
Abstract
Introduction
C
Impairment of redox homeostasis plays a critical role in the genesis of cancer stem/initiation cells, and, thus, targeting redox homeostasis in these cells represents a new approach in cancer therapy. In this setting, we provide evidence demonstrating that fenretinide, a well-known oxidative stress-inducing agent in cancer cells, can effectively induce apoptosis in chronic myeloid leukemia (CML) stem/progenitor cells which are escapable from imatinib therapy. Thus, a combination of fenretinide with imatinib may represent a more sophisticated strategy for the treatment of CML, in which fenretinide targets imatinib-resistant CML stem/progenitor cells whereas imatinib targets leukemic blasts.
Numerous lines of evidence suggest that imatinib resistance and relapse can be largely attributed to CML stem/progenitor cells that are escapable from imatinib therapy (3, 17, 19, 60). These cells are present in small percentages in the leukemic cell mass of CML patients, whereas they are significantly enriched in the primitive CD34+ cells (3), and are indeed able to regenerate CML cell populations in immunodeficient mice (21). In vitro studies demonstrate that these primitive CML cells are insensitive to imatinib (17), dasatinib (5), and nilotinib (26). Accordingly, much attention has been focused on the development of agents and strategies for targeting CML stem/progenitor cells and for potentiating the efficacy of imatinib.
The redox signaling cascade may represent a new target in cancer stem/progenitor cells (37, 39). Mechanistically, PI3K/AKT pathways are abnormally activated by BCR-ABL, which may consequently impair downstream FoxOs, a key regulator in the maintenance of redox homeostasis in hematopoietic stem/progenitor cells (4, 54). It is, therefore, interesting to test whether agents that are able to perturb the redox homeostasis in tumor cells can be effective in targeting CML stem/progenitor cells. In this regard, fenretinide N-4-hydroxyphenylretinamide (4HPR), a well-known cancer chemo-preventive agent that effectively induces oxidative stress in many types of cancer cells, may represent a promising candidate. We have in parallel investigated potential effects of fenretinide on CD34+ cells of acute myeloid leukemia (AML) specimens, and found that this agent preferentially targets CD34+ cells in many of the tested specimens, while having limited cytotoxicity to normal CD34+ cells (58). Since CML is typically considered a hematopoietic stem cell disorder and the maintenance of redox homeostasis in its stem/progenitor cells is potentially impaired by the BCR-ABL signaling, it would be of great interest to investigate whether fenretinide is able to eradiate CML stem/progenitor cells that are refractory to imatinib.
Results
Fenretinide enhances the ability of imatinib for growth inhibition and apoptosis induction in CML-derived K562 cells
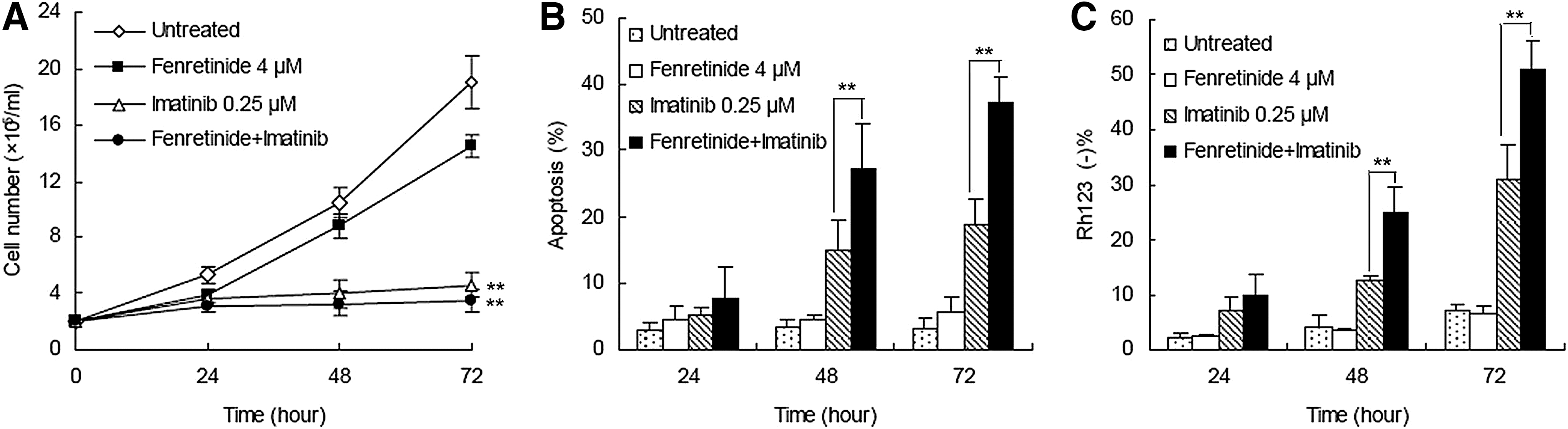
In an attempt to evaluate whether fenretinide was able to potentiate the ability of imatinib for growth inhibition and apoptosis induction in CML, a low dose (0.25 μM) of imatinib (12) and a physiologically achievable concentration (4 μM) of fenretinide (14) were utilized to treat CML-derived K562 cells. Fenretinide alone appeared to exert a minor effect (23.9%±4.4%) of inhibition on cell growth and a minimal effect on apoptosis induction (Fig. 1A–C). In contrast, the combination of fenretinide with imatinib induced significantly more apoptosis than imatinib alone, as evaluated by either Annexin V (37.2%±3.9% vs. 18.8%±4.0%) or Rh123 staining (51.0%±5.3% vs. 31.0%±6.5%). These results indicate that fenretinide is able to potentiate the efficacy of imatinib for apoptosis induction in CML-derived K562 cells.
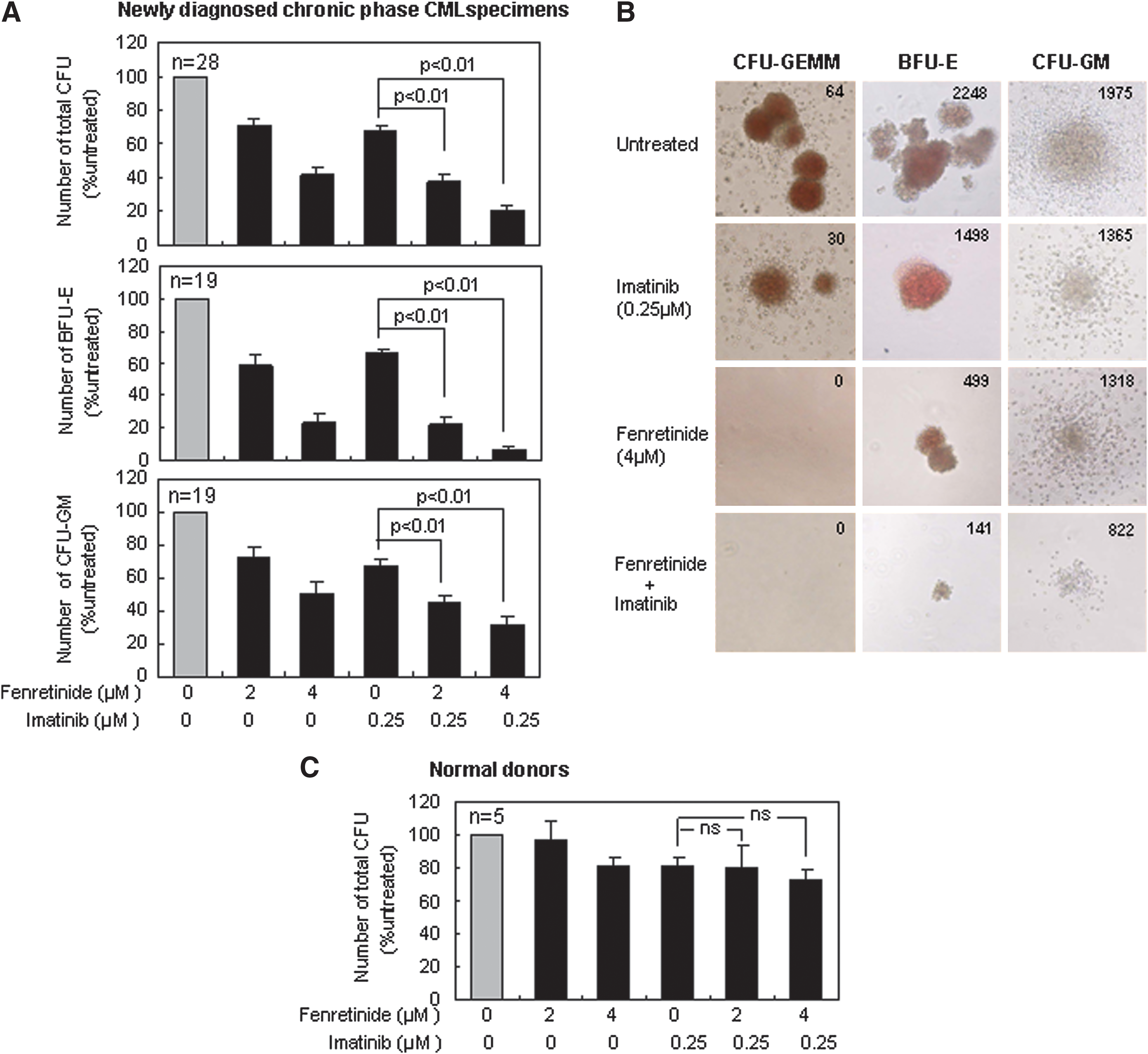
Fenrentinide potentiates the efficacy of imatinib for growth inhibition of colonies derived from CD34+ CML cells
CD34+ cells isolated from bone marrow or leukapheresis products of newly diagnosed CP CML patients (Table 1) were applied to colony-forming cell (CFC) assays on semi-solid methycellulose medium for 14 days with the presence of imatinib (0.25 μM), fenretinide (2 and 4 μM), and the combination of the two, respectively. The median frequency of total colony-forming units (CFUs) from untreated CP specimens was 161 per 1000 cells plated, ranging from 68 to 440. In contrast, the frequency of CFUs on the medium with fenretinide was reduced to 69%±4% (2 μM) and 39%±5% (4 μM) of that from the untreated specimens (the top panel of Fig. 2A). Imatinib was reported to exert a moderate effect on the suppression of primitive CML cells (20). Consistent with this report, the frequency of CFUs on the medium with imatinib was reduced to the level (68%±4%) equivalent to that resulting from fenretinide. With the addition of 2 and 4 μM fenretinide present in the medium, however, frequencies of CFUs dropped to 35%±3% (p<0.01) and 18%±3% (p<0.01), respectively, implicating that fenretinide significantly increases the ability of imatinib to suppress primitive CML cells.
CP, chronic phrase; M, male; F, female; ND, not determined; IM, imatinib; BM, bone marrow; LP, leukapheresis product; –, no mutation found; CML, chronic myeloid leukemia; CP, chronic phase.
CFUs are mainly composed of burst-forming units-erythroid (BFUs-E) and colony-forming units-granulocyte/macrophage (CFUs-GM), respectively derived from erythroid progenitors and myeloid progenitors. As illustrated in the middle panel of Figure 2A, BFUs-E appeared to be highly sensitive to fenretinide or fenretinide plus imatinib. The relative number of BFUs-E was reduced from 59%±7% to 23%±6% (second and third columns), when the concentration of fenretinide was increased from 2 to 4 μM. With the addition of imatinib, such numbers were further reduced to 19%±5% and 6%±3% (fifth and sixth columns), respectively. CFUs-GM appeared to be less sensitive to fenretinide or fenretinide plus imatinib (the bottom panel of Fig. 2A), as compared with BFUs-E. However, significantly (p<0.01) reduced CFUs-GM, as resulting from imatinib combined with 2 μM of fenretinide or 4 μM of fenretinide, suggests that the efficacy of imatinib is potentiated. In addition to the reduction in colony number, a marked reduction in colony size of CFUs-GM appeared to be particularly evident in the medium with imatinib plus fenretinide (Fig. 2B).
Granulocyte, erythrocyte, and monocyte, megakaryocyte-colony-forming units (CFUs-GEMM) are a distinct category of colonies that are derived from more primitive hematopoietic progenitor cells. Although present in a small percentage, they may give rise to a variety of myeloid lineages under certain circumstances. As demonstrated in Figure 2B, a total of 64 such colonies were detected with untreated CML specimens and 30 were observed with the specimens treated by imatinib. Remarkably, no such colonies were detectable with the specimens treated with either fenretinide or fenretinide plus imatinib, indicating that fenretinide and its combinations with imatinib are particularly effective for the growth inhibition of CFUs-GEMM in CML patients.
In addition, we included CD34+ cells from five healthy donors in CFC assays to evaluate the potential cytotoxicity of the agents and their combinations with the indicated concentrations to normal cells. As shown in Figure 2C, the cytotoxicity of the agents and their combinations on these cells appeared to be minor or minimal. Based on CFC assays of thousands of colonies derived from several dozens of CML specimens, we have shown that fenretinide is able to inhibit growth of colonies from multi-lineages of CD34+ CML progenitor cells, particularly with regard to erythroid progenitor and pluripotent/multipotent progenitor cells. In addition, we have shown that this agent increases the ability of imatinib for growth inhibition of various progenitor-derived colonies in CML.
Fenretinide induces apoptosis in CD34+ CML cells that are refractory to imatinib
Next, we conducted apoptosis assays on CD34+ CML cells cultured in serum-free medium (SFM) under physiological growth factor conditions (20) and, respectively, with the presence of fenretinide, imatinib, or combinations of the two for 48 h. CD34+ cells are known hierarchical, and categorized into the minor portion of CD34+CD38− cells and the major portion of CD34+CD38+ cells. CD34+CD38− cells are composed of more primitive progenitors, including hematopoietic stem cells, whereas CD34+CD38+ cells are composed of more mature progenitor cells. Previous reports have implicated that imatinib is ineffective to eradicate CD34+ CML progenitor cells, particularly with regard to those which are more primitive (7, 17). Probably consistent with these reports, apoptotic effects exerted by imatinib on CD34+CD38+ cell were significant, whereas those on CD34+CD38− cells were minor/minimal (Fig. 3A). Interestingly, a four-fold increase in the concentration of imatinib (i.e., from 0.25 to 1 μM) was unable to increase the apoptosis level in either CD34+CD38+ or CD34+CD38− cells. In contrast, a higher concentration of fenretinide appeared to induce more apoptosis in both categories of the cells, in which CD34+CD38− cells were particularly sensitive to fenretinide-induced apoptosis. In the combined treatments, although the apoptosis levels were lower than or equivalent to those in the fenretinide-alone treatments, it was evident that apoptosis rates revealed by the combined treatments were significantly higher than those revealed by imatinib-alone treatments, particularly with apoptosis rates revealed in CD34+CD38− CML cells. In a reversed manner, we examined proportional changes of CD34+CD38− cells under the indicated treatment conditions (Fig. 3B, C). Indeed, percentages of CD34+CD38− cells in the samples treated with fenretinide or fenretinide plus imatinib were reduced significantly (p<0.01), as highlighted by a roughly three- to fourfold reduction. It is reported that cells in the CD34+CD38− subpopulation are particularly refractory to imatinib (17, 20). However, recent in vitro data show that imatinib reduces the number of CD34+CD38− cells to some extent (7). Probably consistent with these data, our in vitro experiments revealed a slight effect of apoptosis on CD34+CD38− CML cells by imatinib, though statistically not significant (Fig. 3A, C). Relatively, CD34+CD38+ CML cells are more sensitive to imatinib (Fig. 3A). Since the absolute number of CD34+CD38+ CML cells or CD34+CD38− CML cells are reduced rather than increased, a proportional increase in CD34+CD38− CML cells on imatinib treatment (Fig. 3B) suggests a proportional decrease in CD34+CD38+ CML cells of the treated sample. Nevertheless, our data add strong evidence that CD34+CD38− CML cells which are largely refractory to imatinib are highly sensitive to fenrentinide. Of note, the results cited earlier may also implicate that the inhibitory effects of fenretinide, as observed in CFC assay (Fig. 2A, B), are likely due to apoptotic mechanisms triggered by the agent. Although imatinib appears to exert some inhibitory effects on the formation of CFUs, CFUs-GM, and BFUs-E as well, mechanisms underlying the effects are possibly different, as apoptotic induction by imatinib is not obvious in CD34+ CML cells, particularly with regard to the cells of the CD34+CD38− subpopulation. As additional controls, CD34+ cells from three healthy donors were included in the assays (Fig. 3D), providing evidence that the apoptotic impacts of fenretinide or fenretinide plus imatinib with the indicated concentrations on normal CD34+CD38−/CD34+CD38+ cells were minimal.
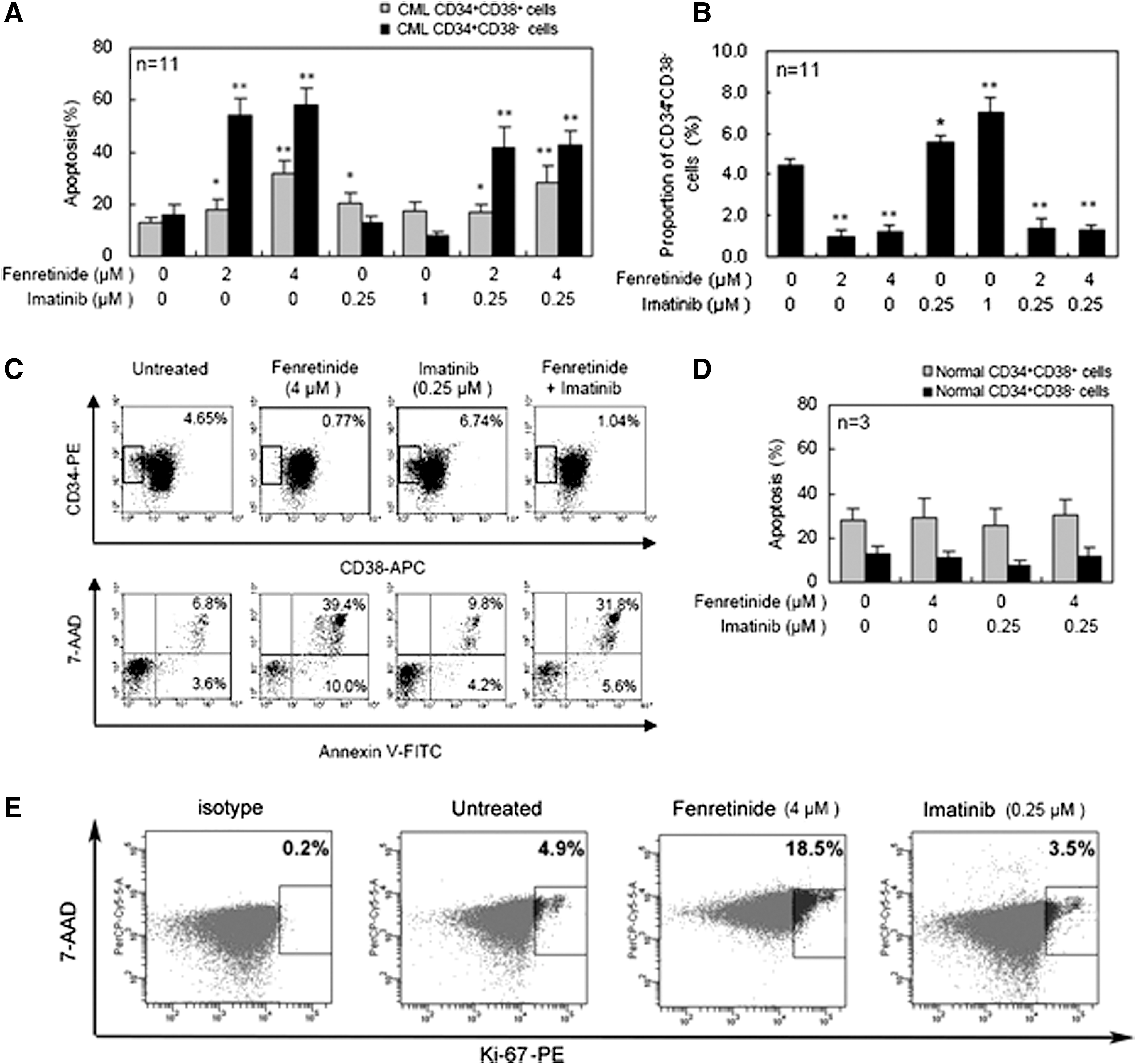
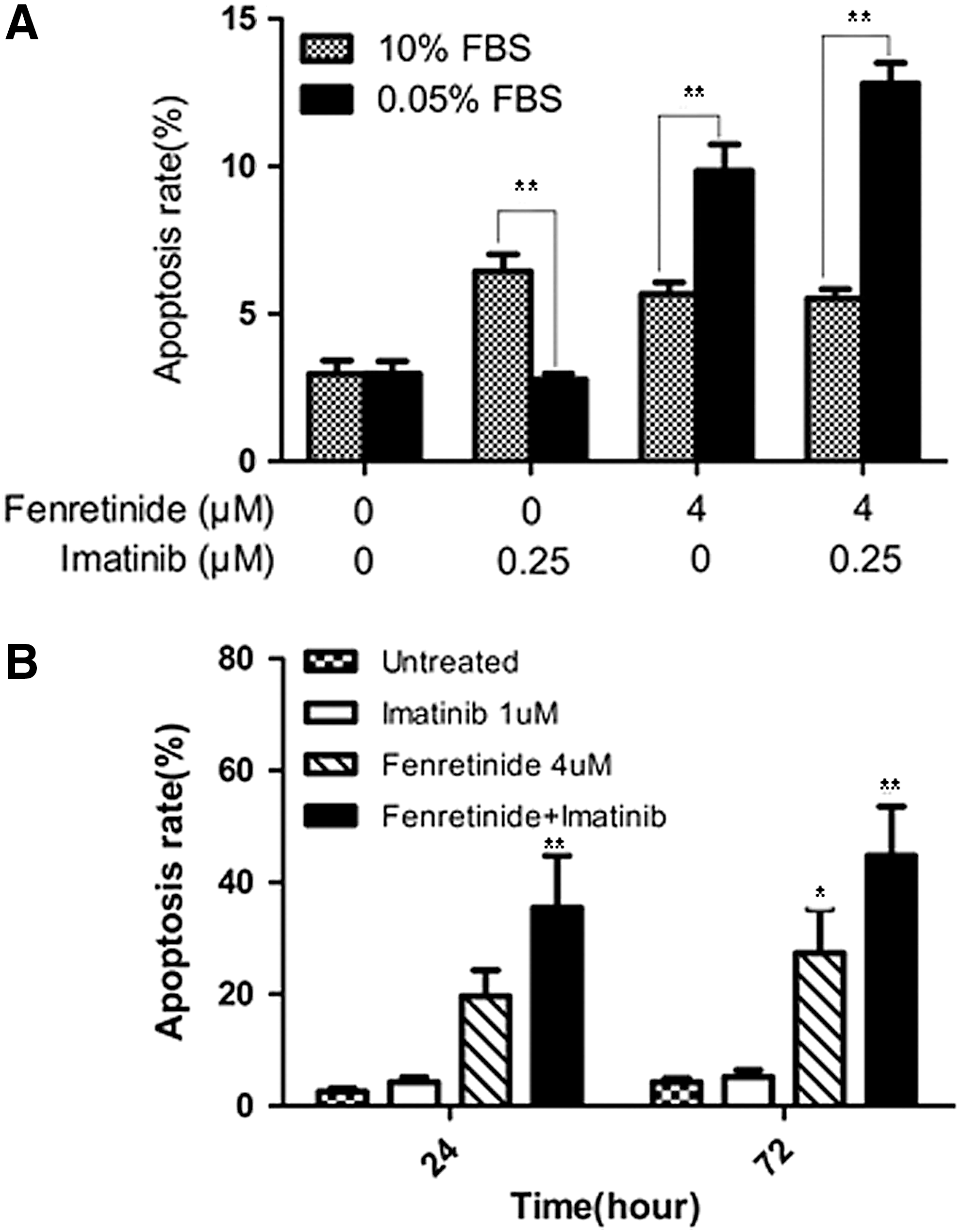
To test whether fenretinide exerts an effect on nonproliferative CML cells in the CD34+ population, we first conducted a Ki-67 expression assay (7) to monitor potential changes of proliferative/nonproliferative cells before and after treatment by fenretinide, as compared with imatinib. Ki-67 is a nuclear protein expressed in proliferating cells. It is expressed in all phases of the cell cycle except G0. The Ki-67 protein is exclusively expressed in proliferative cells and whose negativity is considered quiescence in CML (7). As illustrated in Figure 3E, 4.9% of the untreated CD34+ cells were positive with Ki-67 and a similar percentage (3.5%) of the imatinib-treated cells were positive with Ki-67. In sharp contrast, 18.5% of the fenretinide-treated cells were positive with Ki-67, implicating that the proportion of the nonproliferative cells which were quiescent in fenretinide-induced apoptosis cells were higher. Since the absolute number of CD34+ CML cells are reduced rather than increased after fenrentinide treatment for 48 h (∼30%; Fig. 3A), a sharp increase in the percentage of proliferating cells indicates a considerable decrease of nonproliferative cells in fenretinide-treated samples. In addition, we conducted a serum-starvation assay in K562 cells, which may result in the cell cycle arrest at the G0 phase and, thus, cell quiescence. As illustrated in Figure 4A, serum-starved K562 cells become sensitive to fenretinide even at the 12-h time point. With prolonged exposure to fenrentinide or fenretinide plus imatinib, the apoptosis rate of the serum-starved cells is much faster than that of the cells in the presence of 10% serum. In addition, imatinib-resistant BCR-ABL-T315I Ba/F3 cells were separately treated with imatinib (1 μM), fenretinide (4 μM), or the combination of the two for 24 and 72 h. The combination of fenretinide with imatinib induced more apoptosis than imatinib or fenretinide alone, as evaluated by Annexin V kit analysis (Fig. 4B). Thus, the inclusion of fenretinide for the treatment of CML may represent a strategy that potentiates the efficacy of imatinib in CML patients.
Involvement of oxidative stress responses in apoptosis induced by fenretinide in CD34+ CML cells
Although the CD34+ cell population is hierarchical, possible changes in genes or proteins associated with the apoptosis might be detectable through global approaches. Accordingly, CD34+ CML cells with and without fenretinide treatment were profiled using whole genome expression arrays (Affymetrix HG-U133 Plus 2.0 platform) (patients CML32, CML33, CML34, and CML 35; Table 1). To minimize potential data biases, both treated and untreated cell samples were maintained in culture for 48 h before hybridizations were performed. After data acquisition, the linear models for microarray data (LIMMA) method (15) was used to identify differentially expressed genes (based on the adjusted p-values<0.1; see Materials and Methods section) in a pair-wise manner. Based on four sample pairs, a total of 610 genes (813 probe sets) were identified as commonly regulated genes, of which 357 (478 probe sets) were up-regulated and 253 (335 probe sets) were down-regulated (the left panel of Fig. 5 and Supplementary Table S1; Supplementary Data are available online at
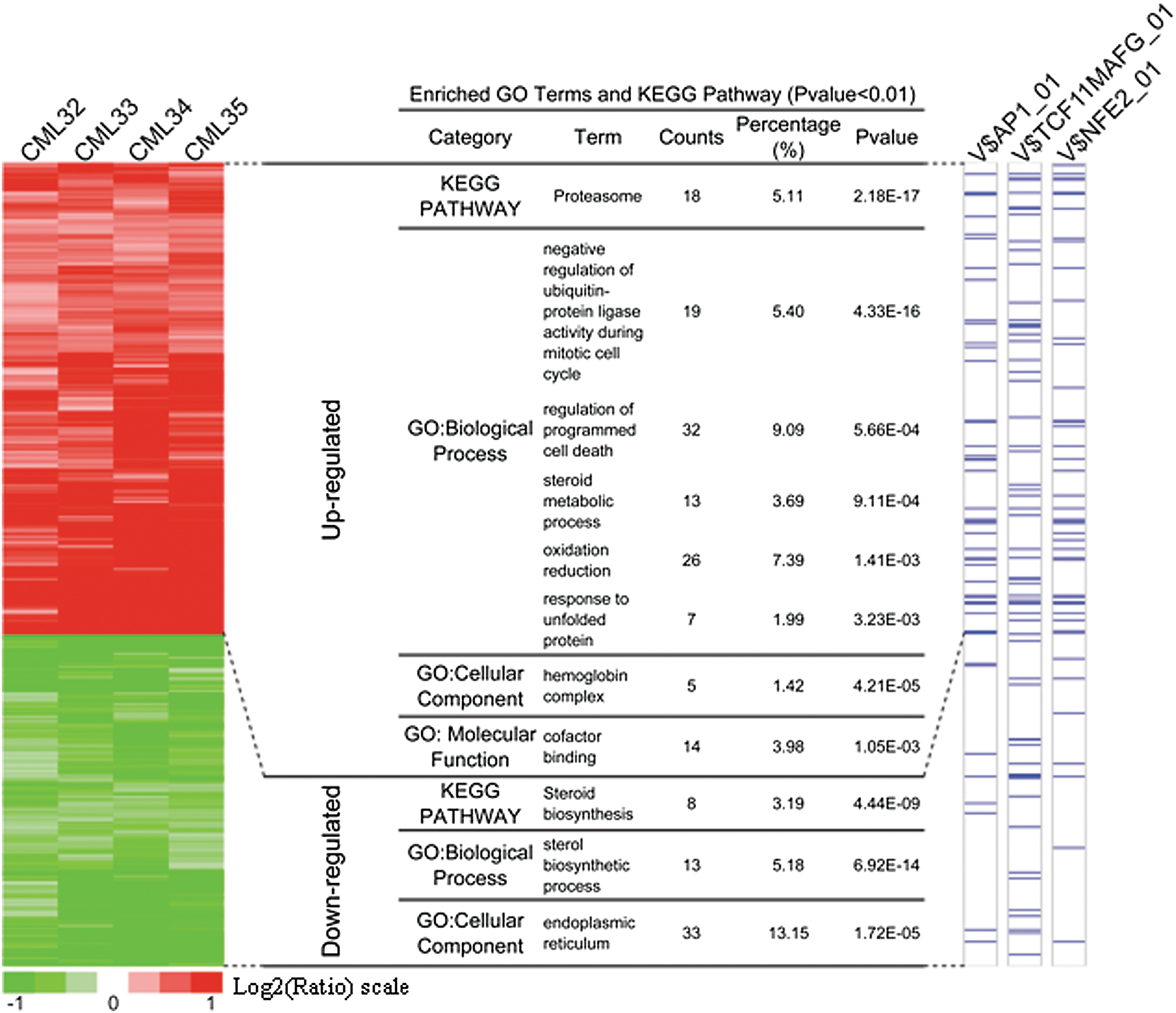
To recognize prominent features inherited to the data, we, respectively, employed UCSC conserved transcription factor binding site (TFBS) (35) and gene ontology (GO) (1, 34) for the enrichment analysis (13). As illustrated in the middle panel of Figure 5, a number of significant GO terms/KEGG pathways were revealed in either the up-regulated or down-regulated gene category. Interestingly, the terms or pathways enriched in the up-regulated category strongly implicate the occurrence of oxidative stress-mediated apoptosis in fenretinide-treated cells, as typically highlighted by genes involved in programmed cell death, redox reactions, and unfolded protein response (UPR). In contrast, the most significant GO terms in down-regulated genes were those associated with endoplasmic reticulum (ER) functions such as steroid biosynthesis. Through hierarchical clustering followed by integration of genomic TFBS information (the right panel of Fig. 5), significantly enriched TFBSs in promoter regions of the up-regulated genes were represented by those of AP-1, TCF11/MafG, and NF-E2 (p-value: p=2.72×10−6, p=6.33×10−6, and p=2.25×10−7, respectively), suggesting that these transcription factors were potentially important for transcriptional changes in response to fenretinide treatment in CD34+ CML cells. AP-1 represents a dimeric complex consisting of various members of the Jun, Fos, and activation transcription factor sub-families, and it, thus, regulates a variety of biological events, including stress response and cell apoptosis (23, 49). TCF11, also known as NRF1, regulates a battery of cytoprotective genes through antioxidant response elements (AREs) in their promoter regions under stress stimuli (41). TCF11 also mediates proteasome homeostasis by inducing proteasome gene transcription, a crucial event in UPR (43). Heterodimerization with the small Maf protein MafG increases the DNA-binding affinity of TCF11, mediating gene transcription that is important for redox signaling and UPR (25). NF-E2 is a key transcription factor in controlling pathways of heme and globin synthesis involved in erythroid cell differentiation (2), thus regulating gene transcription of erythroid and megakaryocytic cells (29, 42). It was known as a negative regulator of erythroid proliferation in erythroleukemias (30), and its regulatory role was largely attributed to the control of its homeostatic concentration, which was mediated by P-JNK (29).
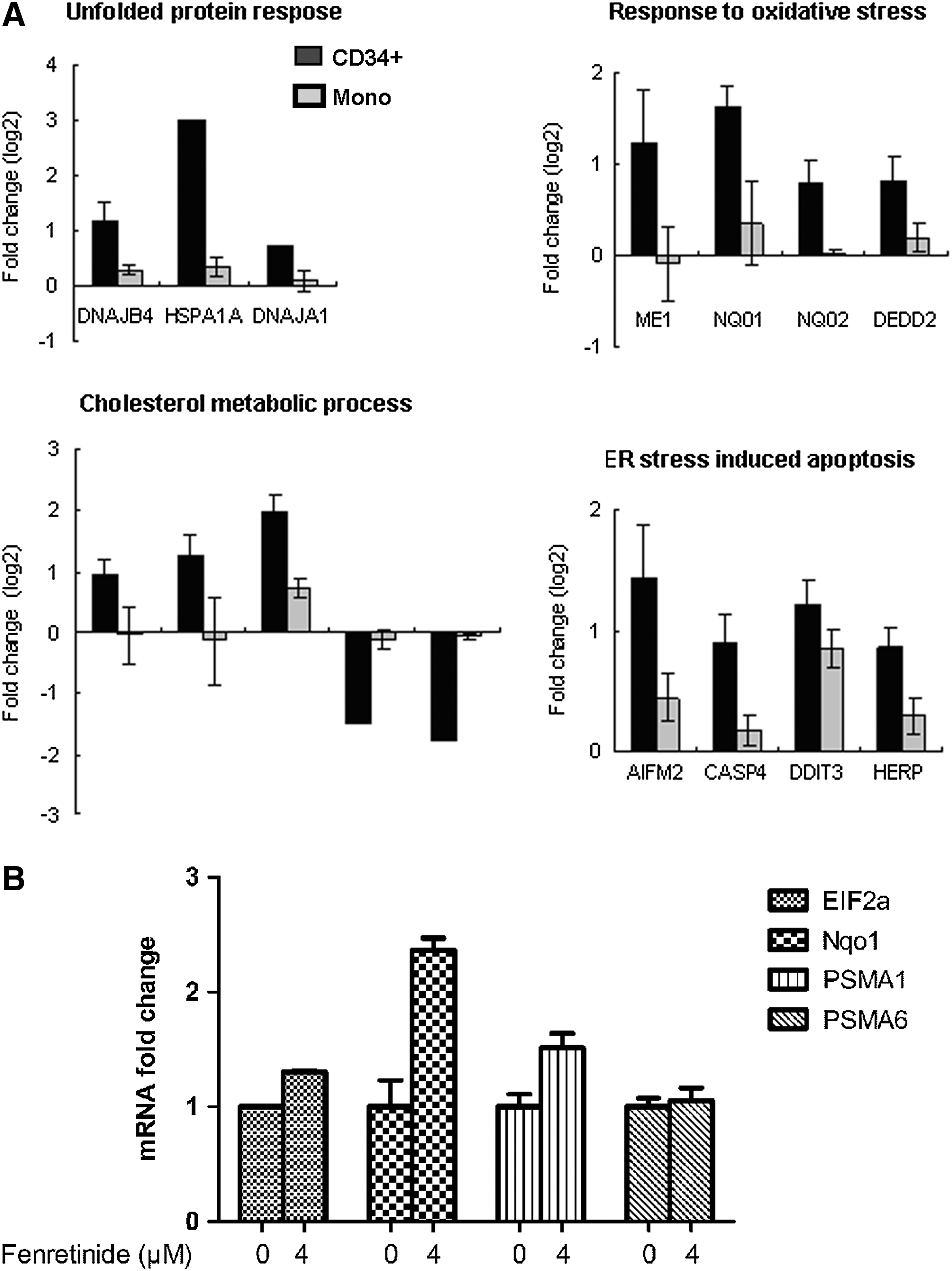
Fenretinide is known as an agent with a primary mode of action to increase cellular levels of reactive oxygen species (ROS) (24, 52, 57). In accordance, prominent features commonly implicated by transcriptome responses of CD34+ cells from the four patients appeared to be indeed relevant to various cellular stress responses, which were probably triggered by ROS. For instance, up-regulation of a number of oxidative responsive genes (e.g., HMOX1, SRXN1, NQO1, NQO2, DDIT3, and GCLM) suggested that anti-oxidative activities occurred in CD34+ cells after fenretinide treatment (Supplementary Table S2). Genes involved in these processes are represented by those encoding heat shock proteins (e.g., HSPA1A, HSPAIB, HSPA6, DNAJB1, DNAJB4, and BAG3) and components of proteasome (e.g., PSMB4, PSMB7, PSMC2, PSMC6, and PSMD14) suggested the occurrence of ER stress and, thus, UPR in the cells. Enhanced steroid metabolic process and reduced steroid biosynthesis, as strongly suggested by the data (Fig. 5), might be relevant to ER stress and UPR as well (28). The markedly induced hemoglobin genes were probably correlated with NF-E2-mediated erythroid differentiation. Taken together, transcriptome changes induced by fenretinide in CD34+ cells of CML suggest the occurrence of a series of stress-responsive events during the apoptosis induced by fenretinide.
Molecular evidence for the involvement of stress-mediated apoptosis in CD34+ CML cells on fenretinide treatment
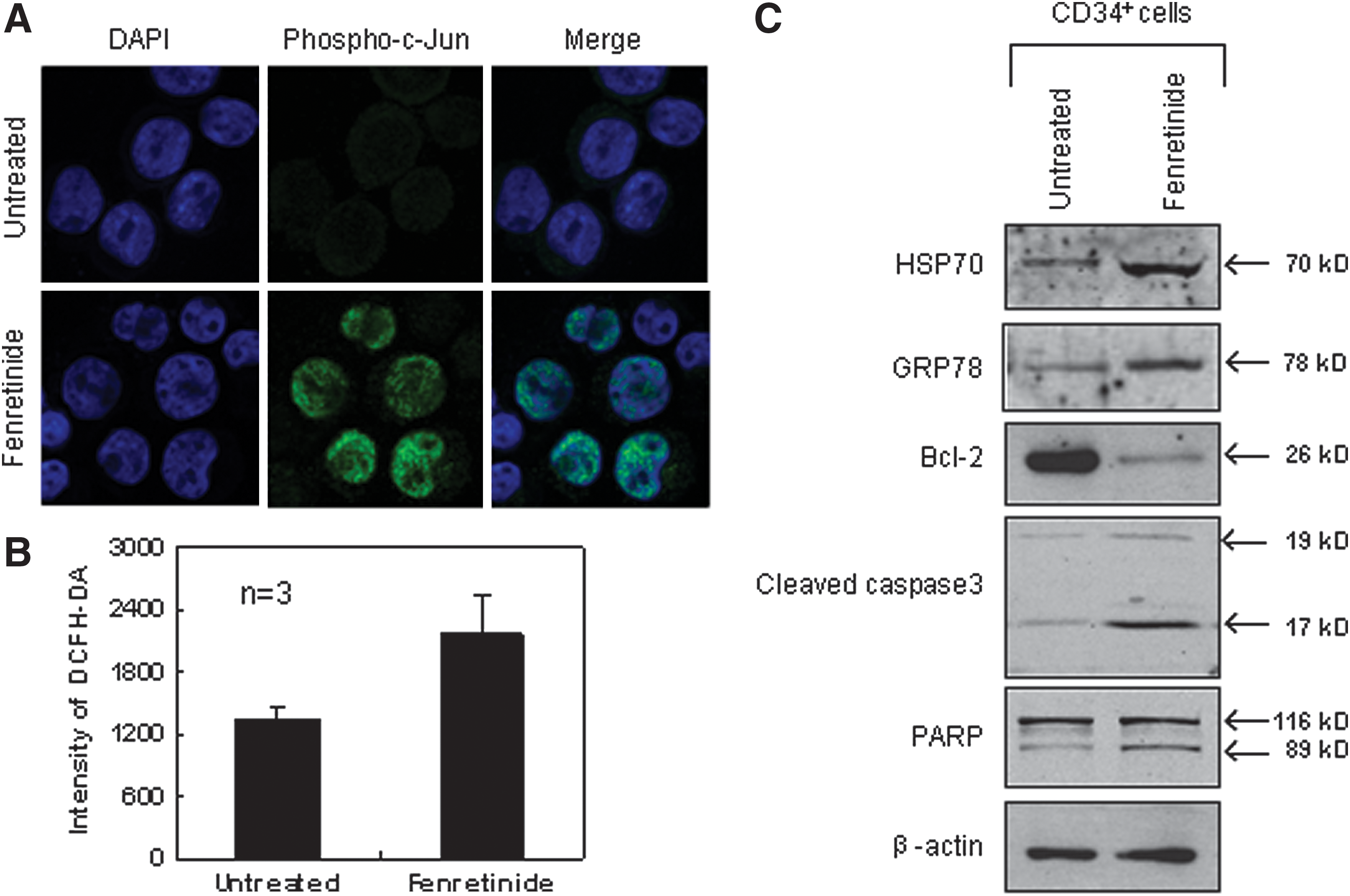
Next, we randomly selected genes, and compared their expression levels between the CD34+ cells and the monocytes after fenretinide treatment of six patients through reverse transcription-polymerase chain reaction (RT-PCR). As shown in Figure 6A, genes involved in oxidative stress, UPR, and ER stress-mediated apoptosis were significantly up-regulated in the CD34+ cells, as compared with the monocytes. In addition, aberrant ER function of steroid metabolic processes appeared to be evident. We also checked expression levels of four ARE cis-element containing genes in the CD34+ cells before and after fenrentinide treatment (Fig. 6B). As illustrated, three out of four revealed obvious changes, adding further evidence that redox signaling is involved in transcriptional regulation of this setting. Since activation of c-Jun, a potent member of AP-1 complex, was considered a pro-apoptotic signal under various stress conditions (50), we performed immunofluorescence assays using specific antibodies for phosphorylated c-Jun to test whether this transcription factor was activated in CD34+ CML cells after fenretinide treatment. As shown in Figure 7A, c-Jun was significantly phosphorylated and translocated into the nucleus, suggesting the activation of AP-1 in the fenretinide-induced apoptosis. To test whether the level of ROS was elevated in the fenretinide-treated cells, flow cytometry analysis by DCFH-DA staining was applied (Fig. 7B), showing that the ROS level was roughly increased by twofold, as compared with the untreated cells (p<0.05). Elevated levels of HSP70 and GRP78 are often used as landmarks for ER stress and UPR. As shown in Figure 7C, both HSP70 and GRP78 were up-regulated in CD34+ CML cells after fenretinide treatment. Bcl-2 is a distinguished protein of anti-apoptosis. The marked reduction of this protein, as illustrated in Figure 7C, indicated the occurrence of apoptosis in CD34+ CML cells treated with fenretinide. Caspase-3 is a downstream executor, and PARP is a common target of caspases. Accordingly, increased amounts of cleaved forms of both the proteins, as detected by the indicated antibodies (Fig. 7C), added additional layers of evidence that apoptosis occurred in cells that were fenretinide treated. Collectively, cellular and molecular data appear to be consistent with transcriptome data that fenretinide induces oxidative stress in CD34+ CML cells, which trigger a series of stress-responsive activities, including anti-oxidative activities, ER stress, and UPR, and, consequently, activate apoptotic caspases.
Discussion
Using imatinib to treat CML represents the first generation of targeted therapy in human malignancy, yielding a remarkable clinical outcome in terms of 5-year disease-free survival. However, late relapse is almost inevitable in imatinib therapy, which is largely attributed to BCR-ABL positive stem/progenitor cells, as these cells are refractory to imatinib and they are able to re-establish CML cell populations. Accordingly, much attention has been now attracted to the development of more sophisticated strategies by combining additional agent(s), which may, on the one hand, target more matured CML cells and, on the other hand, eradicate CML stem/progenitor cells. In this regard, fenretinide may represent a promising candidate. With decades of history in both laboratory and clinical studies, this vitamin A derivative has revealed distinct advantages compared with many other agents for treating tumor and leukemia (24, 36, 40). In addition to its efficacy against a wide range of tumor types, side effects exhibited by fenretinide are minimal, such as impaired night vision adaptation and dry skin, which readily disappear after treatment cessation (32). This agent also appears to be able to synergize with many other anti-cancer agents for apoptosis induction (31, 33, 48). In particular, fenretinide is known as a chemo-preventive agent and is able to reduce the risk of second breast cancer in premenopausal women (56), which has led us to speculate whether this agent possesses the ability to kill cancer cells at early stages. In this setting, we have shown data that fenretinide significantly enhances the efficacy of imatinib, thus offering a promising potential to overcome/reduce drug resistant and probably disease relapse as well in CML patients. Fenretinide markedly improves the ability of imatinib to induce apoptosis in K562 cells (Fig. 1) and exerts similar effects on primary CML cells (Figs. 2 and 3). Through CFC assays, we have shown that colonies derived from CD34+ primitive CML cells are suppressed by fenretinide or fenretinide plus imatinib, particularly with regard to those colonies derived from erythroid progenitors and pluripotent/multipotent stem/precursor cells of CML. In accordance with CFC results cited earlier, apoptosis assessments show that fenretinide induces cell death in both subpopulations of CD34+CD38+ and CD34+CD38− in a dose-dependent manner, whereas the apoptotic induction in the later is more efficient than that in the former, indicating that the stem-cell-enriched CD34+CD38− subpopulation is highly sensitive to fenretinide. Obviously, much remains to be learned regarding the potential of fenretinide for targeting CML stem cells. However, data of this setting have shown that fenretinide may enhance the efficacy of imatinib for the treatment of CML, as primitive CD34+ CML cells are refractory to imatinib. Even in imatinib-resistant cells, fenretinide appears to potentiate the efficacy of imatinib (Fig. 4).
Numerous studies have shown that fenretinide can induce apoptosis in cancer cells via oxidative stress (6, 18). However, mechanisms underlying the apoptosis can be complex and versatile. In this report, our data suggest that mechanisms underlying the apoptosis induced in CD34+ CML cells can be multifaceted, possibly attributed to the heterogeneity of the CD34+ cells. Nevertheless, downstream events of redox signaling that triggers the apoptosis appear to be more definitive, as highlighted by the occurrence of typical ER stress and UPR. In contrast, oxidative stress responses in our recent study of fenretinide on CD34+ AML cells (58) were not as prominent as those in fenretinide-treated CD34+ CML. Possible explanations would include that CML stem/progenitor cells were more close to hematopoietic stem/progenitor cells than AML stem/progenitor cells whereas less oncogenic. Based on TFBS analysis of the prominently regulated genes in this setting, transcription factors, including AP-1, TCF11/MafG (NRF1) and NF-E2, are likely to play important roles in the regulation of genes involved in the stress-responsive JNK pathway. However, transcription factors responding to fenretinide treatment in cancer cells can be versatile and cell type dependent. For instance, fenretinide-induced apoptosis in leukemic NB4 cells appears to be a typical process of oxidative stress-mediated apoptosis, involving a number of stress-responsive events such as ER stress and UPR, and transcriptionally orchestrated by NRF2 and HSF1 (57). In addition, stress-activated signaling pathways in fenrentinide-treated cancer cells can be different to some extent. For example, ceramide signaling is important to oxidative stress-mediated apoptosis in many type of cancer cells; whereas in three fenretinide-treated leukemic cell lines (i.e., NB4, U937, and HL-60), this signaling is only critical to the apoptosis in one of them (i.e., HL-60) (24). It should be noted that mechanistic studies of this setting are based on hierarchical CD34+ CML cells rather than on isolated CML progenitor or stem cells, and, thus, additional efforts are needed to elucidate the detailed mechanisms involved. On the other hand, intracellular redox homeostasis plays a crucial role in various biological processes under both physiological and pathological conditions (59). As byproducts of numerous cellular events, ROS may serve in turn as signaling molecules to regulate cellular events such as differentiation and apoptosis. In hematopoiesis, erythroid lineages and hematopoietic stem cells are highly sensitive to ROS signaling (16, 37). Erythroid precursors synthesize and accumulate hemoglobin, and circulating erythrocytes carry oxygen bound to hemoglobin and, as such, they are prone to oxidative damage. In erythroid cells of healthy individuals, over-produced ROS can be scavenged by antioxidants, protecting the cells from oxidative damage. In erythroid cells of CML, however, the ability to maintain redox homeostasis is probably impaired, which may explicate why erythroid progenitor cells are sensitive to fenretinide treatment in this setting. Hematopoietic stem cells are known to have low levels of metabolic activities, thus reducing the risk of the damage from metabolic products such as ROS. However, increased ROS may cause DNA damage and loss of quiescence, leading to accelerated aging of hematopoietic stem cells or formation of hematopoietic malignancies (16, 44). Since PI3K/AKT pathways are abnormally activated by BCR-ABL in CML cells, which may consequently impair downstream FoxOs, a key regulator in the maintenance of redox homeostasis in hematopoietic stem/progenitor cells (54). Interestingly, although ROS levels of CD34+CD38− CML cells appear to be equivalent to those of CD34+CD38+ CML cells, higher levels of ROS are observed in quiescent CML cells, as compared with proliferative CML cells (38). Such observations may, therefore, explicate why quiescent CML cells are highly sensitive to fenretinide in this setting. Since much of our knowledge about redox perturbation by fenretinide comes from various cancer cell lines, it remains to be elucidated whether this agent induces equivalent changes in ROS in CML stem/progenitor cells in order to trigger efficient apoptosis. However, the nature of fenretinide for inducing apoptosis in these stem/progenitor cells may allow us to address such a complex question in near future.
Materials and Methods
Cell culture, viability, and apoptosis assay
CML-derived cell line K562 cells and serum-starved K562 cells were maintained in RPMI 1640 supplemented with 10% and 0.05% fetal bovine serum (FBS) (PAA), respectively. Imatinib-resistant BCR-ABL-T315I Ba/F3 cells were kindly provided by Dr. Wenli Feng of the Department of Clinical Hematology, Key Laboratory of Laboratory Medical Diagnostics Designated by the Ministry of Education, Chongqing Medical University. BCR-ABL-T315I Ba/F3 cells were separately treated with imatinib (1 μM), fenretinide (4 μM), or the combination of the two for 24 and 72 h. Imatinib was kindly provided by Novartis Pharma, and fenretinide was purchased from Sigma. K562 cells were, respectively, treated with imatinib (0.25 μM), fenretinide (4 μM), and a combination of the two for 24, 48, and 72 h. Serum-starved K562 cells were also treated with imatinib (0.25 μM), fenretinide (4 μM), and a combination of the two for 12 h. Viable cells were counted by trypan blue exclusion. Apoptosis assays were performed using Annexin V-FITC apoptosis detection kit (BD Biosciences PharMingen), and mitochondrial transmembrane potential (ΔΨm) was evaluated using rhodamine 123 (Sigma) and propidium iodide staining, followed by flow cytometry.
CML specimens, CD34+ cell isolation, and culture
Fresh bone marrow cells or leukapheresis products were obtained from 37 CML patients in the newly diagnosed CP and five normal donors with informed consent and approval of Institutional Review Board at the School of Medicine in Shanghai Jiao Tong University (Table 1). Mononuclear cells were isolated by Ficoll density gradient centrifugation. CD34+ cells were enriched using EasySep® Human CD34 Positive Selection kit (Stem Cell Technologies) according to the manufacturer's instructions. The purity of CD34+ cells ranged between 92% and 98% in all samples determined by flow cytometry. CD34+ cells were cultured in a STEMPRO-34® SFM Complete Medium (Invitrogen) supplemented with growth factors (PeproTech) (20). The growth factors consisted of GM-CSF (200 pg/ml), G-CSF (1 ng/ml), SCF (200 pg/ml), LIF (50 pg/ml), MIP-1α (200 pg/ml), and IL-6 (1 ng/ml).
CFC assay
CD34+ cells were, respectively, mixed with imatinib (0.25 μM), fenretinide (2 or 4 μM), and the combinations of the two agents, and a quantity of 1000 CD34+ cells were plated in Methocult H4434 (Stem Cell Technologies). After incubation at 37°C for 14 days, CFUs-GEMM, CFUs-GM, and BFUs-E were, respectively, counted, and all the colony assays were performed in triplicate.
Apoptosis assessment of primitive CD34+ CML cells
CD34+ cells were, respectively, treated with imatinib (0.25 or 1 μM), fenretinide (2 or 4 μM), and the combinations of the two for 48 h in SFM. Then, CD34+ cells were first stained with antibodies against the surface markers CD34-PE and CD38-APC (Beckman Coulter) and incubated at room temperature for 15 min. Cells were then washed with cold phosphate-buffered saline (PBS) and resuspended in Annexin V 1×binding buffer with AnnexinV–FITC (BD PharMingen) and 7-aminoactinomycin (7-AAD) (Molecular Probes). Subsequently, the samples were incubated at room temperature for 15 min and analyzed by flow cytometry using FACSCalibur (BD Biosciences).
Ki-67 analysis
CD34+ cells were, respectively, treated with imatinib (0.25 μM) or fenretinide (4 μM) for 48 h in SFM. Then, the treated CD34+ CML cells were washed twice with wash buffer (PBS with 1% FBS, 0.09% NaN3 pH7.2) and stained for Ki-67 antibody according to the manufacturer's instructions (Beckman Coulter) and 7-AAD staining solution (Molecular Probes), and then tested by flow cytometry analysis using FACS Calibur (BD Biosciences).
Microarray hybridization and data mining
Total RNAs of CD34+ cells from four CML patients with or without 4 μM fenretinide treatment for 48 h were amplified and labeled with biotin according to the standard Affymetrix® protocol. The fragmented and biotinylated cDNA was then subjected to hybridization with the GeneChip® Human Genome-U133 Plus 2.0 array (Affymetrix). Raw expression data were normalized using robust multi-array averaging (RMA) with quantile normalization. The normalized expression data were subsequently imported into LIMMA bioconductor library (15) for the detection of differentially expressed probe sets using paired t-test for the fenretinide versus control comparison across four CML patients. The criteria for identifying the top significant probe sets was based on adjusted p-values (<0.1) corrected using Benjamini and Hochberg procedure. Hypergeometric distribution-based enrichment analyses of biological themes were performed to explore the underlying themes of those differentially expressed genes (p-values<0.1) in terms of biological relevance, for example, functional relevance as revealed by GO (1, 34) enrichment analysis and regulatory relevance as revealed by UCSC conserved TFBS enrichment analysis (13, 35). p-Value was applied to account for multiple hypothesis testing, thus to assess the significance of the biological theme enrichments.
Confocal microscopy
CD34+ CML cells were treated with 4 μM fenretinide for 48 h. Then, cells were fixed, rinsed, and incubated with phospho-c-Jun antibody (Cell Signaling Technology) at 4°C overnight. Cells were stained with goat anti-rabbit Alexa 488 secondary antibodies and Hoechst 33342 (Invitrogen) for nuclear staining. Fluorescence was observed using the×100 oil immersion objective (NA 1.4), further magnified by a×3 zoom, on a Leica SP5 confocal microscope (Leica Microsystems). Images were acquired using Leica confocal software v. 1.8.2 Build 1465.
ROS detection
CD34+ CML cells were treated with 4 μM fenretinide for 24 h. After washing with SFM, cells were incubated in SFM containing 10 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA; Sigma) for 30 min. The elevations of intracellular ROS induced by fenretinide were detected using flow cytometry.
Western blot analysis
Cells were harvested and lysed in 100 μl of ice-cold Triton lysis buffer (0.1% v/v Tx-100). Lysates were cleared by centrifugation, and a sample of the supernatant was removed for protein determination. Equivalent amounts of protein lysate had sodium dodecylsulfate sample buffer added and were boiled for 5 min. Samples were resolved by sodium dodecylsulfate–polyacrylamide gel electrophoresis and blotted onto polyvinylidene difluoride filters. Immunoblotting was performed with specific antibodies for GRP78 (Santa Cruz Biotechnology), HSP70 and Bcl-2 (BD PharMingen), PARP, cleaved caspase-3, and β-actin (Cell Signaling Technology). For detection, blots were incubated with secondary HRP-linked antibodies and visualized using the ECL Plus system (GE Healthcare).
Statistical analysis
Statistical analysis was performed using the Student's t-test and one-way analysis of variance (ANOVA). A p-value less than or equal to 0.05 was chosen to be a statistically significant difference, unless specified otherwise.
Publicly deposited data
The transcriptome profilings of fenretinide-treated and untreated CD34+ cells from CML patients are available at GEO accession GSE17480. The following link is provided for review of record GSE17480 (
Footnotes
Acknowledgments
This work was supported in part by grants from the National Natural Science Foundation (81170503 and 90919059), Chinese Academy of Sciences (KSCX2-EW-Q-1-08), Ministry of Science and Technology of China (2012AA02A505 and 2013CB966802), the Shanghai Commission of Science and Technology (11431922402), and SA-SIBS Scholarship Program (Y.D.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.