
Abstract
Introduction
Innovation
Carbon monoxide–releasing molecules (CO-RMs) are being investigated for potential use in delivering CO to cells and tissues and show promising early results as antimicrobial agents. Since the modes of CO-RM action are probably dissimilar to currently used antibiotics, CO-RMs have the potential for topical or targeted treatment of antibiotic-resistant bacteria. However, rational design and exploitation of CO-RMs requires a fundamental understanding of their activity. We report that Ru(CO)3Cl(glycinate) has complex, time-dependent effects on bacteria, including stimulation and inhibition of respiration, and promotion of transmembrane cation transport, highlighting the potential of CO-RMs for multifaceted, broad-spectrum utilization in clinical microbiology.
Nevertheless, the mode(s) of action of CO and CO-RMs remain unclear. We have shown that CORM-3 enters cells, delivers CO to intracellular oxidases (11), and inhibits bacterial growth. In addition, transcriptome profiling of the response of E. coli to CORM-3 reveals downregulated expression of respiratory genes; however, the modularity and redundancy of bacterial respiration (47) suggests that a functioning respiratory system would persist (11), so the impact on respiration remains uncertain. Evidence for previously unrecognized activity of CO in bacteria comes from the transcriptomic effects on genes involved in metal metabolism, homeostasis, and transport (11). Probabilistic modeling (52) of the microarray data identified the involvement of eight transcription factors (11), only two of which (ArcA and FNR) have direct roles in regulation of respiration. In another transcriptomic study, using tricarbonyl dichlororuthenium (II) dimer (CORM-2), respiratory genes were not major targets (42). Intriguingly, although solutions of CO gas also impair bacterial growth (43), they do not match the effectiveness of CORM-2 or CORM-3 (11).
Here, we describe for the first time direct evidence for “classical” inhibition of respiration by any CO-RM. That the action of CO-RM-derived CO is due to binding of CO to hemoproteins is demonstrated by photorelief of respiratory inhibition and bacterial killing. We also demonstrate transient stimulation of respiration similar to the “uncoupler”-like action of CORM-3 on mitochondria recently reported (22). However, we attribute it not to protonophore activity, but to facilitation of cation transport.
Results
CORM-3 both stimulates and inhibits bacterial respiration
CO release from CORM-3, using dithionite-reduced myoglobin as a CO trap, is complete in <10 min (8,38). However, recent measurements with myoglobin in the absence of dithionite (which greatly enhances CO release) or oxyhemoglobin as a trap for the released CO show that CORM-3 liberated no detectable CO in 1 h (38). A previous study, however, revealed that high CORM-3 concentrations (125–250 μM) dramatically inhibited E. coli respiration within 30 min, whereas 30 μM CORM-3 elicited major changes in transcript levels within 15 min (11).
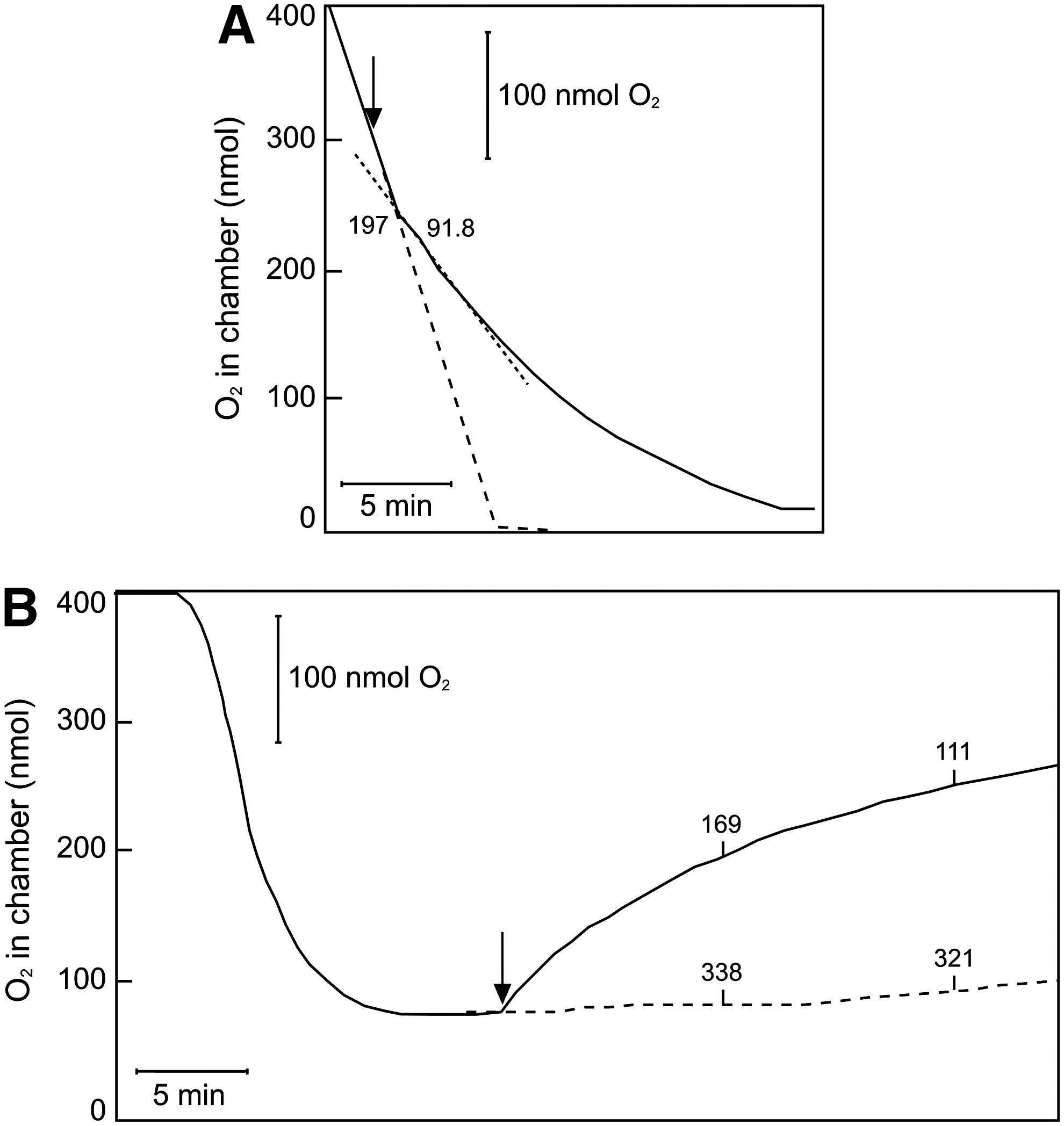
We, therefore, investigated whether CORM-3 inhibits respiration in a timeframe that is consistent with the rapid loss of CO in certain biological experiments. E. coli cells were incubated in an O2 electrode chamber, and CORM-3 was added before O2 exhaustion (c. 100 μM O2; Fig. 1). On anoxia (maximum CO:O2 ratio), the chamber was opened, allowing inward diffusion of air. Under these “open” conditions, the respiration rate is given as follows (13,14):
where v r is the respiration rate; K is a rate constant that is dependent on reaction volume, surface area, temperature, and others; T G is the concentration of O2 in the buffer when equilibrated with the gas phase; and T L is the concentration of O2 in the liquid sample at time t. Thus, at a constant rate of O2 entry from the atmosphere, governed by the stirrer and liquid/air interface, inhibition or stimulation of respiration is indicated by an increase or decrease, respectively, in the measured O2 tension.
Unexpectedly, addition of CORM-3 (100 μM final concentration) in the closed phase of the measurement (Fig. 1A, shaded portion) did not inhibit, but instead stimulated respiration by 36.8% (mean value of 34.7%, SEM 6.46%, four determinations in two biological repeats). After the chamber was opened, stimulation was still observed, here as a depression of the electrode trace (Fig. 1A, curved solid line) when compared with the no-CORM-control (dashed line). However, ∼20 min after CORM-3 addition, inhibition of respiration was observed as the electrode trace increased above the steady-state level of the control. Thus, prolonged respiration measurements in the open system reveal an initial phase of respiratory stimulation, followed by inhibition.
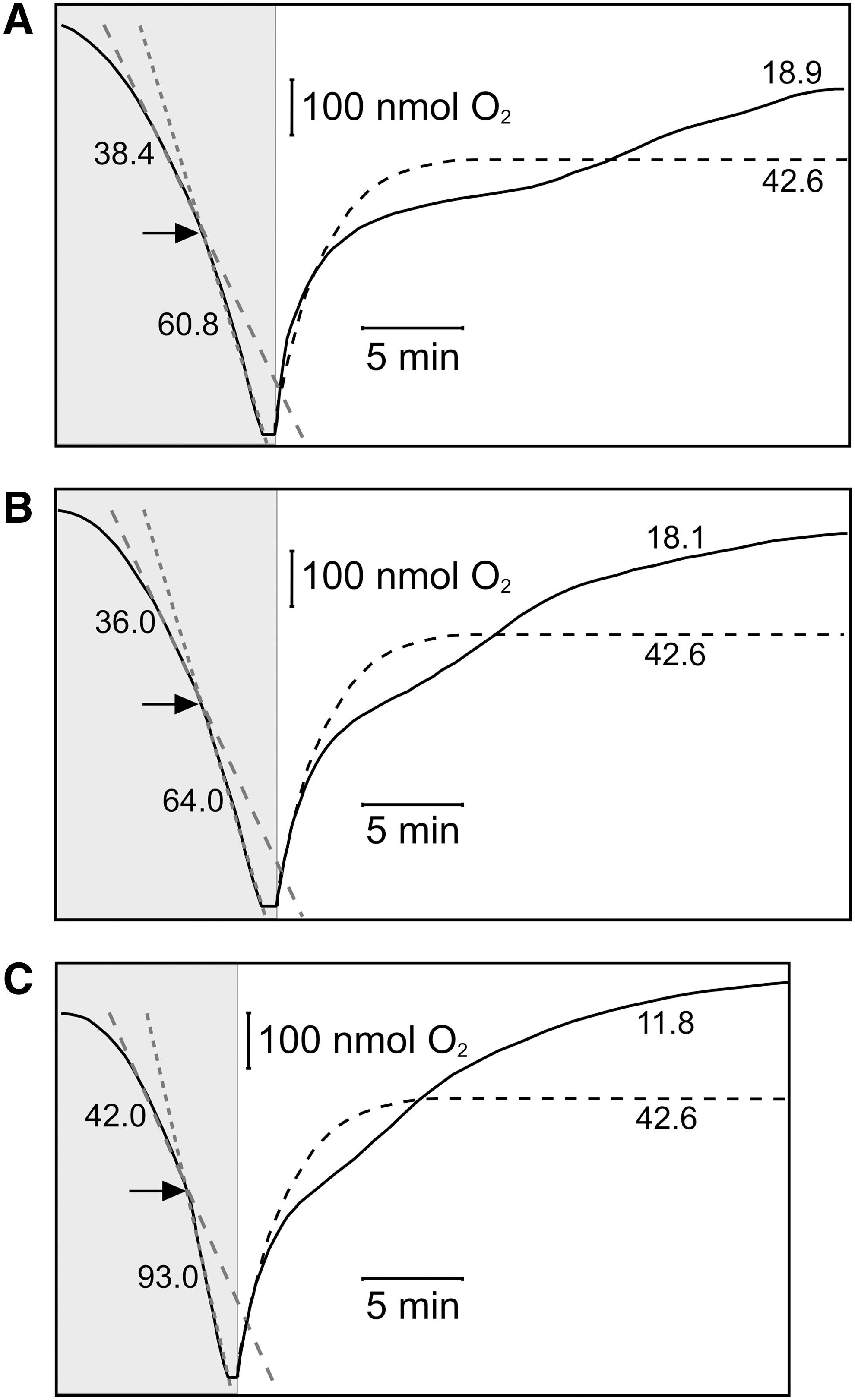
As the CORM-3 concentration was increased to 250 μM (Fig. 1B) then 500 μM (Fig. 1C), the initial stimulation in the closed system increased slightly (up to 49.8% stimulation, SEM 3.53%), and inhibition occurred progressively earlier after CORM-3 addition.
CORM-3 inhibition of bacterial respiration is enhanced by prior anoxia
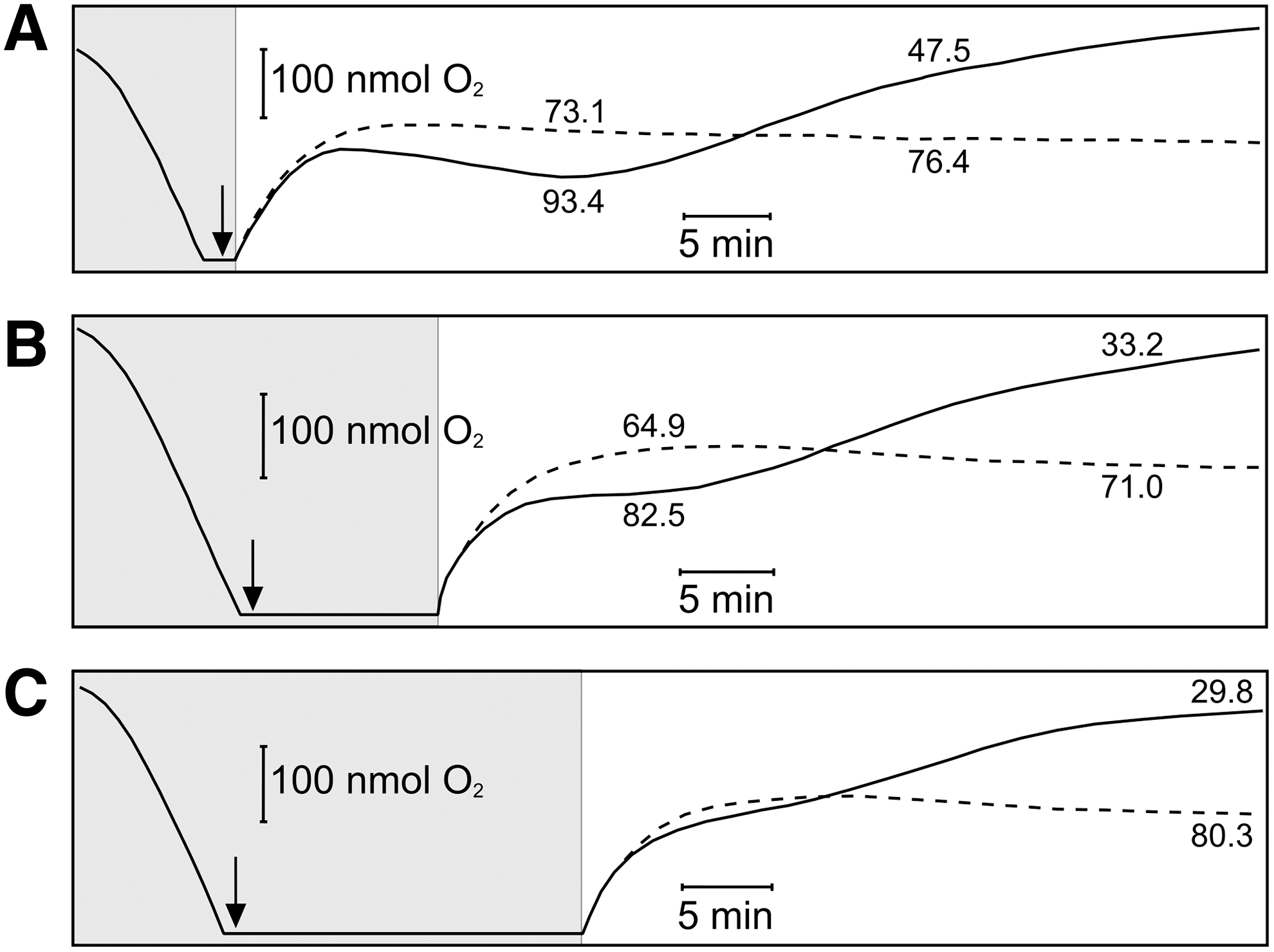
Maximal inhibition of respiration is expected when the CO:O2 ratio is increased, as CO is a competitive inhibitor of O2 binding (28). We, therefore, modified the method to allow increased anoxic contact between the CO-RM and cells. In the absence of CORM-3, opening the chamber after brief anoxia (Fig. 2A, dashed line) results in a steady-state O2 tension, as constant respiratory O2 consumption is balanced by inward O2 diffusion. However, when 100 μM CORM-3 was added at anoxia and the chamber opened immediately (Fig. 2A, solid line), respiration was initially stimulated (O2 trace below the control level), then inhibited (O2 trace above the control level). Increasing the period during which cells were incubated with CORM-3 from 0 min (Fig. 2A) to 10 min (Fig. 2B) or 20 min (Fig. 2C) before re-admitting O2 progressively increased the degree of subsequent respiratory inhibition. Thus, when air was admitted immediately after CORM-3 addition, the mean inhibition was 41.4%±3.58%; after 10 min, this rose to 53.7%±1.46% and, at 20 min, the inhibition was 61.2%±2.63%. These increases in inhibition were accompanied by a less marked phase of respiratory stimulation. The sequence of stimulation then inhibition, only the latter being anticipated from CO biochemistry, is not measurable in conventional short-term O2 measurements.
To test the possibility that the phase of respiratory inhibition is accompanied by diversion of electrons to O2 via non-oxidase routes, thus generating superoxide and peroxide [as in Campylobacter jejuni (59)], we added catalase during the inhibitory phase of an experiment which was similar to that shown in Figure 2A. No perturbation of the O2 trace was observed (Supplementary Fig. S1; Supplementary Data are available online at
CORM-3-mediated “uncoupling” of respiration?
CORM-3 (1–20 μM) “uncouples” mitochondrial respiration (22,50), deduced from the stimulation of respiration in State 2 [i.e., the respiration that occurs on addition of substrate to mitochondria, but limited by lack of adenosine diphosphate (ADP) (41)]. CORM-3 also decreased mitochondrial membrane potential (ΔΨ). Both changes were gradual and slight compared with the instantaneous effects of only 1 μM (or lower) carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (41). Such compounds, generally referred to as “uncouplers,” are better described as protonophores, catalyzing the net electrical uniport of H+ and thus increasing the H+ conductance of the membrane (41). However, the possible protonophore activity of CORM-3 was not directly tested (22,50).
Bacterial membranes are not known to possess the uncoupling proteins or adenine nucleotide transporters implicated in the modest “uncoupling” of mitochondria by CORM-3 (22); so, we sought direct evidence by measuring H+ translocation using Mitchell's oxidant pulse technique (39). Here, a bacterial (or mitochondrial) lightly buffered suspension becomes anoxic by substrate oxidation. Respiration is re-initiated by injecting a known amount of O2, and H+ efflux (driven by H+ pumps or vectorial chemistry) is recorded with a micro-pH electrode. Protonophores such as FCCP and carbonyl cyanide m-chlorophenylhydrazone (CCCP) collapse the measured H+ extrusion.
Figure 3A shows H+ pulses from E. coli cells in a medium containing K+ plus valinomycin (a K+-specific ionophore) and SCN− (a permeant anion) to increase the rate and extent of H+ pumping by neutralizing the thermodynamic back-pressure of the ΔΨ. Successive additions of air-saturated 150 mM potassium chloride (KCl) elicited acidification of the bulk medium due to respiration-driven H+ translocation. H+/O ratios, calculated after calibration with anoxic hydrochloric acid (HCl), were ∼3 (mean 2.65±0.11), that is, within the range of values reported for E. coli cells (33,48). Treatment of cells with the protonophore CCCP (35), which acts similar to FCCP, before O2 additions dramatically reduced the H+ pulses due to rapid H+ exchange across the bacterial membrane (i.e., true “uncoupling,” Fig. 3B).
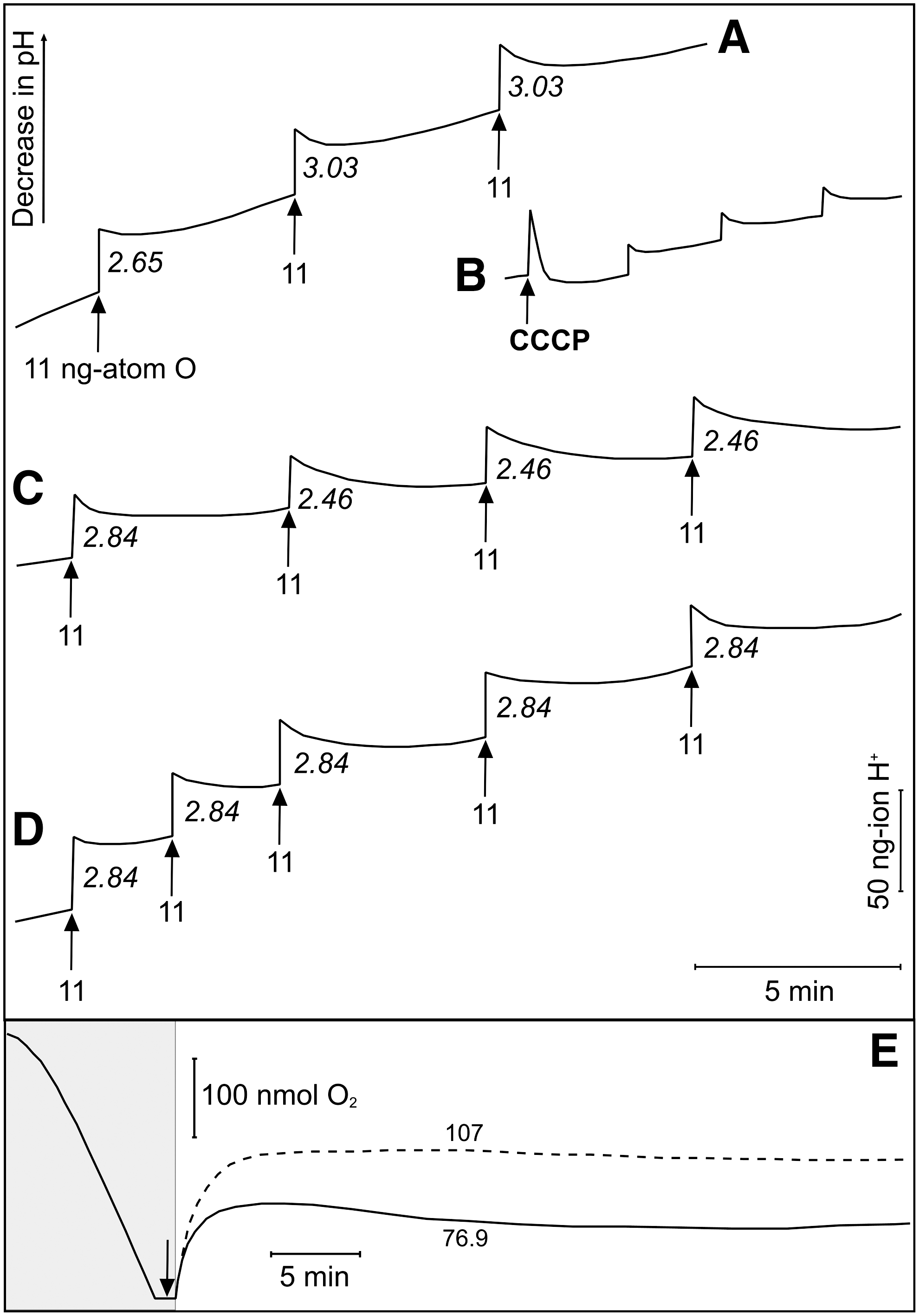
In contrast, previous treatment with CORM-3, followed by pH readjustment to compensate for loss of a H+ from the CO-RM on addition to buffer, resulted in only a modest concentration-dependent reduction in the H+/O ratio. CORM-3 at 20 μM (Fig. 3C) or 100 μM (Fig. 3D) attenuated only marginally the H+/O ratios (mean H+/O=2.51±0.06, H+/O=2.56±0.07, respectively). Inactive CORM-3 [iCORM-3, i.e., CORM-3 that was allowed to lose CO and then flushed with N2 to displace CO from solution (8,22)] was without effect (Supplementary Fig. S2). In addition to measuring H+/O ratios, we measured the rate of H+ backflow after the O2 pulse in the absence and presence of 20 μM CORM-3 and iCORM-3. In both cases, the t ½, measured from semi-log plots of pH changes were not significantly lowered by CORM-3 (1.45 min for CORM-3 and 1.66 min for corresponding control pulses; for iCORM-3 1.47 min and corresponding control pulses 1.57 min) (Supplementary Fig. S3).
Addition of 10 μM CCCP to an open O2 electrode elicited the same downward deflection of the aerobic steady state (i.e., stimulation of respiration; Fig. 3E) as that seen with 100 μM CORM-3 (cf. Fig. 2A). Cell viability was unaffected over 60 min by CCCP. Thus, CCCP, but not CORM-3, is a protonophore in E. coli cells, stimulating respiration rates and collapsing respiration-driven H+ translocation.
Membrane potential is not dissipated by CORM-3
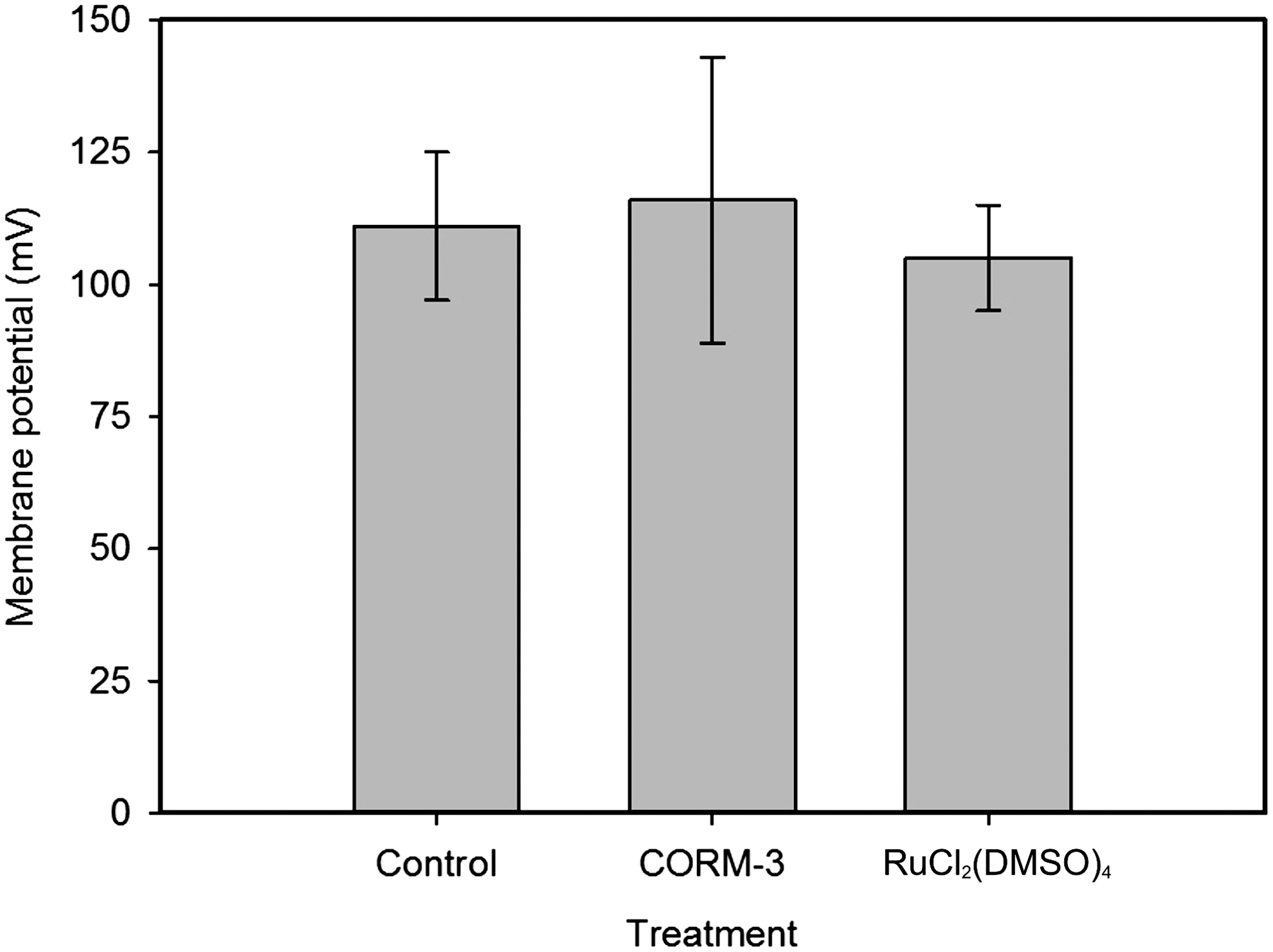
The transmembrane pH gradient (ΔpH) and Δψ together constitute the protonmotive force (pmf). Protonophores shuttle H+ across the lipid bilayer, effectively short-circuiting the transmembrane H+ gradient and transiently stimulating respiration when electron transport is coupled to H+ translocation (41). Protonophores dissipate both the ΔpH and Δψ components of the pmf. To determine whether CORM-3 affects the Δψ of respiring E. coli, CORM-3 (100 μM) was preincubated with cells (for 0–20 min) and the Δψ determined (Fig. 4). Under the experimental conditions used here, the external pH is buffered at pH 7.5, and, therefore, the total pmf is predominantly Δψ. The Δψ of untreated cells was 111±15 mV (Fig. 4). Cells treated with CORM-3 had a Δψ of 116±27 mV (range 95–143 mV); thus, the CO-RM has a mild stimulatory effect on the Δψ, but did not dissipate the Δψ even with 20 min of incubation (Fig. 4). Cells treated with the control molecule RuCl2(DMSO)4 (100 μM) had a Δψ of 105±10 mV. These data demonstrate directly that CORM-3 does not dissipate the Δψ of respiring cells.
CORM-3 does not directly stimulate oxidase activity
Having demonstrated that the stimulatory effect of CORM-3 cannot be attributed to protonophore activity, we investigated other potential mechanisms. Interestingly, low doses of CO (10 μM) transiently stimulate cytochrome c oxidase activity in mitochondria; however, the overall effects on organelle or cell respiration were not reported (49). To eliminate transmembrane ion fluxes from consideration here, respiration of isolated membranes, not bacterial cells, was measured. When bacteria are disrupted by harsh physical methods such as ultrasonication, membranes are fragmented and form “leaky” vesicles of mixed orientation that are unsuitable for transport measurements (41,46). We measured respiration in such vesicles using both open and closed electrode systems (Fig. 5). In the conventional closed system, addition of high concentrations of CORM-3 (400 μM) were required to inhibit initial rates of respiration by >50% (Fig. 5A). In the open system, which allowed more protracted measurements and CO-RM addition at low (35 μM) poised O2 levels (Fig. 5B), respiration was reduced to one-third of the control rate by CORM-3 (100 μM). Importantly, in neither experiment was stimulation of respiration observed, suggesting that this phenomenon is associated with ion fluxes rather than catalytic function of the quinol oxidases.
K+ and Na+, not H+, transport is facilitated by CORM-3
Membrane channels other than those for H+ are modified by CO and CO-RMs in eukaryotic cells. The effect may be indirect, caused by an interaction between critical Cys residues with reactive O2 species generated from CO inhibition of mitochondrial respiration (56) or by a direct interaction between CO and channel proteins (21). CORM-2 is an allosteric inhibitor of voltage-gated K+ channels, reducing the voltage dependence of the opening transition (24). We, therefore, tested whether CORM-3 promotes cation transport using osmotic swelling of spheroplasts (18). Iso-osmotic sucrose (0.5 M) provides support to detergent- and osmotically-sensitive spheroplasts, as shown by constant light-scattering properties (18). For swelling to occur, both cation and anion should enter to prevent charge imbalance; water follows, causing the spheroplasts to swell.
Spheroplast behavior in iso-osmotic solutions of NO3 − salts is shown in Figure 6A; the NO3 − anion freely permeates the spheroplast membrane (18). Iso-osmotic potassium nitrate (KNO3) (0.25 M) provided osmotic support until spheroplasts were treated with valinomycin (Fig. 6A). This effect was mimicked, although more slowly, by 100 μM CORM-3, showing it to have ionophore-like properties (Fig. 6A), but iCORM-3 did not promote spheroplast swelling (Fig. 6A). When spheroplasts were suspended in 0.25 M sodium nitrate (NaNO3), further addition of the Na+ ionophore X-537A (lasalocid) or of CORM-3 was required for swelling (Fig. 6A) despite the facile permeation of the NO3 − anion. X-537A promotes transport of a number of monovalent and divalent cations, including Na+, in an electroneutral exchange with H+ (26,41). A solution of CO gas (final concentration 100 μM) did not promote swelling (Fig. 6A), indicating that CO alone does not act as an ionophore for Na+ ions.
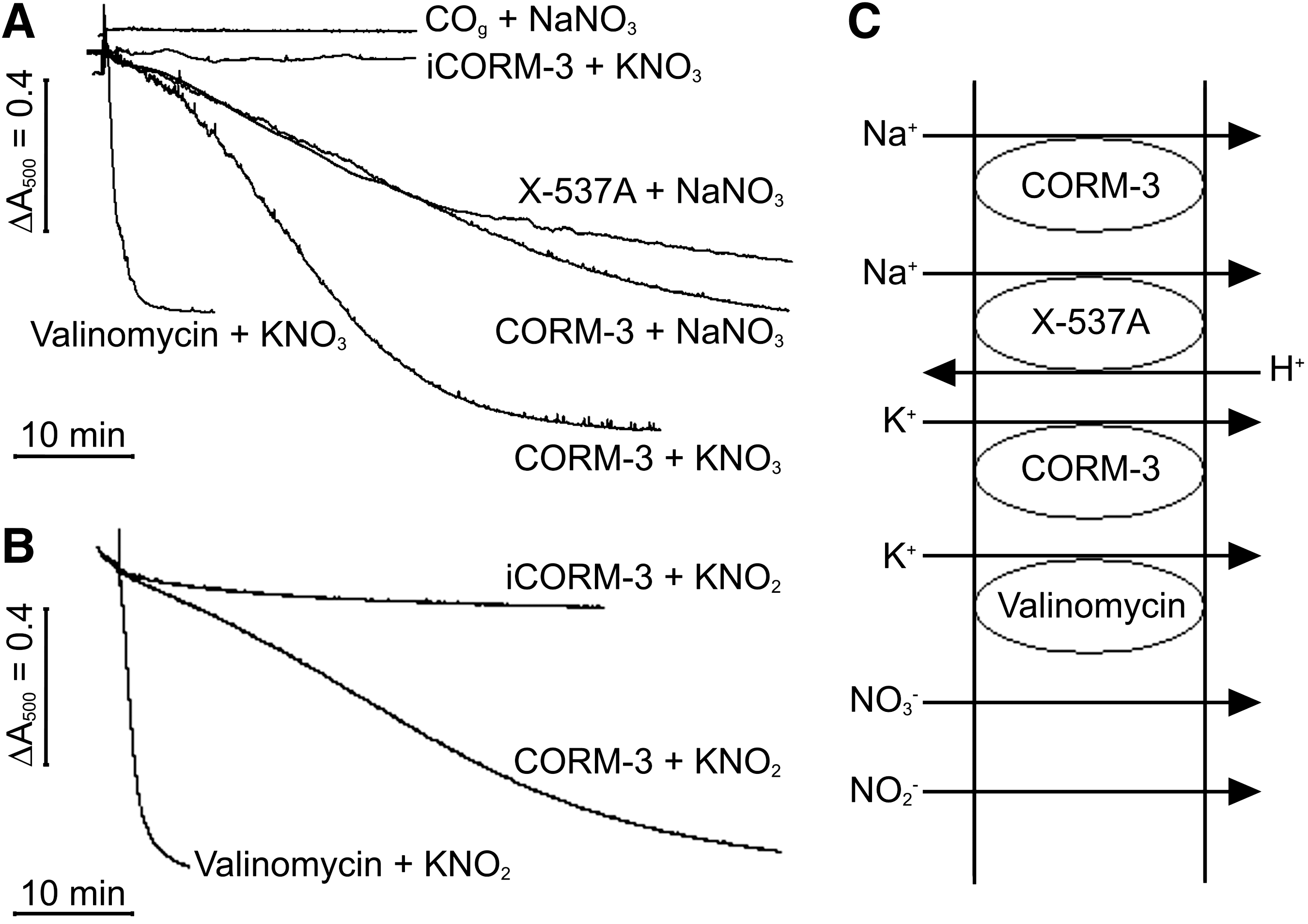
Spheroplast behavior in iso-osmotic solutions of NO2 − salts is shown in Figure 6B; the NO2 − anion also freely permeates spheroplasts (18). Thus, in iso-osmotic 0.25 M potassium nitrite (KNO2), swelling was again promoted by either valinomycin or CORM-3 (Fig. 6B), as both nitrous acid (HNO2) and the NO2 − anion can cross the membrane (18).
The K+, Na+, H+, NO3 −, and NO2 − movements responsible are shown in Figure 6C along with the activities of established ionophores. Based on these data, CORM-3 simulates the effects of K+ and Na+ ionophores.
Ru compounds that do not release CO, or CO gas, do not inhibit respiration
The commonly used CO-RMs have properties other than CO release that may interfere in biological processes. In particular, CORM-3 is a Ru carbonyl, Ru being a metal ion with redox chemistry that does not occur naturally in biology. Further, CO release may also be accompanied by loss of glycinate, chloride, and other unknown processes and interactions. To demonstrate that the stimulatory and inhibitory effects of CORM-3 on bacterial respiration are due specifically to CO release, we used three Ru-containing control molecules. RuCl2(DMSO)4 is a control for CORM-2, a precursor in CORM-3 synthesis (8), in which the CO groups are replaced by DMSO (53). When used at 100 μM (Fig. 7A, solid line), neither stimulation nor inhibition of respiration was observed (compare with Fig. 2A). The same result (Fig. 7B, solid line) was obtained with iCORM-3. Both these compounds are imperfect controls: RuCl2(DMSO)4 lacks the glycinate group of CORM-3, and iCORM has not been exposed to biological species that induce release of more CO than can be achieved by gassing. We, therefore, devised a new control (myoglobin-inactivated CORM-3, miCORM-3) where CO is removed by two treatments with myoglobin [dissociation constant, CO=27 μM −1 (58)]. This preparation, when tested with ferrous myoglobin, gave no detectable CO release (Fig. 7C, inset) and did not elicit the same response as CORM-3 (Fig. 7C, solid line compared with gray dotted line). We conclude that CO release is necessary for the perturbing effects of CORM-3 on bacterial respiration.
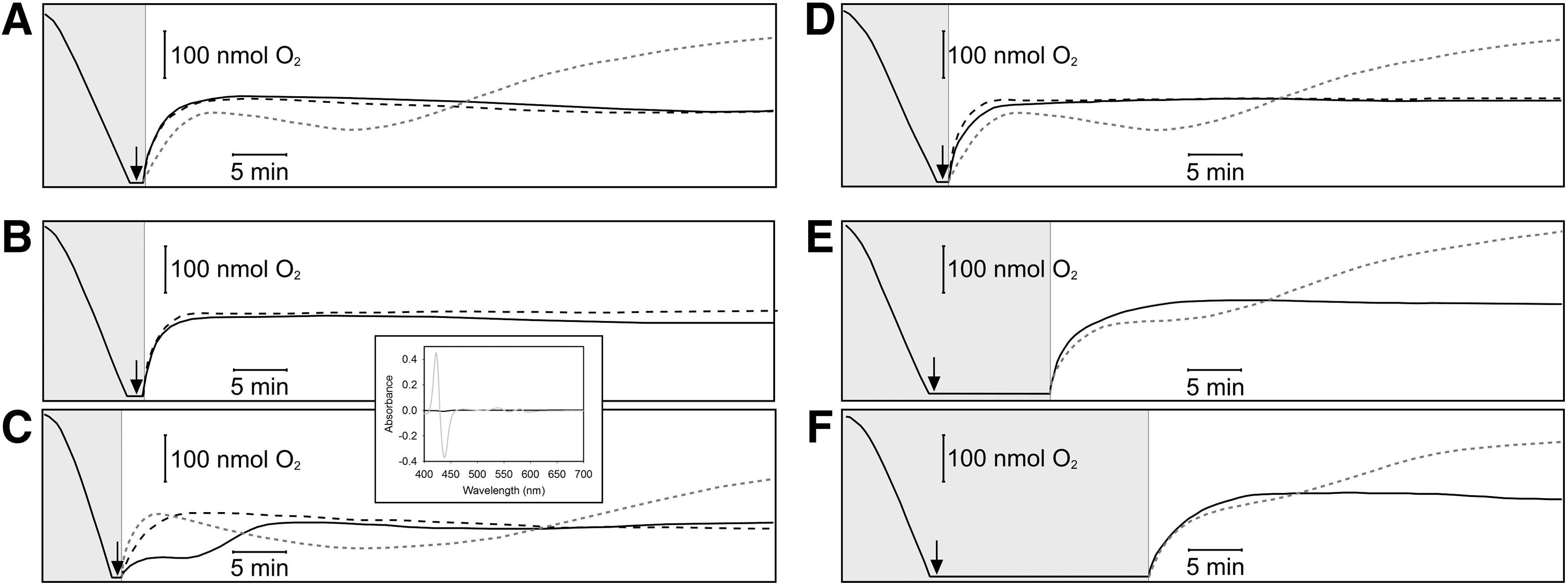
We previously reported (11) that solutions of CO gas were without effect on E. coli, when used at concentrations equimolar with inhibitory levels of CORM-3. Figure 7D–F show that, irrespective of the period (up to 20 min) during which 100 μM CO and cells were incubated anoxically to maximize the CO:O2 ratio, CO neither stimulated nor inhibited respiration (solid lines). In mitochondria, a modest increase in State 2 respiration is reported, but only at grossly nonphysiological (960 μM) CO concentrations (22).
CORM-3 as an antimicrobial agent: viability loss is coincident with effects on respiration
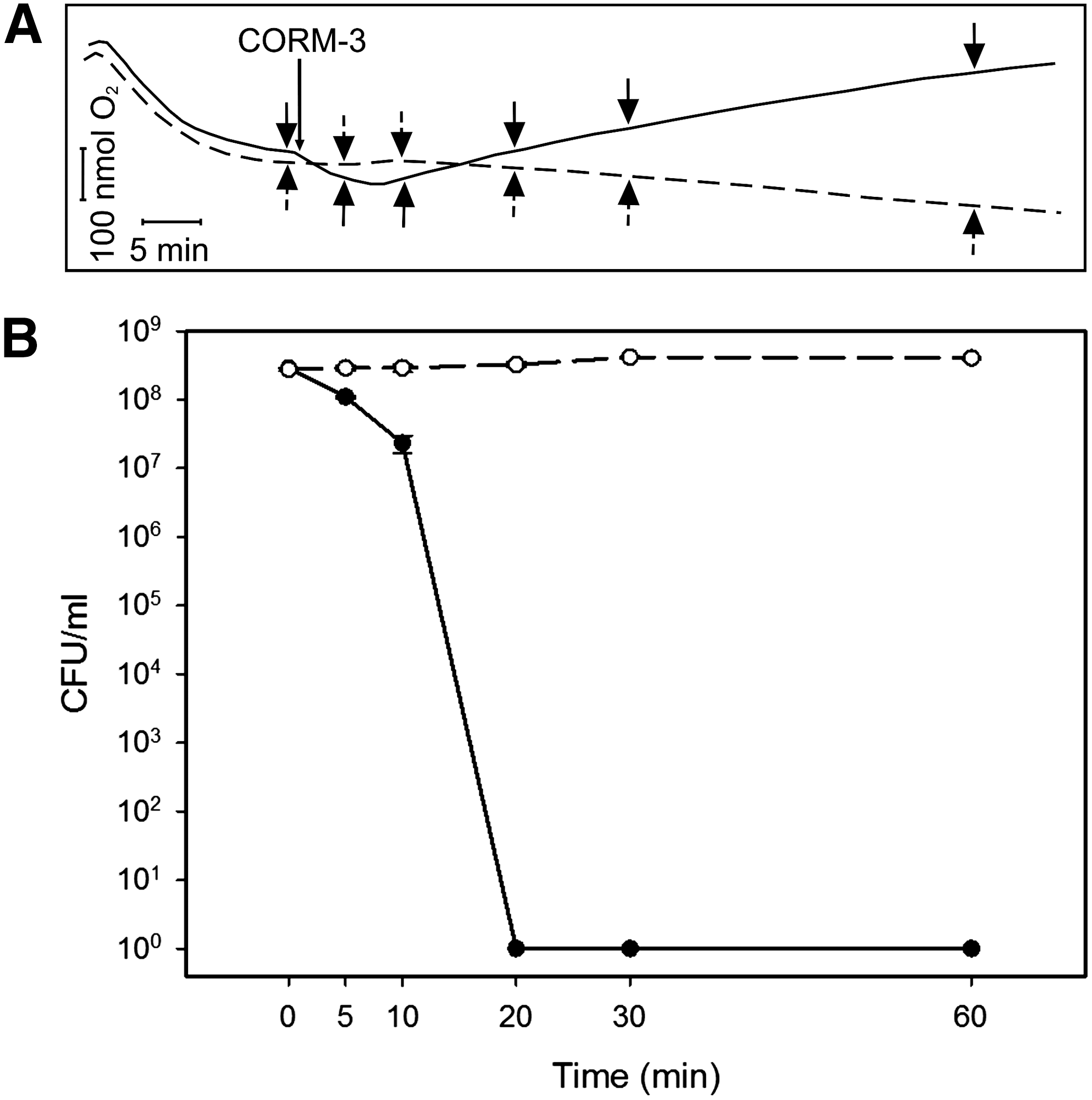
Earlier, we (15) showed that inhibition of respiration preceded loss of P. aeruginosa viability, but did not observe bacterial respiratory stimulation. In the present study, we sampled from an open O2 electrode chamber to simultaneously measure O2 uptake and bacterial viability. On adding CORM-3 to this low-density culture, an initial phase of respiratory stimulation (downward deflection of the O2 trace) followed by inhibition (upward deflection) was observed (Fig. 8A) as before (Fig. 2). Within 10 min of adding 100 μM CORM-3 to a culture, cell viability was reduced by 90% (Fig. 8B) and, within 20 min, the number of viable cells was below the limits of detection (<9×106 cells). These parallel measurements of O2 tension and viability show that loss of viability began and was largely completed during the phase of stimulation, before respiratory inhibition.
Reversal by photolysis of respiratory inhibition protects cells from CORM-3-induced killing
The photoreversibility of CO inhibition of respiration was exploited in the classical photochemical action spectrum approach to identifying terminal oxidases (4). Although CO-RMs inhibit respiration, and the CO binds to terminal oxidases (11,15), it has not been demonstrated formally that respiratory inhibition in vivo is due to CO reactivity with oxidases. To test this hypothesis, we treated membranes in a glass chamber with CORM-3 and measured respiration rates during light–dark cycles (Fig. 9A). Addition of 300 μM CORM-3 inhibited respiration completely when the O2 in the chamber was reduced to ∼45 μM using N2-saturated buffer (Fig. 9A). Exposure to actinic light reversed this effect, and repeated light–dark cycles confirmed that CORM-3 inhibition was reversible by white light, thus identifying heme oxidases(s) as primary respiratory target(s).
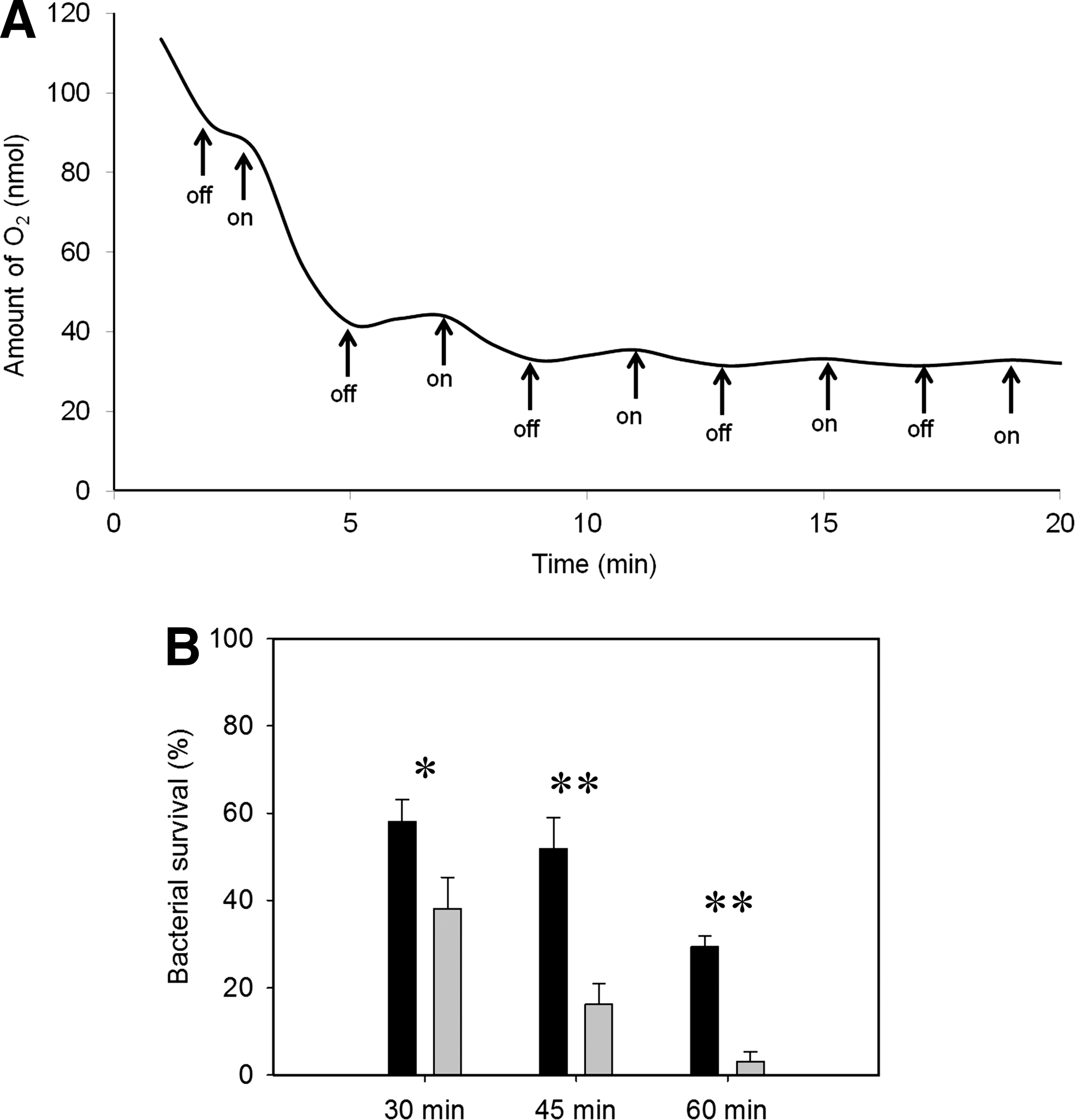
We reasoned that, since cells are killed by CORM-3 during respiratory perturbations (Fig. 8A, B) and since inhibition of respiration is light-reversible (Fig. 9A), then cell viability may be protected from CORM-3 under illumination. This hypothesis is supported by Figure 9B; 30 μM CORM-3 elicited a drop in cell survival after exposure to actinic light, such that, at 60 min, ∼30% of the initial viable counts remained. However, the viability loss was dramatically exacerbated in the dark; 95% of the population was killed in 60 min (Fig. 9B). We could not determine whether cells “recover” when cultures are switched from dark to light because the effects of CO-RMs are not bacteriostatic, but bactericidal as shown in Figure 8 and elsewhere (11,43).
CORM-3 is an inhibitor of respiration in pathogenic bacteria and yeast
CORM-3 (25–100 μM) inhibited respiration not only of E. coli but also of P. aeruginosa (Supplementary Fig. S4A), in which inhibition of growth has previously been reported (15), and Salmonella enterica serovar Typhimurium (Supplementary Fig. S4B). For P. aeruginosa, the inhibition of respiration elicited by 25, 50, and 100 μM CORM-3 was typically 23.7%, 61.7%, and 74.7% ∼30 min after CORM-3 addition. In S. Typhimurium, the inhibition of respiration elicited by 50 μM CORM-3 was typically 74.8% 30 min after addition. Respiratory stimulation was not observed in these species. Control experiments employing one or more iCORM-3 compounds [RuCl2(DMSO)4, iCORM-3, and miCORM-3] showed that only CORM-3 was effective. In S. Typhimurium, for example, 50 μM miCORM-3 was not a respiratory inhibitor (Supplementary Fig. S4B). Although CORM-3 also inhibited respiration of Candida albicans, higher concentrations were required (250–500 μM; Supplementary Fig. S4C) than for bacteria (Supplementary Fig S4A, B). These concentrations typically inhibited respiration by 13.4% and 39.0%, respectively. At lower concentrations (50–100 μM), opening the electrode chamber led to a continuous decline in dissolved O2 due to uninhibited respiration, which was identical to the control (i.e., no CO-RMs added). Again, 250 and 500 μM iCORM-3 were ineffective.
Discussion
Utilizing CO gas as an antimicrobial therapeutic (6) is thwarted by CO toxicity and the systemic delivery methods necessary (40). CO-RMs allow localized CO delivery, but the fate of administered CO-RMs is poorly understood. Although CO-RMs release CO inside bacteria (11,43), we do not understand the biochemical basis of CO-RM action. In the case of CORM-2 and ALF062, a contributing factor is the generation of oxidative stress (61). Here, we demonstrate unequivocally that CORM-3 is a potent inhibitor of microbial respiration, and rapid loss of bacterial viability correlates temporally with the interference of respiratory metabolism (Fig. 8).
CO is widely used as a probe for hemes in biology and for oxidase identification and studies of ligand binding/exchange [e.g., (19)]. We previously demonstrated oxidase–CO adducts in CO-RM-treated cells (11,15) and here show that such complexes are photolabile at ambient temperatures with moderate intensities of visible light; photolysis reverses respiratory inhibition, alleviating the loss of cell viability (Fig. 9). To our knowledge, this is the first evidence that the biological consequences of CO release from a CO-RM can be reversed by light, although CO-RMs that release CO on irradiation have been described [reviewed in (54)]. These findings have important implications for future therapeutic applications of CO-RMs in the light.
Several lines of evidence suggest that the mode of action is more complex than the release of CO at concentrations equal to the administered CO-RMs. First, unlike other inhibitors of bacterial oxidases, for example, cyanide (23,64), inhibition by CORM-3 in whole cells is slow and not readily observed in a conventional, closed O2 electrode. This is presumably because of the need for the CO-RMs to penetrate the cell and interact with cellular species [e.g., (38)] that displace CO. Respiratory inhibition is accelerated by preincubating cells with CO-RMs under anoxic conditions (Fig. 2), thus maximizing the CO:O2 ratio and binding of CO to target hemes, as reported for hypoxic mitochondria (10).
A further complexity is the stimulation of respiratory rates that precedes inhibition (Figs. 1 and 2). This can indicate not only protonophore activity in bacteria as in the case of CCCP (Fig. 3B) or nisin (12) but also the action of any compound that catalyzes electrogenic ion movement. As here, Iacono et al. (22) showed in mitochondria that the effect of CORM-3 was unlike that of protonophores. First, much higher CO-RM concentrations (around 2–50 μM) were needed than for FCCP (0.02–1 μM) to observe stimulation of State 2 respiration, which was more marked and instantaneous with the latter compound (2.4-fold stimulation with 20 μM CORM-3 vs. 6.3-fold with 1 μM FCCP). Second, mitochondrial Δψ was minimally affected by CORM-3 compared with FCCP. The effect of 0.02 μM FCCP was similar to that of 20 μM CORM-3, whereas 1 μM FCCP collapsed the potential instantaneously. No direct measurements of H+ fluxes accompanying respiratory stimulation, the hallmarks of a classical protonophore, were reported (22), although the “recoupler” 6-ketocholestanol did not affect mitochondrial stimulation by CORM-3 (22). Further, in models of renal function and sepsis, low CORM-3 concentrations increased the respiratory control index, a measure of the tightness of coupling (31,51). In the present bacterial case, there was no collapse of the pmf as expected (Figs. 3 and 4; Supplementary Fig. S3). CO gas slightly enhances terminal oxidase activity in mitochondria (1), but not in E. coli (Fig. 7D–F). We suggest that the term “uncoupling,” to describe CO-RM-promoted respiration, is inappropriate, as it implies a dissociation (i.e., uncoupling) between the processes of respiratory electron transfer and adenosine triphosphate (ATP) synthesis (41), as in the “uncoupled” (unc) mutants of E. coli with defects in the FoF1 ATP synthase (16). We conclude that CORM-3 stimulates respiration not by acting as a protonophore but rather by its transient effects on ion transport and pmf.
Despite similarities between the effects of CORM-3 on mitochondria (22) and bacteria, there are important differences. First, the concept of States 2 (ADP-limited) and 3 (ADP-stimulated) (5) to describe the effects of ADP availability on respiration tightly coupled to ATP synthesis is not generally applied to bacteria. Respiratory stimulation in bacteria by ADP, to which bacteria are impermeable, is not easily demonstrated, although the effects of protonophores can sometimes be observed as a stimulation of respiration (45). ADP/ATP exchange cannot be measured in fragmented bacterial membrane vesicles, and intact bacteria are not susceptible to oligomycin used before (22) to block ATP synthase activity. Further, 5-hydroxydecanoate, an inhibitor of mitochondrial ATP-dependent K+ channels, did not reduce CO-RM-elicited respiratory stimulation, whereas, in the present study, CORM-3 was an effective K+ and Na+ ionophore (Fig. 6). The basis of the promoting effect on K+ transport is currently unclear; E. coli possesses several systems for transport of K+, the major intracellular cation, which is maintained at 0.1–0.5 M. The most attractive candidate may be Kch (30), the first prokaryotic K+ channel to be identified with clear homology to those eukaryotic voltage-activated K+ channels that CORM-2 modulates (24). A recent report (49) suggests that CO prevents, not promotes, mitochondrial permeabilization, thereby preventing swelling, but the effect appears unlinked to K+ permeability.
A probable explanation of the greater effectiveness of CO-RMs, compared with CO gas, is that the intracellular concentrations of CO achieved by CO-RM administration are significantly higher than for CO gas, despite the facile diffusion of CO through membranes. Based on Ru analysis, the intracellular level of CORM-3 is >200 μM after adding only 30 μM to growing cultures (11). The preferential release of CO inside cells is consistent with recent data showing that CO release rates from CORM-3 are greatly enhanced by sulfite, but not by a reaction of CORM-3 with globins without such a ligand (38). In the present work, corroborating evidence for the requirement of an intracellular or other complex biological milieu is provided by Figure 7D–F that fails to show effects of CO. Further, CO gas did not promote cation transport (Fig. 6A).
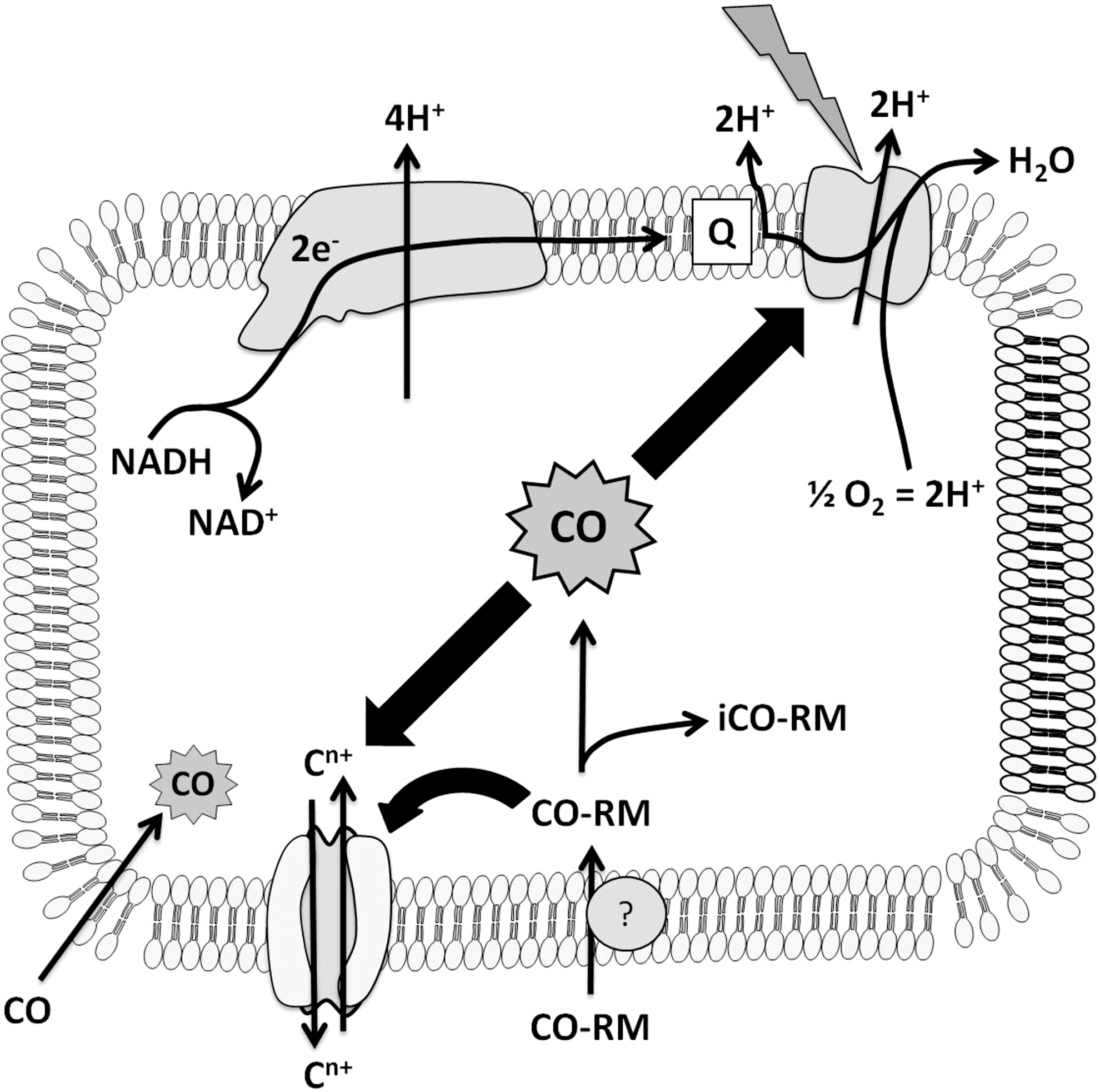
We now propose a “Trojan Horse” mechanism for the antimicrobial actions of CO-RMs to explain the potent effects on respiration that cannot be mimicked by CO gas (Fig. 10). In brief, a CO-RM may lose its CO permanently in the extracellular milieu through, for example, the presence of a species to capture the CO or a change in the CO-RM after CO loss. Alternatively, the CO-RM is transported into the cell and concentrated (11) carrying its CO cargo (Trojan Horse effect), and CO dissociation is promoted by a reaction with intracellular ligands that include sulfite (38). Thus, CO is immediately accessible at relatively high concentrations to membrane-bound heme targets, to which it binds. Respiration is inhibited in a light-reversible manner, and viability is drastically reduced.
Nonheme targets for CO may explain, in part, the diverse effects of CO and CO-RMs (3,34,40). There are many examples of nonheme Fe(II) carbonyls, and ligands from amino acids (S in Cys and N in His) may be targets in voltage-gated (Slo1 BK) channels that are important in vasodilation (21) or in bacterial ion channels (Fig. 6). Other nonheme targets are likely: In Chlamydomonas hydrogenase, CO binds to an Fe-Fe center (60), and CO also binds to binuclear copper sites as in tyrosinase (29) and hemocyanins (17,62). Identifying modes of action is critical if CO and CO-RMs are to be used therapeutically.
Materials and Methods
Microbial strains and growth conditions
E. coli K-12 MG1655 (RKP5416) (2), P. aeruginosa PAO1 (RKP5417), S. enterica serovar Typhimurium (RKP4901), and a clinical isolate of C. albicans (CA3153 302, kind gift from Professor P. Sudbery, The University of Sheffield) were used. E. coli and S. Typhimurium were grown in defined medium with glycerol (54 mM) (11). P. aeruginosa was grown in minimal M9 medium with glucose (11.1 mM) as a carbon source (15). C. albicans was grown in defined medium containing D-glucose (111 mM) and yeast nitrogen base (6.8 g/L). Bacteria were grown aerobically in 20–30 ml medium in 250 ml flasks fitted with side arms for measurements of optical density with a Klett meter (red filter) during shaking at 200 rpm and 37°C.
CORM-3 and control treatments
Aqueous stock solutions of CORM-3 (25) and RuCl2(DMSO)4 (the latter compound supplied by Dr. Tony Johnson, Chemistry Department, The University of Sheffield) at 10 or 100 mM were made fresh each day. iCORM-3 was prepared (8,22) by dissolving CORM-3 in phosphate-buffered saline (PBS), allowing CO liberation to the atmosphere for 48 h at room temperature, and bubbling with O2-free N2 (BOC; GU2 5XY) periodically throughout the incubation to ensure dissociation and loss of CO. miCORM-3 was prepared by treating CORM-3 twice with a twofold excess of ferrous myoglobin, prepared by adding sodium dithionite solution (2.2 mM final concentration in 0.1 M potassium phosphate, pH 7) to 1 mM metmyoglobin. Carbonmonoxy myoglobin was separated from the required inactivated CORM-3 residue by centrifugation in a Vivaspin 20 concentrator (Sartorius Stedim Biotech) with a molecular weight cutoff of 5 kDa. CORM-3 or inactivated CO-RMs were added directly to cells suspended in medium or 50 mM Tris (pH 7.5) in an O2 electrode chamber. CO was added as a solution saturated with the gas by bubbling from a cylinder (BOC) at room temperature. CO release from CORM-3 and control molecules to ferrous myoglobin was assayed (8,38) in a dual-wavelength scanning spectrophotometer (27). Data were plotted as CO reduced minus reduced spectra (63).
O2 consumption
Cultures were harvested at midexponential phase, washed, and resuspended in medium or 50 mM Tris buffer (pH 7.5). Cells were suspended in a stirred Perspex chamber fitted with a Clark-type polarographic O2 electrode (OXY041A; Rank Brothers Ltd.; CB25 9DA) at 37°C (20). Data were recorded on a chart reader (REC112; Amersham Pharmacia Biotech) or a Lab-Trax-4/16 recorder and Data-Trax™ software (World Precision Instruments, Inc.). Protein concentrations were measured using a modified Lowry procedure (37). Respiration was stimulated by the addition of glycerol or nicotinamide adenine dinucleotide (NADH). An “open” O2 electrode system was used in most experiments, in which a stirred sample is open to the atmosphere, allowing continuous O2 diffusion from the vortex surface into the sample; prolonged measurements can be made without O2 depletion (14). To correlate O2 consumption and bacterial viability, cells were added to the chamber at a low concentration that would result in a steady-state level of c. 25% air saturation. Samples were taken and diluted in PBS followed by plating drops (diluted 10−5–10−8) on nutrient agar for viability counts.
Photosensitivity of respiratory inhibition and loss of viability elicited by CORM-3
Bacteria were grown in 1 L Luria broth (LB) medium in 2 L baffled flasks at 37°C with shaking at 250 rpm until late exponential phase. Membranes were prepared (46) and incubated in a glass chamber (RC350 respiration cell; Strathkelvin Instruments Limited; ML1 5RX) with 1.95 ml N2-saturated buffer (50 mM Tris-HCl, 2 mM magnesium chloride, and 1 mM ethylene glycol tetraacetic acid; 37°C), then treated with 300 μM ethylene CORM-3. A microcathode O2 electrode (SI130; Strathkelvin Instruments Limited) located in the chamber lid was connected to an O2 meter (Model 781) and then to a Lab-Trax-4/16 recorder and Data-Trax software as described earlier. Respiration was stimulated with 12.5 mM NADH. A 150 W projector bulb was focused on the wall of the glass chamber using a convex lens, giving an intensity of 175,000 lux at the vessel surface.
To make parallel measurements of respiration rate and bacterial viability, bacteria were grown to midexponential phase (50 Klett units) in defined medium. Samples (2 ml) were transferred to two parallel glass respiration cells, as described earlier, and stirred at 37°C; one was foil-wrapped, and an actinic light beam was focused on the other as described earlier. Samples were taken immediately before addition of 30 μM CORM-3 and at intervals thereafter for viability counts.
Respiration-driven H+ translocation across the bacterial membrane
The apparatus was based on (32,33) incorporating a sealed, stirred Perspex chamber fitted with a Clark-type polarographic O2 electrode (OXY040A; Rank Brothers Ltd; CB25 9DA) at 30°C. The lid of the chamber was modified to support a semimicro calomel combined pH electrode (pHC4000; MeterLab; Radiometer Analytical). The signal from the pH electrode was taken to a pH/ion meter (PHM240; MeterLab; Radiometer Analytical) and then to a potentiometric recorder.
E. coli was grown aerobically at 37°C and 200 rpm in defined medium supplemented with 0.1% casamino acids to midexponential phase. Cells were starved by shaking for 2 h in medium lacking carbon sources, harvested by centrifugation at 4°C, washed twice, and resuspended in cold 150 mM KCl. An aliquot was added to 2.5 ml lightly buffered medium (150 mM KCl, 50 mM potassium cyanide, and 1.5 mM glycylglycine; pH 7; 30°C) in the chamber. Valinomycin (Sigma-Aldrich; 22 μM final concentration, added as a methanolic stock solution) was added immediately after, followed by 1 mM glycerol to stimulate respiration. The experiment commenced after consumption of O2. Additions of an anoxic solution of 5 mM HCl were used to calibrate the apparatus (25–75 ng-ion H+). Pulses of air-saturated 150 mM KCl (30°C) (25 μl, 11 ng-atom O) were added to promote respiration-driven H+ translocation and medium acidification. CCCP (Sigma-Aldrich; 1.6 μM final concentration) was used as protonophore. H+/O ratios were calculated from deflections caused by the acid and O2 pulses.
[3H]methyltriphenylphosphonium iodide accumulation
The Δψ of washed cell suspensions optical density (OD600 ∼1.2) in 50 mM Tris buffer (pH 7.5) was determined by measuring the accumulation of [3H]methyltriphenylphosphonium iodide ([3H]TPMP+, 30–60 Ci/mmol; 10 nM final concentration, NEN™; Life Science Products, Inc.) using filtration assays (0.45-μm cellulose-acetate filters; Sartorius Stedim Biotech) (55) after incubation at 37°C. Filters were washed twice with 2 ml of 100 mM lithium chloride and dried for 60 min at 40°C. Filters were resuspended in 2 ml of scintillation liquid, and counts per minute were determined using an LKB Wallac 1214 Rackbeta liquid scintillation counter. The Δψ was calculated from the Nernst equation (Δψ=62×log [TPMP]in/[TPMP]out), and an intracellular volume of 2.8±0.5 μl per mg of protein was used (57).
Spheroplasts and osmotic swelling measurements
Cells were grown in 400 ml LB, harvested at midexponential phase and spheroplasts were prepared (36). Cells were washed once in 10 mM Tris-HCl (pH 7.4) and resuspended in 20% (w/v) sucrose containing 33 mM Tris-HCl (pH 8) to an OD600 of c. 0.8. The suspension was stirred gently at 4°C with additions of 0.01 ml of 0.1 M ethylenediaminetetraacetic acid (pH 8) and 1 μl of lysozyme (5 mg/ml) (per ml suspension). Osmotic fragility was checked by 10-fold dilutions into water and measuring decreasing turbidity at 500 nm. When spheroplast formation was complete (no further reduction in OD500), they were harvested at 4°C, washed once, and resuspended gently in the same Tris/sucrose buffer using a loose-fitting hand homogenizer. Osmotic swelling was studied (18) by following the change in turbidity at 500 nm, using a Cary 50 spectrophotometer (Varian), following dilution of spheroplast samples in iso-osmotic 0.25 M solutions of KNO3, KNO2, or NaNO3. Valinomycin and X-537A (Sigma-Aldrich; acetonitrile solution) were used at final concentrations of 1 μg/μl.
Footnotes
Acknowledgments
This work was supported in part by the Biotechnology and Biological Sciences Research Council (United Kingdom) and by a Vacation Studentship to K. Naylor.
K.S. Davidge thanks the Royal Society and the Society for General Microbiology for travel funds.
Author Disclosure Statement
B.E. Mann declares a financial interest in Alfama. All other authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.