
Abstract
Introduction
Excitation is initiated by opening of voltage-gated Na channels. The generated current (INa) is large in amplitude (>10 nA). Due to its short in duration (∼10 ms), the amount of Na ions entering the cell is not sufficient to change intracellular Na concentration greatly. Its large amplitude leads to the fast upstroke of the action potential (AP). Fast Na current inactivation and reduced driving force at positive potentials, together with activation of transient outward rectifying K current (Ito), limits AP amplitude and generates the AP notch. During the AP plateau phase, L-type Ca channels open, resulting in ICa, which maintains AP plateau until delayed rectifying K currents initiate repolarization. Mainly during the AP plateau phase, Ca ions enter the cell via ICa into the dyadic cleft very close to the Ca release channel (ryanodine receptor, RyR2) of the sarcoplasmic reticulum (SR). This relatively small Ca influx results in a Ca-induced Ca release from the SR, which is mainly responsible for the transient increase in cytosolic Ca concentration (Ca transient), resulting in myofilament activation and contraction. For Ca removal, two major pathways are involved: SR Ca ATPase (SERCA2a) and sarcolemmal Na–Ca exchange (NCX1) transfer Ca either into the SR or into the extracellular space, respectively.
There is substantial evidence that disturbed Ca handling is central for contractile dysfunction in HF (17). The mechanisms, however, are incompletely understood but involve activation of stress kinases such as cAMP-dependent protein kinase A (PKA), protein kinase C (PKC), and Ca/calmodulin-dependent protein kinase II (CaMKII) (17). Under pathological stress, excessive and/or protracted phosphorylation of target proteins like the L-type Ca channel, phospholamban, and RyR2 appear to contribute to dysregulation of normal intracellular Ca homeostasis. In addition, expression patterns of Ca regulatory proteins are altered.
SERCA2a expression (and activity), for instance, is reduced, which reduces SR Ca content, Ca transients, and impairs systolic contractile function (17). Increased diastolic RyR2 open probability contributes to reduced SR Ca load and increased diastolic Ca (89). Since intracellular Na and Ca handling are tightly interrelated, changes in Ca handling are accompanied by disturbed Na handling. Accumulation of intracellular Na has been observed in HF (105), mainly due to enhanced Na influx through voltage-gated Na channels (135) and Na/H-exchanger (NHE, 12, 13). Increased intracellular Na enhances Ca entry via reverse mode NCX activity during the AP, while it compromises NCX-mediated Ca export during diastole (9, 11, 18, 38, 104, 141 –143). Thus, increased NCX expression as shown in HF (57, 122), together with increased activation upon ROS (52) may partly compensate for decreased SR Ca load by contributing to the systolic Ca transient (18).
However, increased NCX-mediated Ca influx and reduced Ca efflux may also lead to cytosolic Ca accumulation (137).
Intriguingly, HF is also associated with increased oxidative stress defined as excess production of reactive oxygen species (ROS) and/or reduced antioxidative capacity. Mallat and colleagues showed that levels of lipid peroxides and 8-iso-prostaglandin F2α, the major biochemical markers of ROS generation, were elevated in the plasma and pericardial fluid of patients with HF and correlated with disease severity (92). Electron spin resonance (ESR) spectroscopy provided direct evidence for increased ROS production in HF (69).
It is known that ROS can impair the function of Ca-regulatory proteins. The mechanisms, however, are incompletely understood but involve direct modification of target proteins (i.e., ion channels and transporters) as well as activation of serine/threonine kinases. The latter may act as second messengers translating the ROS signal into an altered function of ion channels and transporters (Fig. 1). Both these pathways may be involved in the initiation and progression of HF.
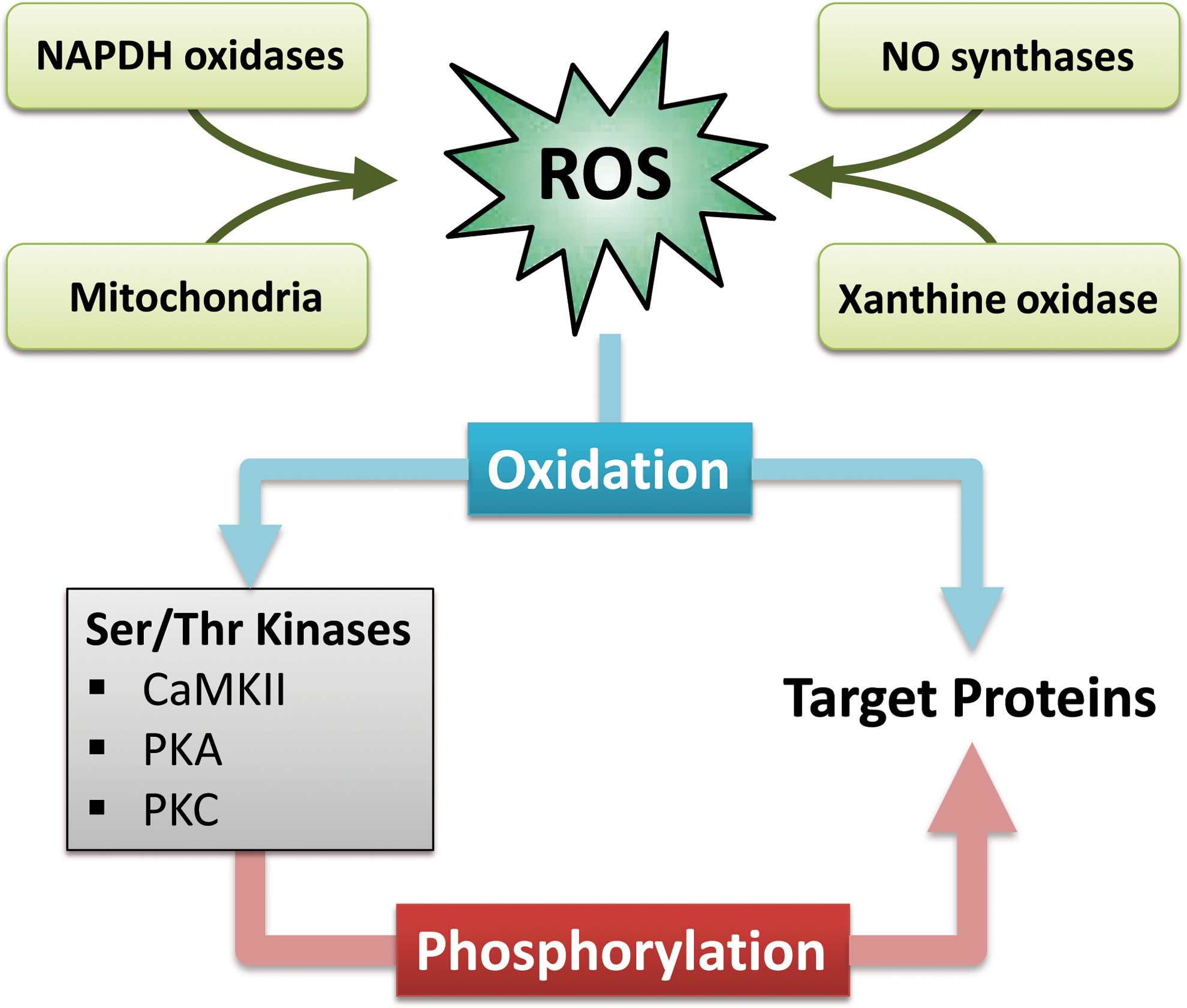
This review will focus on the redox-regulation of intracellular Ca. However, ROS may also play an important role in prohypertrophic and maybe proapoptotic signaling and thus, structural remodeling. Therefore, the reader is encouraged to see references 4, 5, 130, and 131 for a more complete picture of ROS effects on the heart.
The Redox System of the Heart
The redox system of the heart consists of a delicate balance of ROS-generating proteins and antioxidative capacities.
Sources of ROS and antioxidative capacities of the heart
ROS are generated from several intracellular sources including mitochondria, NADPH oxidase, xanthine oxidase, and uncoupled nitric oxide synthase (Fig. 1).
Physiologically, small amounts of superoxide (O2 −) occur upon mitochondrial oxidative phosphorylation but are rapidly inactivated by superoxide dismutase (SOD) into H2O2. O2 − has very limited diffusion distance, in the range of only a few nanometers due to its high reactivity. H2O2, on the other hand, is less reactive and can reach the cytoplasm. H2O2 itself is reduced by gluthathione peroxidase, catalase, and the thioredoxin (Trx) system (Trx reductase and Trx peroxidase) into H2O (73, 127, 148). Gluthathione peroxidase requires gluthathione, the major cytosolic redox buffer. The ratio of reduced to oxidized gluthathione (GSH/GSSG) is usually >10 but can become significantly decreased under pathological conditions (102). Gluthathione peroxidase is present in high amounts in mitochondria and cytosol.
Under conditions of enhanced superoxide production, increased amounts of H2O2 may overwhelm the antioxidative capacities and may lead to the formation of highly reactive hydroxyl radicals (OH−) via Haber–Weiss reaction (with a second molecule O2 −) or via Fenton reaction. Alternatively, superoxide may lead to the generation of peroxynitrite (ONOO−) by reaction with NO (101).
In HF, it has been shown that increased amounts of superoxide are generated in mitochondria (69, 71, 103). In addition, NADPH oxidase (Nox) 2 and 4 are richly expressed in cardiomyocytes, and myocardial Nox activity has been shown to be increased in human HF (59, 87, 119). Stimuli relevant to the pathophysiology of HF (mechanical stretch, endothelin-1 and angiotensin II) are known to induce Nox activity (2, 82). Interestingly, recent evidence suggests that there may be a connection between Nox activation and mitochondrial ROS production. Nox-dependent ROS production may be amplified by mitochondria in a ROS-induced ROS release manner (32 –35). The role of Nox 4, however, is controversially discussed. It was proposed that upregulation and translocation of Nox 4 to mitochondria may augment mitochondrial ROS production and contribute to the progression of heart failure (2, 82). On the other hand, it was shown that genetic deletion of Nox 4 aggravated heart failure development, which was attributed to beneficial effects of Nox 4 on angiogenesis (152).
The third source of ROS, xanthine oxidase, has also been shown to be increased in expression and activity in HF (27). Finally, nitric oxide synthase (NOS) that is exposed to oxidative stress becomes structurally unstable and generates ROS (NOS uncoupling). It was shown that uncoupled NOS contributes to myocardial remodeling upon pressure overload in mice (126). Interestingly, endothelial NOS (eNOS) was shown to co-localize with L-type Ca channels in caveolae, and neuronal NOS (nNOS) was reported to co-localize with RyR2 in the SR (14). This suggests that NOS may play an important role for ROS-dependent Ca handling (25, 154).
Controversy exists with respect to antioxidative capacity in HF. While it was shown in HF following myocardial infarction that antioxidative capacity was reduced (62), others reported increased activity of gluthathione peroxidase in hearts with pacing-induced HF (129). Because of the very short half-life of ROS, there are very different effects on ion channels or ion transporters depending on the source and localization of ROS generation.
Redox-modification of proteins
ROS are known to oxidize sulfhydryl (SH) groups of cysteine residues in proteins, which can lead to the formation of disulfide bonds. The latter affects the tertiary and quaternary structure of proteins, resulting in altered function. Only recently, it was shown that ROS can also oxidize methionine residues, which can also influence structure and function of proteins (42, 74). Many Ca handling proteins have been shown to be subject to ROS-dependent oxidation but the physiological and/or pathophysiological relevance is largely unknown.
Redox Modification of Serine/Threonine Kinases
Since ROS are highly reactive molecules, their intracellular diffusion is very limited. Therefore, ROS generated by endogenous systems can only affect close targets, resulting in very compartmentalized signaling. On the other hand, it has been shown that ROS can have much broader effects on the cardiomyocyte. One explanation for this discrepancy is ROS-dependent activation of serine/threonine kinases. These kinases may translocate, leading to changes in the activity of a broad range of Ca-regulatory proteins by phosphorylation (Fig. 1).
Ca/calmodulin-dependent protein kinase II
In recent years it has become evident that Ca/calmodulin-dependent protein kinase II (CaMKII) is crucial for the regulation of intracellular Ca and excitation–contraction coupling.
CaMKII is a serine/threonine kinase that is robustly expressed in heart tissue. It forms homo-multimeric structures via its association domain (Fig. 2). CaMKII can phosphorylate numerous Ca-regulatory proteins including L-type Ca channel (54, 78), ryanodine receptor (RyR2), phospholamban (PLN, 88). In addition, we have recently shown that CaMKII can phosphorylate cardiac voltage-gated Na channels, which increases Na influx (persistent or late Na current) and prolongs action potential duration (APD, 68, 135).
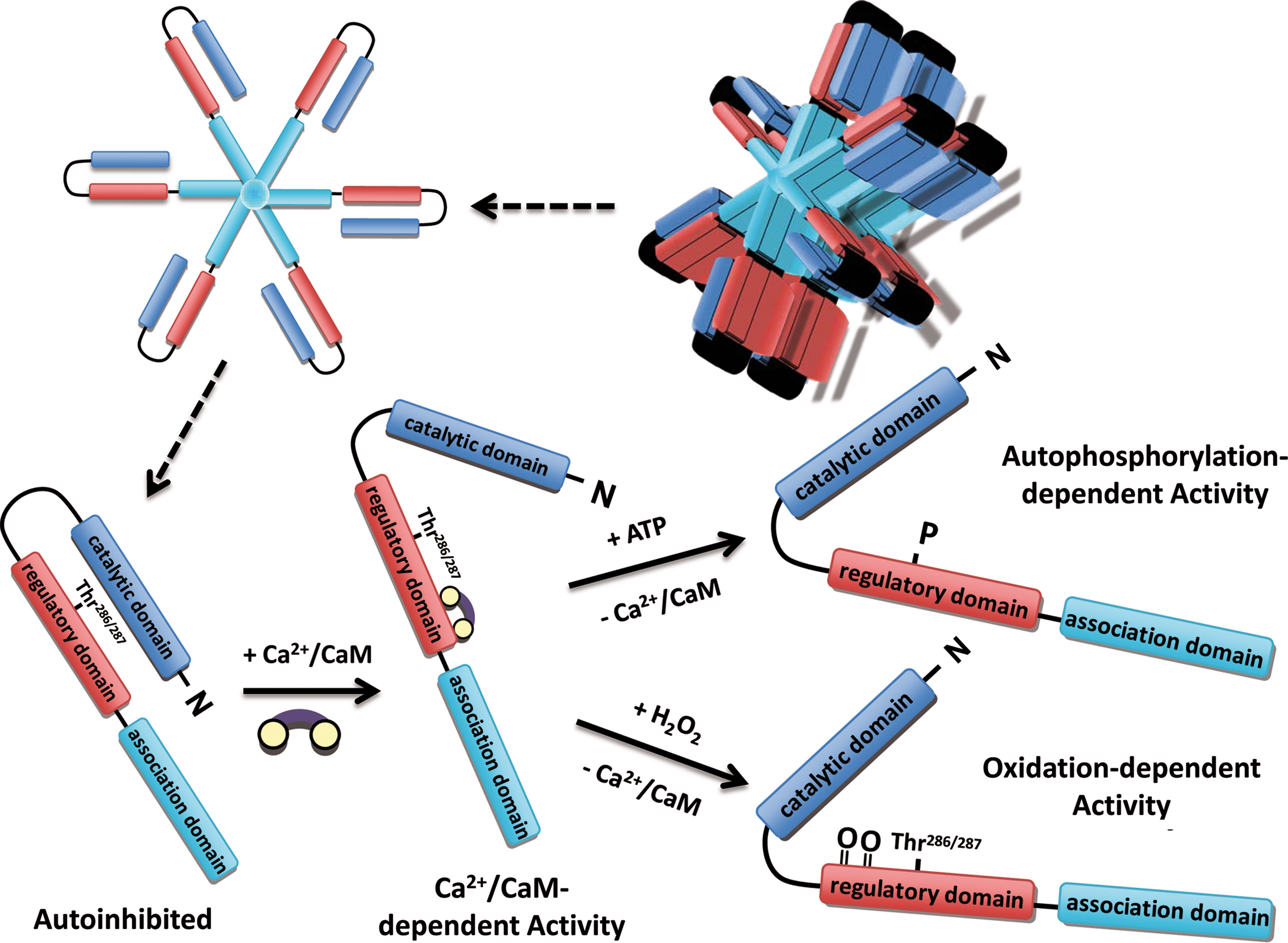
The typical activation is Ca-dependent (116). Active, Ca-bound calmodulin (Ca-CaM) can bind to the regulatory domain of CaMKII, resulting in a conformational change disturbing the interaction of regulatory (autoinhibitory) domain and catalytic domain, which results in ATP-binding and target protein phosphorylation (Fig. 2). Upon activation, inter-subunit autophosphorylation at threonine 286 or 287 (T286 or T287, dependent on species) occurs, which prevents re-association of catalytic and regulatory domain, even when Ca-CaM has dissociated from the catalytic domain (Fig. 2; 81, 117). Therefore, T286 autophosphorylation confers prolonged and autonomous (Ca-CaM-independent) activity. By integrating the relatively fast transient changes in the intracellular Ca concentration, T286 autophosphorylation serves as the Ca “memory” for CaMKII. Beside changes in the enzyme kinetics of inactivation, T286 autophosphorylation is also required for maximal kinetic activity.
Erickson and colleagues described a novel mechanism of Ca-independent CaMKII activation (42). They showed that ROS can oxidize methionine 281/282 (M281/282) in the regulatory domain resulting in an activation mode very similar to autophosphorylation at T286 (i.e., Ca-CaM-independent activity) (Fig. 2). This type of activation can occur, for instance, upon stimulation of endogenous NADPH oxidase 2 (Nox2) by angiotensin II binding to its receptor on cardiomyocytes. Oxidation at M281/282 is a reversible process and oxidized methionine can be reduced by methionine sulfoxide reductase A (42). The pathophysiological relevance of this oxidized CaMKII has recently been verified for the development of sinus node dysfunction (124) and aldosterone-induced heart injury (58). The role of oxidized CaMKII in the cardiovascular system was reviewed recently in detail (43).
Protein kinase A
Catecholamine hormone-binding to the G-protein coupled β receptor stimulates adenylate cyclase, resulting in increased cyclic adenosine monophosphate (cAMP), which activates cAMP-dependent protein kinase A (PKA, 144). Two major forms of PKA, type I and type II, have been described, both of which are organized as tetramers comprising two catalytic and two regulatory subunits. The regulatory subunit can bind to protein kinase A anchor protein (AKAP), which targets PKA to its substrate proteins. Activation of PKA can occur upon binding of two molecules of cAMP to each regulatory subunit. This favors dissociation of the catalytic and regulatory subunit, which results in substrate phosphorylation. It is known that PKA can phosphorylate several Ca-regulatory proteins, including RyR2, L-type Ca channel, and PLN. In addition, PKA phosphorylation of troponin I regulates myofilament Ca sensitivity (109).
There are two types of regulatory subunits (RI and RII) and accordingly, the enzyme is defined as type I or II, respectively. Type I PKA is localized in the cytosol, whereas type II appears to be primarily targeted to AKAP proteins associated with subcellular compartments. There is a large uncertainty about target specificity of the PKA subtypes. Interestingly, peptide substrate enhanced the activation of PKA type I at low, physiologically relevant concentrations of cAMP through competitive displacement of the regulatory RI subunit. This substrate-induced sensitization is not present in type II PKA (133, 134). This suggests that the activity of PKA type I is determined not only by the cAMP level but also by the availability of substrate.
Additionally, it was recently shown that type I regulatory subunit I is subject to oxidation by ROS (24). The oxidation of cysteines 17 and 38 leads to inter-subunit disulfide bond formation (between two regulatory subunits) and dissociation of the PKA holoenzyme complex. The type I PKA translocation (from cytosol to membrane and myofilaments) and activation results in increased cellular contractility without elevations in cAMP. It was suggested that increased PLN phosphoryation and SERCA2a activation underlies the observed increase in contractility (24). The translocation of oxidized type I PKA seems to be beneficial for activation since it favors substrate binding, which is required for full activation.
To date, however, the relevance of this novel PKA type I activation in cardiomyocytes under physiological and pathophysiologocal conditions, especially in comparison to cAMP-dependent activation, is completely unknown.
Protein kinase C
By molecular cloning, at least 12 isozymes of protein kinase C (PKC) have been identified, which are classified by their activation characteristics. The conventional PKC isozymes (α, βI, βII, and γ) are activated by Ca and diacylglycerol (DAG). In contrast, novel isozymes (δ, ɛ, θ, and η) and atypical isozymes (ζ and λ) are Ca-independent but activated by distinct lipids (39). PKCs consist of N-terminal regulatory (autoinhibitory) and C-terminal catalytic domain. When inactive, the regulatory domain is bound to the catalytic domain (63). The binding of the activating factors (distinct lipids and Ca) results in a conformational change, resulting in release of the autoinhibition and activation of the enzyme. Moreover, activation leads to PKC tranlocation and increased membrane association. Recently, intracellular receptor proteins have been described, which bind activated PKC in the presence of phosphatidylserine and Ca (95). The binding site on PKC is distinct from the substrate binding site, and binding was further increased with the addition of diacylglycerol (95). These binding proteins are called receptors for activated C-kinase (RACKs) and may play a role in activation-induced translocation of PKC.
The effects of PKC activation are complex, especially due to parallel activation of several isozymes, isozyme interdependence, cross-talk, and overlapping isozyme effects. The cellular activity of the individual isozymes depends on their expression levels, subcellular localization, and phosphorylation state (91). All these factors, however, vary dramatically between species and cell types. This may explain why experiments using knock-out of single PKC isozymes did not show consistent results. Despite these confounders, it has been consistently shown that G protein-coupled receptor agonists such as isoproterenol and angiotensin II, but also mechanical stretch, induce cardiac hypertrophy through the activation of PKC (20, 125). Also, in various in vivo models of cardiac hypertrophy, it was shown that PKCα and PKCβ are upregulated, PKCɛ is either upregulated or preferentially activated, and levels of PKCδ,λ or ζ do not change (22, 125, 139). During ischemic preconditioning, on the other hand, it was shown that PKCδ and PKCɛ have opposite roles (29, 30, 40). For more information about the complex role of PKC for cardiac hypertrophy, heart failure, and ischemic preconditioning, we refer to the following reviews (30, 39, 40, 91).
PKC has been shown to regulate excitation–contraction coupling. PKCα can phosphorylate inhibitor 1 at Ser67, resulting in increased protein phosphatase 1 activity, leading to phospholamban dephosphorylation and reduced SERCA2a activity (23). PKCα (and maybe also PKCβ) has been shown to phosphorylate troponin I, troponin T, titin, and myosin binding protein C, which leads to decreased myofilament Ca sensitivity (16, 60, 65, 123, 138). Also, PKCα, βI, βII, and γ have been shown to phosphorylate the α1c subunit of the L-type Ca channel (151).
In addition to the Ca and lipid-induced activation, it was shown that mild oxidative stress can activate PKC (53). At high doses of ROS (5 mmol/L H2O2), both catalytic and regulatory domain are oxidized, leading to irreversible inactivation of the enzyme. Low doses of ROS (50 μmol/L H2O2), on the other hand, selectively oxidize the regulatory domain generating a Ca and phospholipid-independent PKC activation (53). The pathophysiological relevance of this novel redox-dependent PKC activation is largely unknown.
Redox Modification of Ca and Na Handling Proteins
It is well established that accumulation of ROS lead to cytosolic Ca overload (49, 120, 128, 134, 136). ROS-induced accumulation of intracellular Na has also been reported (134). Interestingly, it was shown that in failing cardiomyocytes, cytosolic Na overload greatly enhances mitochondrial ROS production (77). This would result in a positive feedback loop greatly augmenting ROS-induced injury. Since intracellular Na and Ca homeostasis are tightly interrelated, changes in Na handling proteins with consequent changes in intracellular Na can have a profound impact on intracellular Ca and contractility. A synopsis of ROS effects on Na and Ca handling proteins is shown in Figure 3 and Table 1.
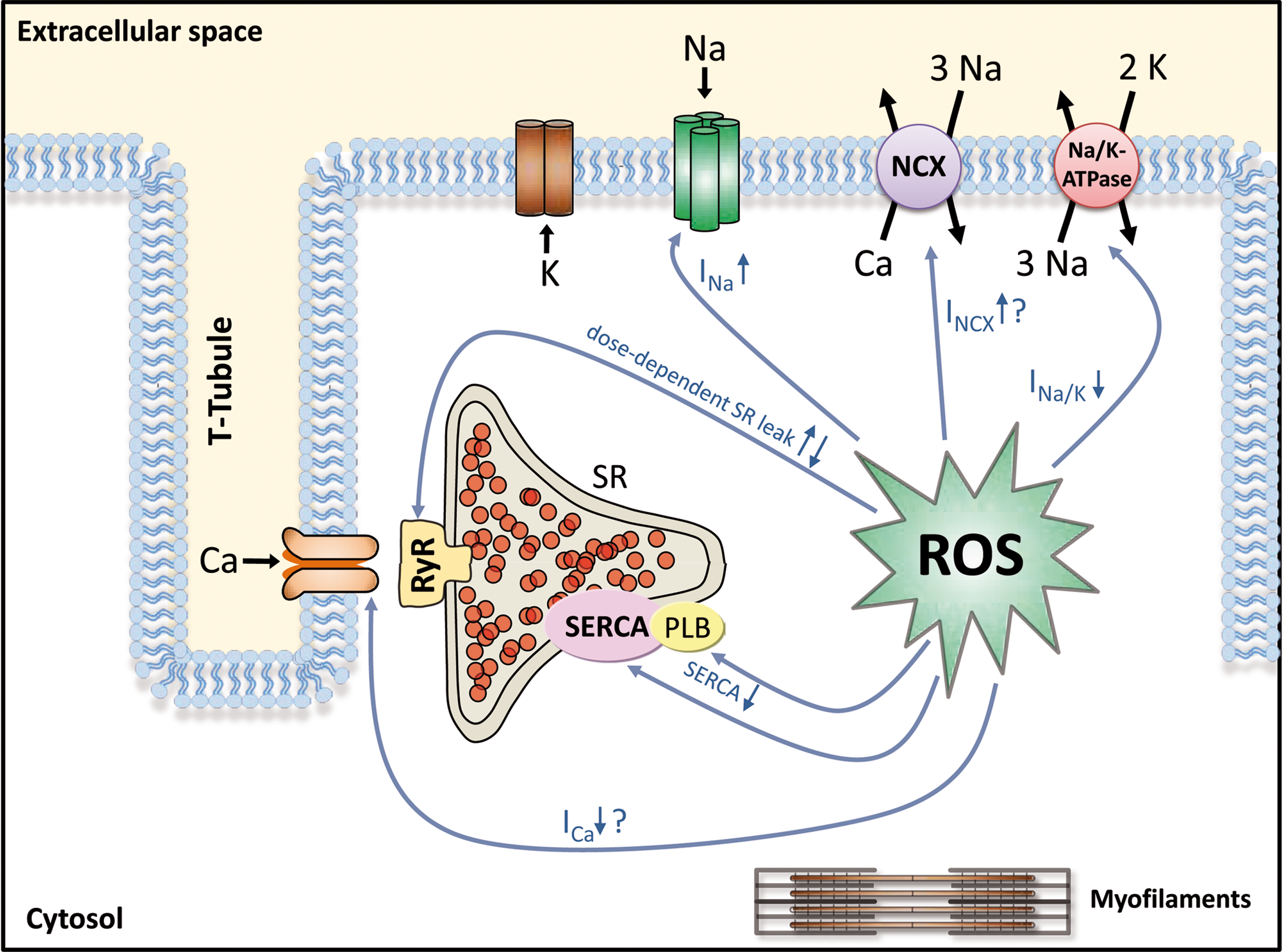
Avail↓, reduced INa availability, enhanced steady-state inactivation; IM, intermediate inactivation; LTCC, L-type Ca channel; NaV1.5, pore-forming subunit of the Na channel encoded by the SCN5A gene; NCX, Na/Ca exchanger; MMP-9, matrix metalloproteinase-9; PLB, phospholamban; PLM, phospholemman; PP1-I1, phosphatase-1 inhibitor-1; RyR2, ryanodine receptor 2.
Voltage-gated Na channels
Cardiac Na channels consist of a pore-forming α-subunit NaV1.5 and a regulatory β-subunit. NaV1.5 contains methionine residues that may be substrate to ROS-dependent oxidation (74). Oxidation of these methionine residues have been shown to impair open-state inactivation (74). ROS also reduce Na channel availability, causing a negative shift in the voltage dependence of inactivation due to enhanced intermediate inactivation and delayed recovery from inactivation. Activation, on the other hand, was unaltered (48).
Interestingly, a novel INa gating mode has been described recently that is also activated by ROS (120). Beside peak INa lasting only for a few ms (ca. 10 ms), there is a late INa component persisting over hundreds of milliseconds (90). Because of its persistent nature, the amount of Na entering the cell via late INa is substantial despite its small amplitude (approximately 1% of peak INa). In fact, it has been suggested that late INa is an important regulator of intracellular Na under pathophysiological conditions (135, 136).
ROS have also been shown to increase intracellular Na, prolong the action potential, and induce EADs, and ROS-enhanced late INa may be involved (15, 48, 120).
In addition to direct ROS-dependent oxidation of NaV1.5, changes in the lipid environment or ROS-induced activation of PKA, PKC, and CaMKII may be involved in the ROS regulation of cardiac Na channels (48, 74, 85, 132, 135, 136).
The α-subunit NaV1.5 is substrate to phosphorylation by PKA, PKC, and CaMKII. CaMKII has been shown to phosphorylate serine 571, serine 516, and threonine 594, which results in a negative shift in the voltage dependence of inactivation due to enhanced intermediate inactivation (10, 68). Although the relevant phosphorylation site remains to be determined, CaMKII has been also been shown to enhance late INa, which leads to accumulation of intracellular Na, AP prolongation and arrhythmias (135, 136). These effects resemble the above described ROS effects on Na channel gating. Moreover, the ROS-induced late INa was not observed in CaMKIIδ knock-out mice, suggesting a crucial role for CaMKII in redox regulation of INa (136).
Beside CaMKII, PKA and PKC have been shown to influence cardiac Na channels. NaV1.5 has two serine residues (serine 526 and 529) in the I-II cytoplasmic linker that can be phosphorylated by PKA. The PKA-dependent phosphorylation of NaV1.5 increases peak INa current density via acceleration of channel trafficking to the plasma membrane (56), but does not change inactivation kinetics (47, 98).
Also PKC can modify INa. Ward and Giles have shown that the H2O2-dependent slowing of INa open state inactivation was blocked in the presence of the PKC inhibitor Bis-indolylmaleimide (140). In heterologeous expression systems (rat and human NaV1.5), however, PKC activation did not change INa inactivation (99, 108) questioning the role of PKC for regulation of INa gating. The most consistent effect of PKC on cardiac Na channels is phosphorylation of serine 1505 in the III–IV linker, which reduces peak INa current density (107).
Although both PKA and PKC have been shown to divergently influence peak INa possibly by changing the number of functional channels in the membrane, they both do not appear to regulate INa gating (i.e., the activation and inactivation process). Therefore, ROS-activated PKC or PKA do not account for the ROS-induced changes in Na channel gating. However, it has been shown that ROS generated from mitochondria (by elevation of cytosolic NADH) may reduce peak Na current density (85) via a PKC-dependent pathway (132).
Sarcolemmal Na/K-ATPase
Physiologically, intracellular Na is heavily controlled by the Na-K-ATPase (NKA). Indeed, computational modeling of rabbit ventricular myocytes exposed to H2O2 revealed that even a 16-fold ROS-induced increase in INa conductance would not result in increased intracellular Na due to dramatic activation of NKA (136). Therefore, a substantial inhibition of NKA activity needs to occur to explain the ROS-induced increase in intracellular Na. Interestingly, ROS have been shown to inhibit NKA function potently, although the underlying mechanism is not known (76, 80, 118, 145). Beside changes in the lipid environment, direct NKA oxidation has been suggested. In this respect it is interesting that the β-subunit of NKA contains a sulfhydryl group that has been shown to be essential for catalytic activity.
Beside direct oxidation, NKA may also be redox-regulated via PKA or PKC. Phospholemman (PLM), which inhibits NKA by reducing its affinity for internal Na, has been shown to be phosphorylated by PKA (serine 68) and PKC (serine 63 and 68). PLM phosphorylation results in dissociation from the catalytic subunit, which activates the pump (37). Since ROS inhibit NKA activity, but ROS-induced activation of PKA or PKC would result in PLM phosphorylation, dissociation and NKA activation, it is unlikely that PKA or PKC are involved in the ROS regulation of NKA.
If a substantial reduction in NKA function is incorporated into the computer model, intracellular Na increases dramatically. Beside enhanced Na influx via INa, enhanced Na-proton-exchanger-dependent Na influx has been suggested, but the pathophysiological relevance in unclear (112).
Intracellular Na is tightly associated with intracellular Ca. The activity of cardiac Na-Ca-exchange (NCX) is a function of membrane potential and trans-sarcolemmal gradients for Na and Ca. Upon prolonged action potential duration and increased intracellular Na, reduced Ca efflux (forward mode) and/or Ca influx (reverse mode) via NCX contributes to intracellular Ca accumulation. Indeed, computational modeling revealed that the Ca entry mode of the NCX was dramatically favored under conditions of increased intracellular Na (136).
Sarcolemmal sodium–calcium exchanger
For cardiac NCX (NCX1), intramolecular disulfide bonds between cysteine residues of different domains have been implicated to be functionally relevant (100). ROS have been shown to activate NCX (52, 75, 110, 114), but the effect was inconsistent with respect to ROS source and ROS level. There is substantial controversy about the role of serine/threonine kinases in the regulation of NCX. No functional change in cardiac NCX has been shown upon application of catalytic subunits of PKA and PKC (61), suggesting that NCX may not be subject to regulation by kinases. On the other hand, it was shown that the intracellular loop of NCX can be phosphorylated by PKA or PKC, which may increase NCX activity and may be responsible for part of the ROS-induced NCX activation (72).
Under conditions of increased intracellular Na, ROS-dependent stimulation of NCX may result in cellular Ca overload. Indeed, it was shown previously that increased expression of NCX, as observed in HF, augmented ROS-induced cellular injury (134), and that pharmacological inhibition of Ca entry via NCX reduced ROS-induced Ca overload and diminishes cellular injury (136).
However, the situation in human HF may be more complex: human failing myocardium was paradoxically more resistant to ROS-induced contractile dysfunction, while human nonfailing was not (86). It was suggested that an increased endogenous activation of mitochondrial KATP channels in human failing myocardium may explain this “stunning paradox” such that failing myocardium may be endogenously preconditioned.
Voltage-gated L-type Ca channel
The pore-forming subunit α1C of the cardiac L-type Ca channel contains more than 10 cysteine residues (94), which can potentially undergo redox modification. It was shown that thiol-oxidizing agents or ROS irreversibly decreased ICa in heterologeous expression systems (HEK293 cells, 46, 64) or cardiomyocytes (50, 51, 83). These results are, however, controversial since others have shown that thiol-oxidizing agents (26) or ROS may increase ICa (121). Part of the discrepancy may be a result of the fact that CaMKII (19, 41, 54, 66), PKA (28), and PKC (151) can activate ICa by phosphorylation, and all three kinases are subject to ROS-dependent oxidation/activation. PKC has been shown to phosphorylate the α1c subunit of the L-type Ca channel at several sites including serine 1928 (150,151). PKA also phosphorylates α1c at serine 1928 (36, 67). However, the key phosphorylation site involved in Ca current modulation is still a matter of debate since phosphorylation of both α1C as well as the auxiliary β subunit (55, 106) can result in increased ICa.
Therefore, ROS can simultaneously induce ICa activation (via serine kinases) and ICa inhibition (via direct cysteine oxidation). The net result depends on the sources and levels of ROS generated. However, since it has been shown that H2O2 causes Ca overload due to Ca release from intracellular stores and Ca entry via NCX but not due to activation of ICa (49, 136), it is likely that the direct redox modification of α1C is the predominant ROS effect.
Cardiac ryanodine receptor
The RyR2 (cardiac isoform) is a large tetrameric complex that contains up to 89 cysteine residues per monomer, of which approximately 21 are free (147).
Thiol-oxiziding agents such as H2O2 activate RyR Ca release after oxidation of more than 7 thiols per subunit (1, 6, 21, 45, 84, 147, 155). Since the RyR2 has multiple sites for regulation by phosophorylation and/or interaction with Ca, Mg, ATP, CaM, or regulatory proteins (FKBP12.6 also called calstabin), oxidation may interfere with these regulators. Indeed, the mechanism of ROS-induced increase in RyR Ca release involves a change in RyR sensitivity to cytosolic Ca and ATP (42, 93), the alteration of the RyR interaction with triadin, which regulates RyR sensitivity to luminal Ca (84), and the disturbance of FKBP12.6 binding (155). For activation to occur, it requires the oxidation of more than 7 thiols per subunit (147). In contrast, the oxidation of less than 5.5 thiols per subunit occurs readily without affecting RyR function (147). Moreover, it has been shown that low ROS levels increase diastolic RyR Ca release (Ca spark frequency), whereas excessive ROS production reduces Ca spark frequency (149). Therefore, the quality of ROS-dependent RyR regulation may be closely related to the amount of oxidized thiols.
Beside direct oxidation of cysteine residues of RyR2, ROS may increase RyR Ca release via activation of serine/threonine kinases (113). It is well known that CaMKII and PKA phosphorylate RyR2 increasing diastolic Ca leak (88). Both kinases can be oxidized (see above) and activated by ROS (24, 42). Therefore, it is conceivable that part of the observed ROS effects on RyR2 are mediated via oxidized CaMKII or oxidized PKA. The relative contribution of each kinase and the pathophysiological relevance, however, is completely unclear.
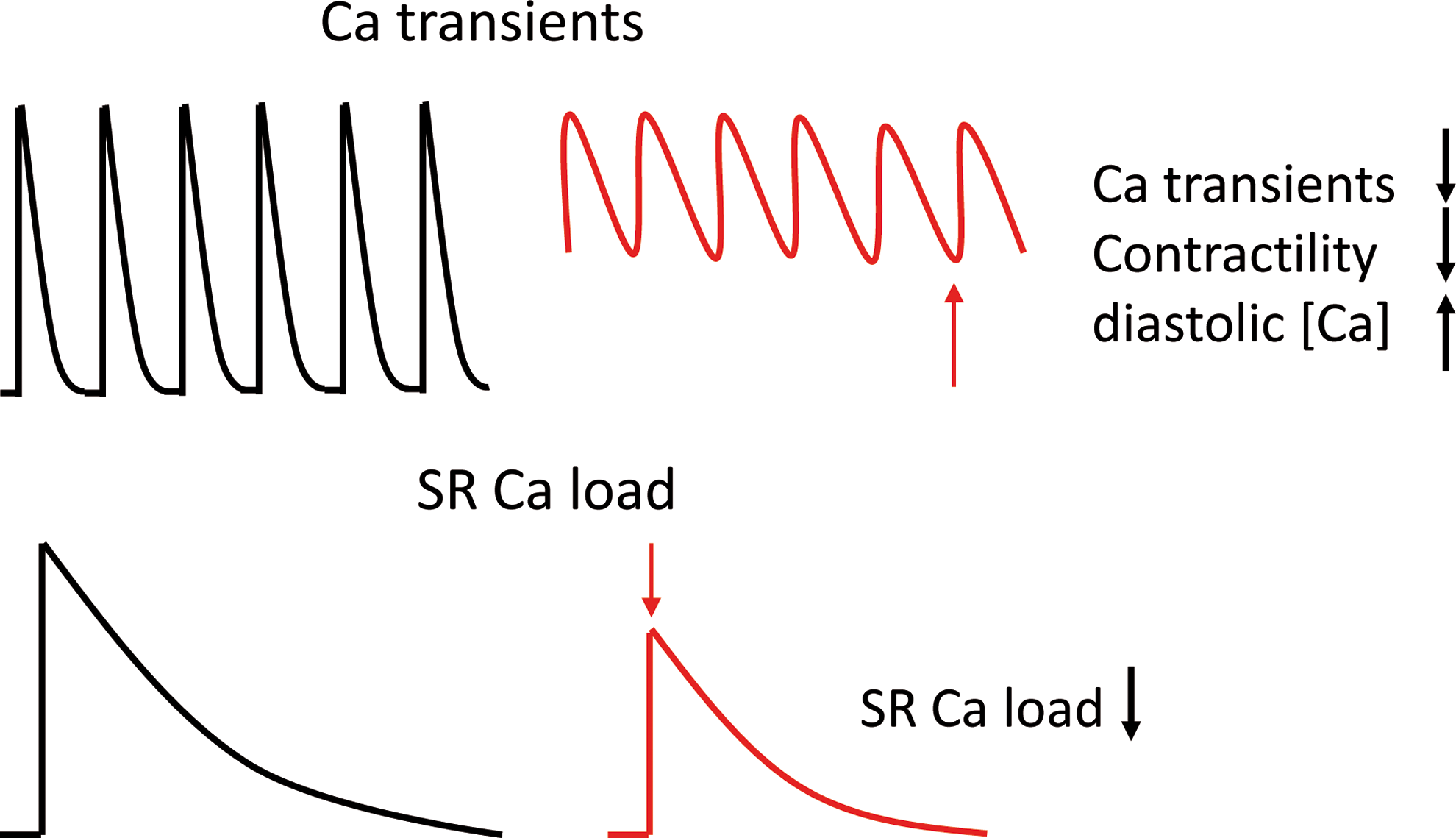
One consequence of increased diastolic Ca leak is reduced SR Ca content, especially if ROS reduce SR Ca ATPase function as well (153). ROS have been shown to reduce SR Ca content (136, 153). This results in smaller Ca transients and reduced contractility (136). Interestingly, due to rapid activation of Na-Ca-exchange in forward mode (Ca exit mode, net positive charge movement into the cell) diastolic Ca leak is also mainly responsible for delayed afterdepolarizations. The ROS-induced increase in Ca spark frequency is therefore associated with increased propensity for delayed afterdepolarizations and arrhythmias (136), especially if Na-Ca-exchange is enhanced via ROS (52).
Sarcoplasmic reticulum Ca-ATPase
SERCA2a and its regulatory (inhibitory) protein PLN are substrate to redox modification. SERCA2a contains 25 cysteine residues, of which only 1 or 2 are essential for enzyme action (97). Thiol-oxidizing agents or ROS have been shown to inhibit cardiac SERCA function (79, 96, 115, 146). Part of this effect may be a result of direct interference with the ATP binding site (146). In addition, PLN is also substrate to phosphorylation by CaMKII (threonine 17) and PKA (serine 16) resulting in PLN dissociation and activation of SERCA (88). The latter effect, however, appears to be less relevant for ROS-dependent SERCA regulation, since ROS have been unequivocally shown to reduce SERCA function. Possibly PKC-dependent signaling is also involved. In contrast to CaMKII and PKA activating SERCA, PKC has been shown to reduce SERCA activity. Hearts of PKCα-deficient mice are hypercontractile, whereas those of transgenic mice overexpressing PKCα are hypocontractile. The underlying mechanism involves PKCα-dependent phosphorylation of protein phosphatase inhibitor-1 (PPI-1), resulting in dephosphorylation of PLN. The reduced SERCA2a activity leads to reduced SR Ca content and diminished Ca transient amplitude (23).
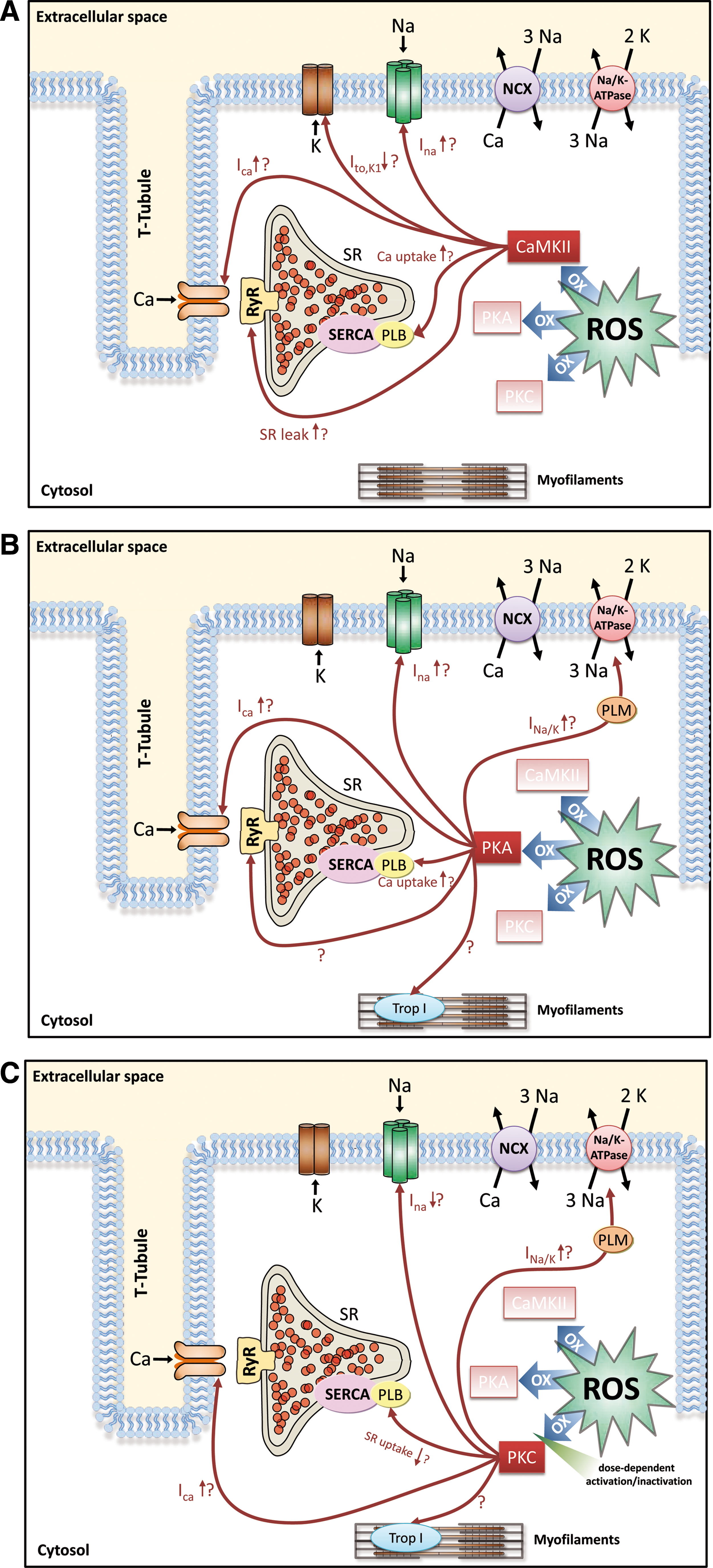
To summarize the discussed ROS effects on Na and Ca handling proteins, Figure 4 and Table 1 give an overview of the effects possibly mediated by ROS-activated CaMKII (Fig. 4A), ROS-activated PKA (Fig. 4B) or ROS-activated PKC (Fig. 4C).
ROS-Associated Arrhythmias
Changes in intracellular Na and Ca handling are associated with electrical instability.
It was shown previously that ROS increase AP duration resulting in early afterdepolarizations (EADs) due to reactivation of ICa (120). Since the selective Na channel blocker tetrodotoxin (TTX) could reverse this ROS effect, enhanced late INa was attributed as underlying mechanism of AP prolongation. Indeed, recently this mechanistic link was confirmed showing that redox-activated CaMKII and enhanced late INa are required for AP prolongation and EADs (68, 136). Depending on the sources and levels of ROS, a ROS-induced enhancement of ICa and transient outward K current (Ito) inhibition by oxidation of SH groups may additionally contribute (111).
On the other hand, cellular Ca overload and ROS-induced diastolic Ca leak predispose to increased transient inward INCX (Iti), which transports the released diastolic Ca outside the cell, generating a depolarizing current and predisposing to delayed afterdepolarizations (DADs). The propensity for EADs and DADs has been shown to be dramatically increased upon ROS exposition but reduced in CaMKIIδ knockout mice (136). The relevance of CaMKII for life-threatening arrhythmias was confirmed in CaMKII-transgenic mice showing a dramatic increase in the propensity for monomorphic and polymorphic ventricular arrhythmias (135). Therefore, redox-modified CaMKII may be crucially involved in the ROS-induced arrhythmogenesis (Fig. 5).
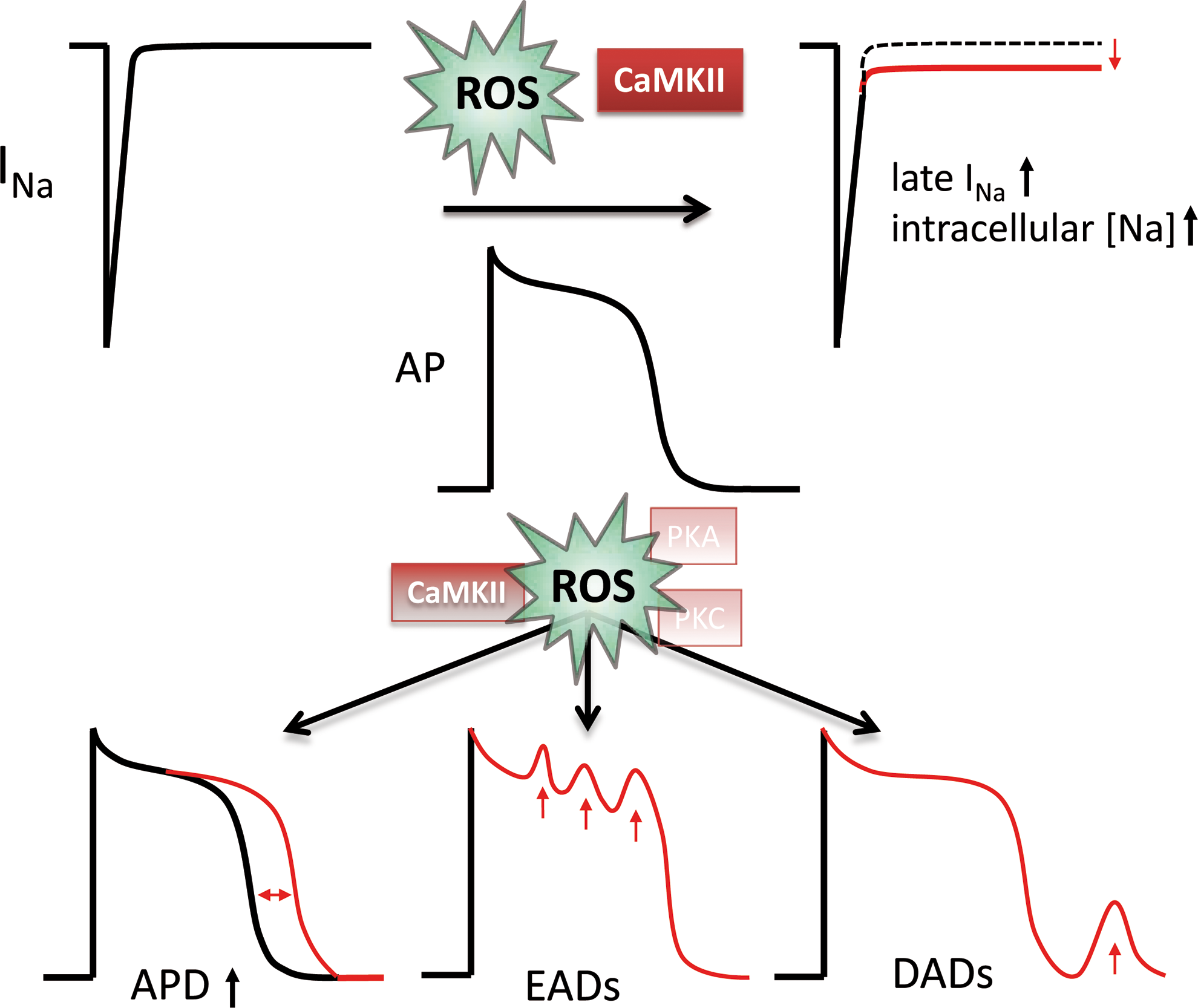
Beside dysregulated Na and Ca handling, ROS-effects on mitochondrial ATP production may also increase arrhythmogenesis (3, 7, 8). It was shown that ROS-induced ROS release from mitochondria results in mitochondrial depolarization, during which ATP production is inhibited and sarcolemmal KATP channels are activated, leading to shortened AP duration and slowed conduction. Due to spatiotemporal differences of this mitochondrial membrane depolarization between different regions of the heart, re-entry and life-threatening arrhythmias may develop.
Conclusion and Perspectives
Here we have discussed ROS effects in Na and Ca handling. ROS have been shown to induce activation of late INa that in the face of reduced NKA activity lead to intracellular Na accumulation and action potential prolongation. As a consequence, Ca entry mode of ROS-activated NCX is favored. On the other hand, ROS increase diastolic leak through enhanced RyR2 open probability, which leads together with dysfunctional SERCA to reduced SR Ca load. The dysfunctional SR cannot compensate the Ca entry via NCX, leading to a dramatic increase in intracellular Ca, reduced SR Ca transients, diminished contractility, arrhythmias, and cellular injury (Fig. 6). Interestingly, all these changes are also present in the failing heart, suggesting that ROS may be involved in the development of the disease.
Low amounts of ROS constitute redox signaling, which may have physiological relevance but may also be involved in the processes leading to myocardial remodeling. Part of these ROS signaling effects is mediated via serine/threonine kinases, translating the short living ROS signal into a signal of longer duration. These kinases are known to be involved in the pathophysiology of heart failure. Excessive uncontrolled ROS generation, however, results in profound myocardial damage, including substantial direct and possibly irreversible redox-modification of Ca handling proteins, which contributes to the progression of HF. Depending on the source, localization, and amount of ROS generated, ROS could have very different effects on Ca handling proteins.
Future treatment strategies interfering with ROS signaling in HF have to deal with these issues.
Footnotes
Acknowledgments
SW was funded by the Forschungsförderprogramm der Universitätsmedizin Göttingen. SW was supported by a travel grant from the Fondation Leducq. AGR is supported by a career development grant from the Fondation Leducq. MEA is funded by NIH Grants R01HL70250, R01HL079031, and R01HL096652. MEA and LSM are funded by a grant (08CVD01) from the Fondation Leducq as part of the 'Alliance for CaMKII Signaling in Heart'. LSM is funded by Deutsche Forschungsgemeinschaft (DFG) Grants MA 1982/4-1, MA 1982/2-2, as well as TPA03 SFB 1002. LSM is also funded by the Fondation Leducq Transatlantic Network of Excellence on 'Redox and Nitrosative Regulation of Cardiac Remodeling: Novel Therapeutic Approaches for Heart Failure.
Author Disclosure Statement
MEA is a named inventor on patents claiming to treat heart failure and arrhythmias by CaMKII inhibition. LSM acknowledges research grants and funding from CVT, GILEAD, and MENARINI/Berlin-Chemie.