
Abstract
The pathophysiology of multiple sclerosis (MS) involves several components: redox, inflammatory/autoimmune, vascular, and neurodegenerative. All of them are supported by the intertwined lines of evidence, and none of them should be written off. However, the exact mechanisms of MS initiation, its development, and progression are still elusive, despite the impressive pace by which the data on MS are accumulating. In this review, we will try to integrate the current facts and concepts, focusing on the role of redox changes and various reactive species in MS. Knowing the schedule of initial changes in pathogenic factors and the key turning points, as well as understanding the redox processes involved in MS pathogenesis is the way to enable MS prevention, early treatment, and the development of therapies that target specific pathophysiological components of the heterogeneous mechanisms of MS, which could alleviate the symptoms and hopefully stop MS. Pertinent to this, we will outline (i) redox processes involved in MS initiation; (ii) the role of reactive species in inflammation; (iii) prooxidative changes responsible for neurodegeneration; and (iv) the potential of antioxidative therapy. Antioxid. Redox Signal. 19, 2286–2334.
I. Introduction
A. Basic facts
M
MS takes different clinical courses in patients (75, 192). The most common is the relapsing–remitting course (progressive-relapsing MS [RRMS]) in which flare-ups of neurological disabilities occur from time to time, followed up by a complete or partial recovery. In some of these patients, neurological disabilities accumulate without proper recovery, that is, MS takes a secondary progressive course (SPMS). In other patients, the progressive course is taken from the beginning of clinically manifested MS, and this course is called primary progressive MS (PPMS). There is also a progressive form of MS that is combined with occasional relapses, that is, PRMS. There are also patients who have rare attacks of the disease and always show complete recovery, and such a course of MS is called benign. Finally, there are cases of clinically isolated syndrome (CIS), that is, one episode of neurological deficits which does not always progress into definite MS. The disease is also heterogeneous according to histopathological findings in MS patients. Based on histopathology and pathogenic mechanisms, four subtypes of MS have been identified (258). We shall address this later in relation to redox changes.
Epidemiological data indicate that both genetic and environmental factors are important for the etiology of MS (184). It is known that some major histocompatibility complex (MHC) haplotypes, as well as some alleles of cytokines and their receptors, increase the risk for MS (174). Environmental risk factors include infections, inadequate exposure to sunlight, and smoking (23, 184). An interaction between environmental and genetic factors in the susceptibility to MS is presumed to be very complex. Still, some recent findings revealed possible scenarios for collaboration between specific factors in MS initiation. For instance, MS risk modulators, including genetic variants in interleukin-7 receptor-α (IL7RA*C), interleukin-2 receptor-α (IL2RA*T), MGAT1 (IVAVT−T) and CTLA-4 (Thr17Ala), and environmental factors affecting vitamin D3 levels, converge in order to alter branching of Asn (N)-linked glycans (323). Importantly, branching reduction results in T-cell hyperactivity and promotes spontaneous inflammatory demyelination and neurodegeneration in the MS animal model (176). N-glycan branching is positively regulated by the Golgi enzymes Mgat1 and/or Mgat5. Down-regulation of Mgat1 by IL7RA*C and IL2RA*T is opposed by MGAT1 (IVAVT−T) and vitamin D3, which optimizes branching and mitigates the risk of MS when combined with enhanced N-glycosylation of CTLA-4. Therefore, various genetic and environmental factors regulate a final common pathway—N-glycosylation—which is highly relevant for MS pathogenesis. In addition, it has been shown that vitamin D directly stimulates the expression of HLA-DRB1*15, MHC class II allele, which is a major MS genetic risk factor (174, 373). It has been suggested that a low vitamin D level in early childhood leads to a decrease in the expression of HLA-DRB1*15 in the thymus and, therefore, to inadequate presentation of self-antigens and inefficient central tolerance (for more details on these processes, see the next section) (186). As a consequence, more autoreactive T cells could emerge in the immune system of people who had low vitamin D in their early childhood. To make the story even more complex, low vitamin D levels may be a consequence of genetic malfunction, for example, nonfunctional vitamin D converting enzyme CYP27B1, which converts 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D (174), or a consequence of an environmental influence, that is, low exposure to sunlight or an inadequate diet. The latter might explain why a higher frequency of HLA-DRB1*15 is detected in MS patients in Scandinavian countries compared with those in Mediterranean countries, or those in some other non-European countries (391).
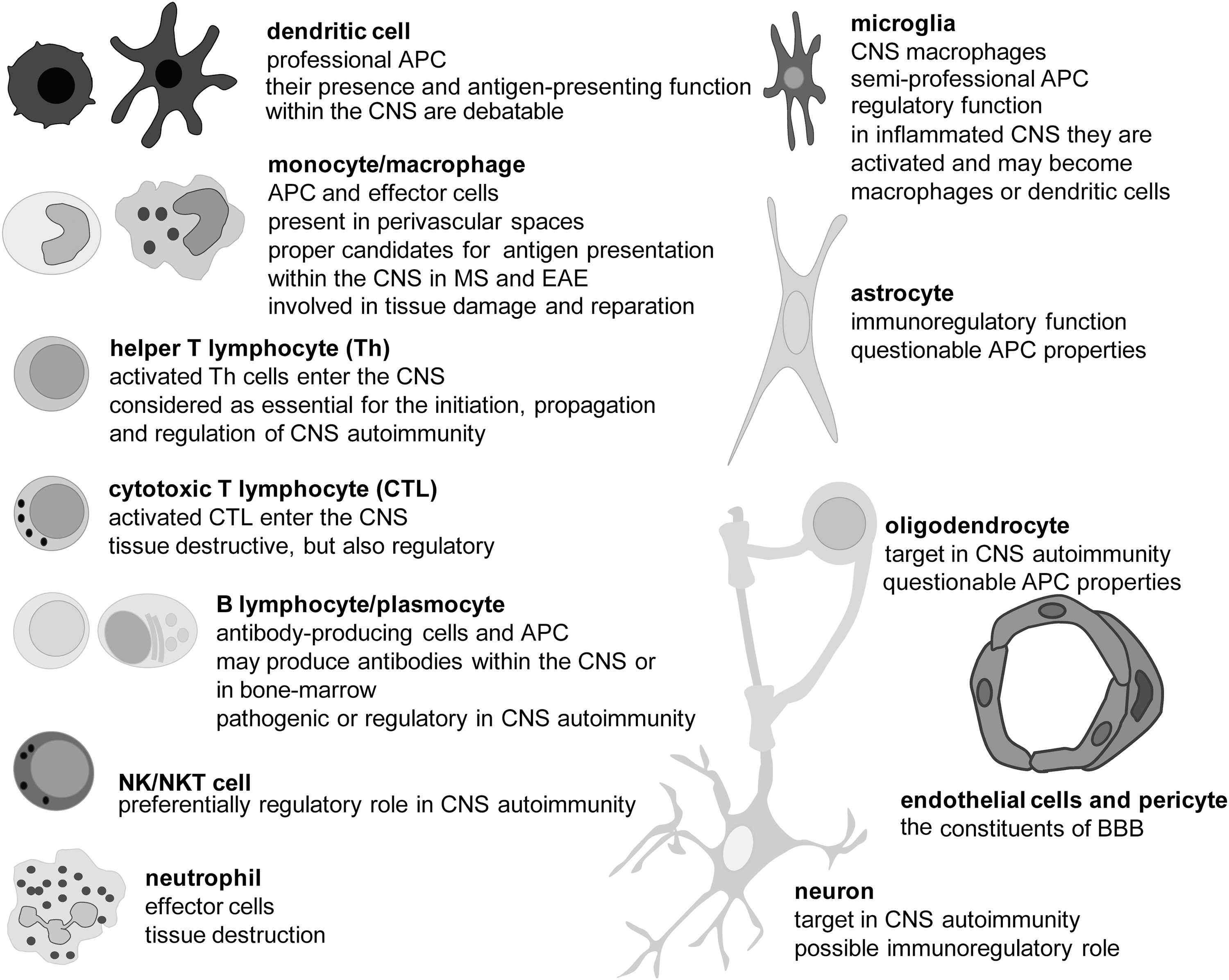
MS investigation is very vivid, as many elements of its complex pathogenesis are still unknown, and (consequently) there are no medications for curing the patients with the disease. Various therapies, which are designed to affect immune response directed against the CNS, are being applied, but they only slow down and/or reduce severity of the clinical course of MS. This disappointing fact emphasizes the importance of other components in MS pathology. Numerous studies, performed mainly in the last decade, suggest that redox processes have a critical role in all stages of MS, particularly in the first—the initiation, and the last—the neurodegeneration. However, before we address the various involvements of redox alterations in MS initiation and progression, we should introduce the readers to the major cell types of the CNS and the immune system involved in the pathogenesis of the disease, as well as to some of their properties that are relevant for MS pathogenesis (details are given in Fig. 1).
B. Autoreactive T cells
T cells, which are essential for MS pathogenesis, are autoreactive, that is, they react with self-antigens of the CNS. Such T cells initiate and propagate an autoimmune response against the CNS and orchestrate cellular and humoral effectors, which attack myelin, oligodendrocytes (ODC), and neurons. They are also one of the important effectors involved in the CNS damaging. The essential signal for T-cell activation comes from the recognition of antigens presented by MHC molecules on the surface of antigen-presenting cells (APC). In order to efficiently present an antigen to a naïve T cell, APC have to express a complex of antigens and MHC molecules, as well as co-stimulatory molecules, and to produce adequate cytokines (95). Dendritic cells (DC) are APC that are capable of activating naïve T cells; hence, they play a central role in the initiation of the adaptive (auto)immune response (98). T cells contact antigen/MHC complexes with their T-cell receptors (TCR) and co-receptor molecules (CD4 or CD8), and in addition, bind co-stimulatory molecules of APC (Fig. 2A). If a T cell expresses CD4, it recognizes MHC class II molecules, and if it expresses CD8, it recognizes MHC class I molecules. Most of the T cells express exclusively CD4 or CD8, and are, therefore, designated as CD4+ T cells and CD8+ T cells, with the former being helper T cells (Th) and the latter being cytotoxic T cells (CTL). In addition, among both populations, a certain proportion belongs to regulatory T cells (Treg). Th cells produce soluble mediators that perform effector and regulatory functions in immunity, while CTL use perforin and granzymes to kill infected, transformed, or damaged cells. Th cells are particularly important for the pathogenesis of MS, because they play a crucial role in the maturation and complete activation of other immune cells. Without the support of Th cells, B cells do not mature into long-lasting memory cells or plasma cells (Fig. 2B). CTL are also not fully activated without the support from Th cells (Fig. 2A). Similarly, macrophages and neutrophils will not be properly engaged in adaptive immunity without the contribution of Th cells. There are several populations of effector Th cells, including autoreactive Th1 and Th17, which appear to be the major pathogenic populations in MS (362). Importantly, in order to perform their effector functions, T cells have to be reactivated once they enter the CNS. It is generally accepted that there are no DC in the healthy CNS (375). Still, other APC are capable of reactivating T cells in the CNS. For instance, perivascular, meningeal, and choroid plexus macrophages are present in healthy CNS (375). Besides macrophages, CNS contains other semi-professional APC (microglia, B cells, and endothelial cells) and non-professional APC (astrocytes), which can reactivate autoreactive T cells (12, 38, 109, 440).
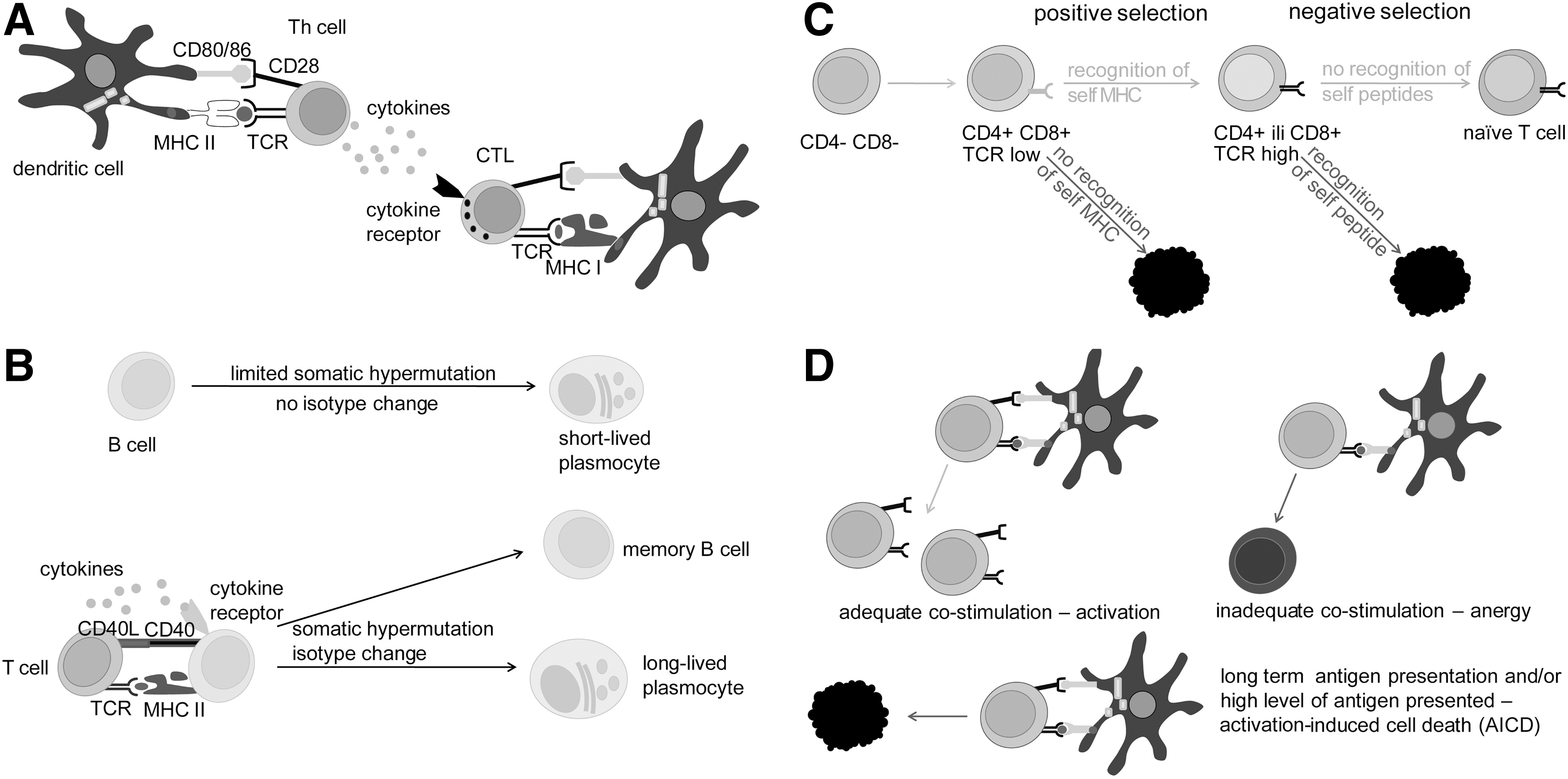
TCR are being generated stochastically by somatic recombinations and, therefore, T cells expressing TCR that recognize self-antigens are produced in the immune system. Still, in the process of maturation, such T cells are eliminated or inactivated by mechanisms of central and peripheral tolerance. Central tolerance is based on the interaction between TCR and MHC. It takes place in the thymus and involves two steps—positive and negative selection (175). Every subject has a highly specific repertoire of MHC molecules that are expressed in the cells. This fact is the base for positive selection, as many of the T cells produced in any subject will not be able to interact appropriately with the MHC molecules that are expressed in the thymus and will, therefore, be eliminated (Fig. 2C). Nevertheless, some of these cells that are capable of recognizing antigens in the complex with MHC molecules present in the thymus will interact very strongly with the MHC-antigen complex. Such T cells will be eliminated in the process of negative selection (Fig. 2C). This is very important for the prevention of T-cell autoreactivity, as antigens expressed in the thymus are self-antigens, and, therefore, the eliminated T cells would have been potential candidates for autoreactivity. Ideally, after positive and negative selection, naïve T cells generated in the thymus should be able to interact with MHC molecules that express foreign, but not self-antigens. However, negative selection is not perfect. Not all of the antigens are expressed in the thymus, and, therefore, some of the T cells with a potential to recognize self-antigens will survive the selection. Typical antigens that are not expressed in the thymus are those whose expression is limited to embryonic development and those which are specific for certain tissues (so-called tissue specific antigens [TSA]). Given that some of the CNS antigens are TSA, T cells that are potentially reactive to CNS antigens are present in our immune system. It can be speculated that CNS antigens are TSA due to the immune privilege of CNS, which developed in order to provide the “control center” with high protection (see the next section), but leave the immune system non-introduced to certain CNS antigens.
At the periphery of the immune system (every lymphoid organ and tissue outside the bone marrow and thymus), T cells that recognize antigens in an inadequate manner could be eliminated or become anergic (Fig. 2D). A naïve T cell can be activated only if it receives simultaneous stimulation through TCR, co-receptors, and cytokines (95). If the co-stimulation is absent, even though primary TCR-antigen-MHC recognition is perfect, a T cell will become irresponsive - anergic. In addition, if the recognition lasts too long and/or is too intense, a T cell will be eliminated in the process of activation-induced cell death (AICD). Typically, self-antigens are presented in the complex with MHC molecules for a long time and in many copies, which makes them perfect candidates for AICD induction. It is also important to note that Treg are actively involved in peripheral tolerance, restraining autoreactivity (96). Findings that suggest disturbances in their development and function in MS patients strongly imply the importance of peripheral tolerance for prevention of the disease (450). As already stated, Th cells are crucial for complete activation of macrophages, neutrophils, CTL, and B cells (Fig. 2A, B). Therefore, if self-tolerance mechanisms fail at the level of Th cells, the road is paved for other populations of immune cells to become autoreactive.
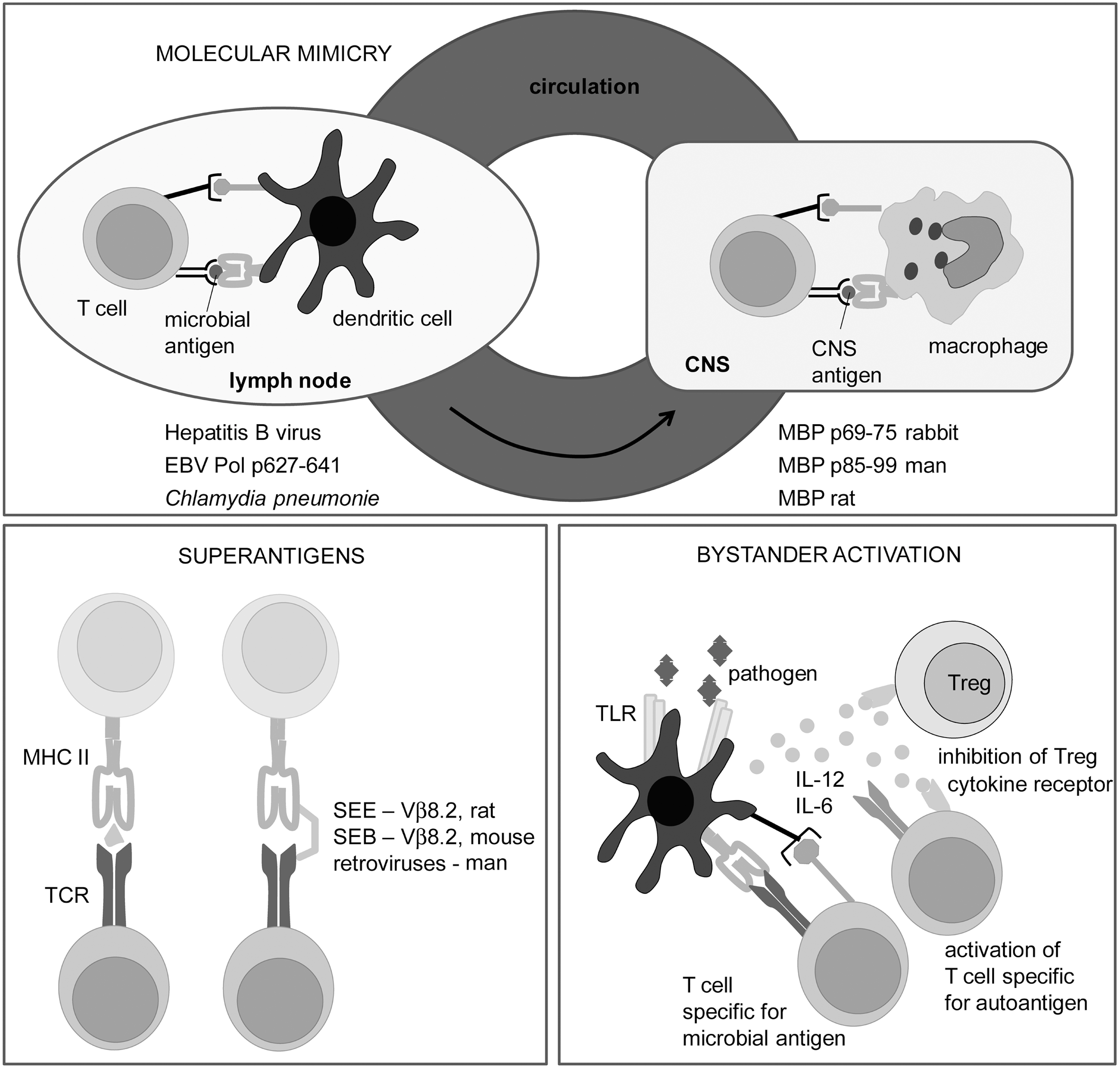
Due to the imperfections of tolerance mechanisms, the activation of self-reactive T cells with self-antigens, specific modifications of self-antigens, or microbes is possible (22, 82). Some of the major molecular targets in the immune response in MS and its models are identified. These are myelin proteins, including myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP), and proteolipid protein (PLP); lipids, such as sulfatide, sphingomyelin, and oxidized lipids; and glycans [e.g., Glc(α1,4)Glc(α)] (114). Axo-glial proteins, such as neurofascin and contactin-2, are also important molecular targets in MS (114). Modification of self-molecule antigenicity as a consequence of redox changes in the CNS might be essential for the initiation of autoimmunity in MS pathogenesis, as discussed in detail in section II.J.1. Finally, microbes might be involved in the primal activation of encephalitogenic T cells. There are at least three mechanisms of activation of self-reactive T cells involving microbes: molecular mimicry, superantigens, and bystander activation. These are described in detail in Figure 3.
C. The immune privilege of the central nervous system
Although autoreactive T cells exist in the immune repertoire, they are no guarantee for the induction of immune response against the CNS components. It was shown, about 30 years earlier, that peripheral blood of healthy individuals contains T cells that are specific for myelin antigens (e.g., MBP) (63). Moreover, the frequency of myelin-reactive T cells in the blood of MS patients is not increased compared with that of healthy controls (44). Bearing in mind the incidence of MS, it is obvious that the activation of the autoreactive cells is more of an exemption than a rule. The base for the impediment of CNS-directed autoimmune reactivity lies in the fact that CNS is not an ordinary system in relation to the immune system, because it is immune privileged (Fig. 4).
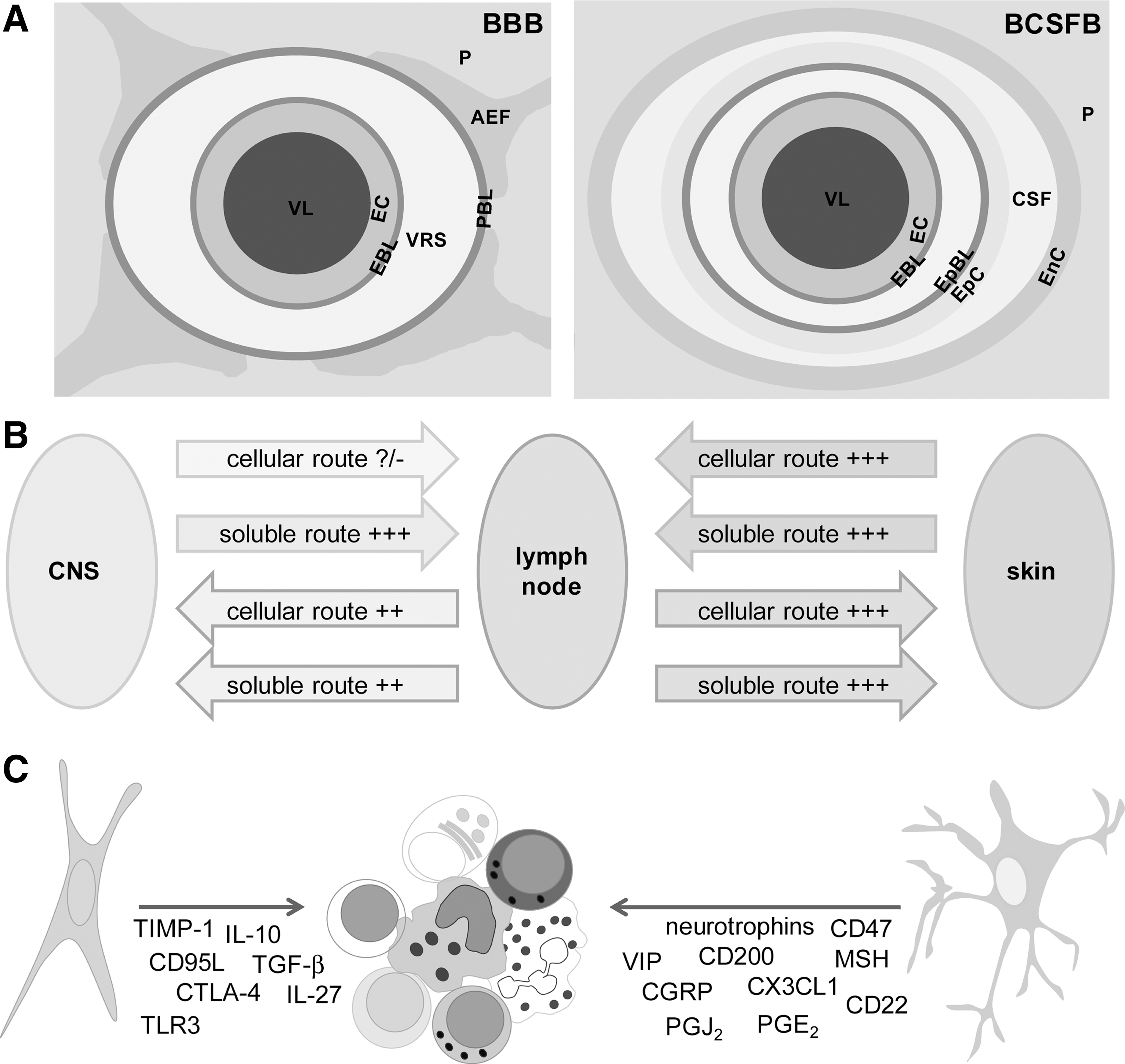
The immune privilege of the CNS is based on the blood–brain barrier (BBB) and blood–cerebrospinal fluid (CSF) barrier (BCSFB), which actively regulate the transport of molecules and cells between systemic circulation and CNS parenchyma (Fig. 4A). BBB is composed of endothelial cells that make strong interconnections—tight junctions (TJ) (132). Astrocytes and pericytes are essential for the formation and function of BBB (465). Postcapillary venules are blood vessels that are relevant to immune cell infiltration into the CNS. There, the immune cells have to cross the endothelial cell layer and endothelial basal lamina and to enter perivascular space (Virchow-Robin space [VRS]) (40). This space is limited by endothelial basal lamina and glial basal lamina. Astrocytic end-feet lie beyond glial basal lamina. Both these make glia limitans. The entrance of immune cells into the VRS is not considered classical infiltration. Instead, immune cells have to pass the glia limitans and enter the CNS parenchyma in order to infiltrate the CNS (40). Although the complex structure impedes immune cell entry into the CNS, macrophages and activated T cells of any specificity are capable of passing BBB and BCSFB and entering the CNS (199, 459). In addition, there are regions of the CNS where BBB is incomplete, such as circumventricular organs (147). These have a specific immune status and have been associated with neuroinflammation (468). Still, in general, the transport of antibodies and cells from immune organs and tissues into the CNS is impaired due to the BBB and BCSFB, and may be promoted in the case of BBB dysfunction and breakage.
Another specificity of the CNS is underdeveloped lymphatic drainage. There are no classical lymphatic vessels that drain brain and spinal cord. Instead, cerebral extracellular fluids drain into the blood across the arachnoid villi and into the lymph along certain cranial nerves and spinal nerve root ganglia. The interstitial fluid of the CNS drains via perivascular channels into the CSF, which is directly reabsorbed into blood via arachnoid villi (375). CSF drains into local afferent lymphatics along cranial and spinal nerves, for example, along olfactory rootlets to the nasal mucosa and then into the deep cervical lymph nodes. While the transport of soluble matters from the CNS into the draining lymph nodes is adequate, cellular transport is heavily impaired (147) (Fig. 4B). There is a special interest in the ability of APC to leave the CNS and nest into lymphatic organs. Although there is no direct connection between CNS parenchyma and lymph nodes, APC can travel from the CNS to the periphery via CSF and blood (201). This is important in relation to initial antigen presentation in MS (83). It is conceivable that antigen presentation to naïve T cells occurs in the lymph nodes, and, thus, DC have either to collect the antigen within the CNS and then to travel to the draining lymphoid tissue, or to trap a soluble antigen which is transported through lymph vessels. The existence of both soluble and cellular drainage from the CNS to lymph organs is essential for the initial steps of inflammation seeded within the CNS. Even if the initial antigen derives from a microorganism or some other external source and APC collect it at the periphery, CNS drainage will still be important for the perpetuation of inflammation and for epitope spreading.
A very important characteristic of the CNS, which contributes to the immune privilege, is the limited presentation of antigens within the CNS. Namely, so far, the presence of DC (the professional APC) in the healthy CNS parenchyma has not been validly documented (375). However, it is known that microglia and macrophages may differentiate into DC-like cells (368, 427). Activated microglial cells share numerous properties with DC, including constitutive expression of MHC II in situ and the expression of MHC I on activation (11). Microglia appears to be essential for the activation of CD8+ T cells. For example, it was shown that transgenic mice overexpressing co-stimulatory molecules on microglial cells develop spontaneous CNS demyelinating disease that is mediated by autoreactive CD8+ T cells (59). Apart from microglial cells that are the only parenchymal macrophage population, several populations of monocyte/macrophage lineage are situated at the vicinity. These cells are capable of taking samples from the parenchyma as a consequence of their position in the CSF flow pathways. Macrophages are potent APC, as they express MHC class II, co-stimulatory, and adhesion molecules. Furthermore, they are capable of initiating antigen-specific proliferative response in resting CD4+ T cells (375). Finally, it has been reported that CD14+ monocytes migrate across the inflamed human BBB and that, under the influence of BBB-secreted transforming growth factor-β and granulocyte-macrophage colony-stimulating factor, they differentiate into CD83+CD209+ DC (216). Importantly, CD209+ DC promote proliferation and expansion of Th1 and Th17 lymphocytes, and they are detected in close association with lymphocytes within active MS lesions (397). Monocytes are also present in the CSF, where they constitute around 5% of the cellular content (375).
Finally, active immunosuppresion performed by CNS resident cells, predominantly astrocytes and neurons, is an important factor of the immune privilege (39) (Fig. 4C). For instance, astrocytes provoke apoptosis in activated T cells in Fas-FasL dependent and independent manner, down-regulate proinflammatory cytokine generation in these cells, and even induce Treg (318). Neurons may also down-regulate microglia/macrophage activity (329).
D. Animal models of MS
Our current knowledge of MS etiopathogenesis is largely based on the studies of its animal models. In this section, we will present frequently used MS models and critically address their place in MS studies.
1. Experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) is the most widely used model of MS. It is an autoimmune disease of the CNS that is actively induced in susceptible animals, including rodents and primates, by immunization with CNS-specific antigens, peptides derived from these antigens, or CNS tissue homogenates. The disease can also be provoked by transfer of encephalitogenic CD4+ T cells, obtained from draining lymph nodes of animals immunized for active EAE induction, into the syngeneic animals (transfer or passive EAE). Depending on the species and strain used in experiments, specific requirements (e.g., adjuvants) for effective immunization are imposed, and divergent clinical courses and severity are induced in animals (314). Since it shares many clinical, histological, immunological, and genetic features with MS, EAE has been used in order to gain important insights into MS pathogenesis and to validate new targets for MS therapy (168, 251). However, the credibility of EAE as an MS model has been increasingly criticized [reviewed in Handel et al. (185)]. In short, criticism can be summarized as follows: (i) mice and rats are used in most of the studies of EAE, and these are phylogenetically distant from humans; (ii) the acute monophasic course is characteristic for many EAE variants; (iii) CNS lesions in most of the EAE models are limited to the spinal cord; (iv) demyelination rarely occurs in EAE; and (v) in most EAE variants, CD4+ cells are the major culprits, while some other immune cells might have pathogenic predominance in MS. Taking into account these and other discrepancies between EAE and MS, we have to be careful when translating data from EAE to MS. This is especially important in attempts to test potential MS therapeutics in EAE. Many agents that proved to be beneficial in EAE are inefficient or even detrimental in MS (36). Still, there are some MS therapeutics, including copaxon and natalizumab, the development of which has been based on EAE studies (460). Importantly, numerous molecular and cellular mechanisms of MS pathogenesis have been discovered in EAE research (36, 91, 251, 460). New EAE variants are also emerging, and they are exploring aspects of MS that were previously unaddressed in animal models, for example, the role of CD8+ T cells in neuroinflammation (14, 225).
2. Theiler's murine encephalitis virus-induced demyelinating disease
Theiler's murine encephalitis virus-induced demyelinating disease (TMEV-IDD) has been intensively used as a model for human demyelinating diseases, including MS (119, 300). TMEV is a positive-stranded RNA virus from the family Picornavirade (genus Cardiovirus). The TO subgroup of TMEV induces acute encephalitis between days 5 and 10 postinjection. TMEV-IDD is of particular interest, because it presents a parallel to a possible scenario in humans, where the initial trigger for CNS inflammation and demyelination is a virus. Importantly, clinical signs of TMEV-IDD are similar to those observed in patients with progressive MS, and they include spasticity, incontinence, weakness of the extremities, and, eventually, paralysis (273). As a parallel to the oligoclonal bands found in the CSF of MS patients, intrathecal antibody production has been observed in this model (351). The general criticism for this model is that the non-human pathogen has been used in a model for a human disease. Interestingly, it has recently been reported that a specific type of encephalomyelitis in humans, Vilyuisk encephalitis, might be caused by a human-TMEV recombinant virus (272). It has been shown that TMEV-IDD is especially important for studies of epitope spreading and molecular mimicry (349). Additional viruses, such as murine hepatitis virus, canine distemper virus, coronaviruses, and some retroviruses, are also being used for induction of MS-like demyelinating disease in experimental animals (257, 295, 352, 441).
3. Cuprizone
Finally, demyelination that is induced in mice with a copper chelator cuprizone is an important tool for MS studies (241, 425). In this model, ingestion of cuprizone by mice leads to early ODC death, the activation of microglia/macrophages, and subsequent reversible demyelination (93, 197). This model is particularly useful for studying demyelination and remyelination, and their relation to axonal loss (241). It is highly relevant for the evolution of type III and type IV MS lesions, where changes in ODC appear to be the primary events in disease pathogenesis. The model is also important for studies of demyelination in specific CNS regions, such as hippocampus, which are affected in MS patients but not in other animal models (345). Besides cuprizone, other toxins, such as ethidium bromide and lysolecithin, are being used for the induction of demyelination in experimental animals (441).
II. Redox Processes in the Initiation of MS
A. The baseline characteristics of initial lesion
The most prominent caveat in our understanding of MS is the primary cause that provokes autoimmunity against the CNS. It is rather hard to detect the seeding changes within or outside the CNS that start the disease pathogenesis. However, this is essential for designing curative MS therapy and preventive tests that would enable early diagnosis of subjects who are at risk of developing MS, that is, subjects who have genetic predisposition for MS. At the moment, there is no combination of biological markers that can predict MS development. The earliest time at which patients can be diagnosed for MS is during the first clinical episode, that is, CIS, which best correlates to a newly developed focal lesion(s) (18). There are not many reports on initial lesions, but most of them concur that their hallmark is the development of ODC death before myelin degradation. This is further accompanied by macrophage-mediated myelin removal, BBB breakage, and potential inflammation (381). Pertinent to this, McFarland and Martin proposed that the initial steps in lesion development can be independent of immune cells (302). This represents a vice versa set of events compared with the processes taking place in the lesions that involve the autoimmune component during MS progression. In those lesions, T cells, which are autoreactive toward myelin-based epitopes, decompose myelin, which, as a consequence, leads to ODC death (33). Studying new symptomatic lesions in patients who died during or shortly after a relapse, Barnett and Prineas observed extensive ODC apoptosis and the presence of reactive astrocytes in early lesions showing generally intact myelin (32). There were also a number of myelin-laden macrophages present, most likely originating from perivascular space or microglia, while lymphocytes were absent. It has been suggested that phagocytic activity in nascent MS lesions represents a response to pathological alternations on myelin sheets developing due to a loss of metabolic support by ODC (33). The authors suggested that these early events in lesion initiation may be followed by the immigration of activated lymphocytes. In another study, Prineas' group documented virtual absence of T and B cells in early lesions. They observed ODC loss and the presence of myelin-laden macrophages, which are activated by the presence of degenerate myelin. It has been speculated that the formation of early lesions “has some basis other than destructive cell-mediated immunity directed against a myelin or ODC antigens,” while subsequent accumulation of innate immune system cells has been recognized as a response to the presence of dead myelin (194). It is important to note that although ODC death in early lesions has been loosely labeled as apoptosis, it is not apoptosis in its classic form. Namely, an integrative component of the apoptosis pathway—caspase-3—is not active (32). This characteristic, accompanied by DNA condensations, may qualify ODC death in initial lesions as apoptosis-like programmed cell death. The death of ODC via this pathway rarely takes place in chronic MS lesions, implying that it represents an early event which is associated with acute lesion formation (32, 33). In an autopsy study, a specific type of lesion (marked as “type I”) has been observed almost exclusively in the earliest MS cases (153). The lesions were formed around vessels, and the number of cells was not increased, implying that there was no inflammation; they were also rich in macrophages enclosing myelin material. Probably the most interesting feature was the lack of BBB disruption. In brief, initial white matter lesions show no evidence of T- or B-cell infiltration. Only CNS innate immune system cells—macrophages and activated microglia—are present. According to these facts, the root cause of MS is not immune deregulation, but a set of very specific degenerative processes (417), which, as we will illustrate here, are tightly, if not predominantly, related to redox alterations.
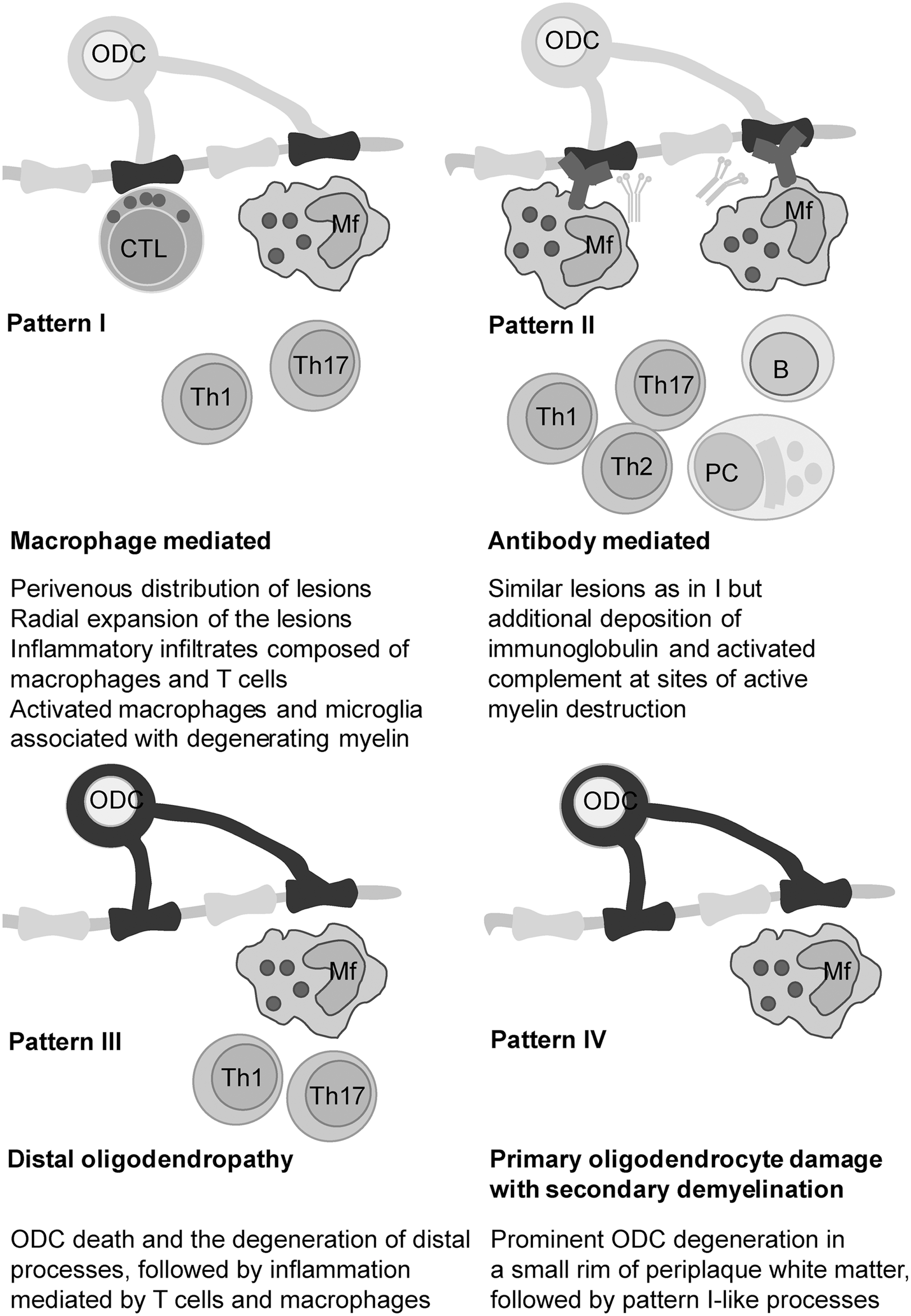
In their milestone study, Lassmann and co-workers determined the presence of four different patterns in MS lesions (283). Patterns I and II involve the autoimmune component and resemble EAE, while patterns III and IV do not comprise autoimmunity, at least not in the early stages of lesion development (Fig. 5). Pattern III lesions appear to be the first to develop. The authors identified pattern III lesions mostly in patients who had a clinical course of less than 8 weeks before the biopsy or autopsy took place (283). Pattern III lesions are localized near the veins or venules. They are characterized by ODC death preceding demyelination, and they show increased expression of redox active enzymes (33). Pattern IV lesions, which are mostly related to degenerative processes and significantly less to inflammation (417), were found in white matter autopsy samples from patients who suffered from PPMS (283, 366). Haider et al. labeled pattern II lesions as complement-associated demyelination, whereas pattern III was described as hypoxia-like tissue injury (181). Several groups proposed that pattern III and IV lesions resemble initial starter lesions (33, 259, 283). In line with this, apoptosis-like ODC death was observed before demyelination in pattern III lesions in autopsy samples of MS patients who died shortly after MS development (some less than a month after). T-cell infiltration was mild until serious demyelination developed, which was followed by a massive infiltration (292). In order to integrate the processes involved in MS initiation, we will rely not only on the scarce data on initial/early lesions, but also on what is known about pattern III and IV lesions.
B. Circulatory abnormalities in pre-MS brain
In order to understand what sets off the formation of initial lesion(s), we should go back to the time before a patient experiences the first signs of MS. A widespread characteristic of MS patients, which appears to be present before the disease onset, is the obstruction of cerebral circulation. A significant role of circulatory defects in MS initiation is implicated by the fact that early lesions (and lesions in general) very frequently develop around veins. For example, Ge et al. documented, using 7T magnetic resonance imaging (MRI), that most lesions form around veins, showing strict perivascular distribution, and following the orientation and course of the vessel (156). Recently, Zamboni and others endorsed and updated so-called chronic cerebrospinal venous insufficiency (CCSVI) hypothesis of MS development, which was proposed by Putnam in 1933 (487). CCSVI describes a vascular condition which is characterized by anomalies of the main extracranial cerebrospinal veins that interfere with normal cerebrospinal outflow, leading to retrograde blood flow. Accordingly, this provokes the lysis of erythrocytes and the formation of “iron sludge,” which activates inflammation, thus contributing to MS pathology (487). However, a number of established research groups found strong evidence against CCSVI (456). For example, Doepp et al. found that only a small population of MS patients has CCSVI according to Zamboni's criteria, and that a large majority of those with CCSVI show no measurable hemodynamic alterations (117). In addition, the absence of increased iron levels in patients with CIS clearly implies that iron accumulations do not precede the development of MS and most likely represent an epiphenomenon (236). Finally, a subgroup of adult and pediatric patients showing MS signs that appear to be provoked by iron deficiency has been identified. The symptoms are usually alleviated by iron supplementation (446). This can not only be easily explained by the high amount of iron needed for normal ODC development and function (159), but also substantiates the fact that iron excess is not responsible for MS initiation. In a nutshell, CCSVI is not a plausible cause of MS. However, CCSVI should not be confused with anomalies in cerebral blood flow and small veins and venules inside CNS tissue, as accumulating data foster further concepts, which involve vascular changes and obstructed circulation in MS initiation (106, 456). Varga et al. reported that cerebral blood flow is decreased in the normally appearing white matter (NAWM) of patients with CIS compared with control subjects (449). Sinnecker and colleagues documented, using 7T MRI, a reduced visibility of periventricular veins in CIS and early MS patients. The authors attributed the results to hemodynamic and vascular alterations (404). In a very recent MRI study, involving 30 CIS patients, Papadaki et al. observed significantly higher values of cerebral blood volume and mean transit time, and decreased cerebral blood flow in NAWM and deep gray matter in CIS compared with healthy subjects. The fact that such changes were not present in RRMS patients implies that they are not secondary (357). Pertinent to this, it has been shown that active lesions in RRMS patients show increased perfusion, whereas inactive lesions show variable levels of perfusion and veins which are ill defined (155). In short, these facts clearly imply that obstructed venous blood flow may be present before the onset of MS. On the other hand, Adhya and co-authors documented reduced cerebral blood flow and blood volume in PPMS patients (2). Another group found a decrease in cerebral blood flow by roughly 50% and more than twofold prolonged mean transit time of blood throughout the NAWM of MS patients (261). However, these changes, observed during the course of disease progression, could be secondary. It is worth mentioning that epidemiological data seem to concur with the proposed important role of circulatory problems in MS initiation: (i) MS shows similar epidemiological features to chronic vein insufficiency, such as higher prevalence in women and in the white non-Hispanic population compared with Hispanic, African-American, and Asian, very low incidence in aboriginal populations of Africa and Australia (42, 81, 100, 108, 320, 372), and a similar age of onset (20–30 years of age) (81, 106, 372); (ii) A decreased blood level of vitamin D, which has been related to an increased risk of MS development (127), may also lead to hypertension and other vascular problems (364). Interestingly, Eskimos and Lapps, who have a rich source of vitamin D in their diet, not only show a low risk of MS development (173), but also rarely develop vascular diseases (433); and finally (iii) one large survey has shown that veterans with MS have a higher prevalence of hypertension compared with the general veteran population (260). However, more correlation studies are needed in order to establish the link proposed here.
C. Blood–brain nitric oxide crosstalk
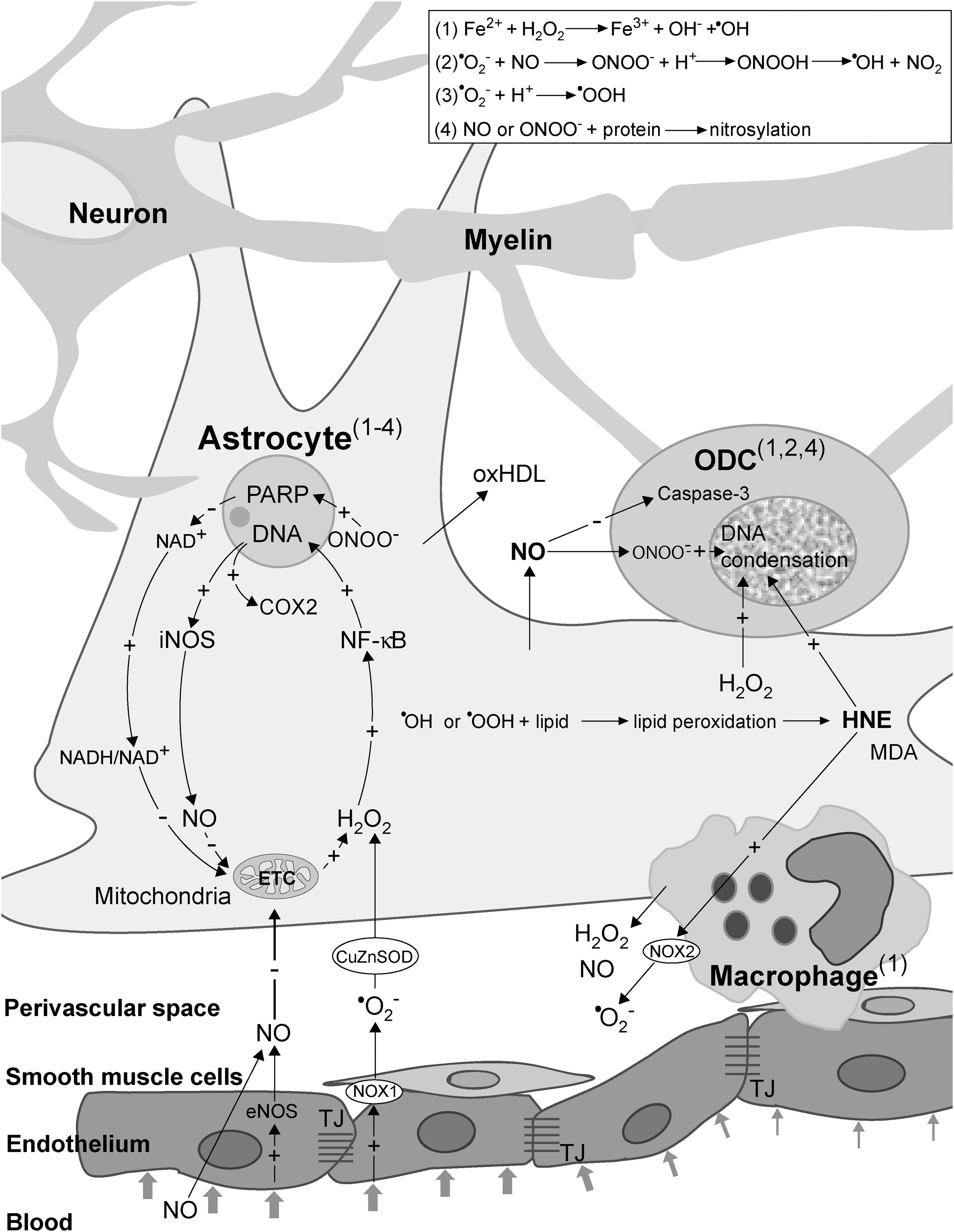
Altered cerebral perfusion may exert a detrimental impact on brain physiology. At the points of blood flow obstruction, shear stress inevitably develops, being reciprocally proportional to the flow (85, 252). This may represent the basis of crosstalk between the circulation and the brain through undisrupted BBB. In response to the shear stress, endothelial cells produce increased amounts of vasoactive agents—nitric oxide (NO) and hydrogen peroxide (H2O2) (189, 277). These two reactive species are uncharged and relatively stable, thus being capable of crossing cellular membranes. From the endothelium, NO and H2O2 may leak into the perivascular space to affect astrocytes, the extensions of which are positioned around the vessels (Fig. 6). The smooth muscle layer is thick and non-homogenous around small postcapillary veins, and it does not represent a significant barrier for NO and H2O2 to reach astrocytes. NO can be synthesized by three distinct types of NO synthase (NOS): endothelial NOS (eNOS), inducible NOS (iNOS), and neuronal NOS. Generally, eNOS activity in endothelial cells results in micromolar concentrations of NO, which might have only physiological effects (266). However, the effects may transcend pathophysiological ones, if the NO concentration exceeds a specific threshold. This may occur in the case of an increased level of NO or its precursor—nitrite—in the systemic circulation. Such a setup may be developed in a potential MS patient due to infection- or injury-related inflammation at remote sites or due to increased core body temperature (234, 382). Pertinent to the later, Uhthoff's phenomenon, the manifestation of MS symptoms and signs when core body temperature is increased by exercise or a hot bath, has been used for decades in MS diagnostics (90). It is important to note that a recent study showed that eNOS expression is, in fact, decreased in early lesions (140). However, one should bear in mind that increased expression of eNOS provoked by shear stress represents a transient effect, which develops before the development of the lesion and is further attenuated by increased NO concentrations (189). The important role of eNOS in MS initiation is implicated by the fact that eNOS-deficient mice exhibit a delayed onset of EAE, which correlates with delayed BBB breakdown (467). Shear stress also increases the activity of NADPH oxidase 1 (NOX1) on endothelial cell membranes (150), which leads to superoxide (•O2 −) production. Fischer et al. reported that this setup develops in initial lesions (140). Since CSF is rich in copper, zinc superoxide dismutase (CuZnSOD) (256), the expression of which may be further promoted by shear stress (189), NOX1-mediated •O2 − generation results in the rise of extracellular concentrations of H2O2.
D. NO and H2O2 from endothelium activate the redox cycle in astrocytes
NO and H2O2 are capable of drastically altering the intracellular redox milieu and of setting a cascade of events that may result in cellular dysfunction or death (15). A number of studies substantiate the development of redox changes in MS lesions, involving nitrosative and oxidative stress. It is particularly interesting that the production of reactive oxygen and nitrogen species (ROS and RNS) in active MS lesions is generally increased only in specific cells, namely astrocytes, macrophages, and endothelial cells, all of which have a direct contact with perivascular space (275, 347, 444), as well as in ODC (181). NO may provoke the inhibition of mitochondrial electron transport chain (ETC) complexes I, III, and IV, leading to increased •O2 − production (333). Mitochondria represent the main site of production of •O2 − and H2O2. The production takes place even under physiological conditions, being drastically increased if ETC is obstructed (333). There are no data on the activity of complexes I and III in early MS lesions, but Mahad et al. observed the inhibition of complex IV in astrocytes and ODC in pattern III lesions, which could be a result of the supraphysiological NO level. It is important to note that inhibition was not present in pattern II lesions (288). The increased production of •O2 − and its derivative H2O2 in astrocytic mitochondria is implicated by the increased levels of mitochondrial antioxidative enzymes—manganese superoxide dismutase (MnSOD) and peroxiredoxin V—in active lesion astrocytes and NAWM (203, 444). Finally, it would have been expected that the inhibition of mitochondrial respiration leads to decreased O2 utilization, and exactly this was reported to develop in the white matter of MS patients (157).
In contrast to •O2 −, which is charged, incapable of crossing mitochondrial membranes, and barely capable of crossing cellular membranes (via Cl− channels), H2O2 can easily enter the cytosol from mitochondria and perivascular space, where it can activate nuclear factor-kappa B (NF-κB) (Fig. 6) (15). Pertinent to MS, Bonetti et al. observed the activation of NF-κB in astrocytes at the edges of active lesions (52). The importance of NF-κB activation in astrocytes is strongly substantiated by the study performed on the cuprizone model of demyelination (371). The authors reported that the depletion of IκB kinase (which is essential for NF-κB activation) in astrocytes, but not in ODC, was sufficient to protect the mice from myelin loss. NF-κB activates the expression of a number of genes, two of which encode redox active enzymes—iNOS and cyclooxygenase 2 (COX2, which has •O2 − as a by-product) (15). The increased level of astrocytic iNOS has been recognized as an important pathogenic feature of MS (318). Under normal settings, the level of active iNOS within the CNS is next to zero. The activation of its expression leads to a drastic increase in NO generation, resulting in millimolar NO concentrations. It is important to note that NO reacts very fast with •O2 − to produce peroxynitrite (ONOO−), which can exert direct negative effects or can be decomposed to dangerous hydroxyl (•OH) and nitrogen dioxide radicals (•NO2) (15). ONOO− and •NO2 may react with Tyr residues in proteins to produce nitrotyrosine. Pertinent to this, Oleszak et al. observed increased levels of iNOS and nitrotyrosine in astrocytes and macrophages in acute monophasic lesions, while this was not the case in chronic lesions (347). Similar results were acquired in other studies, reporting that astrocytes and endothelial cells are positive for nitrotyrosine in active lesions (101, 275). The authors have proposed that astrocyte-derived NO could be important for orchestrating further processes in MS. An abundant expression of iNOS in macrophages early in the course of the disease and before demyelization was also reported (49). The importance of iNOS expression in MS development is further substantiated by the fact that the injection of arginine [which acts both as an iNOS substrate and an up-regulator of iNOS mRNA (263)] into the rat brain provokes demyelination (248). The expression of iNOS in astrocytes most likely represents a key step in MS initiation, leading to further NO-provoked inhibition of ETC and the formation of a self-promoting redox loop (Fig. 6).
E. The decrease of NADH/NAD+ ratio
Probably the most important pathophysiological consequences of the increased NO level are indirectly provoked by ONOO−. Peroxynitrite targets DNA and causes single-strand breakage, which leads to poly (ADP-ribose) polymerase (PARP) activation (15). Veto et al. observed strong PARP immunoreactivity in astrocytes and ODC in pattern III lesions in patients with MS duration from 6 to 60 days. PARP activity was at its highest in very early lesions (453). It has been concluded that PARP activation plays a critical role in MS pathogenesis (136, 453). The importance of the NO/ONOO−/PARP pathway is implicated by the inverse correlation between MS activity and CSF and sera levels of uric acid (123), which represents an ONOO− and •OH scavenger (410). Although the application of inosine (uric acid precursor) did not show significant beneficial effects in an MS trial (170), PARP inhibition has been recently praised as a promising approach in MS treatment (79, 136), primarily because it prevents ODC death (453). However, precaution should be taken in balancing between positive and negative effects of PARP activation in MS. A complete inhibition of PARP function could result in an inability of cells to repair DNA and may even promote MS. For example, PARP-1−/− EAE mice show an earlier onset and develop a more severe EAE when compared with wild-type animals (396).
While the activation of PARP in ODC in MS lesions represents an echo of DNA fragmentation and cellular death, in astrocytes that do not undergo such changes the activation of PARP may represent an important step in MS pathogenesis. PARP catalyzes the cleavage of NAD+, which leads to a decrease in the NAD+ level and an increase in the NADH/NAD+ ratio (15). A high NADH/NAD+ ratio is known to act as the strongest stimulus for •O2 − production in mitochondria (on ETC complex I) (333). The increased production of •O2 − in complex I results in further H2O2 leakage from mitochondria, thus closing another redox loop (Fig. 6). The development of interdependent redox loops in the course of MS pathogenesis is implicated by the fact that nanomolar affinity PARP inhibitor PJ-34 (which delays EAE onset) suppresses iNOS expression (394).
F. Astrocytes and macrophages: centers of oxidative/nitrosative stress in early lesions
In a recent commentary, Smith has proposed that astrocytes are responsible for oxidative/nitrosative damage in ODC and neurons in MS (406). The development of pathophysiological processes that involve positive feedback loops in astrocytes and the activation of macrophages result in these cells becoming the main source of ROS and RNS in early/pattern III lesions. In line with this, astrocytes and macrophages exhibit the activation of the antioxidative system (AOS) and increased levels of the products of oxidation and nitrosylation (275, 347, 444). Van Horssen and co-authors observed that in active lesions only astrocytes and perivascular or myelin-laden macrophages show increased levels of CuZnSOD, MnSOD, catalase, NAD(P)H:quinine oxidoreductase 1, as well as nitrotyrosine and 4-hydroxy-2-nonenal (HNE; a product of lipid peroxidation) (444). It is important to note that the increased level of MnSOD was observed only in astrocytic mitochondria (444). This implies that ETC is obstructed only in astrocytes, while macrophages generate ROS in the intracellular compartment and on the membrane (see section III.A). The expression of many AOS enzymes is regulated by nuclear factor-E2-related factor (Nrf2)-antioxidant response element (ARE) pathway, the activation of which in MS is specifically located in astrocytes (443). Newcombe et al. reported the presence of increased levels of HNE in astrocytes and malondialdehyde (MDA; another product of lipid peroxidation) in astrocytes and myelin-laden macrophages in early lesions (341). The importance of oxidative stress in MS initiation is strongly implicated by the results showing that the level of oxidation markers is more than 8-fold higher in early lesion-resembling pattern III lesions than in pattern II lesions (181). One small study has observed increased levels of lipid peroxidation markers in the CSF of probable MS patients (337). This clearly implies that oxidative changes in lipids take place early in MS development (i.e., before MS can be diagnosed for certain). It is rather intriguing that the only lipoprotein particles present in human CSF—high-density lipoprotein (HDL)-like particles—are specifically secreted by astrocytes and macrophages/microglia (138). It is plausible that oxidized HDL-like particles (oxHDL) could be released from astrocytes and macrophages under prooxidative settings in initial lesions. The particles may integrate into membranes of other cells in order to activate a lipid peroxidation-chain reaction (15), thus disseminating oxidative damage. It has been reported that oxHDL exerts toxic effects on cultured CNS cells and promotes inflammation (138); so, these structures may be involved in the mechanisms of initial lesion formation as well as in disease progression. It is worth mentioning that HDL comprises antioxidative enzymes, the activity of which is decreased by excessive oxidation (138).
The involvement of macrophages/microglia in the development of the initial lesion is strongly implicated by the available data. These cells could promote prooxidative conditions in initial lesions after they recognize cell injury, but the activation may take place even before that. Namely, NO released by endothelial cells and astrocytes appears to act as an independent signal for macrophage/microglia activation (92). Exogenous NO (not produced in macrophages) has been reported to: orchestrate macrophage/microglia migration and accumulation, acting as a chemoattractant (84, 115, 415), induce microglia recruitment to the lesion (115, 122), provoke morphological changes of microglia into ameboid-like cells (115), and provoke hormesis—the exposure to low level NO increases the resistance of macrophages to pro-apoptotic NO overload (481). Activated macrophages/microglia are known to produce and release NO, H2O2, and •O2 − (see section III.A.).
G. Oligodendrocytes death
It has been postulated that the dysfunction of astrocytes precedes ODC degeneration in early lesions (400). NO, H2O2, and HNE may leak from astrocytes and activated macrophages in order to affect other CNS cells, ODC in the first place. For example, it has been shown that astrocytes expressing syncytin (human endogenous retrovirus glycoprotein) provoke the death of ODC by producing excessive amounts of NO on iNOS (19). One may ask since astrocytes represent the source of nitrosative and oxidative stress, why they are not dying while ODC do. In a comparative study, Garthwaite et al. found that the rank order of susceptibility of CNS cell to NO is neurons>ODC>astrocytes (152). The experimental design did not allow work on myelinated neurons, which are known to be drastically less susceptible to NO compared with demyelinated cells (378). This implies not only that the myelin sheet provides axons with good protection from NO excess, but also that the susceptibility under in vivo conditions is, in fact, ODC>(myelinated) neurons>astrocytes. The analysis of biopsy samples obtained from a young patient with early MS revealed the accumulation of nitrotyrosine in ODC, pinpointing it to ONOO−-mediated damage (488). It has been documented that ONOO− does not provoke apoptosis in ODC, and necrosis was proposed to be the main route of cell death (394). On the other hand, astrocytes appear to be relatively resistant to ONOO− (220).
Although ODC are obliged to generate large amounts of ATP in order to maintain high metabolic overturn (424), their sensitivity to NO cannot be explained by NO-provoked ETC inhibition (220). Mature, myelinating ODC successfully survive mitochondrial dysfunction by switching to glycolysis (146). Therefore, NO may act indirectly, by promoting the formation of ONOO−, which is known to provoke DNA damage. In addition, ODC are sensitive to oxidative stress, because they have low antioxidant levels, a high content of iron (redox active metal), and exposed extensive plasma membrane elaborations (33). ODC precursors appear to be even more susceptible to oxidative stress and glutathione (GSH) depletion compared with mature cells (26, 144). They show thrice lower GSH concentrations and 20 times higher iron level than astrocytes (423). Neurons are also known for their weak AOS, but their axons acquire protection from myelin sheets. For example, Peterson et al. found that the number of apoptotic neurons per mm2 was substantially lower in myelinated than in non-myelinated cortex areas in the MS brain (363). This may explain why an oxidative/nitrosative burst unleashed by the astrocytes leads to ODC death, but leaves neurons generally uninjured. Myelin takes the “blow” for the axons because of the spatial arrangement, and because it is rich in iron and lipids (159), which makes it a perfect buffer for reactive species coming from the surroundings. Iron may bind NO in order to form dinytrosil-iron complexes (DNIC) (442), or it may react with H2O2 to produce a highly reactive •OH radical, which is buffered by myelin lipids. It has been shown that ODC in co-culture protect neurons from NO-mediated damage and that this protection is dependent on the function of iron-releasing enzyme heme oxygenase-1 (HO-1) (47). This implies a possible protective role of DNIC formation in CNS against NO overload. Neurons may be affected by NO only at myelin-unprotected points (nodes of Ranvier). Pertinent to this, NO is known to block the conduction in axons (378), which in some cases may lead to the (first) presentation of MS symptoms.
ODC are particularly susceptible to HNE, a major bioactive lipid peroxidation product, which is diffusible and highly toxic (444). HNE level is increased 4-fold in MS active lesions compared with control tissue (463). The reported HNE concentration (∼4 μM) appears to be more than sufficient to kill ODC according to the findings of an in vitro study which showed that HNE provokes ODC death at concentrations as low as 2 μM (151). On the other hand, at this concentration, generally, HNE does not exert significant negative effects on other cells. For example, the first toxic effects on endothelial cells have been observed at 25 μM (435). In addition to ODC death, the accumulation of HNE may obstruct the remyelination by inhibiting the proliferation and differentiation of ODC precursors (151).
Unfortunately for the patients, ODC in MS do not undergo apoptosis, which is the safest way to die if we consider the safety of surrounding tissue. Although nuclear condensation and fragmentation in ODC have been observed in acute MS lesions, caspase-3, which is the main effector of classic apoptosis, was not found to be active (1, 32). This can be attributed to the fact that NO acts as a strong inhibitor of caspase-3 (239). In addition, it has been suggested that HNE, NO, or ONOO− do not provoke ODC apoptosis but most likely necrosis or apoptosis-like programmed cell death (301, 322, 394). H2O2 may also contribute to non-apoptotic death by provoking DNA fragmentation (330).
H. Demyelination
Myelin membranes exceed 100-fold the weight of ODC (424). After ODC death, this huge amount of membrane becomes metabolically unsupported. It is interesting that in initial lesions, the first myelin abnormalities develop at the inner, most distant (compared with ODC body) part of the myelin sheath (381), which is the first to suffer the lost of metabolic support. In addition, S-nitrosylation of myelin proteins (in particular, PLP) may result in myelin decompaction (50). Consequently, the externalization of phosphatidylserine develops, making this dominant mammalian scavenger signal available to macrophage receptors (33). A number of studies documented the presence of myelin-laden macrophages in early/pattern III lesions, most likely originating from perivascular space or microglia (32, 447). The density of perivascular macrophages gradually increases along the vascular tree, from capillaries to postcapillary venules, which represent the most probable site of formation of initial lesions (40). It is worth mentioning that the loss of metabolic support may lead to myelin fragmentation even in the absence of macrophages (32).
Myelin-laden macrophages principally represent an anti-inflammatory acting component of the immune system. However, when exposed to specific stimuli, such as pronounced oxidation, their myelin-removing function may become hampered, while the activity may switch from anti- to pro-inflammatory mode (35). It appears that in MS lesions, macrophages get exposed to excessive oxidation, according to the increased levels of catalase, CuZnSOD, and HNE (444). In addition, it seems plausible that macrophages may be paying the price of their own function. Since myelin sheets contain high levels of iron, myelin-laden macrophages in MS lesions accumulate this transition metal (267), which may promote Fenton reaction-mediated oxidation in the intracellular compartment (Fig. 6). The oxidation-provoked shift in macrophage function may have the incomplete removal of myelin debris as a consequence, which could promote the development of autoimmunity.
I. BBB breakdown
The outlined set of events may exert a tremendous impact on BBB, provoking its dysfunction and breakage via several different mechanisms. First of all, astrocyte foot processes are placed around the blood vessels, contributing to the formation and function of BBB. Astrocytes induce and regulate TJ, balanced redox milieu, high mitochondrial content, and other intrinsic properties of neural endothelial cells that constitute BBB (318). The activation of redox processes resulting in ETC inhibition (and consequent ATP shortage) and pronounced oxidation/nitrosylation may lead to the dysfunction of astrocytes. Such astrocytes may not be capable of properly supporting the integrity of BBB at the site of lesion formation. In addition, altered redox settings may lead to a release of a number of TJ/BBB-affecting agents into perivascular space (242), such as matrix metalloproteases (MMP), HNE, H2O2, NO, and •O2 − (Fig. 7). The stability of endothelium–astrocyte configuration requires matrix adhesion, which may be altered by MMP (113). The expression of MMP-2 and −9 has been detected in astrocytes in MS lesions (102), and it is most likely related to COX2 activity (74). In addition, exposing the endothelial cells and smooth muscles to H2O2 or •O2 − is known to promote MMP-2 activity (43, 437), whereas HNE is known to provoke increased MMP-9 release from macrophages (264). Shear stress is known to induce an increase in the level of MMP-2 and −9 in venous tissue (359). Finally, the exposure of endothelial cells to MBP may affect BBB, and it may promote the production of MMP-2 (105).
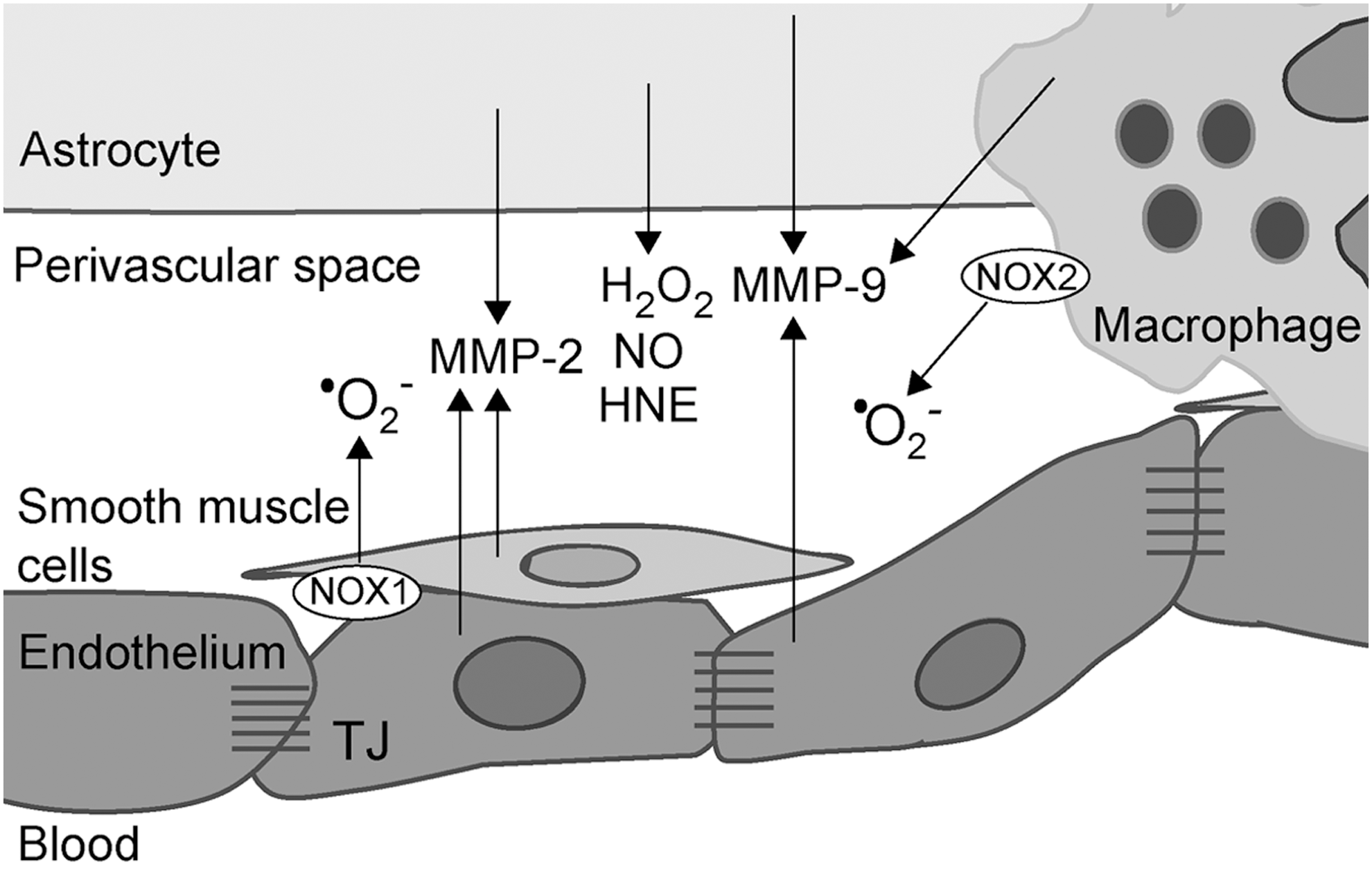
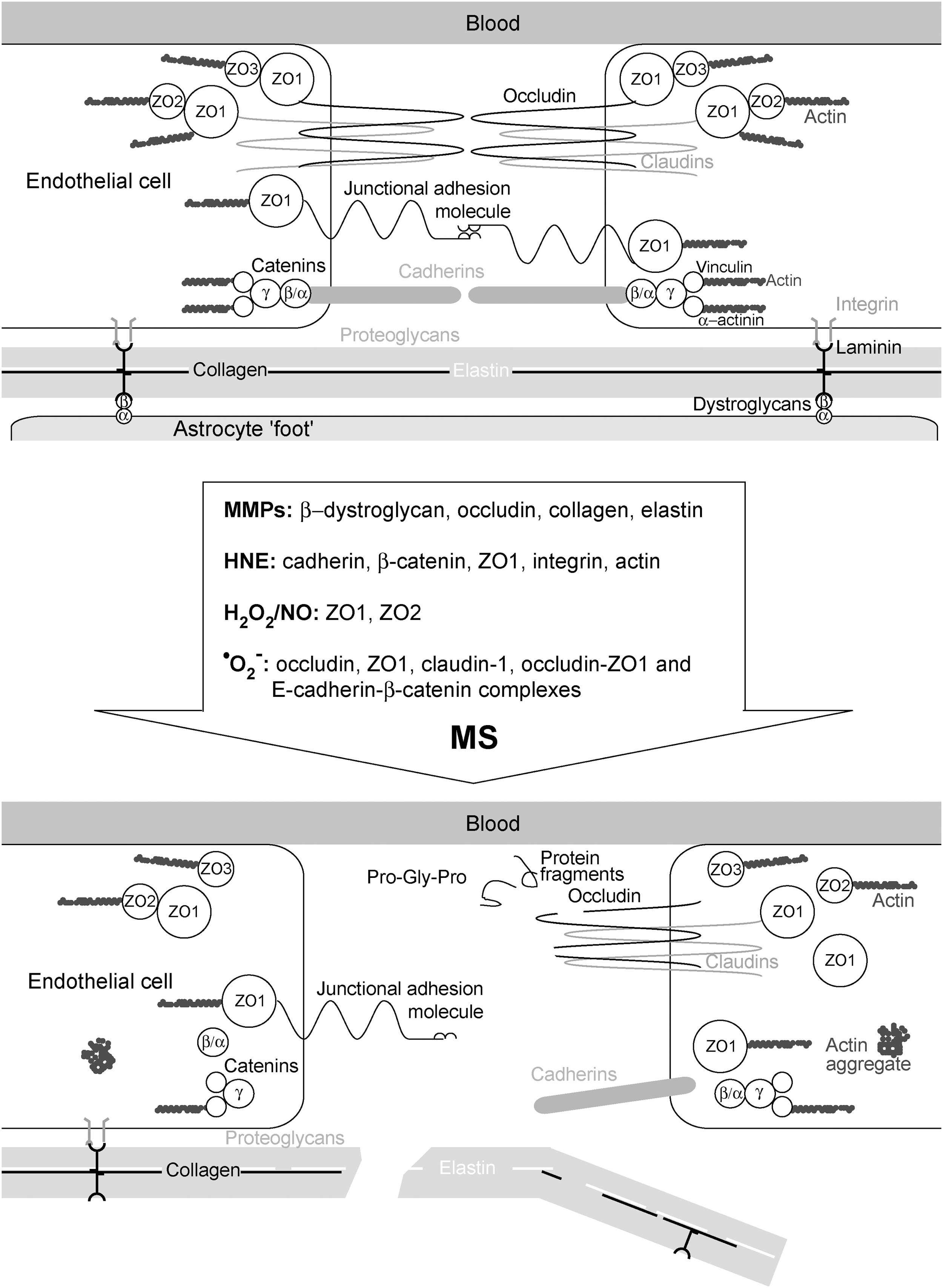
Figure 8 shows TJ before and after the attack of a set of agents that are involved in MS pathogenesis. MMP-2 and MMP-9 cleave occludin and dystroglycan receptors, which leads to the destabilization of TJ and astrocyte end-feet anchorage to parenchymal membrane (4, 163). In addition, MMP participate in the degradation of collagens and elastin (290). This results in the formation of different protein fragments, some of which may act as chemoattractants or immunomodulators, for example, the cleavage of collagen may produce bioactive tripeptide Pro-Gly-Pro, which recruits neutrophils (457). HNE affects the permeability of endothelium by changing the distribution of cadherin, β-catenin, and zonula occludens 1 (ZO1), and by causing a decrease in the surface integrin. In addition, HNE provokes dissolution of peripheral actin bands and the formation of actin aggregates in the cytosol. These and some other changes result in disturbed cell–cell adhesion contacts, intercellular gap formation, and transendothelial cell migration (435). At least some of these effects may be attributed to HNE-provoked ROS production by the mitochondria in endothelial cells and by NOX2 in macrophages (435, 483). It has been shown that H2O2 affects BBB by changing the localization of ZO1 and ZO2 (262). Consistent with this, H2O2-removing enzymes—catalase and GSH peroxidase—are known to protect BBB from disruption (178). H2O2 has been shown to increase solute permeability of endothelium, the effects being pronounced by the co-supplementation of a NO donor, and completely suppressed by NOS inhibition (346). This implies that the mechanisms by which H2O2 affects endothelial permeability overlap or depend on NO-provoked changes. Peroxynitrite may also be involved, as the application of ONOO− scavenger prevented BBB permeability changes in an EAE model (206). Finally, •O2 − may also have its place in BBB disruption. Huppert and colleagues postulated that IL-17-induced BBB disruption involves NOX- and xanthine oxidase (XO)-mediated •O2 − production and consequent down-regulation of occludin (213). The intracellular •O2 − production in endothelial cells appears to suppress the expression of junction adhesion molecules, ZO1, claudin-1, and occludin (476). Pertinent to this, TJ in the brain of MS patients shows a lower level of expression of junction adhesion molecule A (353). Rao et al. showed that extracellular production of •O2 − induces tyrosine phosphorylation of occludin-ZO1 and E-cadherin-β-catenin complexes, resulting in their redistribution, dissociation (376), and, finally, in TJ disruption (Fig. 8). It should be noted that TJ of postcapillary brain venules are less complex than those of brain capillaries, which is probably one more attribute that helps the development of lesions around venules.
The process of MS initiation might stop right here. No autoimmune response has to develop. A specific part of CNS suffered demyelination, which can be reversible, and the patient may go on with his/her life for years, without experiencing further disabilities. This benign MS appears to develop in approximately 30% of MS patients (365). However, in many patients, the process transcends to the next phase, which involves antigen formation and the development of autoimmunity. Pertinent to this, it has recently been proposed that MS pathogenesis involves primary degenerative changes that may (or may not) result in a host's aberrant immune response (a so-called inside-out model of MS) (417). A gate opens to the damaged tissue emitting specific signals and showing epitopes that the immune system is not familiar with, inviting the invasion of immune cells and ending up in MS taking its inflammatory/autoimmune course, as we will discuss in the following section. It should be emphasized that MS may take a benign course even after the development of the autoimmune component (94, 284).
J. The development of autoimmunity
1. Antigens
A combination of ODC's non-apoptotic death and macrophage dysfunction may have a crucial influence on the further steps in MS initiation, that is, induction of autoimmunity. Non-apoptotic and hence not fully controlled death of ODC may go hand in hand with macrophages and microglia's decreased capability of removing myelin, resulting in the accumulation of myelin fragments and vesicles at the site of an early lesion (153, 191). It has recently been reported that in a homeostatic process, microglial cells constantly take up exosomes from ODC, which are unable to recycle membrane components from myelin sheaths that are distant from their somas (141). As a consequence of inflammation, microglial assistance to the recycling is reduced, and myelin-filled exosomes in interstitial fluid might reach CSF (375). Pertinent to this, recognizable myelin fragments have been frequently detected in CSF sediments taken at the time of acute MS attacks (196). It is noteworthy that the phagocytosis of necrotic cells promotes the maturation of DC (389).
The presence of not one but a number of different products of myelin that become antigens in MS is substantiated by the work of Zhang et al. (491) and Bahbouhi et al. (28). They showed that there is a population of T cells memorizing a wide-spectrum of myelin-related antigens in the blood of MS patients. This is in line with the recent assertion that, instead of the classic “single-epitope-targeting” approach, a “multi-epitope-targeting” strategy is required for effective immune-specific therapy for MS (230). A multi-epitope setup may explain why several decades of effort failed at identifying one (or few) specific antigens that are responsible for the autoimmune component of MS pathophysiology. It is interesting that a number of reactive species and products of oxidation in MS lesions may participate in the formation of epitopes (Fig. 9). MDA may form an adduct with MOG, which is taken up more effectively by APC than MOG. In addition, the exposure of mice to MDA-MOG led to increased expression of IL-23, IL-12, and IL-12R in APC, resulting in differentiation of Th17 and Th1 cells (454). Pertinent to this, CSF of MS patients shows increased presence of MDA and MDA adducts on lysine residues in proteins and increased levels of antibodies for MDA-low-density lipoproteins (LDL) compared with controls (161, 172). HNE forms stabile adducts with lysine, histidine, or cysteine (Cys) residues in proteins, which are accumulated in MS lesions (463), and may elicit a strong immune response (7). Some other epitopes, derived from lipid peroxidation, may also act as pathogenic factors in MS. In a recent study, Gonzalo and colleagues found increased levels of carboxymethylated and neuroketal-modified proteins in the CSF of MS patients. The level of neuroketal-modified proteins showed a positive correlation with the amount of antibodies against lipid peroxidation-modified proteins in CSF, which implies that this adduct may have a role of antigen in MS (172). The presence of anti-SNO-Cys antibodies that target S-nitrosylated proteins was documented in the sera of MS patients and in an EAE model (54). Duleu et al. documented an increased presence of circulating antibodies directed against a variety of NO- and ONOO−-modified (NO2-tyrosine) self-antigens in MS patients (124). Nitrotyrosine residues showed the ability to evade central tolerance and to elicit a robust T-cell immune response (46). Discerning whether, when, and which myelin products are to be recognized as antigens, in combination with the characteristics of T-cell populations that target myelin products, may represent the key for development of different MS subtypes. It is important to note that many neurodegenerative diseases are associated with the release of nervous tissue-based antigenic structures, yet the patients do not show signs of MS. It has recently been shown that ODC death (induced by diphtheria toxin in diphtheria toxin receptor-expressing ODC) does not elicit anti-CNS immunity even though the fragments of myelin have been detected in CNS-draining lymph nodes, and not even when the immune system is strongly prestimulated (279). This is in line with the previous assertion that ODC death does not provoke an immune response in multiple system atrophy (461). These findings strongly imply that the fragments of nervous tissue or myelin are not sufficient to provoke an autoimmune response. It appears that some MS-specific myelin-based highly-antigenic molecules, such as MDA-MOG, HNE- and neuroketal-adducts, nitrosylated-peptides, or citrullinated-MBP (citrulline being the product of iNOS activity) (172, 417), are necessary in order to push MS over the edge of autoimmunity.
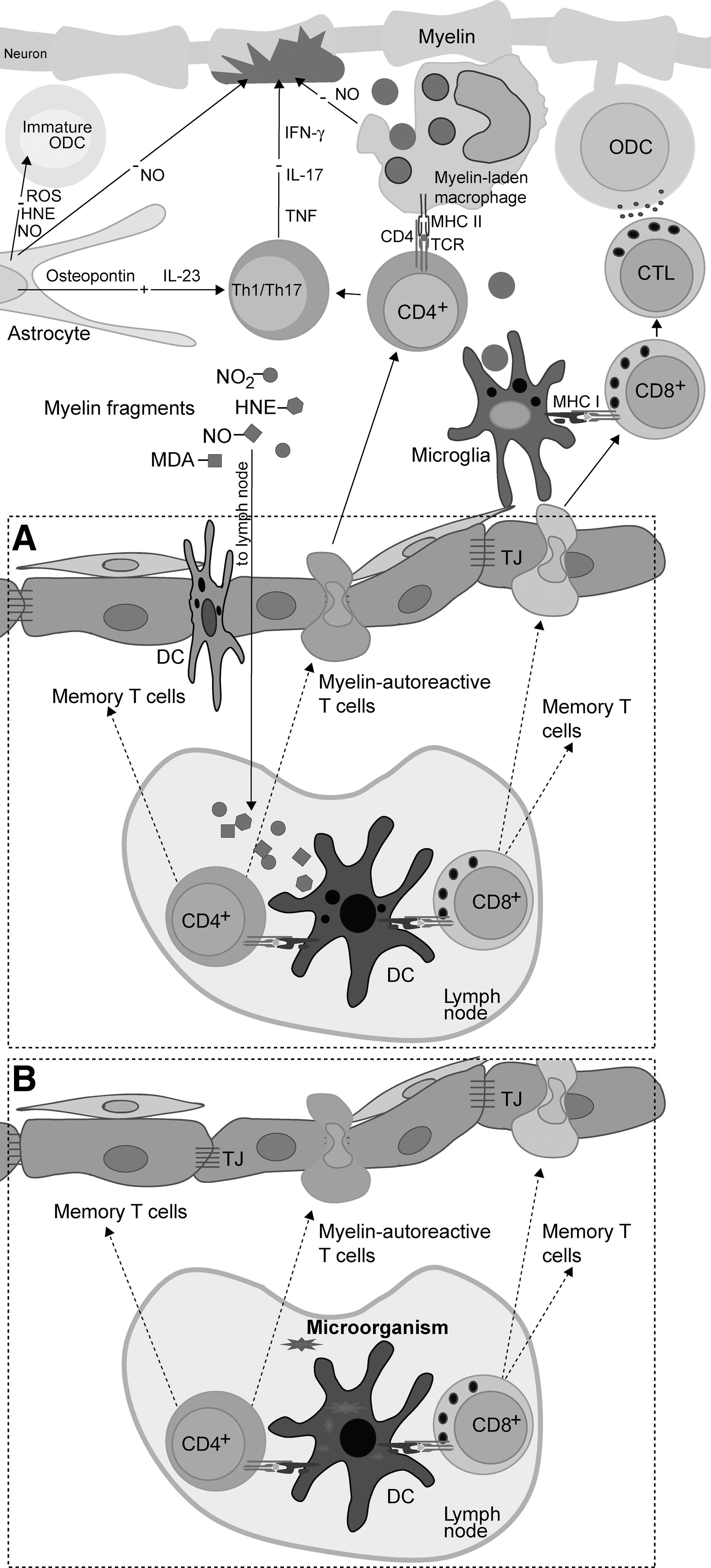
2. The activation/reactivation of T cells
The initiation of autoimmunity is a consequence of an efficient presentation of CNS antigens to T cells by DC in lymphoid organs (Fig. 9A). DC may collect the antigen within the CNS and then go to the lymph node, or they may reside in the node and trap the antigens in the lymph draining from the CNS. It seems plausible that both, the migration of DC and the leakage of soluble antigens from the CNS, could be facilitated in the case of BBB or BCSFB breakage. DC present antigens in the form of peptides in the context of MHC class II molecules to naïve CD4+ T cells. The presentation of antigens by DC to naïve autoreactive CD8+ T cells and their reactivation in CNS by microglia depends on the process of antigen cross-presentation. Generally, molecules of MHC class I present intracellular antigens, while molecules of MHC class II present ingested antigens. In order for APC to present an ingested antigen to CD8+ T cells, it has to engage MHC class I molecules. Antigen processing and presentation by MHC class I and MHC class II molecules are spatially distinct in cells. However, leakage of antigens processed by MHC class II machinery into MHC class I pathway occurs in the process of cross-presentation (310). Activated T cells will differentiate into effector and memory T cells.
After initial activation, T cells migrate into the CNS, where they are reactivated. As a consequence of redox-imposed damage to nervous tissue, CNS-resident phagocytes are activated in order to present antigens on MHC class II molecules. Autoreactive CD4+ T cells are reactivated after interacting with these cells (34, 231). After that, they differentiate into effector interferon (IFN)-γ-producing Th1 cells and/or IL-17-producing Th17 cells (129). The stimulated effector Th cells locally release proinflammatory cytokines, thus initiating an inflammatory reaction, which may result in demyelination (38, 469). It is important to note that NO, which is excessively generated by iNOS in the initial lesion, may have anti-inflammatory effects at this point. NO is known to suppress the expression of MHC II in macrophages and endothelial cells (188), thus hampering the reactivation of T cells. Primary neuronal and/or oligodendroglial damage activates microglia, which, in turn, up-regulates MHC class I expression and releases proinflammatory mediators. Activated microglia is capable of cross-presenting antigens and of efficiently reactivating CD8+ T cells within the CNS (37, 310). Moreover, proinflammatory mediators released by microglia compromise immunosuppressive activity of the CNS resident cells and stimulate ODC and neurons to increase the expression of MHC I (310). Consequently, activated CTL that are specific for CNS antigens both directly and indirectly target myelin, ODC, and neurons via lytic mechanisms (Fig. 9A). It should be noted that activated CD8+ T cells also produce various cytokines and other mediators which sustain neuroinflammation (310, 390).
An additional/alternative pathway of autoimmunity initiation, which also involves redox misbalance within the CNS, is plausible. We have elaborated that redox instability leads to ODC and myelin damage and BBB dysfunction. In addition, we have discussed the possibility of activation of CNS-reactive T cells by molecular mimicry or bystander activation, that is, by microorganisms. A combination of these two spatially distant processes might significantly increase the possibility for the initiation of autoimmunity within the CNS. In short, T cells are activated in an immune response to a microorganism. As a consequence of redox-induced changes in the CNS, their passage through BBB is facilitated, as well as their encounter with myelin and other CNS antigens. As a consequence of redox-induced damage of myelin and/or ODC, microglia and macrophages are stimulated and ready to reactivate T cells. Reactivated T cells perform effector functions that establish inflammation and perpetuate damage in the CNS (Fig. 9B).
In some cases, such as in aggressive Marburg's type of acute MS, the inflammation develops very fast (49). On the other hand, Lucchinetti et al. showed that, within an average of 27 days of potential MS symptom onset, at which point the biopsy samples were taken, around 50% of examined patients showed signs of cortex inflammation and/or demyelination, involving the activity of CD8+ T cells and myelin-laden macrophages. Fortunately, even the development of the autoimmune component of MS is not irreversible. At the mean follow-up of 3.5 years after the initial symptoms, around 20% of patients at first showing the signs of cortex demyelination/inflammation were diagnosed for CIS and did not develop MS (284). Correale et al. observed the presence of newly formed inflammatory lesions in benign MS cases at 10+years' follow-up (94). This indicates that the inflammatory component may be persistent but non-aggressive in benign MS. The probability and the tempo by which the autoimmunity develops in MS are most likely related to genetic predisposition of the immune system to react with the released antigens (417).
K. Redox regulation of T-cell migration and infiltration
In order to enter the brain, T cells have to migrate, recognize the site of entrance, and infiltrate. It has been shown that activated T cells are first captured to the vessel and that they then crawl on to the vascular endothelia. T-cell-endothelium interactions are mediated by adhesion molecules, chemokines, and their receptors (131). It appears that the key integrin receptor-integrin interaction in MS is established between VCAM-1 and VLA-4. Monoclonal antibodies against VLA-4 stop the crawling and transmigration of myelin-reactive CD4+ T cells into the CNS parenchyma of model animals and restrict BBB transmigration of both non-effector and effector memory CD8+ T cells acquired from the CSF of MS patients (34, 215). The transmigration of T cells across BBB also involves their interaction with pericytes, which is established through VCAM-1/VLA-4 pair (451). The importance of VCAM-1/VLA-4 interaction in MS pathogenesis is clearly delineated by the efficacy of anti-VLA-4 monoclonal antibody (natalizumab) in the treatment of MS patients (214). It has been recently demonstrated that this antibody also down-regulates the production of soluble VCAM-1 in endothelial cells (319). The expression of VCAM-1 in endothelial cells is regulated by NF-κB and promoted by oxidative stress (294). It should be emphasized that effector T cells transmigrate more readily across BBB than non-effector T cells, and that endothelium promotes the selective recruitment of effector T cells (215). Endothelial and brain cells may release soluble signals (chemokines) to “invite” T cells. After the initial engagement of chemokine receptors in T cells by chemokines, inside-out signaling pathways induce integrin activation in these cells (240), and, therefore, efficient interaction of integrins and their receptors is allowed. Chemokines may act either by creating a soluble chemotactic gradient or by being associated with extracellular matrix components or they may bind to cell-surface proteoglycans of endothelial and other cells (204). The most important chemokines in MS appear to be CCL2 (receptor—CCR2), CCL3, CCL4, CCL5 (share the common receptor—CCR5) (434), CCL20 (receptor—CCR6) (60), CXCL10 (receptor—CXCR3) (408), and CXCL12 (receptors—CXCR4 and CXCR7) (13) (Fig. 10A). All these chemokines are released from activated astrocytes (13, 55, 304, 434). Regulation of their expression involves NF-κB (55, 237, 304, 462, 492). This implies that in the initial lesion, prooxidative processes and up-regulated NF-κB expression in astrocytes and other cells may promote the infiltration of T cells (Fig. 10). Pertinent to this, the release of CXCL12 and CCL20 from astrocytes in MS lesions has been documented (13). Th17 cells show the most robust chemotaxis toward CCL20 (60). However, non-effector myelin-reactive CD4+ T cells (activated in the lymph node; see Fig. 9), which may be present in CSF or blood, also express CCR6 and respond to CCL20 (332, 475). This may be crucial for the inflammation of the initial lesion, which further results in the development of a fully active autoimmune system. CCL3 is known to promote the chemotaxis and infiltration of activated CD8+ T cells into CNS (420, 430). It has been reported that CCR5 is involved in the modulation of CD8+ T-cell activity, but that it is not essential for the entry into the CNS (166, 167). The affected expression of CCR5 in humans does not prevent MS development, although it appears to be related to a milder course of disease, while the severity of EAE is not reduced in CCR5-knockout animals (434). CXCR3 receptor is present in both CD4+ and CD8+ T cells. The presence of this receptor in T cells is higher in MS patients than in healthy subjects, and shows a significant decrease during remission in MS patients (436). CCL2 appears to promote cerebral infiltration of both CD4+ and CD8+ T cells (77). A very recent study showed that the exposure of endothelial cells to MBP enables them to produce CCL2 (105), which may be related to their role as semi-professional APC. The severity of EAE is reduced in CCR2-knockout animals (434). In addition, it has been shown that MBP induces CXCL12 expression in astrocytes but not in endothelial cells (72). Altogether, this implies a very direct involvement of myelin fragments in the promotion of inflammation in (initial) lesions.
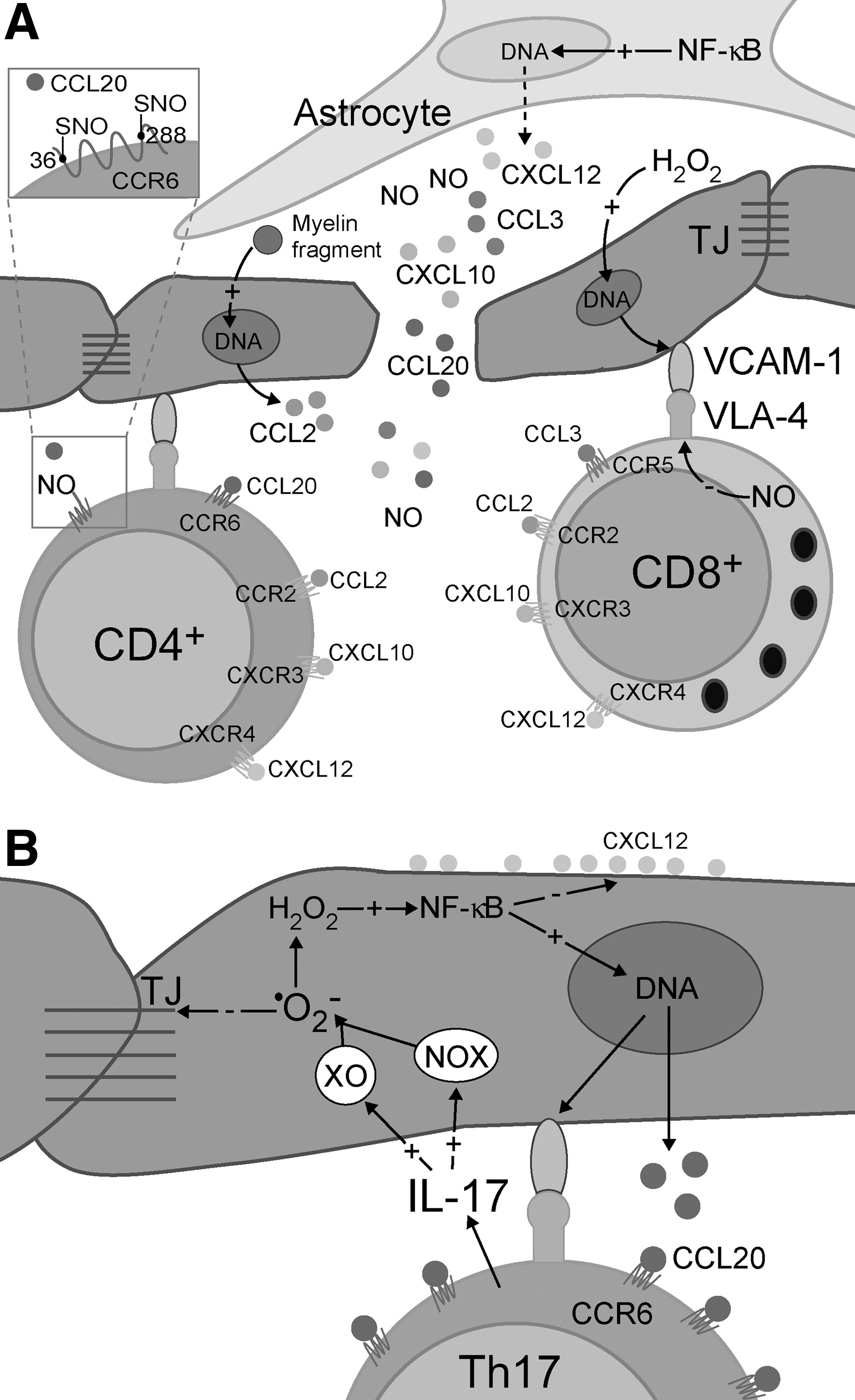
It has been reported that NO inhibits transendothelial migration of T cells (414, 474). This could be at least partially related to NO-provoked down-regulation of β1-integrin (VLA-4 subunit) expression in T cells (418). However, one additional mechanism is possible. It has been shown that some chemokine receptors include extracellular Cys residues with free thiol groups (e.g., Cys36 and Cys288 in CCR6), which are important for ligand binding. The mutation of these residues in CCR6 drastically suppresses chemotaxis of T cells (6). It has been proposed that thiol groups in chemokine receptors may be important for fine tuning the signaling properties of the receptors (374). They can be altered by S-nitrosylation or oxidation, the first process being probably more important for decreasing the infiltration (Fig. 10A), due to a larger diffusion radius of NO in comparison to H2O2. However, this remains to be further elucidated.
The infiltration can be drastically promoted when the Th17 phenotype is reached, as CCR6 is highly expressed by Th17 cells (475). In addition, Th17 cells also secrete the “prooxidative” cytokine IL-17 (213). IL-17 promotes •O2 − and consequent H2O2 production in endothelial cells, thus activating canonical NF-κB signaling pathway (471). This stimulates the release of CCL20 and further infiltration of Th17 cells (60) (Fig. 10B). Even more, canonical NF-κB signaling inhibits basal CXCL12 expression in endothelial cells (286). Under physiological conditions, CXCL12 expression is localized on the surface of endothelial cells facing CNS parenchyma. This specific spatial presentation appears to have a role in retaining T cells, which entered the perivascular space, from advancing toward the brain tissue (326). The decreased expression or redistribution of CXCL12 in endothelial cells results in a more severe EAE (299, 317). It is also known that CXCL12 shifts Th1 cells toward Treg cells (309), and that it induces apoptosis in activated T cells (298). Importantly, we have discovered that NO inhibits CXCL12 expression in astrocytes and endothelial cells (unpublished observation). The ability of Th17 cells to activate IL-17-provoked BBB disruption (213), to self-invite themselves by inducing CCL20 production in endothelial cells, or even to produce CCL20 by themselves (Fig. 10B) (60, 475) may be particularly important for the development of new inflammatory lesions in the previously unaffected brain regions during the course of MS. Furthermore, it has been shown in the EAE model, that as the disease progresses, Th17 infiltration becomes CCR6 independent (377), which may be related to a more general and severe loss of BBB integrity. It should be noted that epithelial cells of choroid plexus constitutively express CCL20, which may be one of the causes for the presence of CCR6+ and CD4+ cells in the CSF of patients early in MS (377). Hence, IL-17-promoted CCL20 release may be particularly important for cortical infiltration (see the next section). Altogether, these facts may explain the notoriety of Th17 cells in MS pathology. It is worth mentioning that a subpopulation of effector memory CD8+CD161+ T cells which release IL-17 and express CCR6 receptors has recently been detected in the peripheral blood of MS patients (17).
L. The outside-in initiation of MS
Currently, there is a heavy debate within the MS research community regarding the chain of events that initiate clinically determined MS. Generally speaking, there are those who favor the “inside-out” hypothesis, that is, damage to the brain comes first followed by an autoimmune reaction (which has been outlined in detail in previous sections), and those who believe that CNS-specific autoreactive lymphocytes are the initial pathogenic entities (“outside-in”). The latter hypothesis is corroborated by EAE models, which involve the activation of T cells at the periphery, resulting in pathological processes within the CNS. EAE studies, especially those using transfer EAE models, clearly show that autoaggressive T cells infiltrate the CNS in order to induce MS-like pathology in experimental animals. Accordingly, the association of immune cells with MS lesions is evident. However, until recently, there have been no reports showing significantly increased presence of lymphocytes in early MS lesions. The key study came from Ransohoff's group, which performed histopathological examinations of cortex biopsies obtained in passing during diagnostic procedures targeting white matter lesions, within days or weeks after MS presentation (284). This is the first study adequately substantiating previous assertions about the development of cortical lesions early in MS (107, 111, 363). Of the 138 patients, 38% had cortical lesions showing signs of inflammation, demyelination, and neurodegeneration. Diffuse meningeal inflammation was significantly and strongly associated with cortical demyelination. The presence of CD3+ cells (CD3+ is a specific immunohistochemical marker for T cells in general) and CD8+ T cells has been documented in a majority of lesions, whereas the data on CD4+ T cells have not been obtained (284, 369). Reboldi et al. also found CCR6+ T cells in early cortical lesions, which implies that T cells infiltrate CSF using choroid plexus/CCL20 route (377) and may proceed to the cortex by provoking IL-17-dependent redistribution of CCL20 on the walls of blood vessels (103). It is important to note that meningeal aggregates of T cells are frequently formed around blood vessels in pia mater (34, 284). This could be explained by the fact that the cells coming from CSF find themselves on the CCL20-presenting side of the endothelium. Lucchinetti et al. proposed that macrophages carrying fragments of myelin may leave the cortex and spread epitopes to lymph nodes, which may result in T-cell activation (284, 369). Alternatively, T cells could be activated due to molecular mimicry, which seems to be a more probable cause (Fig. 11). It has been proposed that memory T cells may enter CSF in the subarachnoid space across the choroid plexus epithelium (244) or via transvascular route (34). The presence of CD4+ and CD8+ memory T cells has been identified in the CSF of MS patients, non-inflammatory neurological disease patients, and even in healthy CSF (130, 222, 245). Analogously, myelin-reactive memory T cells may also circulate in CSF. At some point, these cells may be reactivated by meningeal macrophages and become effector cells, with the interaction being mutually stimulatory (284). Pertinent to this, it has been shown, using the EAE model, that the first myelin-reactive T cells in the CNS appear before the onset of clinical EAE. The arriving cells are restricted to subarachnoidal space and remain strictly in close association with pia mater, crawling on the surface and on the outer face of blood vessels, performing a scan. There, T cells encounter APC meningeal/perivascular phagocytes, with the reactivation being followed by the escalation of the number of T cells (34).
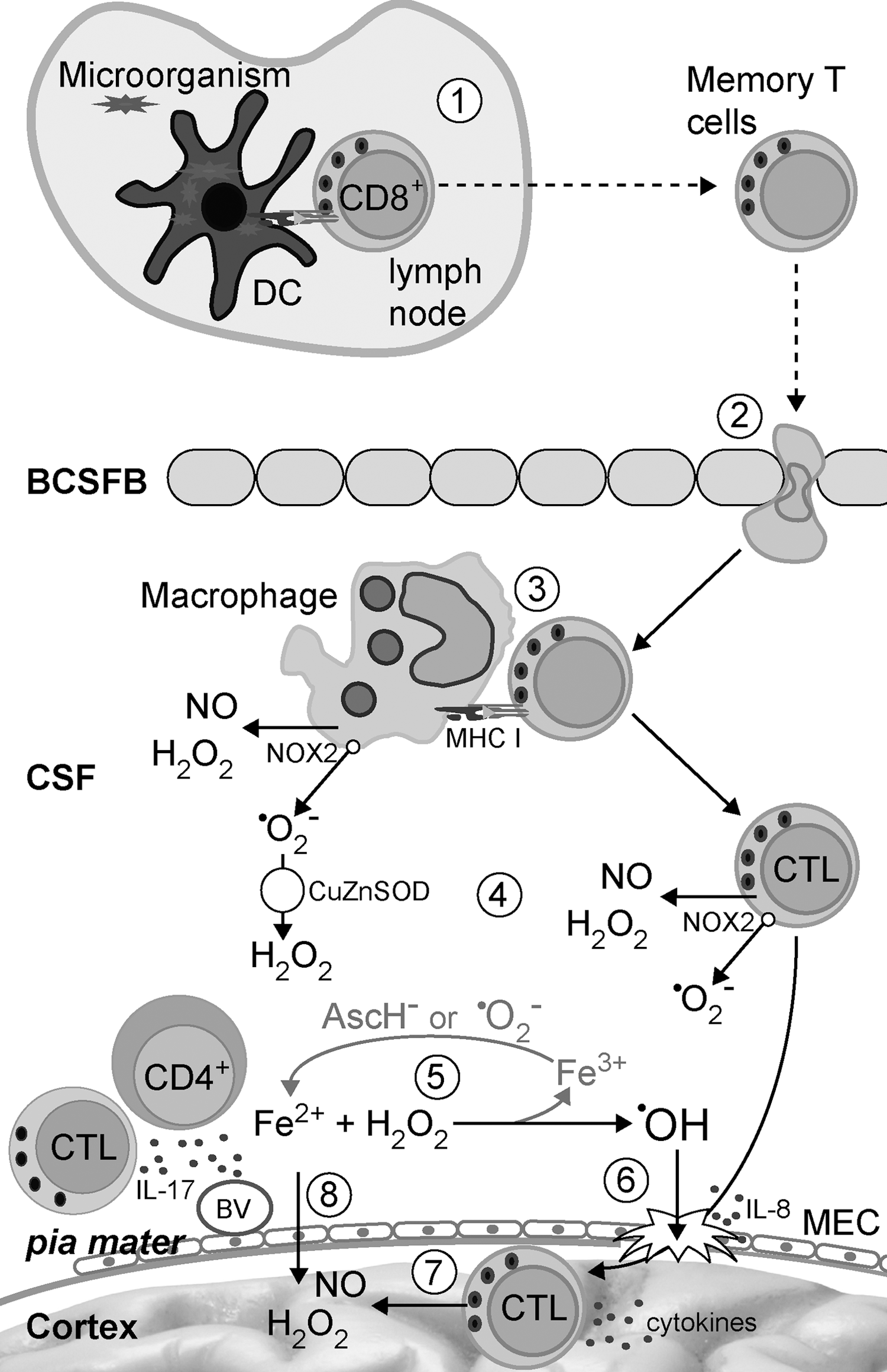
At this point, redox changes may become crucial (Fig. 11). After activation, CTL and macrophages release •O2 −, H2O2, and NO (details presented in sections III.A. and B.). H2O2 and NO along with cytokines can pass through pia mater, a delicate membrane that separates the cortex from CSF. This may result in initial cortex damage and microglia activation, which has been related to overlying meningeal infiltrates (287), followed by BBB breakdown, epitope spread, and perivascular T-cell infiltration (284). The involvement of T cells, which promptly emit high amounts of IL-17 on activation (103), may be particularly important. IL-17 has been reported to induce •O2 − production in brain endothelial cells, resulting in BBB disruption (213). However, an alternative scenario is also plausable, taking into account that (i) both perivascular and submeningeal infiltration of T cells into the CNS of an EAE model have been observed (34); (ii) subpial cortical lesions are more frequent in early MS compared with the lesions formed around vessels (284); and (iii) CSF is significantly different fluid when compared with plasma, showing to be a more fertile ground for prooxidative processes. Namely, CSF shows lower levels of iron-binding proteins and several-folds higher concentrations of ascorbate (Asc) in comparison to plasma (78, 190, 409). The reaction of iron with H2O2 (Fenton reaction) can be easily boosted during meningeal inflammation by •O2 − (via Haber–Weiss reaction) or by Asc (62, 409), and some CSF metabolites also (see section IV.C.) (Fig. 11). This results in excessive production of the most reactive species in the human body—•OH. This radical can damage pia mater, thus enabling the direct infiltration of CTL into the cortex, resulting in the fast development of inflammatory lesions. Pertinent to this, it has been documented that even mild oxidative stress drastically impairs the function and affects the viability of meningothelial cells (MEC), which line the surface of pia mater and are connected by TJ (472). It can be speculated that IL-17 may also affect TJ in this cell layer. It appears that MEC not only act as covering cells, but may also be involved in the inflammation, as, when exposed to oxidative stress, they release IL-6 and IL-8 (a chemotactic factor) (135). It should be noted that meningitis-provoking pathogen Streptococcus pneumoniae, which has been associated with the highest frequency of neuronal damage, employs H2O2 to damage meninges (202).
We propose that the early formation of cortical lesions involves different mechanisms compared with white matter lesions due to specific redox- and immune-relevant settings: (i) According to the presence of T cells in healthy CSF, BCSFB appears to be easier to cross than BBB; (ii) the cortex is the CSF-adjacent tissue, separated from the T-cell containing medium (CSF) only by one thin membrane (pia mater). This makes the cortex a site that may be more prone to the soluble mediators of inflammation (ROS, RNS, and cytokines) and to T-cell infiltration comparing with white matter, which is shielded by a complex and thick BBB. (iii) In comparison to blood, CSF represents a more prooxidative environment, which may favor oxidative damage of pia mater under the inflammatory settings. It is tempting to further speculate what comes first, white matter lesions initiated by inside-out mechanisms or cortical lesions initiated according to the outside-in model. We believe that this varies from patient to patient, and that it may further reflect in different clinical courses of MS. For example, it has been previously shown that white matter lesions in PPMS show pattern IV (283, 366), which is mostly related to degenerative processes and may involve only a weak inflammatory component. However, a very recent study performed by Choi et al. has shown that diffuse meningeal inflammation and cortical lesions represent a hallmark of PPMS. The level of demyelination and neuron loss in the cortex was associated with the degree of meningeal inflammation (86). The progressiveness of symptoms in PPMS and the formation of cortical lesions appear to be closely related (86, 284). So in this case, white matter lesions may represent a reflection of global changes in the brain that are initiated by cortical lesions (369). However, white matter lesions represent the prevailing form of lesion in MS; so we believe that, in most cases, cortical lesions develop later, during progressive phases of MS (86), for example, during relapses in RRMS (66) or in SPMS (254, 358), which are characterized by meningeal inflammation (207).
III. Redox Processes in Inflammation
A. Redox processes in macrophages
In order to elaborate redox changes that are directly related to inflammation, we will focus on pattern II lesions. In pattern III lesions, the effects of prooxidative activity of the inflammatory component are overshadowed by the production of reactive species in astrocytes and other CNS cells, and, therefore, they would be hard to tell apart. It has been reported for pattern II lesions that the relative distribution of T cells and macrophages/microglia resembles the one seen for oxidative damage markers. In addition, the number of T cells and macrophages shows statistically significant correlation with the level of oxidation of DNA and lipids (181). However, pattern II lesions show approximately an 8-fold lower level of oxidation and 1–2 orders of magnitude lower PARP activity compared with pattern III lesions (181, 453). This implies that inflammation-unrelated processes involved in early/pattern III lesion development represent the main route of oxidative damage in MS. Inflammation appears to be significantly less responsible according to the comparatively modest redox changes in pattern II lesions.
In general, inflammatory stimuli are known to activate the expression of iNOS and COX2 in macrophages (29), and this has been observed in inflammatory, demyelinating (pattern II) lesions comprising catabolites of MBP and live ODC (384). The authors contemplated that prostanoids (COX2 products) and ONOO− may contribute to cellular death in the lesions. Nitrotyrosine depositions have been observed near, while the ingested nitrotyrosine positive material has been found inside the iNOS-positive inflammatory cells (384). iNOS and COX2 show a very interesting interplay (Fig. 12), which has been examined in macrophages and other cell types. NO is known to promote COX2 catalytic activity via ONOO−-mediated nitrosylation of specific Cys residue, but at high concentrations, NO inhibits COX2 activity and expression (30, 112). On the other hand, prostaglandin PGD2 (COX2/isomerase product) can be metabolized to 15d-PGJ2, which is known to suppress NO production in microglial cells (120), while H2O2 that is produced by COX2/CuZnSOD activity may promote the expression of iNOS. These interconnected positive/negative feedbacks may act as switches in the regulation of inflammation. An important role of COX2 in the inflammatory component of MS is indirectly implied by a well-documented fact that cytosolic phospholipase A2-deficient mice are resistant to EAE (3). This enzyme produces arachidonic acid, which is the substrate of COX2. In addition, activated macrophages induce iNOS-dependent acyl chain nitration of a complex lipid cholesteryl linoleate. The product—nitro-cholesterol linoleate (CLNO2) suppresses iNOS expression and inflammatory responses in macrophages (30). Therefore, a key mediator of inflammation—NO—is involved in its eventual resolution. It should be noted that PGD2 and 15d-PGJ2 have been shown to induce the apoptosis of ODC precursors (470); hence, the increased COX2 activity may result in hampered remyelination. Finally, excitotoxicity may be related to COX2 activity in macrophages, as prostanoids stimulate glutamate release (384). Another important enzyme that is expressed in macrophages in pattern II lesions is myeloperoxidase (MPO) (292). MPO generates hypochlorous acid (HOCl), which is capable of provoking serious damage to biomolecules. However, due to its high reactivity, HOCl is considered as having a very limited diffusion radius in biological milieu. Therefore, only a small fraction (if any) of HOCl produced in macrophages may find its way out and affect the surrounding tissue (379).
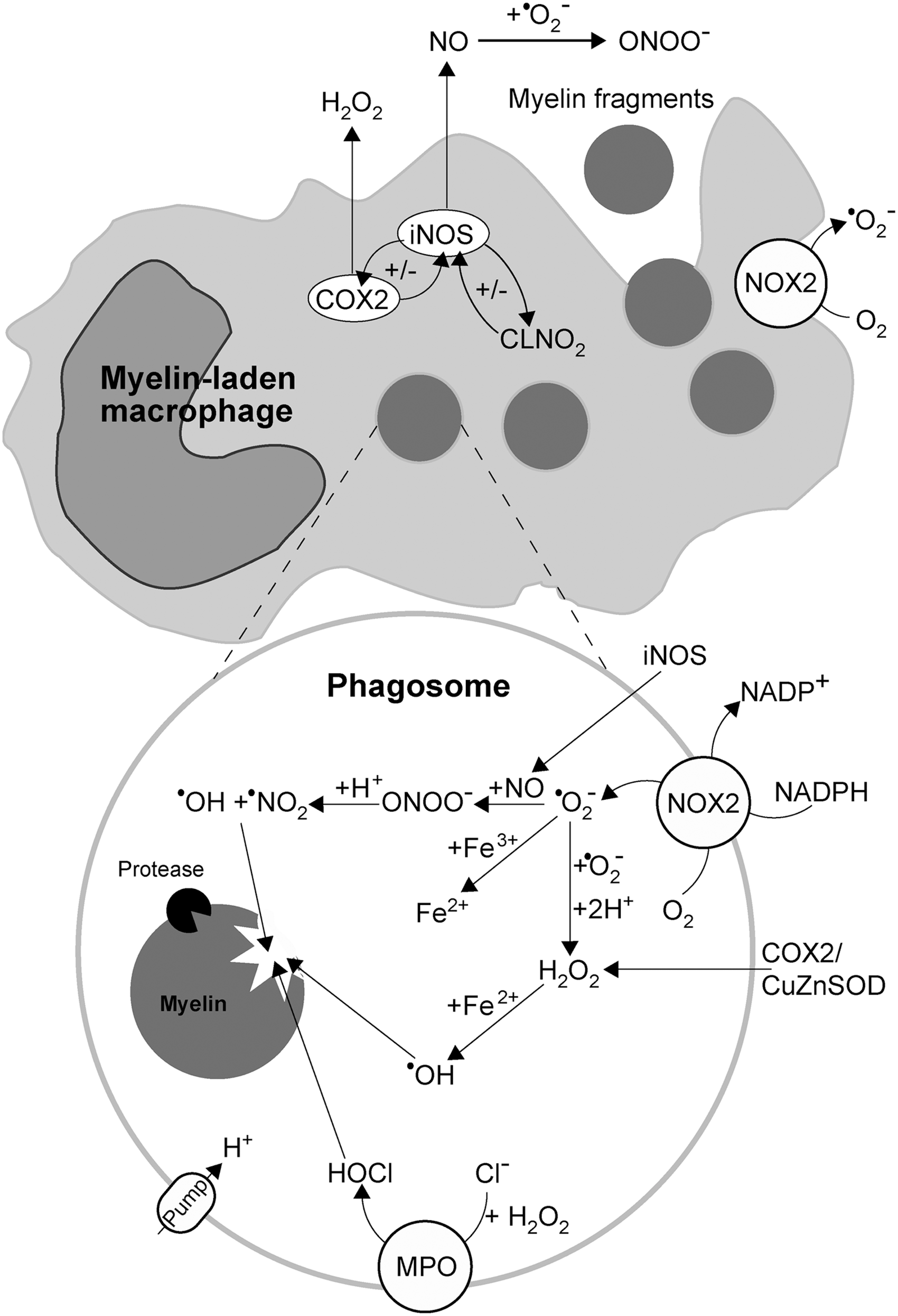
Macrophages have to produce •O2 − and H2O2 in order to conduct the process of myelin phagocytosis. Inactive macrophages show a very low ROS production, which is boosted approximately 10-fold, when myelin is present, but returns to the baseline level in the presence of NOX2 inhibitors (diphenyleneiodonium or apocynin). NOX2 is a multi-subunit enzyme that is dormant in resting phagocytes. After the activation by phagocytosis or some other stimulus, the complex is brought together on phagosome membrane, and it becomes active, delivering large quantities of •O2 − into phagosomal interior (395). In a set of experiments, van der Goes et al. incubated macrophages with myelin and specific ROS scavengers: catalase, CuZnSOD, and mannitol. The removal of H2O2 by catalase, and the scavenging of •OH by mannitol suppressed macrophage-mediated myelin phagocytosis, whereas •O2 − removal by exogenous CuZnSOD did not show any such effects (438, 439). It is important to note that while mannitol can cross cellular membrane, the first two agents are relatively large proteins, which raise up the question: “How do these enzymes find themselves in a position to affect intracellular processes?” The only plausible answer is that in this experimental setup, catalase and CuZnSOD were taken up into phagosomes along with myelin fragments. The lack of effects of CuZnSOD implies that •O2 − is not a direct mediator of myelin degradation. Two superoxide radical anions can be dismutated to O2 and O2 2−, with the latter being protonated to H2O2 (phagosomes show low pH values, which are maintained by the activity of H+ pump). In addition, •O2 − reacts with Fe3+ from dead myelin/ODC to produce Fe2+ and O2. H2O2 is further involved in •OH production via a Fenton reaction (which is promoted by low pH) and in MPO-mediated HOCl production. The main mediator of oxidative damage exerted on myelin fragments in phagosomes appears to be •OH, as implied by the inhibitory effects of mannitol and catalase (438, 439). Superoxide may also react with NO that is generated by iNOS in the cytosol, to produce ONOO− and its derivatives—•OH and •NO2 (Fig. 12). The latter suppresses the inhibition of proteases and binds to proteins, which become more susceptible to protease activity. This could be very important for myelin degradation, as excessively oxidized proteins appear to be poor substrates for proteases (432). When it comes to the oxidative/nitrosative effects of macrophage activity on the surrounding cells, the main redox mediators are most likely NO and H2O2, both of which are produced in the cytosol by iNOS and COX2/CuZnSOD activity, because it is hard to believe that any significant amount of H2O2 could escape phagosomes. In addition to phagosome membrane, NOX2 is also present on the macrophage's cellular membrane (41) Therefore, NOX2-mediated •O2 − release into the extracellular compartment may also contribute to prooxidative effects of inflammation. Superoxide may be dismutated by CuZnSOD in the extracellular fluid (256), or it can react with NO in order to produce ONOO−. It should be noted that macrophages/microglia are highly resistant to damaging effects of ONOO− (220), which implies that these cells use ONOO− for their function.
B. Redox processes in T cells
The activation and proliferation of T cells require a tightly regulated redox microenvironment, which T cells sense by their cell-surface thiol redox switches. The activation also involves ROS production by T cells, and the activity of APC (DC in particular) and Treg cells (477). The stimulation of TCR activates CD4+ and CD8+ T cells to express NOX2, dual oxidase 1 (Duox1) and eNOS (on cellular and outer mitochondrial membrane) (221, 255, 336, 422). The mechanisms behind this are not yet fully understood, but a few milestone papers provided a glimpse of some basic details. In the study performed on highly purified mature T cells, Williams' group observed three distinctive phases (pathways) of ROS production: (i) rapid (2–4 min after stimulation) and transient production of H2O2, independent of NOX2; (ii) NOX2-dependent, sustained H2O2 production; and (iii) NOX2-independent •O2 − production (221). In a subsequent study, the same research group found that the early H2O2 production (first phase) in stimulated CD4+ T cells is mediated by intracellular Duox1 (255). Yet another study on CD8+ cells showed that TCR stimulation leads to H2O2 production and calcium influx. The H2O2 production developed 2 min after stimulation, and was partially suppressed by rotenone, the inhibitor of ETC complex I (480). This implies that in addition to Duox1 activity, H2O2 leakage from mitochondria may account for the first component of ROS production in a stimulated T cell (221). The results showing that rotenone, a well-known promoter of •O2 − production on complex I (333), suppresses the emission of H2O2 from mitochondria are rather interesting and deserve further explanation. TCR stimulation-provoked calcium influx prompts mitochondria to work at a high capacity (336). In such a setting, mitochondria may lack ADP to continue ATP production, which results in increased proton gradient across the inner membrane and a high level of reduced co-enzyme Q. This leads to reverse electron transport from complex II toward complex I and to promoted •O2 − production (and increased H2O2 release), which is known to be abolished by rotenone (333). Calcium has also been reported to activate protease, which converts xanthine dehydrogenase to XO, resulting in •O2 − production (413). This could potentially account for the third phase of ROS production in stimulated T cells, in addition to the prolonged promotive effects of calcium on respiration (336) (Fig. 13). It is worth mentioning that NO production in T-cell-APC conjugates is twice as high at cell–cell contacts than in the cytoplasm, and it represents a checkpoint in T-cell activation (336).
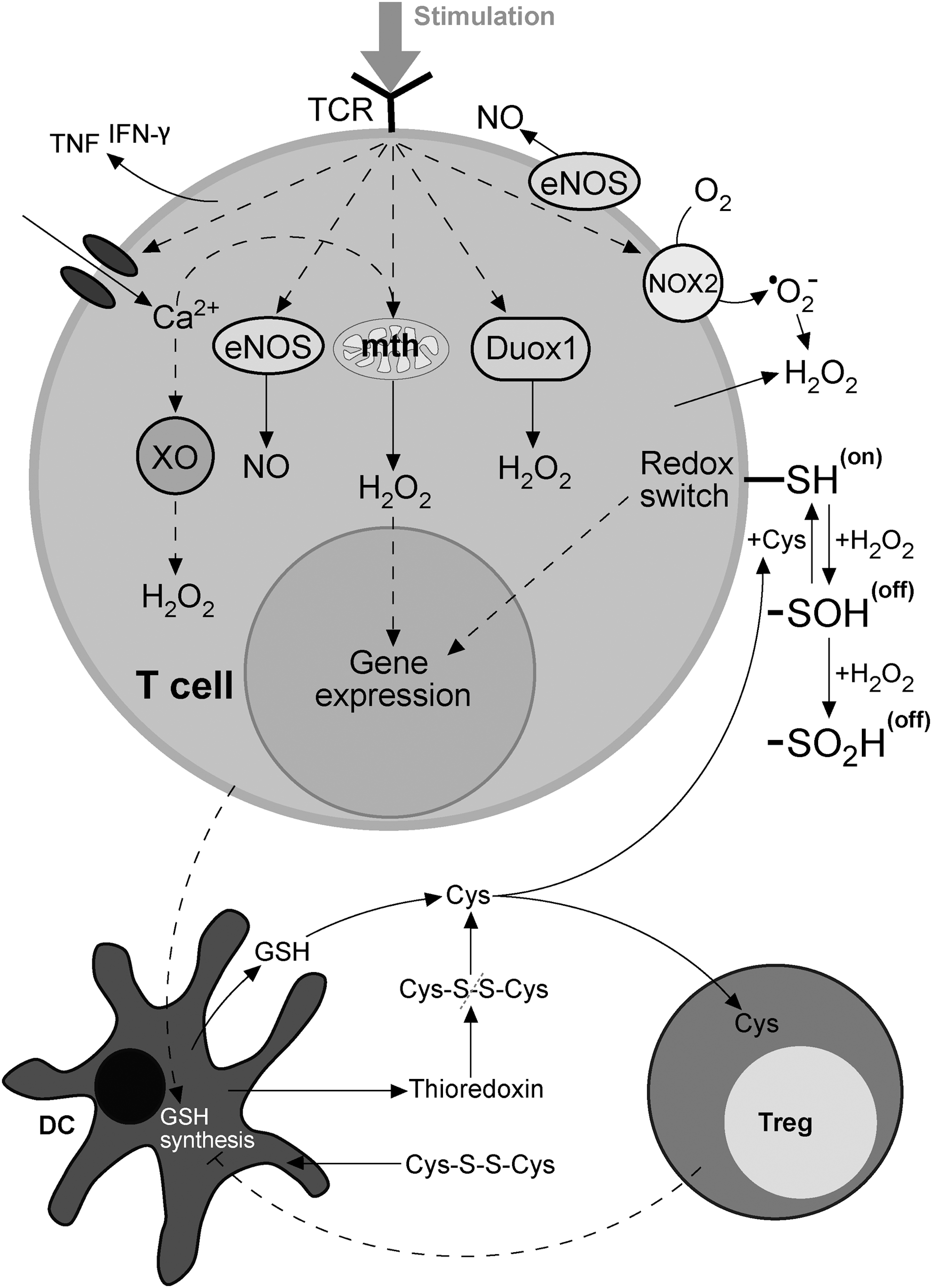
The intracellular production of H2O2 is needed for T-cell activation. For example, it has been reported that the radical scavenger N-acetylcysteine (NAC) can inhibit T-cell activation (336). It should be noted that, on stimulation, GSH peroxidase-deficient CD4+ T cells proliferate at a faster rate compared with wild-type cells, while their differentiation is biased toward Th1 and Th17 cell development (466). Rotenone provokes a decrease in T-cell division and drastically suppresses the release of TNF and IFN-γ from TCR stimulated (effector and memory) CD8+ cells (480). The application of a cell-permeable catalytic antioxidant (manganese (II) tetrakis (N-ethylpyridium-2-yl)porphyrin) on stimulated CD8+ T cells not only slows down the cell proliferation, but also suppresses the production of IFN-γ and TNF (405, 480). Similar results have been observed for Duox1-knockdown CD4+ T cells (255). Therefore, the release of TNF and IFN-γ from T cells appears to be promoted by H2O2, which is in line with the results obtained on some other cell types (331, 338).
On the other hand, the suppression of effector T cells and the induction of Treg cells depend on extracellular ROS production, mainly by NOX2 (128, 250). Pertinent to this, stimulated CD4+ and CD8+ T cells are known to generate extracellular •O2 − by cellular membrane-bound NOX2, most likely for the purposes of self-regulation (422). Gelderman et al. showed that NOX2 activity increases the threshold of reactivity and suppresses proliferation of T cells by provoking oxidation of specific thiol groups (-SH) on T-cell membranes (158). GSH treatment lowered the threshold of T-cell reactivity and promoted the proliferation of T cells, implying that cell surface redox -SH switches play a central role in the regulation of T-cell activity. However, the treatment with oxidized GSH (GSSG), which leads to decreased number of reduced thiol groups on the membrane surface by forming -S-SG moieties, results in T-cell suppression (158). The reaction of thiol group with H2O2 results in the formation of sulfenic acid (-SOH) residue (104). GSH or Cys may reverse -SOH back to -SH. In two elegant studies, Yan et al. showed that after the contact with T cells or antigens, DC promote the activation and proliferation of T cells by releasing GSH, which is then cleaved to Cys (478, 479). Extracellular Cys increases the level of reduced thiol groups on the exofacial membrane proteins of T cells, thus turning the redox switch on (Fig. 13). In a co-culture with effector T cells, DC show enhanced GSH synthesis and the uptake of oxidized Cys (cystine; Cys-S-S-Cys). DC and T cells even release thioredoxin, which could recycle extracellular cystine to Cys (16, 247). On the other hand, Treg cells suppress DC-promoted inflammation by diminishing GSH synthesis in DC (by suppressing the expression of γ-glutamylcysteine synthetase), and by consuming extracellular Cys. It is important to note that the exposure to H2O2 excess may result in the apoptosis of T cells (419), which may be related to irreversible oxidation of -SH switches to -SO2H and -SO3H. T-cell redox switches may also respond to other stimuli, such as heavy metals and carbonyl compounds that bind to thiols (8). Finally, it has been shown that myelin-laden macrophages inhibit TCR-triggered proliferation of myelin-reactive T cells in an antigen-independent manner by iNOS-mediated NO production (51). This implies that S-nitrosylation of thiol switches could be an important mechanism for regulation of the activity of T cells. The nature of redox switch(es) on T cells remains to be further elucidated.
It is clear that ROS and ROS-generating enzymes are involved in a complex signaling network which shows both pro- and anti-inflammatory effects. The concentration of H2O2 appears to be the key factor that defines the outcome (110). Figure 14 shows hypothetical curve of the dependence of effector T-cell activity/proliferation on H2O2 concentration. At the lower end, redox switches are on, but intracellular processes have not been initiated. At rising H2O2 concentrations, intracellular signaling is activated, while redox switches remain on. In this medium phase, the activity/proliferation is at its highest. A further increase in H2O2 level first turns redox switches (irreversibly) off, thus suppressing activity, and finally provokes apoptosis.
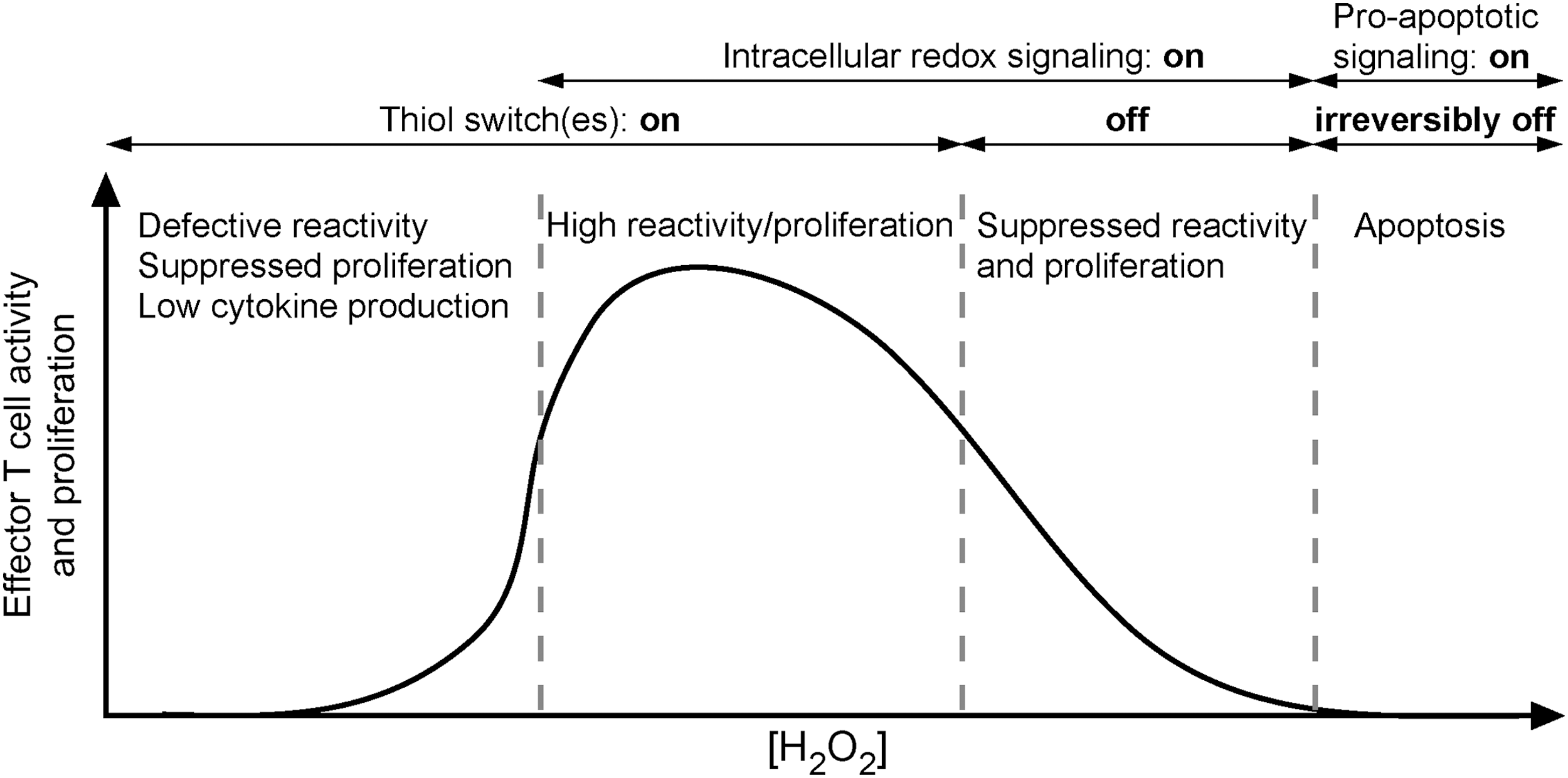
C. Cytokines and the intracellular oxidative stress
One of the important characteristics of MS is a massive release of cytokines within the CNS (270). The production of cytokines may modulate redox settings and vice versa. Taking into account the number of cytokines and the complexity of their effects and interactions, which could be a candidate for more than one review paper, here we will only illustrate redox/cytokine dependence on the example of IFN-γ and IL-17. These are the signature cytokines of the pathogenic CD4+ populations in the CNS autoimmune response, that is, Th1 and Th17 cells, respectively. Th1 proliferation and differentiation may be hampered by NO-promoted development of Treg cells (342), and by NOX2-related inhibition of IL-12 release from DC (224). However, experimental models of MS suggest that IFN-γ plays an antioxidative role in neuroinflammation, as there is more oxidative stress in the CNS of EAE-immunized IFN-γR knockout mice than in controls (133), and as transgenic mice with suppressed ODC responsiveness to IFN-γ develop EAE with an accelerated onset, associated with enhanced early inflammation and markedly increased ODC apoptosis. Accordingly, it was reported that IFN-γ pretreatment of mature ODC in vitro has a protective effect against oxidative stress (31). Moreover, IFN-γ stimulates microglia to uptake glutamate and, therefore, protects neurons (399). The differentiation of Th17 cells may be promoted by ROS generation in mitochondria (494). IL-17 collaborates with TNF in the induction of oxidative stress-dependent death of ODC (354). IL-17 induces BBB disruption through the induction of NOX- or XO-dependent ROS production (213). Thus, it appears that IL-17 has a positive relationship, while IFN-γ has a negative relationship with ROS and oxidative stress in neuroinflammation. It is tempting to speculate that the different effects of IFN-γ and IL-17 on oxidative conditions might be, at least partially, responsible for the observed variability in histopathological findings in EAE induced by Th1 and Th17 cells (223, 253).
D. The contribution of oxidative stress to relapses and progression
Relapses in MS may be related to newly emerging redox-initiated processes in the CNS regions that are distant from the initial lesion (in the case of pattern III and IV lesions), or to significant changes in the immune system (in pattern I and II lesions); for example, to the occurrence of immune-compromised CNS and autoreactive memory T cells. Activated T cells differentiate into effector and memory T cells and later subdifferentiate into central memory T cells and effector memory T cells (387). Central memory T cells retain a high capacity for proliferation, while effector memory T cells retain a high effector capacity in response to antigenic challenge. Similarly, memory B cells subdifferentiate into classical memory B cells and plasma cells, which correspond to central and effector memory T cells, respectively (387). Therefore, rechallenging the immune system with an antigen will elicit a fast and strong response due to activity of memory T and B cells. It is fast, because cells are ready to perform effector functions, and strong, because there are antigen-specific cells that can intensively multiply in a short period of time. This concept is valid for MS pathogenesis, as activated memory T cells invade the CNS and induce novel neurological sequels or sustain those that have already been expressed. Indeed, the number of memory CD4+ and CD8+ T cells is increased in the peripheral blood of MS patients, and these cells readily infiltrate CNS and are found in MS lesions (25, 180, 243). Memory B cells are also implicated in MS pathogenesis, not only through the production of autoreactive antibodies, but also through antigen presentation and cytokine generation, which are highly relevant for T-cell functions (219, 308).
Although CNS-reactive memory cells are present in the immune system of MS patients, there should be a trigger that activates them and releases new attacks against the CNS. Here again, as in the initial autoimmune response, antigens might be released from the CNS that is damaged by oxidative/nitrosative stress. Pertinent to this, it has been reported that plasma levels of oxidation markers are higher in MS compared with healthy subjects (348), and that there seems to be a link between MS exacerbation and lipid peroxidation (426). It is known that the level of lipid peroxidation products, including oxidized low-density lipoproteins (oxLDL), is increased in the plasma and CSF of MS patients. Several different proinflammatory effects of oxLDL were reported (138). For example, oxLDL and ROS stimulate the generation of osteopontin (297), which plays a major role in MS pathogenesis (416), shifting Th differentiation toward pathogenic Th1 and Th17 phenotypes (177). Further, oxLDL increases Treg sensitivity to Fas-mediated apoptosis (307), thus provoking facilitated activation of autoreactive T cells. In addition, ROS and RNS heavily impact the effects of vitamin D by blocking its receptor function (253). Vitamin D3 is important for the optimization of branching of Asn (N)-linked glycans. Reduced branching results in T-cell hyperactivity and provokes inflammatory demyelination and neurodegeneration (176, 323). These facts implicate that oxidative/nitrosative stress may result in less restringent activation of autoreactive cells and their hyperactivity in MS.
ROS and RNS production in MS patients may be promoted by bystander infections. Bacterial and viral infections have been implicated in the induction of relapses and the acceleration of progression, whereas a vaccination against communicable diseases is considered beneficial for patients (280). Microbial stimulation of APC function and oxidative stress-imposed hyperactivity of effector T cells might combine and lead to the activation of autoreactive encephalitogenic T cells and, thus, to relapses and MS progression. It should be noted that redox changes in inflammation result in epitope spreading, which is appreciated as highly relevant for chronicity in autoimmunity (448). Therefore, even if the initial autoreactivity is regulated and annulated, novel antigens still perpetuate autoimmune process. Importantly, it is not only the completely new antigens that emerge in epitope spreading, but also the (oxidative/nitrosative) modifications of existing antigens (22), which gives autoimmunity in MS a dynamic profile.
E. Vitagene network, heat shock proteins, and inflammation
The interplay between redox settings in CNS and inflammation may be indirectly mediated, by factors released from neurons and other cells in response to oxidative stress. Under stressful conditions, cells activate a vitagene transcriptional network (vitagenes encode for heat shock proteins (Hsp) Hsp32 (also known as HO-1) and Hsp70, the thioredoxin and the sirtuin protein systems), and the expression of other Hsp and small Hsp (sHsp; such as Hsp27 and HspB5 [αB crystalline]). It has been well documented that this takes place in MS lesions (20, 335, 445). Integrated mechanisms controlled by vitagenes, Hsp, and sHsp operate in the brain in helping neuronal survival by performing folding and repair of damaged proteins, preventing aggregate formation and prooxidative/cytotoxic effects of free heme, and conducting protein triage. It should be noted that the vitagene network in lymphocytes is up-regulated in MS (361), most likely in order to prevent self-inflicted oxidative damage. However, in addition to their functions in the intracellular milieu, Hsp may be released into the extracellular compartment either by passive release from necrotic cells or via active release, including secretion in the form of exosome (e.g., the secretion of sHsp most likely represents altruistic delivery of supporting and stabilizing factors from one cell to another) (67 –71). Released Hsp may act as immunomodulators, and some of them most likely play a rather important role in MS. The most relevant appears to be Hsp70, whose role in MS is still controversial. In a recent review, Mansilla et al. proposed that Hsp70 overexpression in CNS of MS patients has neuroprotective function. However, if this fails, and large number of neurons and ODC undergo necrosis, a certain amount of Hsp70, sufficient to promote or exacerbate inflammation, could be released. Proinflammatory effects of Hsp70 are most likely related to its activity as danger signal (Hsp70 promotes APC maturation and innate immune response) and adjuvant (Hsp70 binds to peptides and damaged proteins and creates new epitopes, which could potentially initiate endogenous molecular mimicry) (291). Hsp27 appears to act in a similar fashion, as some authors attribute the increased levels of this sHsp in MS to its neuroprotective effects, whereas others outline that it may act as an autoantigen. It is important to note that the level of Hsp27 in MS patients is increased during the phase of disease progression (80). Furthermore, the induction of T-cell tolerance for Hsp60-derived epitope has been reported to ameliorate EAE (45). The abnormal expression of Hsp90 in lymphocytes appears to be responsible for the unresponsiveness of some MS patients to glucocorticoid therapy, as Hsp90 prevents the translocation of glucocorticoid receptor into the nucleus (296). In MS patients, Hsp90 is expressed at the surface of ODC precursors, which seems to promote their depletion in lesions (87). Although much work still has to be done, the general principle of “good inside CNS cells, potentially bad outside” may turn out to be applicable to most Hsp that are involved in MS pathology.
It has been proposed that HspB5 represents a special case, but it appears to us that there are just sufficient amount of data on this specific sHsp for elaborating all important aspects of its activity. The local concentrations of HspB5 may determine the balance between protective innate responses and destructive adaptive responses. In developing MS lesions, HspB5 is selectively induced as a protective protein in ODC and astrocytes, from which it is released in order to trigger microglia activation. The former appears to have neuroprotective effects at first, most likely as activated microglia remove debris. Pertinent to this, animals that do not express HspB5 show worse EAE. However, activated microglial cells also release cytokines and act as APC, which may ultimately overwhelm the protective microglial activity (445). This implies a very important role of HspB5 in transcending degenerative events in MS initiation to inflammation. The previous assertions that HspB5 may act as an autoantigen have been recently discredited by the finding that HspB5 binds to antibodies due to its chaperone characteristics (385). It seems logical to propose that other sHsp, the release of which is controlled, may not act as autoantigens. However, fragments of sHsp may act as autoantigens. For example, it has been shown that MMP-9 may cleave HspB5 in order to produce a promiscuous T-cell epitope (412).
IV. Neurodegeneration in MS
A. “Slow burning” of demyelinated axons
Neurodegeneration of demyelinated axons is a major cause of irreversible neurological disability in MS. Partially or completely demyelinated neurons find themselves unprepared for a hostile environment in the CNS of MS patients, which shows increased levels of ROS, RNS, and iron. With their own weak AOS and without antioxidative and metabolic support, which would have been provided by a thick myelin sheets and glial cells (200), demyelinated neurons are highly susceptible to externally exerted oxidative/nitrosative stress (378) and to metabolic stress in general. However, the main cause of degeneration seems to come from within the demyelinated axon, and it may be viewed as “the triumph of function over survival.” It is important to note that unshielded axons may be remyelinated by maturing ODC progenitors before serious damage occurs. However, the remyelination capacity is diminished in chronic lesions, so the axons remain permanently demyelinated.
Healthy neurons are energetically efficient cells. This is based on the insulating properties of myelin sheets, which enable fast, saltatory conduction of action potentials from one node of Ranvier to another. On demyelination, action potentials are propagated in a “wave-like” fashion, which requires increased expression and redistribution of Na+ channels along the entire axon (97). The restoration of conduction comes at the cost of greater Na+ influx and increased activity of Na+/K+ ATPase, which is used for pumping out Na+, and it represents the largest ATP consumer in CNS even under physiological conditions (355). This increases the demands for ATP (288, 464), which are met by increased number of mitochondria (Fig. 15). It has been reported that demyelinated axons show a 2–3-fold increase of mitochondrial density compared with normal tissue (288, 464). More mitochondria means more H2O2, as there is a constant outflow of H2O2 from mitochondria into the cytosol. It has been reported that demyelinated neurons show increased levels of complex I and IV. While the activity of complex IV increases correspondingly, the activity of complex I, the main site of •O2 − production in mitochondria, is impaired (464). According to this, NO may not play a direct role here, as it inhibits both complexes (333). However, even at lower concentrations, NO may, in combination with promoted •O2 − generation, result in a significant production of ONOO−, which is known to inhibit complex I, but shows significantly less pronounced inhibitory effects on complex IV (334). As a result, mitochondria in demyelinated axons experience oxidative stress, as implied by MtHsp70 chaperone overexpression and oxidative damage of mitochondrial DNA (464). Due to mitochondrial dysfunction and probably because of the decreased O2 availability (428), demyelinated neurons turn to increased glycolysis turnover in order to meet the high demands for ATP. Increased levels of lactate (glycolysis metabolite) have been detected in active MS lesions using MR spectroscopy (355). In addition, Regenold et al. showed that the CSF of MS patients contains increased levels of metabolites of glycolysis and polyol pathway (380). Since NADH is produced in both these metabolic pathways, their up-regulation results in increased NADH/NAD+ ratio and consequent fulmination of •O2 − production on complex I. The importance of this particular sequence of events is strongly implicated by NAD+ metabolism and its pharmacological effects in MS (360). It has been reported that the increase in NAD+ level in EAE animals shows neuroprotective effects (226), most likely by decreasing NADH/NAD+ ratio. Brain in MS appears to try to compensate for NAD+ decrease by de novo NAD+ synthesis, which leads to a decrease in the level of tryptophan (the precursor of niacin and NAD+) in CSF and serum (350). Pertinent to this, a small group of MS patients were treated with tryptophan and they were showing symptoms alleviation (360).
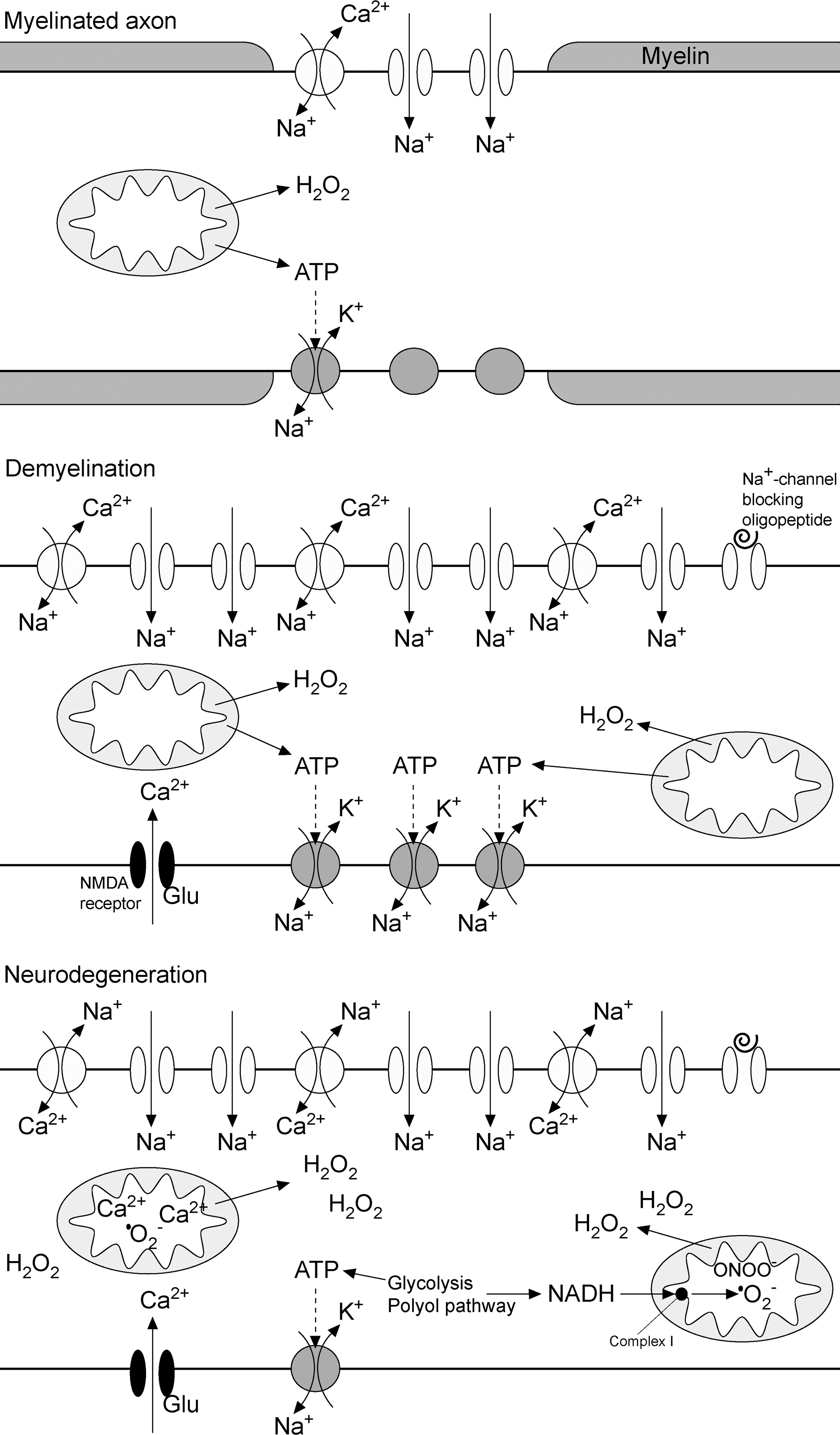
At the end, the increased number of mitochondria and promoted glycolysis are not enough to meet energy demands of chronically demyelinated axons in the long run. Without enough ATP to power Na+/K+ ATPase, neurons suffer from Na+ accumulation, which at higher concentrations leads to reverse action of the Na+/Ca2+ exchanger (97), Ca2+ import, and Ca2+-induced cell death (355). It should be noted that Na+ channel blockers and the inhibitors of Na+/Ca2+ exchange may provide protection from degeneration (228). Another factor that may lead to Ca2+ overload is the increased level of excitotoxic amino acids—glutamate and aspartate—in the CSF of MS patients (171). Calcium overload is known to activate NOX2 in neurons (57), as well as ROS production in mitochondria (121). At the final stage, demyelinated axons in chronic lesions show decreased presence of Na+/K+-ATPase (Fig. 15), which may represent an attempt to slow down the ATP expenditure (482). However, hand in hand with increased number of Na+ channels, this results in a persistent ion imbalance, water influx, axon swelling, and degeneration (482). One MRI study documented that Na+ level is up to twice higher in MS lesions compared with healthy tissue (218). Craner et al. showed that demyelinated axons try to fight Na+ overload by choosing to express Nav1.2 channels, as the persistent Na+ current produced by Nav1.2 is significantly smaller compared with the current through Nav1.6 channels, which represent the predominant isoform in nodes of Ranvier (97). In addition, a small Na+-channel blocking oligopeptide (600–800 Da) was identified in the CSF of MS patients (24), most likely representing an adaptive factor aimed at decreasing Na+ inflow into demyelinated axons (Fig. 15).
B. Neurodegeneration in acute lesions
1. White matter
The traditional view was that the inflammation is the cause of degeneration. Although it has been challenged by a number of studies (145), this concept may not be wrong, but distinction should be made between the neurodegenerative processes and the role of inflammation in neurodegeneration, which take place in the white matter and in the cortex (Fig. 16). In the white matter, the majority of axons degenerate by “slow burning” in inactive chronic lesions with depleted remyelination capacity. Nevertheless, it is clear that inflammation may propagate neurodegeneration in white matter (403, 407). Some axons may not survive acute inflammatory assault, and they undergo degeneration immediately after or even before the demyelination due to the impact of ROS, RNS, cytokines and glutamate released by T cells, activated microglia, and macrophages. Pertinent to this, accelerated deterioration of demyelinated neurons in active lesions in response to inflammation-generated NO has been documented (289). CTL have been reported to transect non-myelinated axons in an MHC I-dependent manner (306). Finally, direct interactions between myelin-reactive Th17 cells and demyelinated axons resulting in damage have been observed in EAE (403). On the other hand, acute axonal injury has been shown to be partially independent of demyelination (48). In EAE model and MS biopsy samples, Nikić et al. observed a sporadically reversible process that they named “focal axonal degeneration,” which is characterized by focal swellings that progress to axon fragmentation, in both de-myelinated and myelinated axons. The process was related to the intra-axonal dysfunction of mitochondria that was provoked by macrophage-derived H2O2 and NO (343). In myelinated axons, the initial disruption typically took place at the nodes of Ranvier, which is in line with our concept that myelin represents a good barrier for ROS/RNS, while unprotected areas of axons represent weak spots.
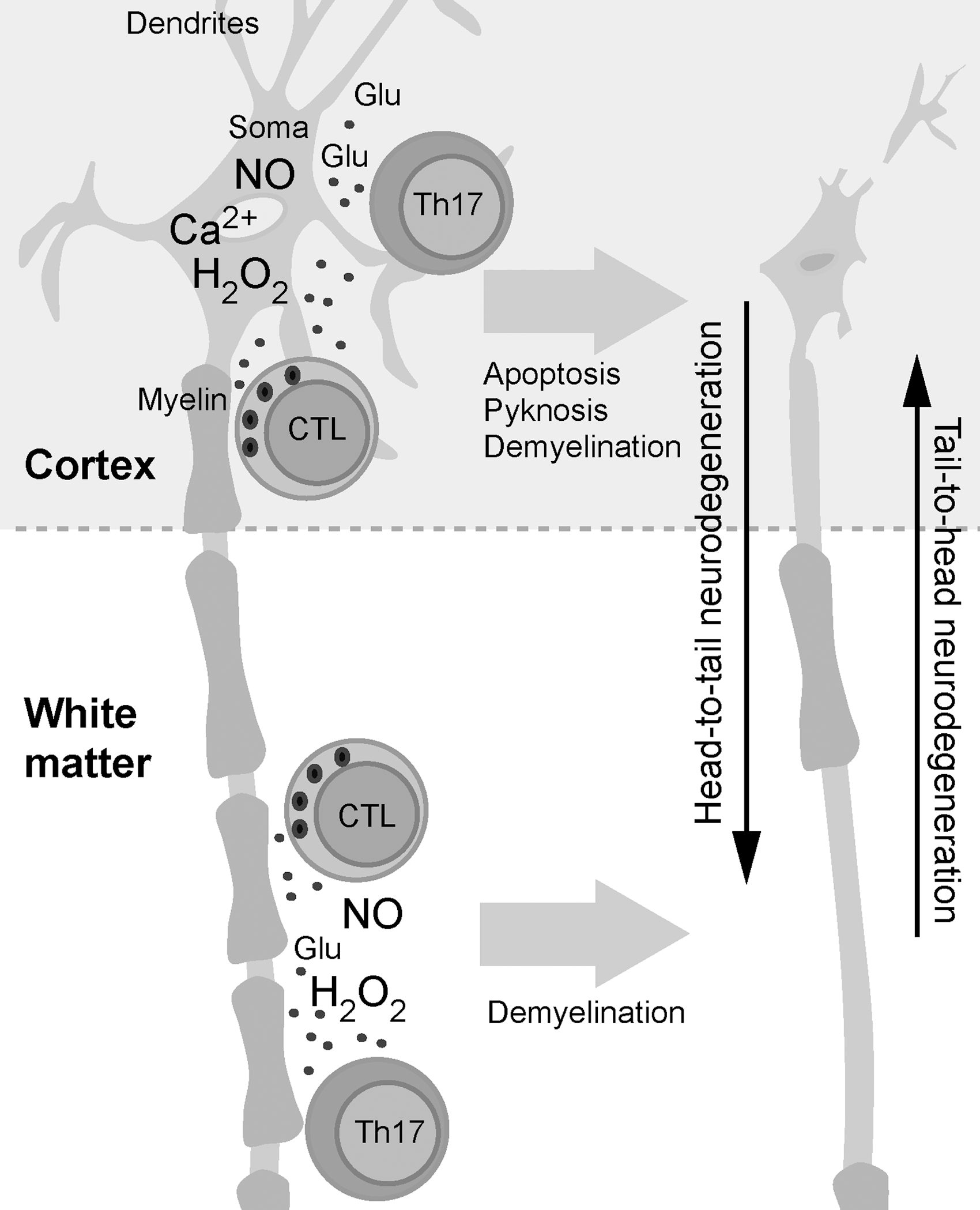
2. Cortex
Cortical demyelinating lesions are widely present in MS patients, and may even represent the initial lesions in some cases. Importantly, cortical damage correlates with MS progression as well as with cognitive and other disabilities (86, 284, 458). While the white matter is mainly composed of myelinated axons, cortex comprises both myelinated axons and unprotected neuron somas and dendrites. This difference may be reflected in the process of neurodegeneration, as neurons (neuron parts) that are unprotected by myelin are highly sensitive to RNS and ROS (152) and directly exposed to cytokines and glutamate. Neuronal injury, morphology changes, pyknosis and apoptosis, dendritic transection and the loss of dendritic arborization, as well as decreased density of neuronal somas and synapses are substantial in cortical lesions (284, 358, 363, 369, 370, 458). Papadopoulos and co-workers found a strong positive correlation between neuronal loss and inflammation in archaeocortex autopsies of MS patients, whereas axons appeared to be spared (358). In addition, phagocytic macrophages and lymphocytes are predominantly positioned close to somas and dendrites, while activated microglia do not ensheath the terminal ends of transected axons (axon “healing” may be futile if somas are affected) in inflammated cortical lesions (363, 369, 403). Finally, working on an EAE model, Pomeroy et al. found the presence of cortical lesions and the loss of cortical thickness that is not related to demyelination (367). All this implies that an inflammatory assault on neuronal bodies takes place in active cortical lesions. T cells attack cortical myelin, as it is documented by the close relation between inflammation and demyelination in the cortex of MS patients (284). However, soluble mediators of inflammation may also affect nearby somas and dendrites [collateral bystander damage (407)], which are clearly more susceptible to damage compared with myelinated axons. Even more, dendrites can be cleaved directly by CTL that bind to MHC I on the neuron membrane and release soluble mediators (306). Giuliani et al. showed that polyclonally activated CD4+ and CD8+ T cells align along soma and (non-myelinated) axons of cultured human neurons and provoke neuronal death via antigen- and MHC-independent mechanisms. It is rather interesting that these effects were not reproduced by stable soluble products such as cytokines or when neurons and T cells were physically separated by a filter (165). This implies that the effects are based on direct cell–cell interactions and/or the involvement of short-living species with a shorter diffusion range compared with cytokines, that is, ROS and RNS. The importance of NO in cortical degeneration is implied by the decrease in the functional activities of ETC complexes I and III in mitochondria isolated from motor cortex of MS patients (126). There are some other circumstantial evidence with regard to the involvement of ROS and RNS in cortical neurodegeneration, such as increased expression of endoplasmatic reticulum stress-associated molecules in gray matter lesions (303), but this subject requires further investigation. T cells' binding to neurons most likely involves LFA-1/ICAM-1 interaction (165), and/or it is related to the fact that neurons express some myelin proteins (306). Zipp's group has shown that myelin-reactive Th17 cells provoke soma and axon degeneration by directly interacting with neurons and inducing glutamate-mediated excitotoxicity (403). Similar results were obtained in another study. It has been shown that activated T cells directly interact in an MHC-independent manner with soma and dendrites and provoke Ca2+ oscillations. This leads to a lethal increase in neuronal Ca2+ levels, which can be prevented by blocking glutamate receptors (344). It is important to note that in neurons, excitotoxicity and oxidative stress promote each other mutually (410).
The death of soma directly results in axon dysfunction and slow disintegration (we propose the term “head-to-tail” neurodegeneration), which is a vice versa process compared with retrograde degeneration starting from axons in white matter lesions and resulting in cell death at the end (“tail-to-head” neurodegeneration) (Fig. 16). According to this, the development of cortical lesions may have a more pronounced and straightforward impact on the patient's condition, as there are no mechanisms for damage reparation analogous to remyelination in white matter. It is important to note that axon–myelin or neuron–ODC interactions and metabolic support are bidirectional. For example, ODC need axons in order to efficiently switch to glycolysis in the case of ETC inhibition (e.g., by NO) (146). Therefore, the damage of neuronal body and axon may promote demyelination (431).
C. Inappropriately chelated iron
The mechanisms of neurodegeneration in MS, similar to neurodegenerative diseases, may involve oxidative pressure exerted by •OH production in the Fenton system that takes place in the extracellular fluid and CSF, which are interconnected and poor in antioxidants (21). In MS, accumulated iron is present in the form of deposits in brain tissue (27, 383, 487). The amount of iron in CNS positively correlates with the number of lesions, whereas the level of deposited iron shows a positive correlation with the neuro–psychological condition of MS patients (154), implying that iron may play a role in axon degeneration. In contrast, CSF levels of the two most important mediators of the Fenton reaction—iron and copper—are not increased in MS (313). However, the prooxidative activity of iron depends more on chelation and solubility than on the total concentration. This fact has recently been brought to attention by the concept of inappropriately chelated iron (233, 410). In brief, iron's prooxidative activity in the physiological milieu is limited by low Fe3+ solubility (it readily forms insoluble complexes with hydroxyl and phosphate ions), and is buffered by a number of chelating proteins. However, specific ligands may catalyze redox cycling of iron by increasing iron's availability to H2O2 and reducing agents, in particular Asc, which is accumulated in CNS reaching concentrations that are several-fold higher compared with plasma (78, 190). Iron and Asc represent a well-known “combination to avoid,” as Asc promotes Fenton reaction (62, 410). Typically, iron can coordinate six ligands in an octahedral arrangement; so, molecules that are capable of binding to all six sites (e.g., transferrin) completely inactivate iron. By contrast, bidentate or tridentate chelators, which bind only to two or three chelation sites, increase the solubility and redox activity of iron (233). Generally, ligands that favor Fe3+ involve oxygen in iron binding, whereas ligands that bind Fe2+ involve nitrogen and sulfur (118). Therefore, biomolecules that are capable of binding iron in both redox states most likely comprise O and N and/or S in their structure in a configuration that allows the formation of two or three coordinate bonds. Such ligands can be rather dangerous, as they promote an •OH-generating chain Fenton reaction (Fig. 17A). CSF metabolome is significantly altered in MS compared with physiological settings, and there could be a number of potential iron accomplices in neurodegeneration, showing increased levels in the CSF of MS patients and meeting structural criteria. It is worth mentioning that we found increased prooxidative activity of inappropriately chelated iron in the CSF of patients suffering from a neurodegenerative condition (217).
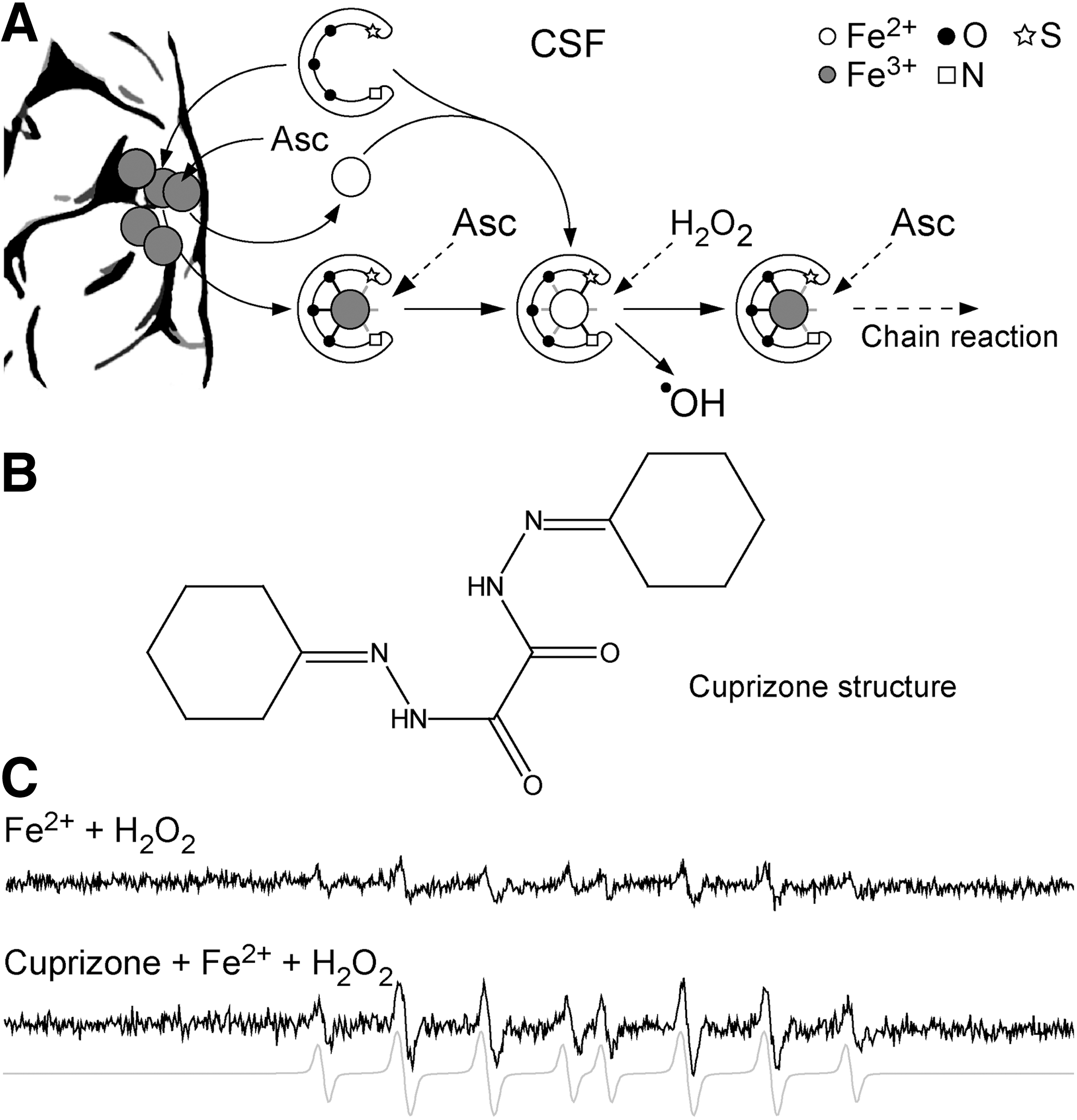
Cuprizone is a copper-chelating agent which is applied in order to provoke MS-similar symptoms in animal models that are highly relevant for the investigation of pattern III and IV lesions (241). The mechanisms of cuprizone-activated demyelination are not fully understood, but they appear to involve metabolic stress in ODC and the activation of CXCR2+ neutrophils (276). Although it has been speculated that cuprizone promotes iron-related oxidative stress in the CNS (282), it has not been suggested to date that it may act by creating inappropriately chelated iron complexes. The structure of cuprizone (Fig. 17B) seems to allow the formation of such complexes. In addition, it has been proposed that cuprizone promotes Cu2+/Cu3+ redox cycling (179), which further substantiates our hypothesis. We performed a brief study using the CSF of patients with neurological conditions that are unrelated to iron metabolism (five lumbar and cervical disc herniations). Using electron paramagnetic resonance (EPR) spin-trapping spectroscopy, the production of •OH radical in the CSF supplemented with Fe2+ and H2O2 was compared with the CSF co-supplemented with cuprizone. In all samples, the presence of cuprizone resulted in two- threefold increase of •OH production (Fig. 17C), implying that cuprizone may form an inappropriately chelated iron complex. Since cuprizone primarily targets ODC (241), it is plausible that similar mechanisms may take place inside these iron-rich cells, resulting in oxidative stress and cellular death. Finally, inappropriately chelated iron complexes may play a role other than provoking oxidative stress. For example, carbonyl–iron has been reported to promote not only oxidation (148), but also the immune response. Actually, it has been used as an adjuvant for induction of extremely harsh EAE (315). We speculate that some inappropriately chelated iron complexes may act as natural adjuvants (149).
V. Redox Therapy in MS
A. General strategy
The development of effective (antioxidative) therapy for MS patients is intricated by the heterogeneity of the pathophysiological mechanisms involved. The redox therapeutic approach could target (i) degenerative component (ODC dystrophy being the hallmark), which predominates in initial and pattern III and IV lesions; (ii) inflammatory/autoimmune component, which is crucial in pattern I and II lesions; and/or (iii) neurodegenerative component, which develops as a result of the first two and involves the collapse of chronically demyelinated neurons. The main problem is that antioxidative therapies targeting different processes in degenerative and neurodegenerative component in MS pathophysiology may be futile or even show negative effects when it comes to autoimmunity, as the activation and proliferation of T cells require a tightly regulated redox microenvironment. In other words, the activity of T cells could be promoted, if supplemented antioxidants provoke a certain level of reduction. We will see later that the most promising agents currently available are able to target both degenerative and inflammatory components. The most accurate way to determine which component predominates in each MS patient is to determine the pattern of lesions. The therapeutic relevance of patterns is best documented by the findings of Keegan et al., who showed that only patients with pattern II lesions exhibit a significant functional neurological improvement after antibody depleting therapy (therapeutic plasma exchange); while this is not the case with pattern III and I patients (232). Currently, the patterns can be identified by means of biopsy/immunocytochemistry (283) or potentially MRI (53, 398). It is important to note that most authors concur that one specific pattern is consistently observed in the samples from the same patient (232, 283). Some MS presentations are indicative of a specific pattern, but no significant correlation has been established between patterns and clinical manifestations (366).
Redox therapy should not be uniformly applied, and it should be developed to target specific mechanisms (patterns). For example, while many antioxidants delivered (variable) promising results in EAE studies, some, such as flavonoids, proved to be detrimental (76, 452). In addition, there is no substantial proof that any of the standard nutritional antioxidants (vitamins C and E, lipoic acid, and others) have any effects on MS development or its progression (137, 490). It is about time for the scientific community to embrace the fact that antioxidants are not all the same and that they are not panacea. They act via specific mechanisms that may or may not interfere with the processes which are responsible for a particular disease (15). Here, we will mostly focus on redox-active compounds that have been involved in clinical research and showed substantial benefits.
B. Fumarates
Probably the most exciting news for MS patients in a while came from the 64th Annual Meeting of The American Academy of Neurology, where the results of CONFIRM trial phase III have been presented (209). In phase II, oral application of dimethyl fumarate in RRMS patients for 24 weeks resulted in a reduced number of new lesions by more than 50% and a decreased annual relapse rate by more than 30% (229). The treatment was prolonged to two years in phase III (involving more than 1400 RRMS patients), showing a reduction in annual relapse rate by around 50%, and a decrease in the number of new lesions by ∼70% (143). The rationale for these trials was antioxidative/neuroprotective and anti-inflammatory effects of dimethyl fumarate observed in vitro and in animal models (229).
Oddly enough for compounds that deliver antioxidative effects, fumarates seem to base their activity on binding to thiols via a Michael-type addition (393). Experiments performed on astrocytes and neuronal cells showed that short-term exposure to fumarates results in decreased intracellular GSH level and short-lived oxidative stress (10, 268, 392). However, long-term exposure showed quite the opposite effects, resulting in increased GSH levels and activated AOS (10, 268). Dimethyl fumarate stabilizes Nrf2 and promotes Nrf2-dependent transcriptional activity. The first metabolite, monomethyl fumarate modifies Nrf2 inhibitor—Kelch-like ECH-associated protein 1 (KEAP1) at Cys151. As a result, fumarates protect neurons and astrocytes from oxidative stress by activating Nrf2-controlled set of enzymes: NAD(P)H:quinine oxidoreductase 1 and GSH-related enzymes (271). Another study on astrocytes exposed to inflammation showed that fumarates reduce nuclear levels of NF-κB p65 subunit, prevent the loss of IκB-α (NF-κB inhibitor), and drastically suppress iNOS expression (268). It should be stressed that the translocation of p65 subunit (as well as p50) and the activity of IκB kinase can be inhibited by thiol-reactive compounds targeting specific Cys residues (73, 116, 356) (Fig. 18). With regard to anti-inflammatory effects of fumarates, it has been reported that dimethyl fumarate provokes T-cell apoptosis both in vitro and in vivo (in cuprizone model of MS) (324, 429). The underlying mechanism most likely involves the irreversible binding of fumarates to thiol redox switches on T-cell membranes (Fig. 18). The modification resembles the effects of excessive oxidation (478). Finally, Ghoreschi and co-authors showed that fumarates provoke the activation of type II DC, which inhibit Th1/Th17 cells differentiation via a pathway involving impaired production of IL-12 and IL-23 (162).
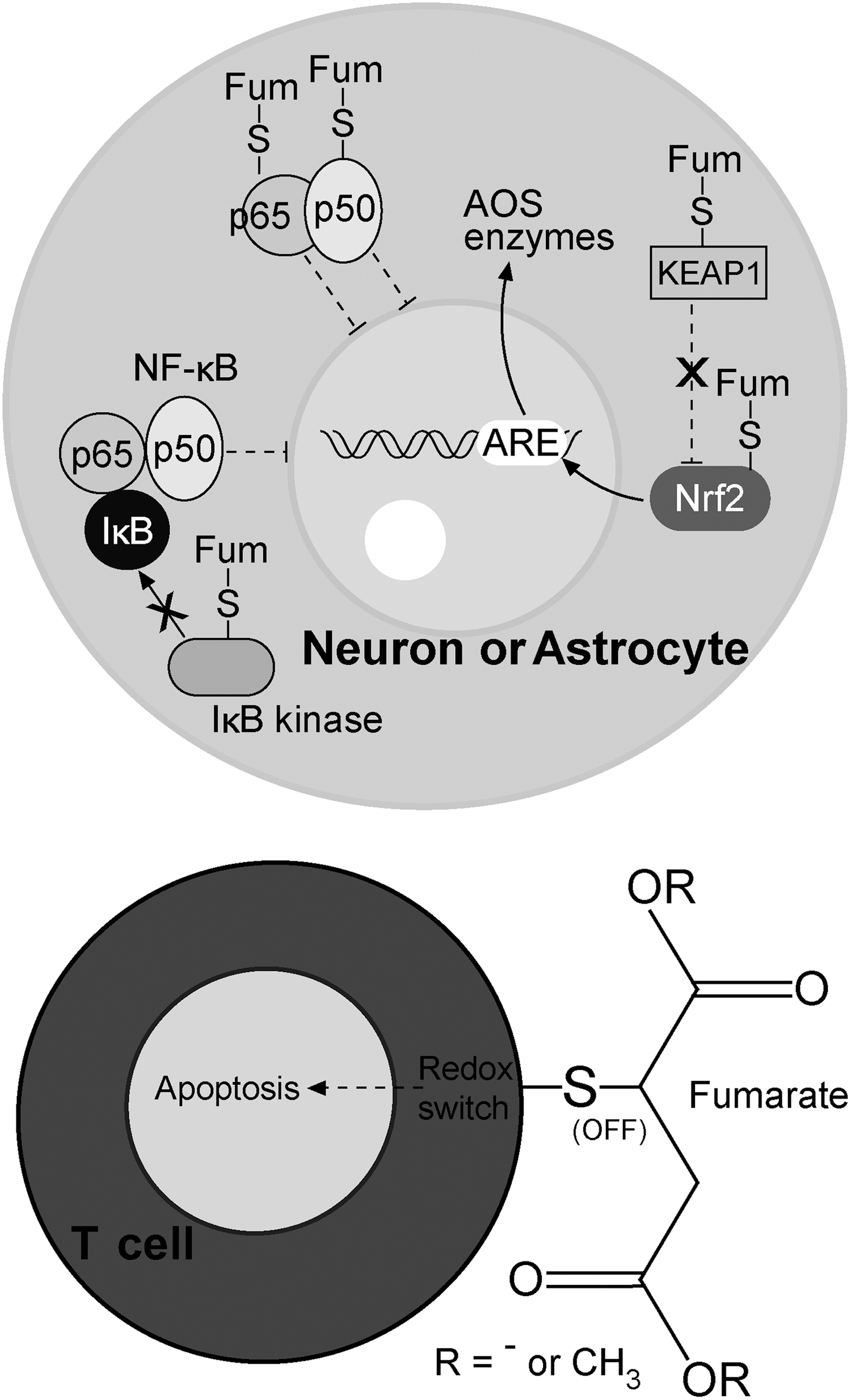
Another compound that forms adducts with thiols, and, as we believe, may be of interest for future MS trials, is pyruvate. It shows many similarities with fumarate, and it is available as a dietary supplement, while its more stabile derivative—ethyl pyruvate—is approved for human use (227). Pyruvate shows neuroprotective effects against oxidative damage (455). Ethyl pyruvate exerts anti-inflammatory effects via GSH depletion, activates Nrf2 in astrocytes, inhibits NF-κB-dependent transcription by targeting p65 subunit, and suppresses the expression of iNOS (183, 227, 401). In addition, pyruvate attenuates the effects of ONOO− (227). Its imunomodulatory effects should be further investigated.
C. Cannabinoids
A number of clinical trials have tested the ability of different cannabinoids or cannabinoid-containing products to alleviate MS symptoms. The beneficial effects on MS-related pain and sleep disturbances are well substantiated (486). A large long-term follow-up study (CAMS) indicated that cannabinoids may slow the progression of disability, increase mobility, and decrease relapse rate (485). However, preliminary results of another large trial (CUPID), presented at the annual meeting of The Association of British Neurologists in 2012, imply that tetrahydrocannabinol does not significantly slow the progression of disability in the general MS population, but may show beneficial effects in patients at the lower end of the disability scale (212). In relation to this, we speculate that cannabinoids do not target the neurodegenerative component of MS, which is responsible for the promotion of neurological problems later during the MS progression, but may affect the degenerative and/or autoimmune component of MS pathophysiology.
The anti-inflammatory activity of cannabinoids involves the inhibition of proliferation and activity of effector T cells, the induction of Treg cells, and the inhibition of macrophage activity (388). In addition, cannabinoids are known to be potent antioxidants. In a comparative study, cannabidiol showed higher antioxidative capacity compared with butylated hydroxytoluene and vitamins E and C (182). Being highly lipophilic, cannabinoids most likely act as membrane-based antioxidants, and in MS, they could prevent HNE and MDA production. Some results imply that cannabinoids show iron-chelating properties (235). It should be noted that a recent study showed that cannabidiol may ameliorate EAE in a direct, receptor-independent manner (249).
Two types of cannabinoid receptors (CBs) have been described: CB1 and CB2. CB1 is widely present in neurons, while CB2 is mainly found in immune cells (486), as well as in ODC precursors and astrocytes (160, 325). Neuroprotective effects of cannabinoids against oxidative stress are not mediated by CB1 receptors (293). On the other hand, the stimulation of CB2 receptors inhibits NF-κB activation and iNOS and COX2 expression in astrocytes (265). In this way, cannabinoids may stop redox-cycle development in astrocytes in early or pattern III lesions. In addition, an in vivo study showed that cannabidiol suppresses neuroinflammation-provoked iNOS expression (134). Finally, cannabinoids affect ODC precursors via CB2 stimulation, promoting their survival and differentiation (169, 325). Cannabidiol is known to protect ODC progenitor cells from H2O2-mediated oxidative stress and inflammation-provoked apoptosis by decreasing the expression of endoplasmatic reticulum apoptotic effectors (305). It seems that future research on the application of cannabinoids in MS treatment should focus on cannabidiol, rather than tetrahydrocannabinol, given that the former showed more promising preclinical results; it is not psychoactive, and unlike tetrahydrocannabinol, its neuroprotective effects are not susceptible to tolerance development (193). In order to achieve the best results, cannabinoids should be applied early in MS.
D. The inhibition of NF-κB and iNOS
Some drugs that are approved for therapy of MS, as well as some of those which have shown beneficial effects in clinical or preclinical trials, are known to affect NF-κB-regulated expression and iNOS activity, which may represent an important component of their mechanisms of action. Mitoxantrone (DNA-reactive agent), which is used for the treatment of patients suffering from SPMS, PRMS, or RRMS worsening, is known to suppress NF-κB DNA-binding activity and NO production in stimulated astrocytes (64). Teriflunomide (the inhibitor of pyrimidine de novo synthesis), which is used for oral treatment of patients suffering from RRMS (211), inhibits iNOS in astrocytes (316). Methylprednisolone (synthetic glucocorticoid), which is widely used for acute treatment of MS relapses, inhibits NF-κB activation in inflamed CNS tissue and iNOS-mediated NO production in ODC (205, 473). Minocycline (tetracycline antibiotic) has been shown to decrease the development of new lesions and the relapse rate in MS patients (312, 484). This may be related not only to its immunomodulatory effects, such as suppressed antigen presentation and reduced T-cell activity (246, 386), but also to the ability of minocycline to inhibit iNOS and to down-regulate MMP-9 (484). The former effect is most likely mediated by NF-κB inhibition and may transpond to COX2 expression (65). Fluoxetine (antidepressant; trade names Prozac and Sarafem) has been shown to reduce the development of focal inflammatory lesions in MS patients (328). Importantly, it is effective in inhibiting microglia activation, including NF-κB-dependent generation of NO by iNOS (274). Natrexone (opioid receptor antagonist) improves mental health and decreases spasticity of MS patients (99, 164). It appears to act by preventing ODC death via the reduction of iNOS activity and ONOO− generation (5). Methylthioadenosine (naturally occurring sulfur-containing nucleoside), which showed good results in preclinical studies, prevents the degradation of IκB-α, resulting in impaired NF-κB activation and suppressed iNOS activity (327). Epigallocatechin-3-gallate (green tea phenol) exerts protective and regenerative effects in neurons exposed to inflammation (195), which may be related to its ability to block the activity of 20S/26S proteasome complex, resulting in the accumulation of IκB-α, the suppression of NF-κB activation (9), and probably inhibited iNOS expression (269). Mycophenolate mofetil (antimetabolite immunosuppressive prodrug), which, according to phase II trial, suspends MS progression and reduces relapse rate, is known to attenuate iNOS expression (281). Laquinimod (immunomodulatory agent) exhibited beneficial effects in the phase III trial on RRMS patients, slowing down the progression of disability and reducing relapse rate (89). The mechanisms of action may involve the protection of CNS cells from cytokine- or H2O2-provoked oxidative stress (311), and the suppression of NF-κB-regulated gene transcription (including iNOS) in astrocytes (61). Euphol (alcohol tetracyclic triterpene) not only attenuates the progression and severity of EAE, most likely by suppressing myelin-specific Th17 cells infiltration and cytokine production, but also attenuates NF-κB-regulated iNOS and COX2 expression in the CNS (125). Very similar effects in an EAE model were observed for metformin (antidiabetic drug) (339). Some studies showed that the application of selective iNOS inhibitor aminoguanidine leads to a significant decrease in the clinical expression of EAE (58, 278, 402, 493), implying that iNOS inhibition may represent an important component of MS treatment. It appears that aminoguanidine may be worth trying in the treatment of early/pattern III MS. It is tempting to speculate that the inhibitors of NF-κB and iNOS could provide the best results if applied early in MS.
E. Other agents
1. Iron chelators
Following several EAE studies, which delivered variable results, iron chelation strategy using deferoxamine was applied in a small trial on SPMS patients with no recognizable effects (411). However, the dose and the interval of application of deferoxamine were limited by a set of safety issues, and deferiprone could have been a more effective choice (321). Some interesting novel strategies have been proposed in iron chelation therapy, such as the development of chelators that are activated by oxidative stress (233). On the other hand, prooxidative activity of iron mainly relies on molecules that form inappropriately chelated iron complexes. Hence, the targeting of those molecules (which are yet to be identified) may represent the future of neurodegeneration treatment, including such processes in MS.
2. N-Acetylcysteine
NAC has been reported to attenuate oxidative stress in CNS of an EAE model (278). In addition, NAC attenuates HNE-provoked redox changes in endothelial cells and prevents pathological modification in TJ protein network (435). However, NAC appears to prevent T-cell apoptosis and activity suppression induced by different agents (56, 198), most likely by reducing the redox switches. According to this, NAC should be preferentially applied in pattern III patients. One phase II trial on RRMS patients has been conducted, but we are still waiting for the results to be published (208).
3. Ketogenic diet
A very recent EAE study showed that ketogenic diet slows down the development of motor disability and the loss of spatial learning and memory. The suggested mechanisms behind these effects involve amelioration of immune response and oxidative stress (238, 340). Ketogenic diet is known to inhibit •O2 − production on mitochondrial complex I by promoting NADH oxidation, which results in decreased NADH/NAD+ ratio (285).
4. Vitagene network activators
The results of several studies have shown that carnitine supplementation may improve the quality of life and reduce the symptoms of fatigue in MS patients (421), and a trial (FACTSEP) is underway that examines this issue (210). The positive effects of L-carnitine/acetyl-L-carnitine may at least partially be attributed to their capacity to activate neuroprotective vitagene network (67 –71). A similar explanation may also be valid for recently published results, showing that natural polyphenols protect neurons in the chronic mouse model of MS and EAE animals (142). Finally, the activation of sirtuin protein system, which is encoded by vitagenes, has been shown to prevent the loss of neurons and axonal density and improve neuronal function in EAE (489). In relation to proinflammatory properties of Hsp70, it seems that the activators of vitagene network should be applied in the remittance phase in order to target neurodegeneration in chronic lesions.
VI. Concluding Remarks
MS is a heterogeneous disease. A number of different components—vascular, redox, inflammatory/autoimmune and neurodegenerative, and various pathological patterns and phenotypes, combined with an astonishing amount of intricate data—may create an impression that this is an insolvable problem and an intractable disease. It is our belief that the extensive research should be accompanied by a comprehensive/integrative approach, in order to reach the true understanding of what happens in the body/CNS of an MS patient. The current review represents our humble attempt to do so. We have focused on redox processes, carefully trying not to over/underestimate their role in each step of MS pathogenesis, from initiation to neurodegeneration, and paying appropriate attention to other components, the autoimmune in particular. We have noticed some very important issues that should be addressed in greater detail in the near future: (i) the redox regulation of immune cells, especially T cells and APC; (ii) the involvement of redox processes in the regulation of chemokine network, and the differences between activated and reactivated T cells in their response to chemokines; (iii) the role of redox mechanisms in MS propagation and the remission–relapse shift; and (iv) the design of new MS models that are based on redox changes which are intrinsic to the CNS. Eventually, the results of redox studies could lead to new therapies in MS, as well as learning about lifestyle adjustments that would contribute to the patients' well-being.
Footnotes
Acknowledgment
This study was supported by the Ministry of Education and Science of the Republic of Serbia, grant numbers OI173014 and OI173035.