
Abstract
Introduction to the Phagosomal NADPH Oxidase
Particulate stimuli can mediate phagocytosis using different types of receptors, including FcγR (binding IgG-opsonized particles), CR (recognizing particles opsonized by complement) (28, 50), and a set of glycoconjugate receptors (interacting with bacterial lectins) (60). Engulfment and ingestion of particles are accompanied by phagosome-granule fusion before or concomitantly with the localized activation of the NADPH oxidase on the phagosomal membrane and the subsequent release of reactive oxygen species (ROS) in the lumen of the phagosome that ultimately destroy the invading microbes (5).
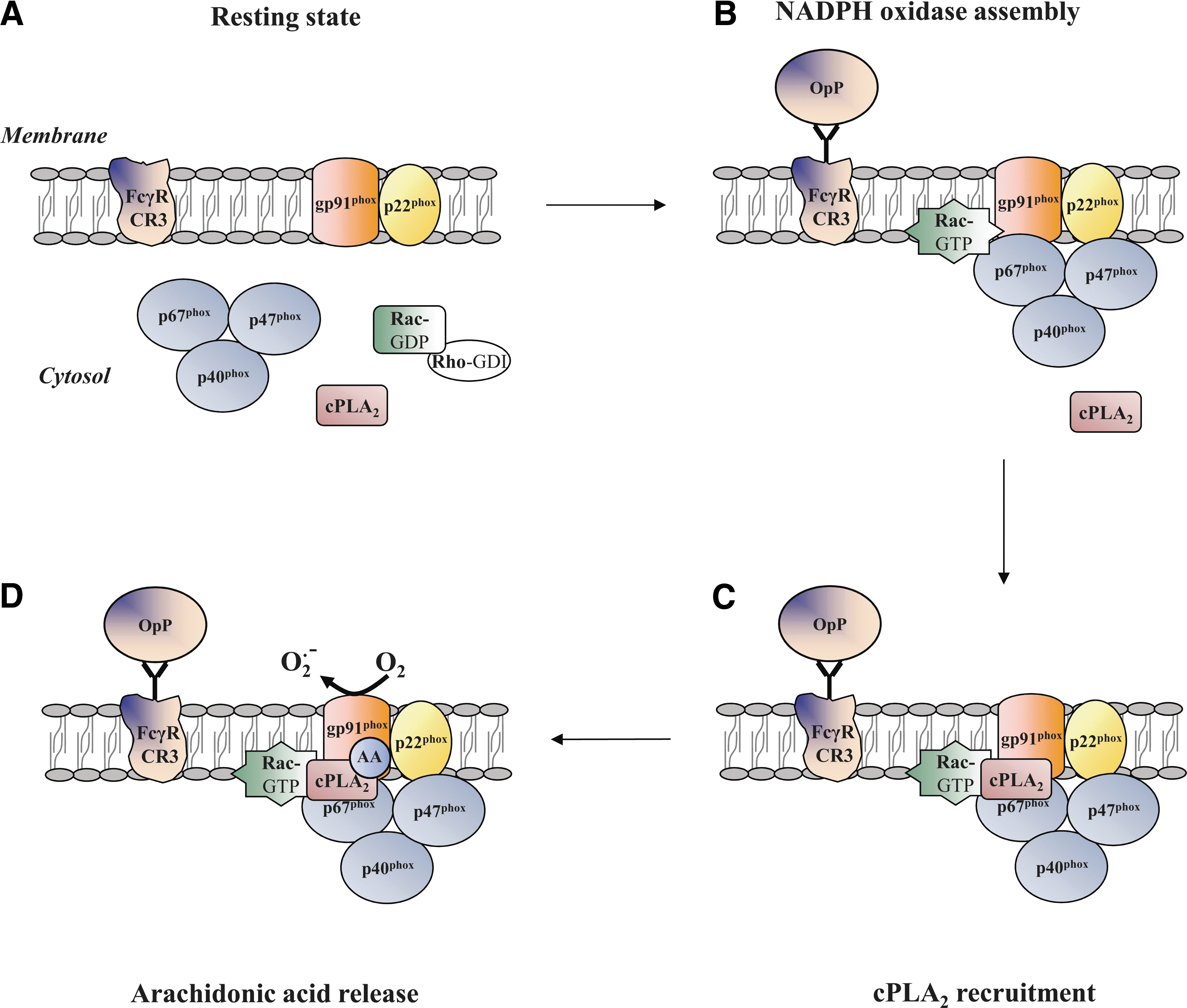
NADPH oxidase is a multicomponent enzyme system that is composed of two integral membrane-associated proteins and several cytosolic proteins. In the resting state, gp91phox (also referred to as Nox2) is associated with p22phox at the membrane of specific granules and, to a lesser extent, at the secretory vesicles, gelatinase granules, and the plasma membrane (102). Both proteins constitute the heterodimeric flavocytochrome b 558 that contains, at the cytosolic side, a putative NADPH binding site, FAD, and two heme groups which are required to transfer electrons from NADPH located in the cytosol to molecular oxygen across the phagosomal membrane. Phagosomes that contain engulfed particles rapidly acquire cytochrome b 558 that has been released by specific and gelatinase granule membrane fusion to the phagosome (58). The activity of cytochrome b 558 is regulated by interactions between the cytosolic proteins, p47phox, p67phox, and p40phox, and the small GTP-binding protein Rac1/2. On stimulation by an opsonized particle, the cytosolic subunits are recruited to the phagosomal membrane where they assemble with the cytochrome b 558 to form the active NADPH oxidase (5, 6). Sustained phagosomal NADPH oxidase activity requires a continuous translocation of cytosolic components to the phagosome (120). The control of NADPH assembly and activation in nonphagosomal intracellular membranes is beyond the scope of the present review and has been discussed elsewhere (11, 119).
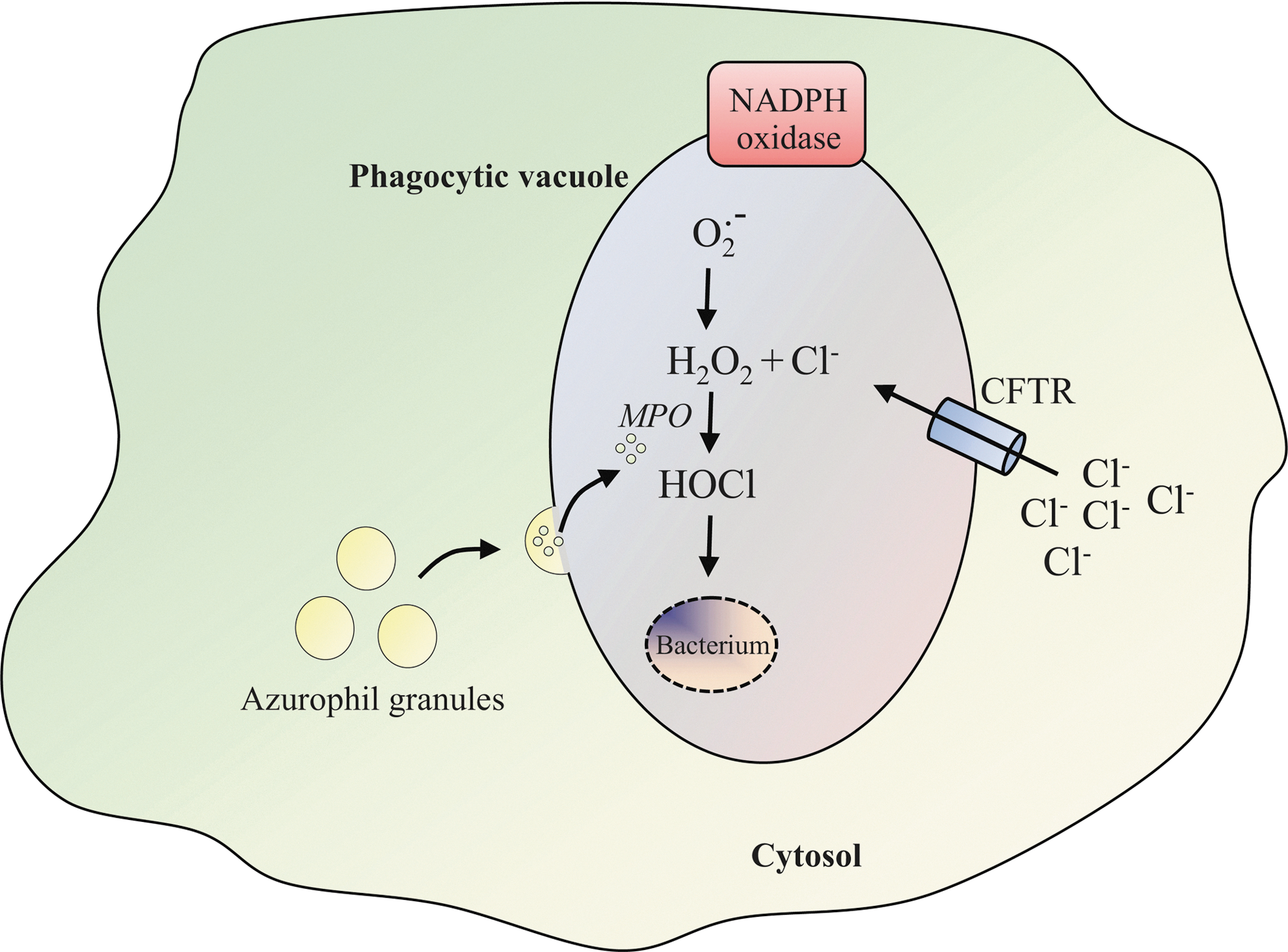
Superoxide anion radicals (O2 .−), generated by the NADPH oxidase inside the phagosome, have a very low bactericidal potency and rapidly dismutate into hydrogen peroxide (H2O2). H2O2 is then further converted into even more toxic compounds. The reaction between H2O2 and chloride ions (Cl−) generates hypochlorous acid (HOCl), which is considered the major oxidative weapon used by phagocytes. The formation of HOCl is catalyzed by myeloperoxidase (MPO), which is released after fusion of primary azurophil granules with the phagosome in proximity of the ingested pathogens (Fig. 1) (78). The physiological importance of NADPH oxidase-generated ROS in host defense is illustrated by a severe immunodeficiency called chronic granulomatous disease (CGD). The CGD immunodeficiency in which defective killing of phagocytosed pathogens caused by NADPH oxidase dysfunction results from mutations in any of the five NADPH oxidase component genes (Table 1) (12, 24, 48, 72, 109) leads to a predisposition to recurrent bacterial and fungal infections.
for normal subunit expression; −for reduced subunit expression; °for absence of subunit expression.
The role of the phagocyte NADPH oxidase is not only limited to the direct destruction of phagocytosed microbes via ROS production. Intraphagosomal luminal pH variation is stringently controlled by NADPH oxidase activity and drastically differs among neutrophils as compared with the well-known gradual acidification observed in macrophages (51). Indeed, luminal pH remains almost neutral for several minutes, after which the mature phagosomes eventually acidify. The acidification that is potentially established by the action of the vacuolar-ATPase delivered by fusion of neutrophil granules to the phagosomal membrane which pumps protons into the lumen of the phagosome is required for the optimal activity of proteases and hydrolases involved in pathogen killing (33).
Regulation of the phagosomal NADPH oxidase during phagocytosis is a complex and not completely understood process. The involvement of different signaling pathways is likely to vary depending on the types of receptors that are engaged (Table 2) (39, 50, 79) and the size of target particles. In this review, we consider the recent progress underlying phagosomal NADPH oxidase activation at the molecular level. We focus on the most-widely studied model of FcγR and CR3-dependent ingestion by neutrophils, which rely heavily on ROS production to mediate antimicrobial defenses within the phagosome. This review summarizes the upstream signaling pathways that lead to downstream phagosomal NADPH oxidase activation. In particular, those mechanisms by which Ca2+ and phospholipid metabolism might modulate NADPH oxidase activity are discussed. Since conclusions regarding the regulation of the plasma membrane NADPH oxidase may be extrapolated to its regulation at the phagosomal membrane, we also discuss recent findings that highlight not only the role of these two signaling pathways but also additional regulatory events.
IP3, inositol 1,4,5-trisphosphate; S1P, sphingosine 1-phosphate; [Ca2+]c, cytosolic free Ca2+ concentration.
Regulation of the Phagosomal NADPH Oxidase Activity
The activation of the NADPH oxidase is tightly regulated by reversible protein–protein and protein–lipid interactions. When phagocytes encounter foreign particles, binding to cell-surface receptors triggers the engagement of multiple signal transduction pathways that are involved in the stimulation of protein kinases/phospholipases. Activation of these enzymes leads to the production of lipid second messengers and Ca2+ release, which ultimately progress to the formation of a functional NADPH oxidase complex on the phagosomal membrane (5). Current understanding of the phagocytic-coupled activation of the NADPH oxidase is mostly based on experiments of phagocytosis in macrophage-like cells [for review, (73)]. However, the idea that innovative techniques have also provided insights into the functional assembly and into the targeting at the phagosome of the NADPH oxidase on neutrophil particulate stimulation should also be considered (77).
Regulation of Phox Proteins by Phosphorylation
Phosphorylation events are key regulatory steps in NADPH oxidase assembly and activation. NADPH oxidase assembly is accompanied by phosphorylation of the cytosolic components p47phox, p67phox, and p40phox. These proteins are phosphorylated by protein kinase C (PKC) isoforms, the ERK/p38MAPK-dependent pathway, cAMP-dependent protein kinases, and protein tyrosine kinase-dependent pathway (18, 31). The current state of knowledge regarding the cytosolic phox protein phosphorylation and subsequent protein–protein interactions involved in the regulation of the NADPH oxidase assembly has been extensively obtained from neutrophils stimulated with soluble physiological agonists such as tumor necrosis factor-α or granulocyte macrophage–colony-stimulating factor (17), only rarely with particulate stimuli (13, 19), and it will be not detailed here [for reviews, see (31, 32, 42)].
Importance of cytochrome b558 phosphorylation
The phosphorylation of cytochrome b 558 in neutrophils is poorly documented, although the phosphorylation of the p22phox subunit has been observed a long time ago (40). Later, McPhail and coworkers demonstrated that p22phox phosphorylation is correlated with NADPH oxidase activity, suggesting that the phosphorylation of p22phox regulates multicomponent enzyme activation (94). p22phox phosphorylation does occur on the threonine 147 residue (64), but its precise role in NADPH oxidase activation has not yet been identified. A recent publication indicated that gp91phox phosphorylation by PKC enhances NADPH oxidase activity as well as increases the binding to Rac2, p47phox, and p67phox (90).
Role of cytosolic phox protein phosphorylation during phagocytosis
The best example of subunit phosphorylation and its physiological significance remains the well-understood intracellular signaling pathways involved in p47phox phosphorylation that induce conformational changes of p47phox (32, 53) to a state which is accessible for p22phox binding, allowing for NADPH oxidase activation (1). The phosphorylation of p47phox provides a control mechanism that modulates NADPH oxidase assembly and/or activity and, thus, prevents inadequate activation. The recent study conducted by Marcoux et al. demonstrated that the phosphorylation of three serine residues in the auto-inhibitory region (AIR) of p47phox is critical for the release of the phox homology (PX) domain of the protein and its subsequent interaction with p22phox at the phagosomal membrane (70). Although the PX domain of p47phox is only a passive requirement for phagosomal superoxide production, experiments using phosphorylation/activation-mimicking p47phox mutants indicated that adequate serine phosphorylation is required for p47phox-induced, p40phox-independent, FcγR-mediated NADPH oxidase activation (65, 117).
The recent discovery of a CGD patient expressing a mutation in the PX domain of p40phox has firmly established the importance of p40phox in oxidase activation (72). Controversies over the role of p40phox phosphorylation exist depending on the experimental approach. In a cell-free system, p40phox phosphorylated on threonine 154 was shown to inhibit oxidase activity (66). Conversely, the PKCδ-mediated phosphorylation of threonine 154 in p40phox (but not serine 315) as well as p47phox translocation to phagosomes is required for full oxidase activation in mouse neutrophils (14). The involvement of NADPH oxidase activity-mediated p67phox phosphorylation still remains unclear.
It is of interest to investigate whether the phosphorylation of specific phox proteins has a role in modifying the affinity of the subunits for each other or with cytochrome b 558 and/or influencing the stability of the complex. Identification of the phosphorylated sites, determination of the phosphorylation status, and site-directed mutagenesis studies should continue to illuminate the significance of phox protein phosphorylation in the regulation of NADPH oxidase during phagocytosis.
Regulation of Phox Proteins by Lipid Metabolism
In addition to phosphorylation, accumulating evidence demonstrates that membrane lipids play a crucial role in the spatiotemporal regulation of NADPH oxidase activity.
Considerable efforts have been undertaken to understand the mechanisms that underlie the lipid-protein interactions and lipid-mediated protein-protein interactions involved in NADPH oxidase activation. It is well documented that the production of diacylglycerol by phospholipase C (PLC) can induce the activation of PKC isoforms (68), which may mediate the phosphorylation of p47phox [for reviews (32, 36)]. The activation of cytosolic phospholipase A2 (cPLA2) is also required for NADPH oxidase activity, but its complete role remains relatively unclear. cPLA2 has been found to translocate to the membrane fractions (45) and to catalyze arachidonic acid (AA) production in opsonized zymosan-stimulated human neutrophils (98). cPLA2 translocates from the cytosol and targets the phagosome membrane during phagocytosis by mouse macrophages (41). On opsonized zymosan stimulation, substantial evidence for the essential requirement of cPLA2-generated AA for the activation of neutrophil NADPH oxidase has been derived from the work of Levy's group that was based on differentiated PLB-985 cells which are deficient in cPLA2 (15, 86) or PLA2 inhibitors (16). Other groups have corroborated these results (122). It is likely that cPLA2-generated AA is not required for the membrane translocation of the NADPH oxidase cytosolic factors (105). Actually, cPLA2 is anchored to the plasma membrane by the assembled NADPH oxidase and releases AA (Fig. 2), promoting NADPH oxidase activity (105). It appears that the target site for AA is located within the gp91phox N-terminal domain where it has a role in the activation of electron transfer through the FAD reduction center of NADPH oxidase (86). Indirect evidence supplied by both cell-free systems and intact neutrophils as well as from different technical approaches (e.g., electron spin resonance, atomic force microscopy) (25, 26, 37, 81) provided an alternative hypothesis, stating that AA may affect the gp91phox N-term domain by inducing conformational changes in cytochrome b 558. In fact, several studies in cell-free systems suggest that AA promotes the translocation of p47phox (104) or conformational changes in both p47phox and p67phox (44, 84), thus increasing NADPH oxidase activity. However, the in vitro model may not reflect the entire suite of complexities of NADPH oxidase activation and, thus, probably cannot faithfully recapitulate the biology of whole neutrophils.
Role of phospholipid and p47 phox/p40 phox interactions
Phospholipase D (PLD), which cleaves phospholipids to form phosphatidic acid (PA), is also an important modulator of NADPH oxidase activity. Regier et al. (95) suggested that p22phox phosphorylation is mediated by PLD-dependent and -independent mechanisms (Fig. 3). PLD-dependent phosphorylation of p22phox could be activated by a not-yet-identified PA-activated protein kinase, whereas the PLD-independent mechanism is mediated by conventional PKC isoforms (94). The significance of p22phox phosphorylation may be the promotion of the p22phox-p47phox interaction at the membrane (46).
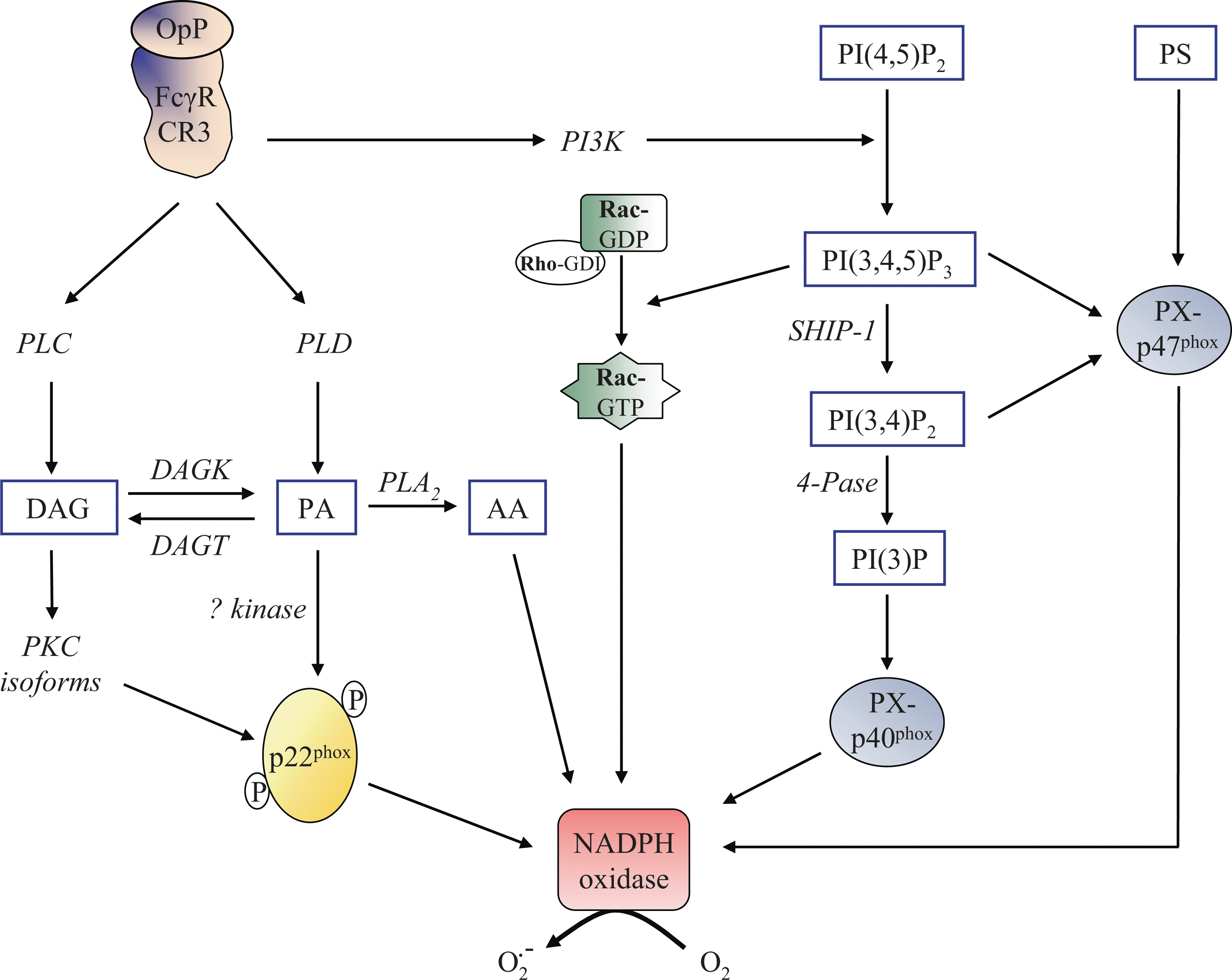
3′-phosphoinositides (PI) are also considered pivotal regulators of NADPH oxidase activation by serving as site-specific membrane signals or by modulating cytosolic localization and/or other biological properties of effector proteins. Direct or indirect PI interactions with the subunits p40phox and p47phox regulate the NADPH oxidase assembly. Both proteins contain a lipid-binding domain, specifically the PX domain that binds PI with a broad specificity. Studies of PX domain structures by X-ray crystallography have revealed the charged binding sites for stereospecific recognition of their cognate PI (10, 57). The PX domain of p40phox specifically binds phosphatidylinositol 3-phosphate (PI(3)P), a lipid product of class III phosphoinositide 3-kinase (PI3K), and is required for p40phox accumulation at the phagosome and assembly/activation of the NADPH oxidase (Fig. 3) (111). Initial evidence regarding the importance of PI(3)P in NADPH oxidase regulation was provided in vitro in semi-recombinant cell-free assays and COSphox systems (35, 111). Generation of the p40phox mutant mice expressing the p40phox which carries a mutation in the PX domain corroborates in vivo that intact binding of PI(3)P to the intact PX domain of p40phox is required for FcγR-mediated ROS production by professional phagocytes (9). The group of Saito (117) proposed that p40phox is able to acquire PI(3)P-binding capacity through conformational changes induced by H2O2 and functions as a carrier of the cytosolic p47phox- p40phox-p67phox complex. Binding of PI(3)P to p40phox PX domain has a differential impact on the downstream signaling pathways, depending on the phagocytic receptor subtypes and the type of target particles (34).
The PX domain of p47phox has two distinct lipid binding pockets: one with preferential affinity for phosphatidylinositol 3,4 bisphosphate (PI(3,4)P2) and phosphatidylinositol 3,4,5 trisphosphate (PI(3,4,5)P3), while the other nonspecifically binds anionic phospholipids such as PA and phosphatidylserine (PS) (Fig. 3) (55, 108). The PI-dependent membrane-binding mechanisms of PX domains and their involvement in NADPH oxidase activation have not been clearly identified. However, it has recently been suggested that p47phox allows p40phox to acquire PI(3)P binding on targeted membranes that act in cooperation with p47phox as an adaptor of NADPH oxidase assembly on the phagosome (117).
The role of the p47phox PX domain in phagosomal NADPH oxidase activation may be selective and may vary greatly depending on the nature of the stimulus. Mutations in the p47phox PX domain that result in the loss of PI(3,4)P2 binding does not impair phagosomal membrane recruitment and the normal activity of NADPH oxidase during phagocytosis of a variety of particles in mouse neutrophils. In contrast, plasma membrane NAPDH oxidase activity is reduced in neutrophils expressing p47phox mutants (65). Thus, although the formation of the phagosomal NADPH oxidase complex is considered similar to the plasma membrane, it seems clear that the regulation of the plasma membrane NADPH oxidase activity differs in terms of signaling pathways from that of phagosomal NADPH oxidase.
Charge-dependent localization of Rac proteins
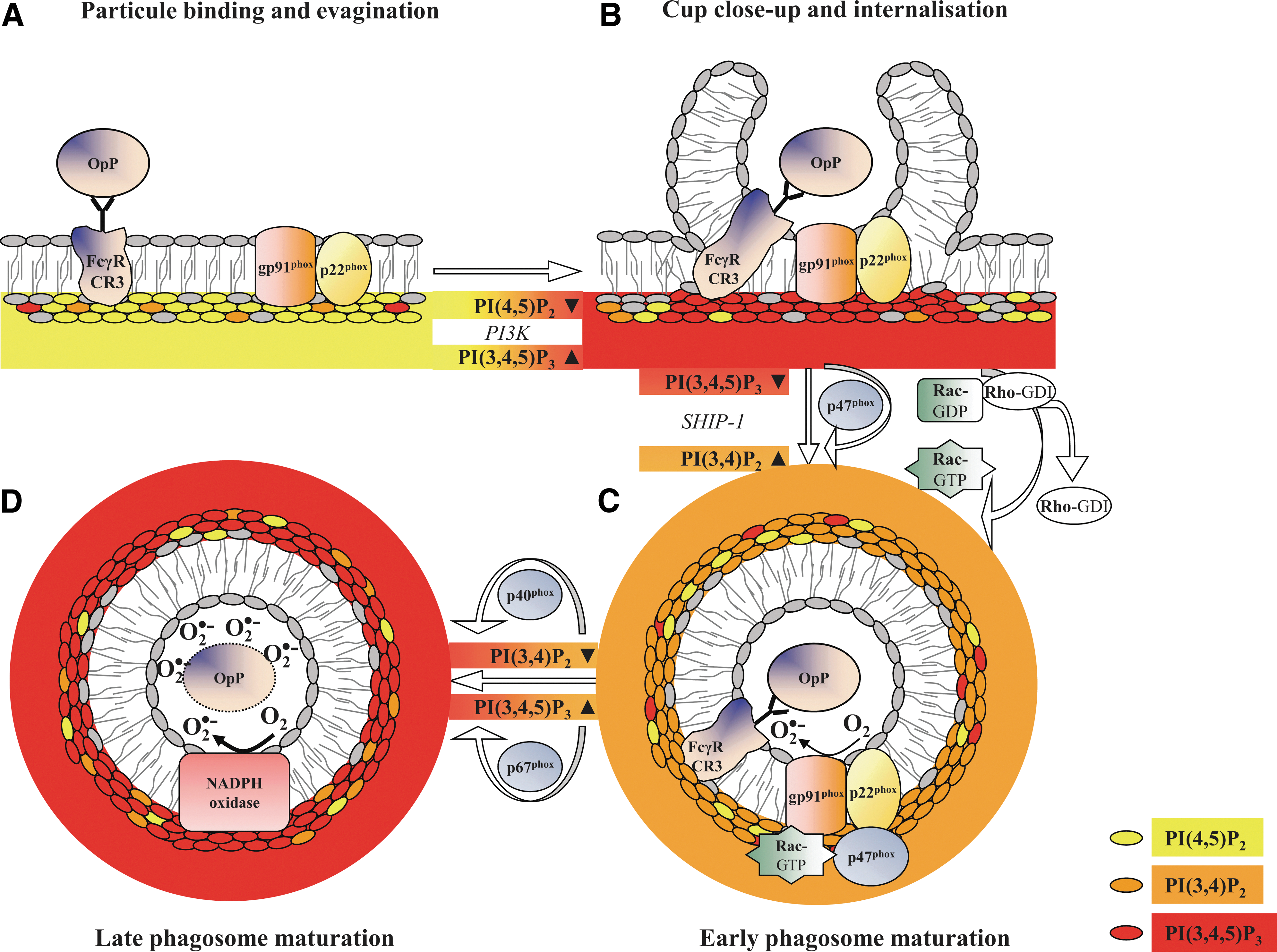
As for p40phox and p47phox, the translocation of positively charged small GTPases to the membrane may involve direct interactions with anionic lipids, mainly PI, PA, and PS. Lipid metabolism regulates the net membrane charge, which is responsible for the selective recruitment of Rac proteins that rely heavily on their own inherent net-positive charge. Based on alterations of lipid metabolism during phagocytosis, a charge-shift mechanism dictates localization patterns to distinct membrane compartments of Rac1 and Rac2 (112). On stimulation, Rac proteins dissociate from RhoGDI, exchange GDP against GTP, and translocate to the membrane where they bind to p67phox (4). They may either act as an adaptor to ensure correct positioning of p67phox toward NADPH oxidase or participate in the electron-transfer reaction. The electrostatic forces between Rac1 and the plasma membrane are higher for Rac2 than for the plasma membrane. The higher electrostatic forces result in a preferential association of Rac1 with highly charged actin-rich pseudopods and primarily the localization to the plasma membrane. During phagosome maturation, membrane lipid composition is altered by the partially localized depletion of specific membrane phospholipids (PI(4,5)P2, PI(3,4,5)P3) and by the partial decrease in PS at the base of the phagocytic cup and phagosomal membrane (Fig. 4). This generates a membrane environment with a moderate negative charge that correlates with the preferential localization of active Rac2 with the intermediately charged phagosome membrane (69).
Contribution of lipid rafts in phagosomal NADPH oxidase activation
Lipid rafts are sphingolipid and cholesterol-enriched insoluble membrane microdomains that are associated with signaling molecules such as receptors. They have the ability to constitute platforms on cell stimulation. These lipid domains have emerged as an important factor in the regulation of NADPH oxidase activity, probably through the control of the efficacy of NADPH oxidase assembly. Indeed, Shao et al. (103) established that NADPH oxidase subunits localize to lipid rafts, which mediate the efficiency of NADPH oxidase coupling to FcγR. A marked negative impact on NADPH oxidase assembly is observed when lipid rafts are disrupted. Later, the work of Jin and co-workers (52) supported this conclusion by highlighting the fact that redox-signaling platforms formed by lipid rafts provide an important driving force for assembling the NADPH oxidase subunits and subsequent NADPH oxidase activation. In addition to the lipid rafts, a member of the Src family tyrosine kinases, which are responsible for the phosphorylation of FcR ITAM motifs and downstream activation of PI3K, has been identified as a regulator of NADPH oxidase recruitment to the phagosomes in rodents (56). This result opens up a new potentiality for our understanding of the mechanisms involved in the regulation of phagosomal NAPDH oxidase activity. In the future, it will be interesting to confirm that Lyn ensures the same function in human neutrophils.
Role of Ca2+ in the Regulation of Neutrophil NADPH Oxidase Activity
Over the years, it has become evident that an elevation of cytosolic-free Ca2+ concentration ([Ca2+]c) in the periphagosomal region participates in NADPH oxidase activation during opsonized zymosan particles-mediated phagocytosis (27, 67). Recent progress in understanding molecular mechanisms linking these two phenomena is highlighted next.
Source of Ca2+-mediated phagosomal NADPH oxidase activity
There is no consensus on the source of Ca2+, and the question of whether phagosomal oxidase activity requires Ca2+ release from intracellular stores or Ca2+ influx remains unanswered. Changes in Ca2+ are global and not restricted to the phagosomal region but are temporally correlated with NADPH oxidase activity (22). Hallett and co-workers research (22) has given rise to speculation that extracellular Ca2+ entry is a rapid event which contributes to [Ca2+]c elevation during C3bi-opsonized zymosan particles-mediated phagocytosis. The fact that knockdown by the specific siRNA of Ca2+ release-activated Ca2+ modulator 1 (Orai1), an essential pore subunit of Ca2+ channels, decreases ROS production on FcγR activation tends to confirm that phagosomal NADPH oxidase activity is dependent on extracellular Ca2+ entry (110). However, Ca2+ influx per se does not exert a sufficient signal that ensures an optimal NADPH oxidase activation during phagocytosis but instead acts in synergy with other events. Given the importance of phospholipids in phagosomal NADPH oxidase activity, it is reasonable to assume that inositol and Ca2+ signaling pathways are interconnected. Observations in B cells that SHIP-1 plasma membrane recruitment on FcγR activation decreases the PI(3,4,5)P3 level and block Ca2+ signals suggest that PI(3,4,5)P3 is a critical regulator of Ca2+ signaling (99). Further, restricting PI(3,4,5)P3 formation by LY294002, a PI3K inhibitor, has been shown to impair both global Ca2+ signal and ROS production triggered by CR3 activation in neutrophils (23). The exact mechanisms linking PI(3,4,5)P3 formation to Ca2+ signals are not yet known. However, this phospholipid may initiate Tec kinase activation, resulting in tyrosine phosphorylation of PLCγ (99) and leading to the formation of inositol 1,4,5-trisphosphate (IP3), the second messenger responsible for Ca2+ release from intracellular stores via the opening of IP3 receptors during FcγR-mediated phagocytosis (110). Depletion of intracellular Ca2+ stores induces activation of the intraluminal Ca2+ sensor STIM1 (stromal interaction molecule 1), which, in turn, interacts with Orai1 to allow extracellular Ca2+ entry (Fig. 5). This mechanism is known as store-operated Ca2+ entry and has been well detailed in research published over the past 20 years (89).
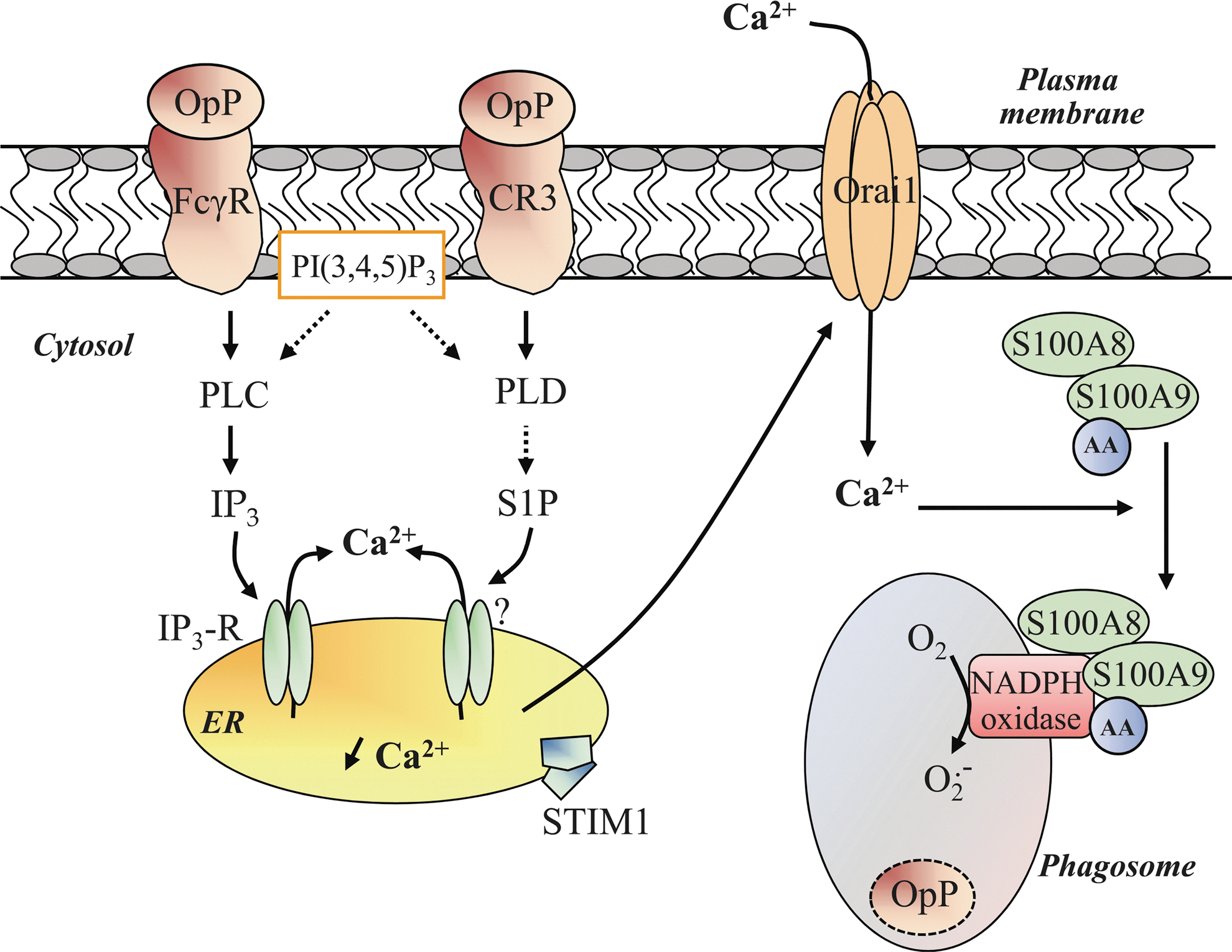
The role of IP3 is the subject of much controversy. On the one hand, its role might be dependent on individual FcγR and CR3 for IgG subclasses and complement activity, respectively. On the other hand, as observed in platelets, PI(3,4,5)P3 may sustain NADPH oxidase activity during neutrophil phagocytosis without an increase in PLC activity but through another lipid-based signaling pathway (85). CR3 could preferentially activate the PLD-dependent cascade, triggering the depletion of Ca2+ from intracellular stores via sphingosine-1 phosphate and subsequent STIM1/Orai1-mediated extracellular Ca2+ entry (Fig. 5) (79).
In addition to Orai1, Hv1 voltage-gated proton channels could regulate phagosomal Ca2+ turnover by preventing the depolarization generated by NADPH oxidase activity, therefore, enhancing the driving force for extracellular Ca2+ entry and sustaining NADPH oxidase activity. Initially, proton channels responsible for H+ efflux were described as a part of the gp91phox subunit (46). Later, convincing evidence demonstrated that channels nonrelated to gp91phox constitute voltage-gated proton channels (21). In this sense, Hv1 channels also preserve physiological membrane electroneutrality by compensating the charge generated by the electrogenic transfer of electrons from NADPH to O2 .− in the cytosol and hydrolysis of NADPH [for review, see (33)] (Fig. 6).
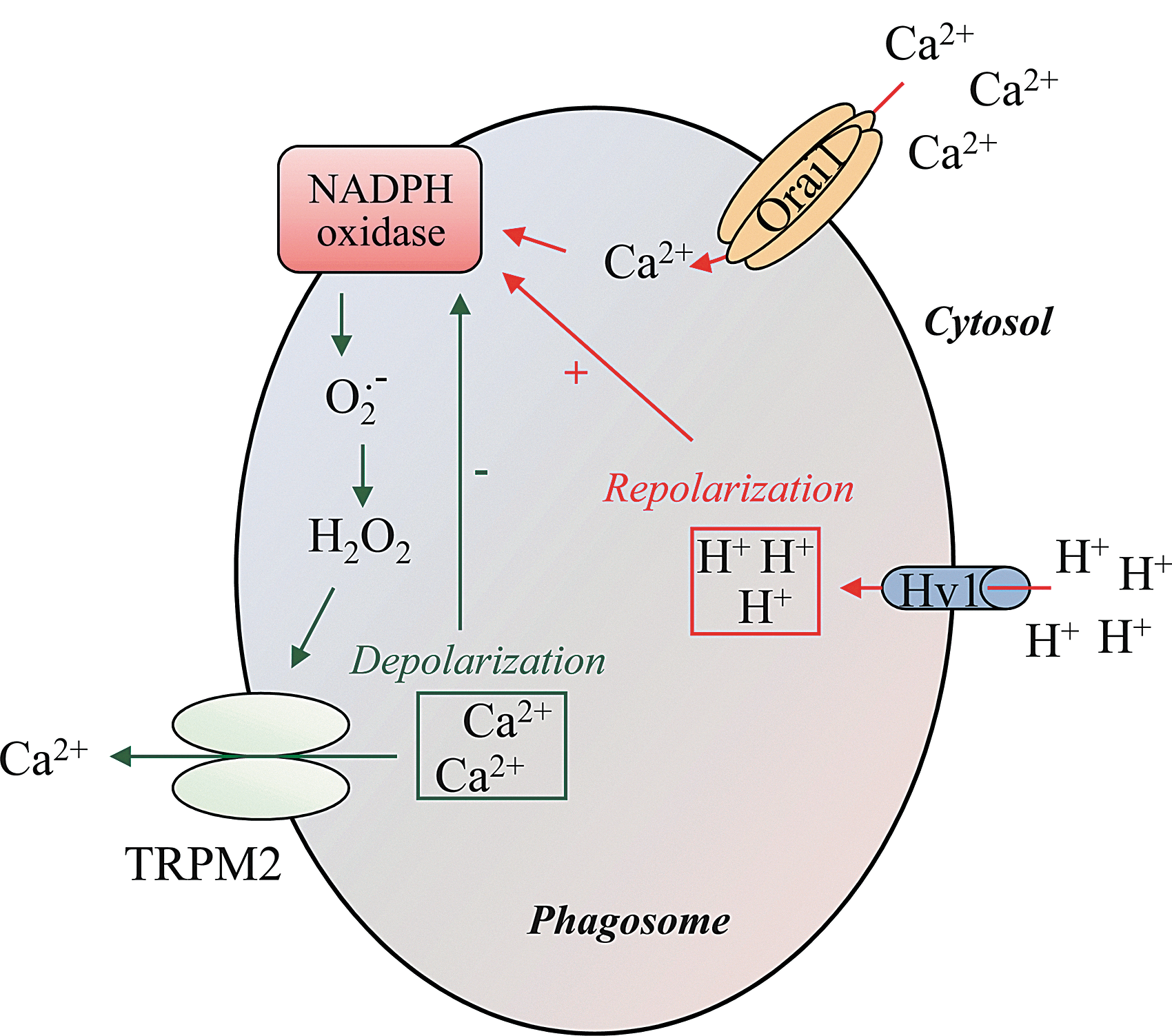
At this point, it is interesting to emphasize that a feedback mechanism could exist between Ca2+ homeostasis and NADPH oxidase activity at the phagosomal level. Indeed, it has been shown that extensive membrane depolarization dependent on ROS generation results in a marked decrease of the driving force for Ca2+ influx (91, 115). TRPM2, a nonselective and ROS sensitive channel cation found to be expressed in lysosomes (61), might be a putative ROS target linking the redox state of the membrane to Ca2+ homeostasis (114, 121). Moreover, TRPM2 possesses the ability to increase [Ca2+]c and the bactericidal activity of neutrophils (49). A model is proposed in Figure 6 that depicts the feedback mechanism for the inactivation of ROS production.
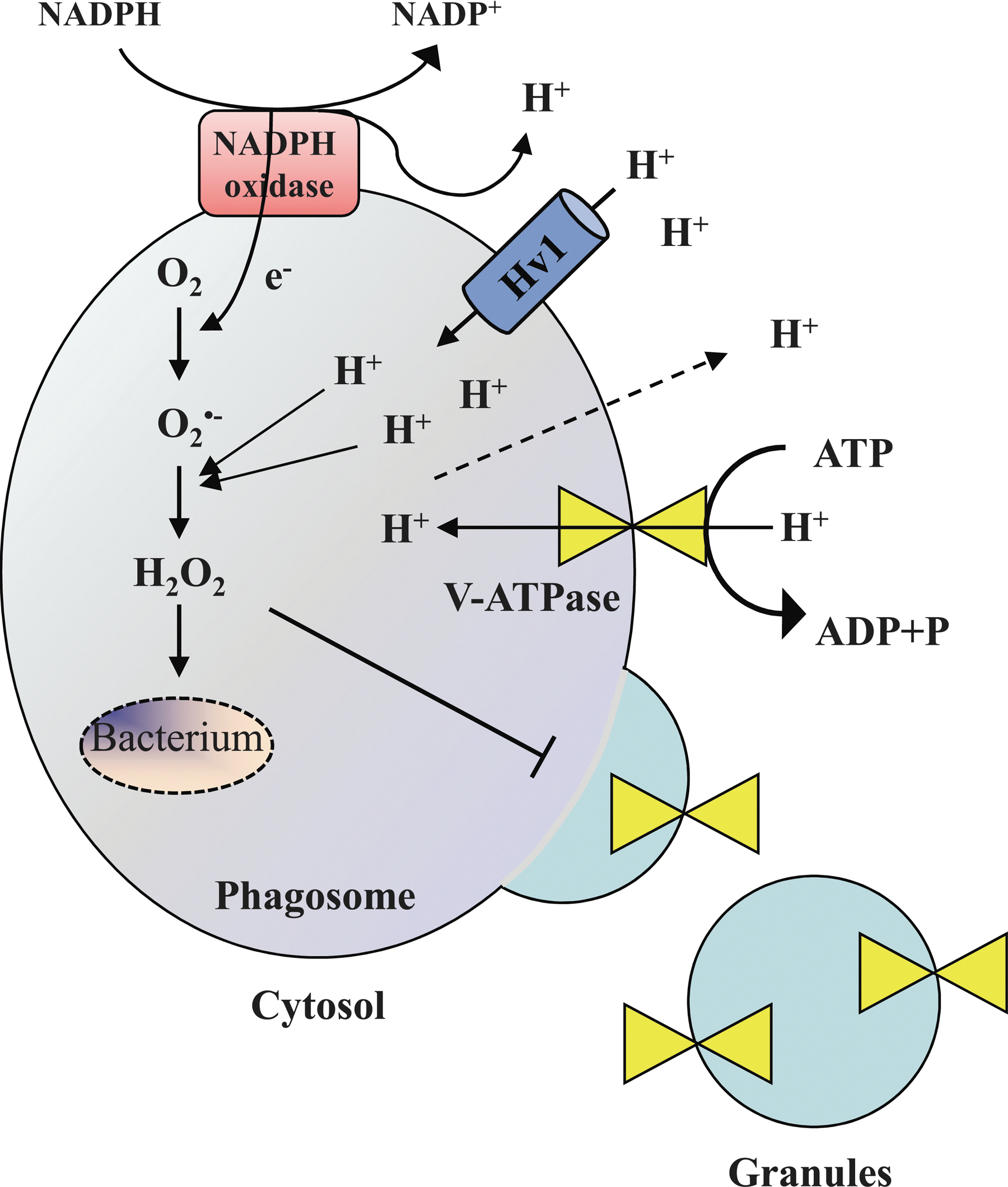
There is a close cooperation between the NADPH oxidase and the Hv1 proton channels to adjust intraphagosomal and cytosolic pH variations. Hv1 proton channels are present in specific granules and accumulated in phagosomes along with the oxidase during FcγR-dependent phagocytosis (80). It has been recently shown that NADPH oxidase-dependent bacterial killing was significantly reduced in Hv1-deficient mice neutrophils in vitro (87, 93). During phagocytosis-mediated NADPH activation, the consumption of NADPH produces H+ that acidifies the cytosol (74). The acidification and the electrogenic translocation of electrons into the phagosome are perfectly compensated by Hv1 proton channels, as they are activated by intracellular acidification and depolarizing voltages (75). Surprisingly, the intraphagosomal pH remains near neutral for several minutes, possibly due to several mechanisms such as the net consumption of luminal H+ during the dismutation of O2 .− to hydrogen superoxide, a large passive proton leak to the cytosol, preventing accumulation in the lumen and a reduced insertion of V-ATPases in the phagosome membrane, resulting in lower rates of H+ efflux (Fig. 7) (33, 51).
S100A8/A9: the link between Ca2+ and NADPH oxidase
Over the last decade, the phagocyte-specific Ca2+-binding S100A8 and S100A9 proteins have been proposed as essential regulators of the plasma membrane NADPH oxidase activity. These proteins are abundantly expressed in the cytosol of neutrophils and are able to form Ca2+-dependent heterocomplexes, with heterotetramers being a probable prerequisite for their biological activities in myeloid cells (62). S100A8 and S100A9 have been proposed as essential regulators that exert their role through interactions with NADPH oxidase subunits (8).
The addition of Ca2+-loaded S100A8/A9 to the reconstituted assembled NADPH oxidase complex prepared with neutrophil cytochrome b 558 and B lymphocyte cytosol is able to increase the constitutive activity of cytochrome b 558 in the absence of any stimulus. Initially, S100A8/A9 has been designated as a positive allosteric effector of NADPH oxidase activity that interacts preferentially with p67phox and increases its affinity for cytochrome b 558. However, the increased affinity of p67phox for cytochrome b 558 as inferred by S100A8/S100A9 was confirmed by Berthier and co-workers (8) in a semi-recombinant cell-free system, whereas S100A8/S100A9 appear to also interact directly with cytochrome b 558. Observations conducted on structural conformation changes by atomic force microscopy underlie the fact that the S100A8/A9 complex is able to enhance or induce a transition from an inactive to an active conformation state of cytochrome b 558. Preincubation of S100A8/S100A9 in the absence of Ca2+ led to an interaction with cytochrome b 558 but not to a conformational change, allowing ROS production. Thus, the relevant role for S100A8/A9 in NADPH oxidase activity is dependent on Ca2+ (7) and is probably mediated via S100A8/A9 translocation to the membrane where NADPH oxidase is activated (97, 100, 101). An elevation of [Ca2+]c is necessary for S100A8/A9 redistribution to the plasma membrane; thus, intracellular Ca2+ store depletion appears to be substantially responsible for this phenomenon (100).
From these observations, it is logical to question whether S100A8/A9 could also constitute the link between [Ca2+]c elevation and phagosomal NADPH oxidase activity. Different research groups have provided indirect evidence for such a speculation. Kumar et al. (59) showed that phagocytosis was associated with a rapid reduction of cytoplasmic immunostaining of the S100A8 homodimeric form, and a Ca2+ dependence of S100A8/A9 translocation in cytoskeletal structures was reported (97). Direct evidence that S100A8/A9 constitute the molecular switch between [Ca2+]c elevation and phagosomal NADPH oxidase activity has been provided by a recent work of Steinckwich et al. (110). These authors established that S100A8/A9 siRNA knockdown decreases intraphagosomal ROS production. Further, on FcγR-induced [Ca2+]c elevation, endogenous S100A8/A9 is recruited to the phagosomal membrane at the beginning of the formation of the phagocytic cup and persists throughout the zymosan internalization process. Therefore, extracellular Ca2+ entry-mediated S100A8/A9 phagosomal membrane recruitment serves as a determinant for the NADPH oxidase activity. Via a mechanism similar to that proposed at the plasma membrane level (8, 81), S100A8/S100A9 could deliver AA to the phagosomal membrane (Fig. 5), increasing its local concentration and favoring the conformational change of cytochrome b 558. This hypothesis requires confirmation, but it may provide a foundation for further investigation of the link between S100A8/A9 and phagosomal NADPH oxidase. In the near future, it will also be of interest to (i) identify phagosomal NADPH oxidase subunits involved in direct interactions with S100A8/A9 and (ii) characterize the additional signaling pathways leading to S100A8/A9 phagosomal membrane recruitment with an emphasis on phospholipid and phosphorylation signaling cascades.
The pro-inflammatory role of S100A8/A9 is not restricted to the intracellular level. Neutrophils are known to secrete S100A8/A9 that are involved in autocrine/paracrine regulatory mechanisms underlining the inflammatory process (30, 92). In a variety of cell types, this dual action is mediated through the engagement of Receptor for Advanced Glycation End (RAGE) products signaling (47). This signaling contributes to the activation of p38 MAPK and the downstream effector NF-κB (29). S100A8 and S100A9 have recently been identified as target genes of NF-κB (76), which regulate their expression, thus allowing them to fulfill their intracellular roles (Fig. 8). In parallel, exogenous S100A9 may have the ability to induce NADPH oxidase activation, although it mainly stimulates neutrophil microbicidal activity by promoting phagocytosis (106). Further, other RAGE ligands have been associated with a modulation of bacterial destruction by neutrophils and activation of NADPH oxidase. Definitely, the prototypical RAGE ligand AGE has been found to increase bacterial killing by neutrophils and activation of NADPH oxidase, likely through the phosphorylation of the p40phox subunit. In contrast, high mobility group box 1 diminished the ability of neutrophils to destroy pathogens and NADPH oxidase activation (113).
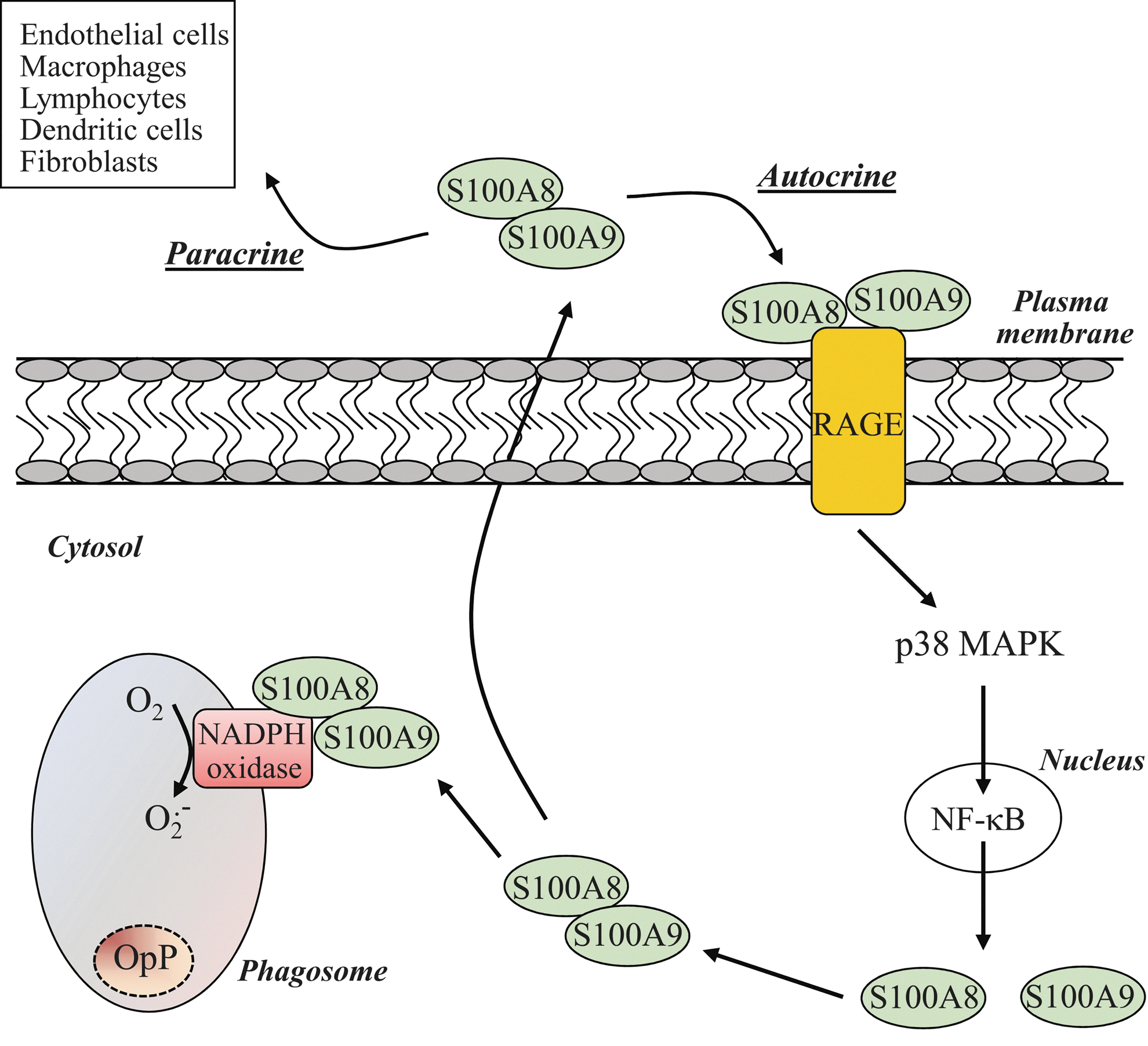
In light of these results, an intriguing possibility arises that S100A8/A9 secreted by neutrophils on opsonized zymosan stimulation bind to RAGE and regulate neutrophil phagosomal NADPH oxidase activity (Fig. 8). Kumar et al. (59) have hypothesized that the rapid loss of cytoplasmic immunoreactivity for homodimeric S100A8 was the consequence of S100A8 release by active secretion on opsonized zymosan stimulation. Using the same line of evidence, Guignard et al. (43) showed that S100A8 and S100A9 were recruited to the plasma, underlining the eventual and subsequent S100A8/A9 secretion after opsonized zymosan. However, these authors have provided no clear indication for a secretion of S100A8 and S100A9 proteins. While the release of S100A8 and S100A9 upon soluble stimuli is well established, to our knowledge, no direct evidence for the release of neutrophil S100A8 and S100A9 in response to particulate stimuli has yet been convincingly provided. Further investigations are, thus, compulsory. Understanding the S100A8/A9-RAGE-NADPH oxidase axis, in an autocrine/paracrine fashion, potentially offers some interesting perspectives for providing a more comprehensive mechanism for the regulation of the phagosomal NADPH oxidase.
Monitoring of phagosomal ROS production and Ca2+ mobilization
To understand the relationship between intracellular Ca2+ mobilization and NADPH oxidase activity, quantitative concomitant measurement of phagosomal NADPH oxidase activity and [Ca2+]c changes with sufficient resolution is not only necessary but also a challenge. One of the issues is the difficulty of distinguishing the contribution of [Ca2+]c elevation for the phagosomal oxidative event from earlier phagocytic events (chemotaxis, phagocytic cup formation, engulfment, and phagosome closure). Commonly used techniques for following intraphagosomal ROS production are provided in Table 3 [for a more detailed description, see (38, 96, 116, 123)]. An original technique was developed by Dewitt and co-workers (22) in which C3bi-opsonized particles labeled with an oxidant-sensitive probe (2′,7′-dichlorodihydrofluorescein [DCFH2]) were presented to neutrophils at a defined time and location using micromanipulation. Later, Steinckwich et al. (110), using fluorescence light microscopy, which allows for the detection of intraphagosomal ROS production, measured simultaneously [Ca2+]c elucidating the relationship between phagosomal NADPH oxidase activity and changes in [Ca2+]c. Classical indicators used to monitor [Ca2+]c changes remain fluorescent dyes such as fluo-4 or fura-2, but genetically encoded probes are being developed to follow Ca2+ amplitude and kinetics at specific subcellular locations and improve Ca2+ imaging resolution [for review, see (20)].
DCFH2, 2,7-dichlorodihydrofluorescein; DHR-123, dihydrorhodamine 123; HyPer, genetically encoded fluorescent indicator; MPO, myeloperoxidase; NBT, nitroblue tetrazolium; SNAP-tag, organelle-targetable fluorescent probes; +and – indicate advantages and inconveniences of each technique.
To overcome the lack of sensitivity, and problems with auto-oxidation and quenching observed with the fluorescein derivative DCFH2, research has been devoted to the development of a specific probe to improve the detection and measurement of intracellular H2O2 production. Genetically encoded probes, which can be targeted at specific subcellular locations (96), represent potentially useful tools for monitoring H2O2 production within the phagosomal compartment.
Hypothetical Involvement of N-Ethylmaleimide-Sensitive Factor Attachment Protein Receptor-Mediated Exocytosis in Phagosomal NADPH Oxidase Activity
One point of regulation could occur at the level of proteins collectively termed soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNAREs). SNAREs are key regulators in all fusion events occurring in the exocytosis pathway, which lead to protein trafficking between intracellular compartments. SNAREs comprise a series of proteins that can be divided into vesicular (v-SNAREs), including VAMP family members and target (t-SNAREs) including syntaxins and SNAP-23/25 family members. These proteins mediate the formation of extremely stable complexes between adjacent membranes, bringing the membranes into close apposition (107).
Several authors have identified a number of SNAREs in neutrophils, including the t-SNARE SNAP-23. SNARE proteins and their regulators have been involved in the process of cytosolic secretory granule fusion and exocytosis of their contents (71). SNAP-23 has been described as being a mediator of specific granule secretion (71) and recently, Uriarte and co-workers (118) demonstrated that the SNAP-23-mediated neutrophil granule exocytosis contributes to ROS production during phagocytosis. Thus, the assumption can be made that SNAP-23 participates in the appropriate localization of cytochrome b 558 by specific and gelatinase-granule release in phagosomes. Further studies are needed to determine the mechanisms by which SNARE proteins are precisely involved in the regulation of the phagosomal NADPH oxidase activity.
Conclusion
Phagosomal ROS production is critical for efficient host innate defense against infections through direct microbial killing. In addition, ROS control spatiotemporal phagosomal proteolytic processes, through modification of the luminal redox environment of phagosomes, which are also required to mediate pathogen destruction. It is becoming increasingly evident that variations in the types of engaged receptors trigger different downstream signaling cascades. The challenge will be to define the contribution of the distinct molecular mechanisms involved in phagosomal NADPH oxidase activation during individual receptor-mediated phagocytosis. Ca2+ signals control many pathways, and growing evidence shows that [Ca2+]c elevation exerts an effect on NADPH oxidase activity through interconnections with other regulatory pathways, including lipid metabolism. However, despite many decades of intensive research regarding the vast numbers of pathways and molecules that are involved in phagocytosis, the regulation of the phagosomal NADPH oxidase remains poorly understood.
Some bacteria have developed highly sophisticated mechanisms compromising the bactericidal function of phagosomal ROS production and contributing to long
Footnotes
Acknowledgments
The authors would like to thank Dr. Tonie van Dam and Dr. Fabrice Tolle for a critical reading of the article. This work was supported by the University of Luxembourg.