissues at pO2>10 torr (38). It is estimated that sulfide presents ∼20% H2S/80% HS− ration in extracellular fluid (37). Under the present experimental condition, pO2 of the perfusate in the chamber is estimated to be ∼140 torr, equilibrating rapidly with environmental dry air. Thus, based on earlier consideration, actual concentration of H2S at the tissue level after the administration of 300 μM NaHS could be lower than 60 μM at most or much lower in the current study.
Animals
All experiments conformed to the guidelines on the ethical use of animals set by the U.S. National Institutes of Health and were approved by the institutional Animal Care and Use Committee of the Graduate School of Veterinary Medicine, Hokkaido University. All efforts were made to minimize animal suffering and to reduce the number of animals used. Adult SPF male Wistar rats (200–250 g) purchased from Clea Japan (Tokyo, Japan) were housed under a controlled environment at an ambient temperature of 22°C and a 12:12-h light-dark cycle. Animals were deprived of food overnight before the experiment but had free access to water.
Solutions
Normal Ringer's solution, used throughout acinar isolation and experimentation (Standard HEPES-buffered solution), contained (mM): sodium chloride, 138.0; potassium chloride, 4.7; calcium chloride (CaCl2), 1.3; magnesium chloride, 1.13; disodium phosphate, 1.0;
Preparation of pancreatic acini
The animals were anesthetized by CO2 inhalation and euthanized by exsanguination. Pancreatic acini were obtained from the rat pancreas by collagenase digestion according to the method previously reported (32,33). Briefly, pancreata were removed, freed from fat and lymph nodes, and 5 ml of standard solution containing 60–75 U/ml collagenase and 0.1 mg/ml soybean trypsin inhibitor was injected into the interstitium of the pancreatic tissue, which was incubated in a conical flask at 37°C under vigorous shaking for total of 60 min. After 30 min of incubation, 5 ml of new collagenase was added to the flask. Mechanical disruption of the tissue was performed by gentle suction through pipettes with decreasing orificial size. The acinar suspension was then filtrated through 150 μm nylon mesh, rinsed thrice, pelletted (×60 g), and resuspended in a suitable amount of the standard solution. Acinar cell viability was virtually 100% when assessed by the trypan blue exclusion test (32,33).
Optical imaging of Ca2+ and NO
Imaging was carried out as previously reported (32). Briefly, isolated acini were resuspended in the normal Ringer's solution containing 10 μM Fluo-3/AM or 10 μM DAF-2/DA and incubated at 37°C with constant mild shaking in the dark for 60 min. After incubation, the acini were washed in the standard solution, transferred to a recording chamber, on the bottom of which a cover glass previously coated with Cell-Tak was attached, and left for at least 5 min. The chamber was placed on the stage of an inverted microscope. Imaging was performed using a confocal laser scanning microscope (Fluoview FV500; Olympus, Tokyo, Japan). The acini were continuously perfused with normal Ringer's solution at a flow rate of 1 ml/min before and throughout the experiments. Acini were illuminated at 488 nm with a krypton/argon laser, and the emission light (>505 nm) was guided through a×40 water immersion objective to a pinhole diaphragm. Photodamage was minimized by attenuating the laser intensity by interposing a neutral density filter into the illumination path (usually 1% transmission was sufficient to obtain fluorescence). Confocal images of acini were taken at 10 s intervals. The time courses of changes in fluorescence intensity at the ROI were analyzed using Fluoview 5.0 with Tiempo. The change in the fluorescence intensity was expressed as the percent of basal fluorescence intensity by setting the prestimulated fluorescence just before the application of chemicals at 100% (baseline). In addition, the degree of the change of [Ca2+]i was estimated by calculating the SFC above the baseline during stimulation as previously reported (32).
Statistical analysis
The results are reported as means±standard error, with n denoting the number of cells in the acinus analyzed. Statistical significance was determined using Student's t-test and ANOVA, with a value of p<0.05 being considered significant.
Footnotes
Acknowledgment
The authors are grateful to Goho Life Science International Foundation for their generous research support.
Author Disclosure Statement
No competing financial interests exist.