
Abstract
Introduction
U
We provide data that support a novel hypothesis for the role of mutant Plasmodium falciparum chloroquine resistance transporter (PfCRT) in the chloroquine resistance phenotype. The data indicate that mutant PfCRT facilitates the redistribution of glutathione (GSH) from the cytoplasm to the digestive vacuole. This results in GSH-dependent removal of heme-binding sites, both directly by occupying them and indirectly through causing heme breakdown, which is a significant contributor to the decrease in chloroquine susceptibility.
CQR correlates with reduced accumulation of drug in the parasite due to diminished drug–heme binding in the DV, although other interpretations have been recently suggested (5, 7). CQR is associated with polymorphisms within the DV transmembrane protein PfCRT (11, 17, 18, 42). A K76T mutation in PfCRT is conserved in all CQR parasites and is considered key in mediating the CQR phenotype (18). However, there is a wide variation in the response to CQ of cloned parasite lines containing identical mutant pfcrt alleles, which indicates the involvement of additional mechanisms contributing to CQR. The multidrug-resistant homolog gene pfmdr1 is probably one (12, 32, 37), but it is likely that other factors also modify susceptibility to CQ.
Research reported during the last three decades has pointed to glutathione (GSH), the major antioxidant thiol in the parasite (2), playing some part in CQR (1, 13, 14). One possible mechanism would be that GSH reduces heme-binding sites for CQ in some way. We postulated that different concentrations of membrane-impermeant GSH in the DV of CQR and chloroquine-sensitive (CQS) parasites could be the basis of such a mechanism. To investigate this, GSH concentrations have been compared between the CQS and CQR lines. The data generated, however, have been difficult to interpret, as isolates with different genetic backgrounds were used (30). It is thought that such lines have variable transcriptional and translational regulation of oxidative defenses and efflux transporters such as PfMRP1, which affects GSH levels (23, 31, 36). Thus, we have now analyzed in this study isogenic parasite lines, which differ only in their substituted pfcrt allele (Table 1) (42). We have confirmed that GSH is generated in the cytoplasm and demonstrate that artificially increasing GSH levels has little effect on CQ sensitivity unless the parasites also harbor mutant alleles of pfcrt. Moreover, we show that mutant PfCRT can transport GSH. This newly discovered function facilitates GSH redistribution from the cytoplasm into the DV selectively in the CQR lines. The data generated have allowed us to postulate a CQ resistance mechanism in which GSH is selectively transferred into the DV via mutant pfcrt, where it competes with CQ for heme binding and results in destruction of heme, and thereby protects the parasites from the prooxidant activity of the CQ–heme complex.
CQ, chloroquine.
Results
Localization of GSH biosynthesis enzymes
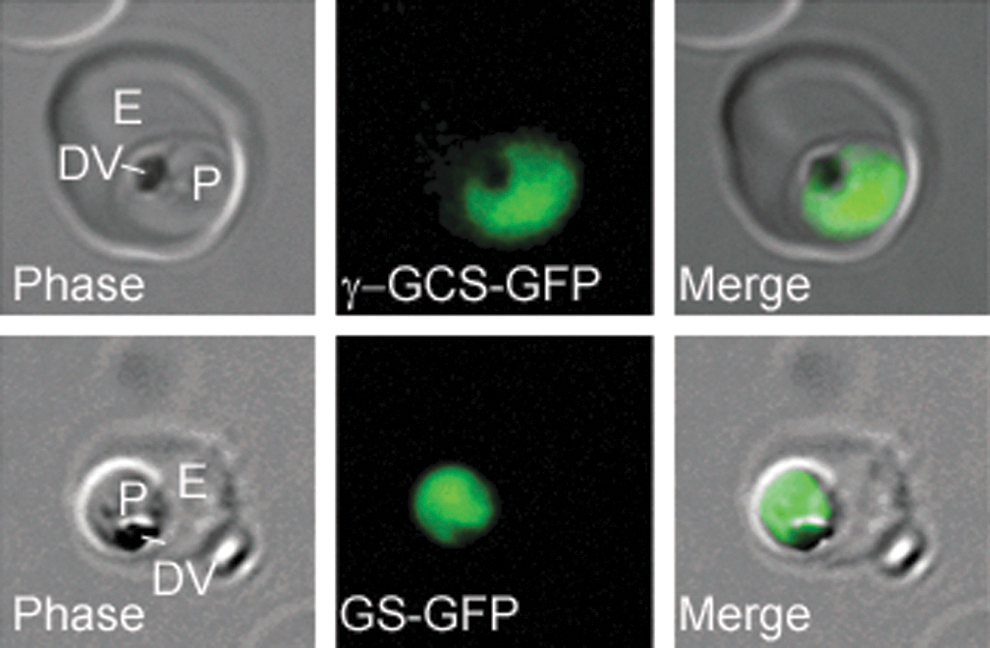
P. falciparum possesses genes encoding γ-glutamylcysteine synthetase (PfγGCS) and glutathione synthase (PfGS), required for GSH synthesis (24, 29). C-terminally tagged green fluorescent protein (GFP) variants of each gene were transfected into P. falciparum, and the resultant line expressed proteins were both localized to the parasite cytosol (Fig. 1).
GSH levels in isogenic lines and their susceptibility to GSH-depleting agents
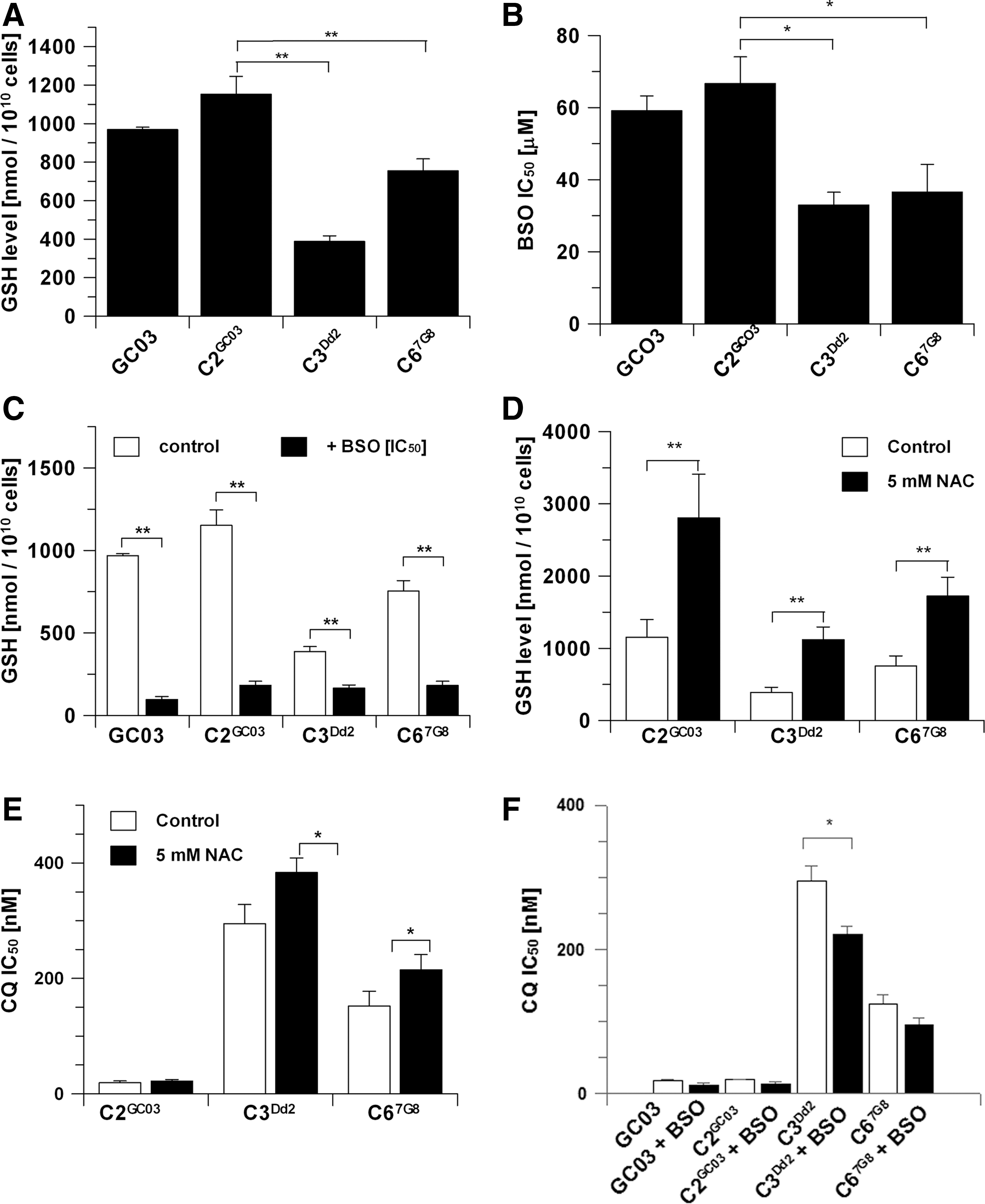
Total GSH levels of isogenic parasite lines GC03, C2GC03, C3Dd2, and C67G8 (Table 1) were determined using a validated HPLC method (42, 51). Transfected parasites carrying the Dd2 or 7G8 pfcrt CQR alleles contained significantly less GSH than the lines carrying the sensitive HB3 wild-type pfcrt allele (Fig. 2A). We also determined the GSH levels of the nonisogenic untransformed CQR Dd2 standard laboratory parasite line (see Table 1) and found no significant difference in GSH content between this line and the isogenic CQS parasites whose genotype is that of GC03 (see Table 1) (Dd2, 984±184 nmol/1010cells; C2GC03, 1152±93 nmol/1010cells). These data highlight the importance of our use of the isogenic parasite lines, because this allowed us to probe specifically the effect of mutations in crt on GSH levels and avoided the problems associated with the confounding effects of other compensatory changes that would be likely to have occurred in untransformed CQR isolates.
To elucidate whether the isogenic lines were differentially affected by the specific γGCS inhibitor
Susceptibility of parasite lines to CQ and the effect of N-acetylcysteine
Incubating the isogenic parasites with 5 mM N-acetylcysteine (NAC) for 16 h increased intracellular GSH levels in the CQS C2GC03 line by more than twofold (Fig. 2D) without causing a significant change in the response to CQ, the IC50 ratio being 1.1 (Fig. 2E). In contrast, increasing GSH levels in the CQR parasites C3Dd2and C67G8 (Fig. 2D) led to statistically significant increases of the CQ IC50 values of both lines (Fig. 2E). These data support the case that elevated GSH levels in the presence of the mutant crt gene mediate a further increase in the resistance to CQ. Additional supporting evidence for the involvement of GSH and CRT was provided by the demonstration that a decrease in GSH levels by preincubation of parasites with BSO resulted in a significant reduction in CQ IC50 in C3Dd2 (Fig. 2F).
Effect of moderating intracellular GSH levels on CQ accumulation
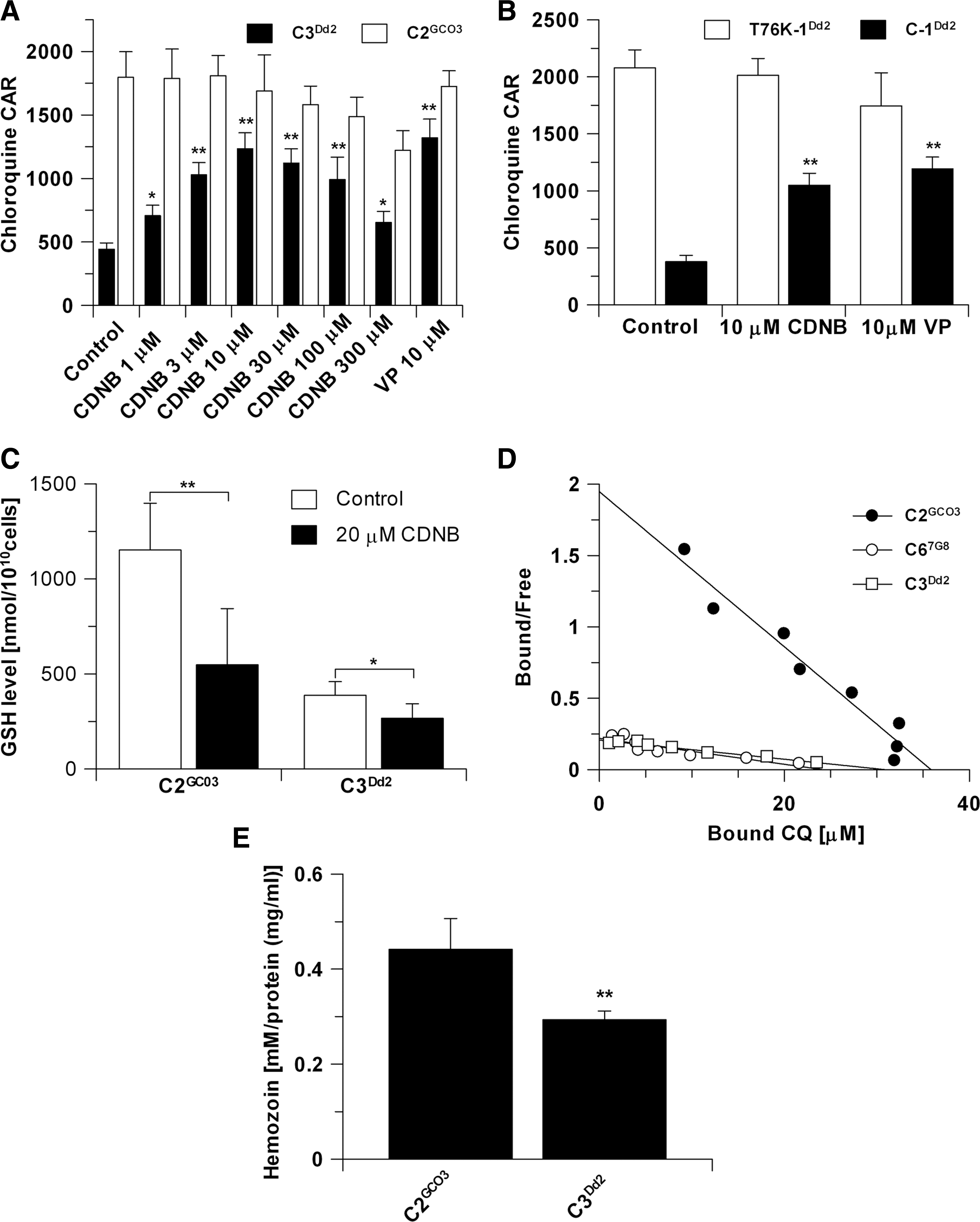
The C2GC03 and C3Dd2 lines were treated with 1-chloro-2,4-dinitrobenzene (CDNB), a substrate for glutathione S-transferase (GST) that leads to the formation of 2,4-dinitrophenyl-S-glutathione adducts, which are excreted, and thus result in a decrease of intracellular GSH levels (22, 28), and their effect on accumulation of CQ was measured. Increasing concentrations of CDNB to just above the IC50 concentrations (8.5±4.5 μM and 9.5±1.1 μM for C2GC03 and C3Dd2, respectively) significantly stimulated the accumulation of [3H]-CQ in the CQR parasite line C3Dd2, but not in the CQS line C2GC03 (Fig. 3A). The CDNB effect on [3H]-CQ accumulation in CQR parasites was similar to that caused by 10 μM verapamil (VP) (Fig. 3A), which is an L-type calcium channel blocker known to interact with mutated forms of PfCRT (18, 26, 33, 42). In contrast, [3H]-CQ accumulation in CQS C2GC03 was unaffected by CDNB or VP (Fig. 3A). Qualitatively similar results were obtained comparing a CQS back-mutant parasite line derived from a CQR parent line (Dd2) called T76K-1Dd2 (Table 1) with the CQR C-1Dd2 (Table 1) (Fig. 3B) (18). The effect of 10 μM CDNB and VP was negligible in the CQS T76K-1Dd2, whereas the effects on the CQR line C-1Dd2 were similar to that observed with C3Dd2 (Fig. 3B). It was confirmed that incubation with 20 μM CDNB for 20 min (the same incubation time as in the cellular accumulation ratio for chloroquine experiment described above) resulted in significantly reduced GSH levels (Fig. 3C). These data show that the reduced GSH levels resulting from CDNB treatment impact on CQ accumulation, but only in parasites carrying the K76T mutation in the pfcrt allele. An alternative explanation could be that the 2,4-dinitrophenyl-S-glutathione adduct generated itself interferes with CQ movement through mutant PfCRT in a similar way to VP.
CQ equilibrium binding studies and the concentration of hemozoin
The data detailed above suggested that cytoplasmic GSH may have access to the DV in CQR parasites and thus be able to interact with the free heme there to form a GSH–heme complex, possibly with the neutral thiol serving as an axial ligand to heme iron as suggested by Shviro and Shaklai (41). This interaction would scavenge free heme and slow down the rate of hemozoin formation, but as the GSH–heme complex is nontoxic (41), it would not damage the parasite, and would also compete with CQ for binding to the heme target and so reduce the toxicity of CQ. In addition, it is possible that some of the heme could be spontaneously destroyed by GSH, liberating iron and GSSG (1, 41), which would also result in reducing CQ binding and so its antimalarial effectiveness.
For these reasons, we postulated that the amount of free heme in the DV of CQR parasite lines should be reduced. It is difficult to measure directly the concentration of free heme in the DV of P. falciparum, but its concentration can be reliably estimated from analysis of the CQ equilibrium binding experiments performed on intact infected erythrocytes (4, 5, 9). The basis of this assay is that CQ readily forms a complex with free heme and so prevents its biomineralization into hemozoin (3, 5). Our analysis of the apparent affinity of CQ binding to heme demonstrated that it is greatly reduced in the CQR lines C3Dd2 and C67G8 compared to the C2GC03 CQS line (Fig. 3D), with both reduced apparent affinity and also reduced heme-binding capacity. Least-squares analysis of the CQ-binding isotherms indicates that CQ equilibrium binding capacity is lower in C3Dd2 and C67G8 isolates (30±0.8 μM and 29±1.7 μM, respectively) compared to C2GC03 (35±1.1 μM). This is consistent with free heme availability being less, although only by about 15%, in these two CQR lines and potentially linked to mutations in PfCRT. The decreases are also consistent with the 15%–20% reduction in CQ-binding capacity reported in the CQR progeny of a genetic cross [reported as Jmax in Sanchez et al. (40)].
We also determined hemozoin levels in the CQS and CQR lines, and showed that they were significantly lower (p<0.001) in CQR C3Dd2 compared with CQS C2GCO3 (Fig. 3E). Consistent with the model, we propose that GSH in CQR parasites scavenges or destroys free heme. It should be noted that a report by Gligorijevic and colleagues (15) concluded that there was no difference in the hemozoin content between untransformed GC03 and transformed C3Dd2. In that study, hemozoin quantity was determined indirectly from the volume of the malaria pigment as measured microscopically, whereas we used a quantitative chemical assay. This may suggest that pigment volume and hemozoin content are not linearly correlated.
Expression of GSH biosynthesis and GSH-dependent enzymes
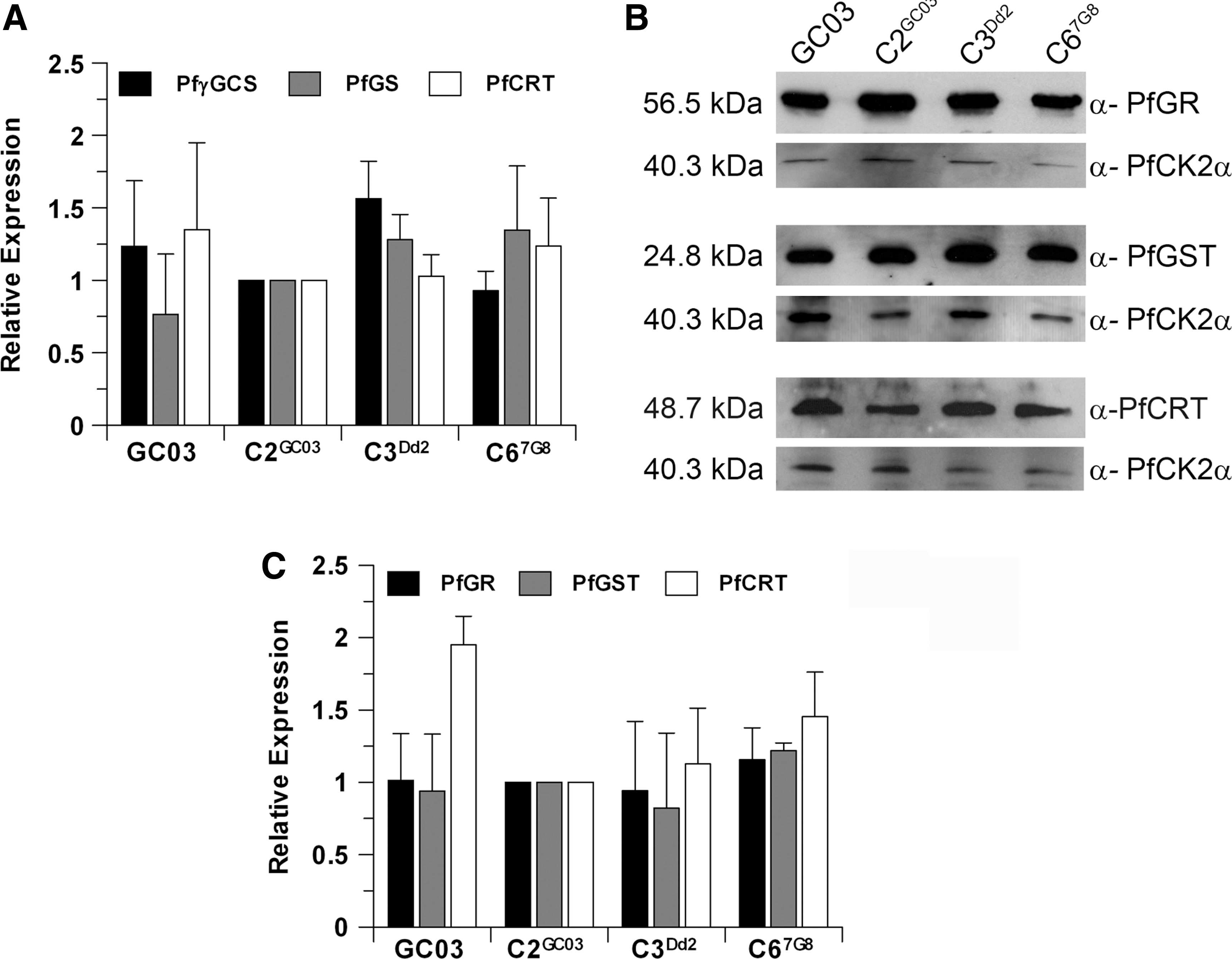
To investigate whether the lower levels of GSH observed in the untreated lines with the mutant CRT could result from differential expression of GSH biosynthetic genes, real-time quantitative polymerase chain reaction (RT-PCR) of pfgs and pfγgcs was undertaken. This revealed only slight variations in the expression of these genes between parasite lines (Fig. 4A), ruling out that the different GSH levels were a consequence of reduced GSH biosynthesis in the CQR parasites. Moreover, the levels of expression of pfcrt mRNA were equivalent in the four parasite lines. In addition, PfCRT protein expression, as determined by western blotting, was similar in the three isogenic lines C2GC03, C3Dd2, and C67G8, while the parent line GC03 contained 1.5-fold to 2-fold higher levels of PfCRT protein (Fig. 4B, C), which is consistent with previous reports by Sidhu and colleagues (42). The levels of P. falciparum glutathione-S-transferase (PfGST) and P. falciparum glutathione reductase (PfGR) proteins were similar in all parasite lines (Fig. 4B, C), suggesting that the CQR parasites preserve their capacity to maintain their redox status through the action of PfGR and to generate conjugates with GSH through the activity of PfGST. One caveat is that we cannot exclude the possibility that PfGST activity might be negatively affected by the lower GSH levels in the CQR parasite lines.
GSH transport activity of PfCRT expressed in Xenopus oocytes
The data above demonstrate that the presence of mutant pfcrt leads to a reduction in intracellular GSH levels in CQR parasites without any alterations in GSH metabolism. Further, the mutant allele is required to demonstrate an effect of CDNB on CQ accumulation and to show an increase in CQ IC50 values in the presence of NAC. We thus hypothesized that these effects are mediated by selective transfer of GSH to the DV, the site of CQ action, possibly via transport by mutant PfCRT that is located in the DV membrane (49). This hypothesis was tested using the X. laevis heterologous expression system to express mutant and wild-type forms of PfCRT. This system has been previously validated experimentally as being suitable for measurement of GSH uptake in a study of a family of PfCRT-like proteins occurring in Arabidopsis thaliana (AtCLT), which are GSH transporters (27). Further evidence for the suitability of the assay system was provided by measuring the uptake of [3H]-CQ into oocytes expressing the PfCRTDd2 variant (26).
We expressed several PfCRT variants in Xenopus oocytes: PfCRTHB3 as a representative of the CQS wild-type allele of pfcrt, as carried by C2GC03; PfCRTDd2 as the predominant CQR mutant from Southeast Asia and the PfCRT isoform found in C3Dd2; PfCRTK76T as it carries only the single K76T mutation, but is otherwise wild type (a form that has no corollary in parasite isolate lines); A. thaliana CLT1 (AtCLT1) as a positive control. Indirect immunofluorescence was performed using anti-PfCRT as the primary antibody (11). The images demonstrated that PfCRTHB3, PfCRTK76T, and PfCRTDd2 were all expressed in the oocytes injected with the cRNA of their respective genes (Fig. 5). Immunofluorescence is visible at the plasma membrane and also in the cytoplasm at comparable levels for all three PfCRT variants. There is no fluorescence signal in the water-injected control group.
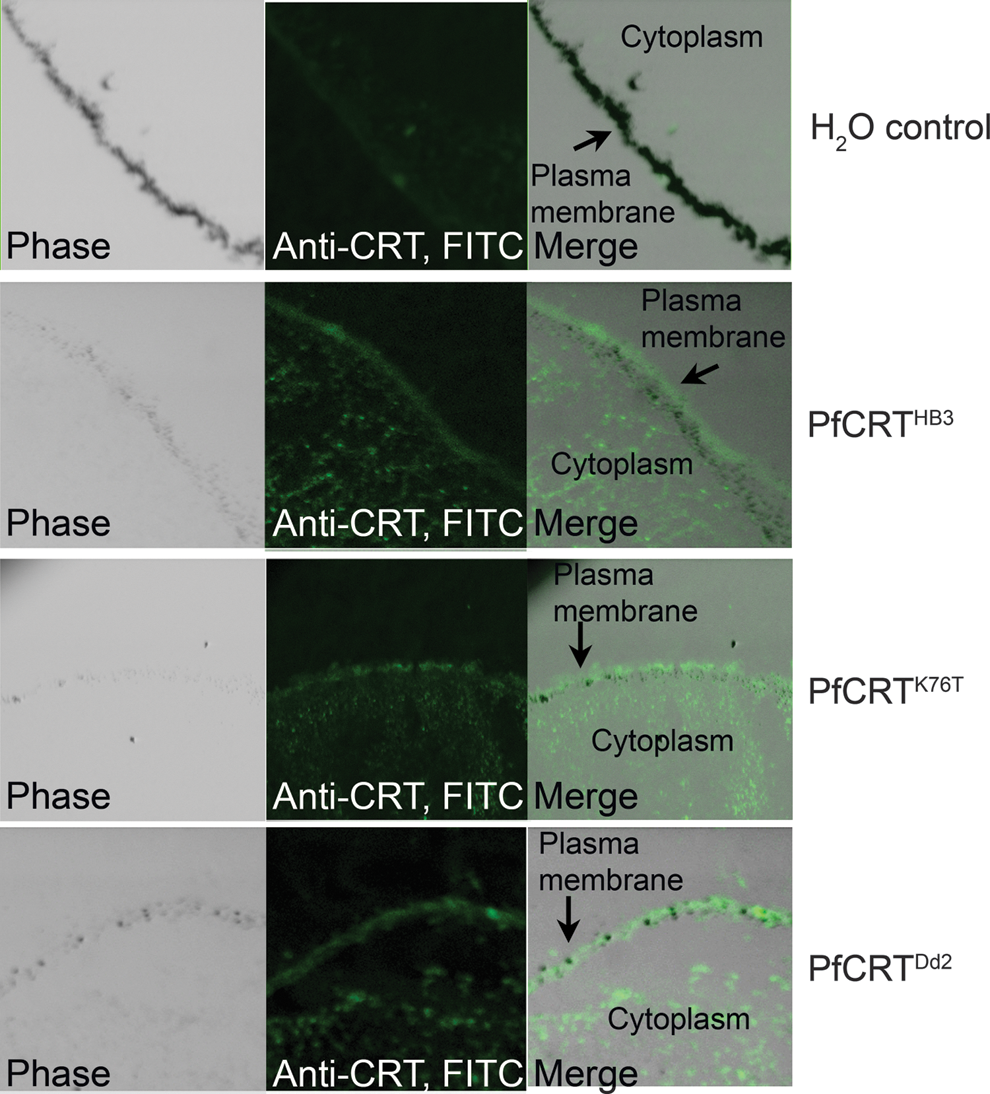
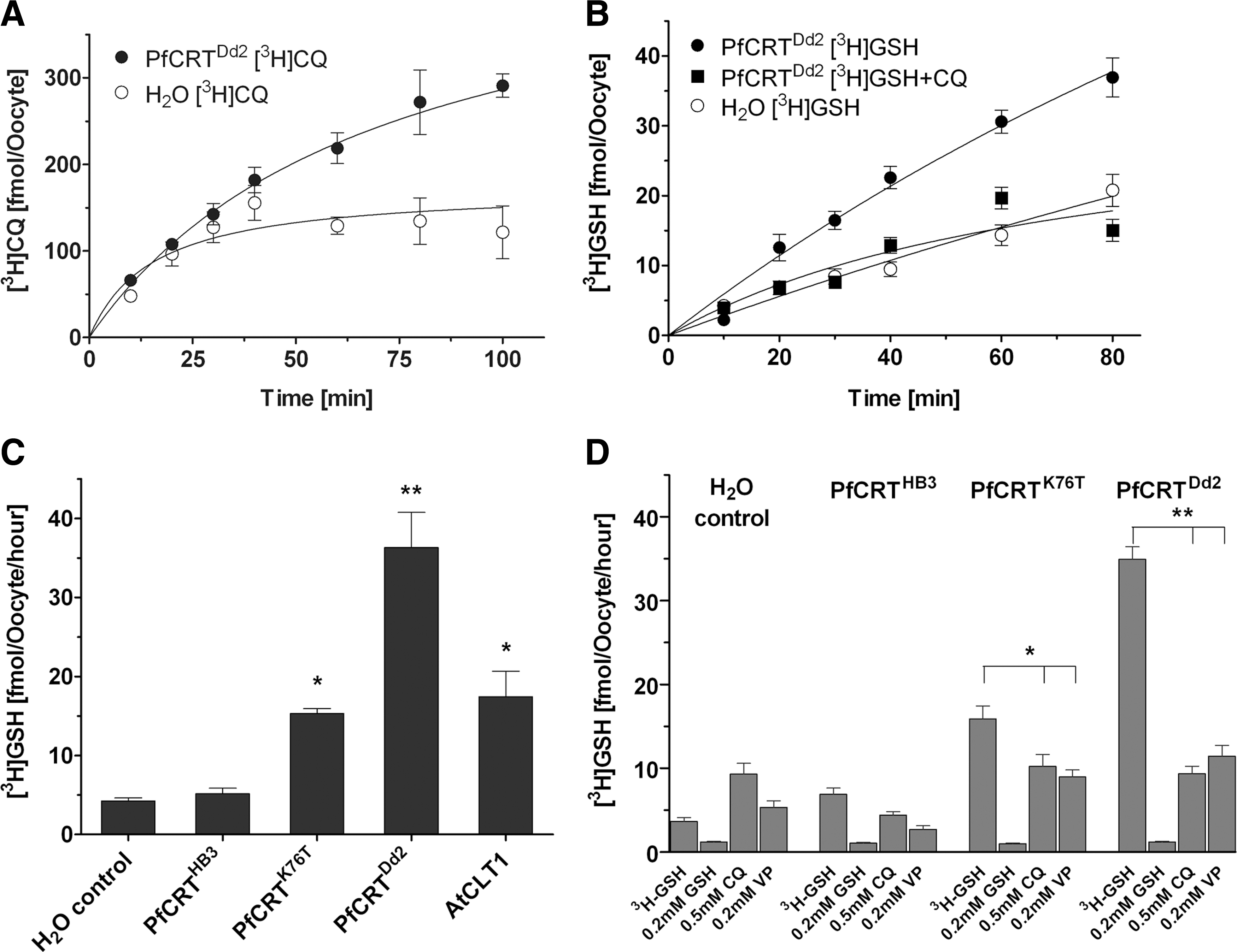
We confirmed previous reports that oocytes expressing PfCRTDd2 demonstrated a two-threefold increase in [3H]-CQ uptake compared to water-injected controls, an effect that was time dependent (Fig. 6A) (26). The X. laevis oocytes possess a low endogenous ability to take up GSH, which has been observed in previous studies (21, 27). The expression of PfCRTDd2, however, conferred time-dependent transport of [3H]-GSH, which was reduced to basal levels in the presence of CQ (Fig. 6B). PfCRTDd2 and PfCRTK76T showed six- and twofold higher rates of GSH membrane transport than water-injected controls. [3H]-GSH uptake into oocytes expressing PfCRTDd2 or PfCRTK76T was specific, with transport inhibited by saturating concentrations of unlabeled GSH, CQ, and VP (Fig. 6D). No transport of [3H]-GSH was detected in oocytes expressing PfCRTHB3, showing that only the CQR isoforms of PfCRT transport GSH (Fig. 6C). Thus, these findings strongly support the proposal that the mutant PfCRT occurring in CQR parasites transport GSH.
Discussion
It is widely acknowledged that resistance to CQ is primarily conferred by mutations in the P. falciparum transporter gene pfcrt (11, 17, 42, 49), although other factors, including the multidrug-resistant transporter homolog pfmdr1, also contribute to the level of CQR (32, 37). This current study re-evaluated the hypothesis that GSH plays a part in CQR (14) and has revealed a previously unconsidered role for the CQR alleles of pfcrt. GSH was reported previously to scavenge free heme, forming a complex in which the thiol group is linked to heme iron, and provides a mechanism to protect membranes from oxidative heme damage (41). CQ also binds to heme, inhibiting the detoxification of heme into hemozoin crystals and causing a build-up of a drug–heme complex (4, 9, 18, 20, 50). This CQ–heme complex is toxic, in contrast to the GSH–heme complex, promoting lipid peroxidation and destruction of parasite membranes and mediates parasite killing by CQ (41). Thus, it can be envisaged that GSH could interfere with CQ action simply by competing for binding with heme. It has also been reported that GSH degrades heme in vitro (1, 41); if this happens in situ, then GSH would interfere with CQ action by destroying its target. A third possibility whereby GSH could play a role is that it may protect parasites from enhanced heme-induced oxidative stress (2). Our data showing that CQR P. falciparum contains reduced levels of hemozoin are consistent with free heme being either scavenged or degraded by GSH in these parasites. However, until now, it was not clear how modulating the levels of GSH, which is synthesized in the cytoplasm of the parasites, can influence the sensitivity to CQ, which acts within the DV of the parasites where heme is generated.
Our findings in this study resolve this issue and reveal a new role for mutant PfCRT in transporting GSH between the cytoplasm and the DV, presumably leading to a change in the overall distribution of the tripeptide. We demonstrate direct and specific transport of GSH by two CQR isoforms (K76T and Dd2) of PfCRT expressed in X. laevis oocytes. Further evidence for mutant PfCRT-driven GSH transport is provided by selective inhibition of GSH uptake by both CQ and VP. GSH transport by CQR alleles of pfcrt is in agreement with a previous report that the CQR mutant PfCRTDd2 is capable of transporting small peptides as well as CQ (26). In fact, we found that a single-point mutant PfCRTK76T was sufficient to allow uptake of [3H]-GSH, although not to the same level as the mutant PfCRTDd2 allele. In contrast, we could not demonstrate any [3H]-GSH transport activity in oocytes expressing the CQS allele of pfcrt. The only other study in which pfcrt has been expressed in oocytes is that reported by Martin and colleagues (26) studying CQ transport. In that study, the pfcrt sequence had to be modified extensively to achieve functional expression of the protein in the oocyte system. This was not necessary in the current study where we see adequate and functionally relevant PfCRT protein in the oocyte plasma membrane as well as the oocyte cytoplasm using the unmodified pfcrt codon sequence. A further discrepancy between the data presented here for GSH transport and CQ transport reported by Martin and colleagues (26) is that they found the single-point mutant of PfCRTK76T incapable of CQ transport. Our conclusion is that the K76T mutation in PfCRT generates structural changes that are sufficient to allow GSH transport, but not CQ transport. It is not clear what orientation PfCRT takes when inserted into the oocyte membrane; our results suggest that the majority of the protein has an orientation that is equivalent to that in the DV, or alternatively, the transporter can move GSH in either direction, depending on the prevailing concentration gradient.
Mutant PfCRT has been postulated to act either as a channel, permitting the mediated and fast transport of diprotonated CQ across the DV membrane, or as an outwardly directed slower CQ carrier (6, 26, 35, 39). The transport of GSH is time dependent, but appears to be nonsaturable, which could suggest that the transport is not carrier mediated. Another possibility is that the tripeptide is metabolized by X. laevis oocytes, and thus a thermodynamic equilibrium cannot be reached. In either case, the inhibition of GSH transport by CQ and the inhibition of CQ transport by small peptides (26) suggest that CQ and certain peptides, including GSH, share the same translocation sites within mutant PfCRT.
The functional relevance of mutant PfCRT-mediated GSH transport is revealed when GSH levels are elevated in both CQR and CQS parasite lines using NAC, leading to a significant increase of the CQ IC50 values only in CQR lines (Fig. 2). Increased cytoplasmic GSH appears to only access the DV compartment in the CQR parasites, which is consistent with the data presenting GSH transport by mutant PfCRT in situ.
Conversely, the GST substrate CDNB is known to reverse CQR, and it was suggested that this was directly caused by a reduction in GSH (14). We show here that incubation of parasites with CDNB as well as reducing intracellular GSH levels also cause CQR parasites to accumulate more CQ (Fig. 3), indicating a clear role for GSH in modulating CQR. However, direct competition between the DNP-SG conjugate and CQ for PfCRT-mediated efflux cannot be completely excluded.
Substitution of mutant pfcrt alleles in otherwise isogenic backgrounds of P. falciparum has a marked effect on the cellular level of GSH, with CQR isogenic parasites having significantly lower levels of GSH, which is also reflected in their increased susceptibility to BSO (Fig. 2). Our data demonstrate that this is not attributable to changes in the expression of the enzymes involved in the synthesis, conjugation, and reduction of GSH (Fig. 4). Thus, the differences observed in GSH levels are most readily explained as a direct result of the different genotypes of pfcrt. We suggest that cytoplasmic GSH is transported into the DV in parasites expressing mutated pfcrt and that this is an important feature of CQR.
The CQR parasites contain less GSH than CQS counterparts, which superficially seems strange if this is the mediator of resistance. However, the key factor is not the level of GSH, but its location; it is the GSH in the DV that has the protective effect through binding to heme. This binding itself would reduce the amount of free GSH in the parasite overall, and also there are a multitude of peptidases in the DV, and it seems likely that they eventually destroy much of the DV-located GSH through proteolysis. Proteolysis of GSH and GS-X adducts (which could include GSH–heme) by carboxypeptidases has previously been shown to occur in plant vacuoles, but a fuller investigation of this possibility in Plasmodium was beyond the scope of this study (52).
Our findings clearly show that high GSH levels alone are not sufficient to affect CQR; it is the location of the GSH that is central to resistance. This is reiterated by our demonstration that GSH levels in untransformed Dd2 were found to be comparable with those of the CQS lines GC03 and C2GC03. This is also supported by a previous study where two nonrelated isolates, CQS 3D7 and CQR Dd2, were compared, and higher GSH levels were reported in the resistant parasites (30). Discovery of the crucial role of the mutant PfCRT in transporting GSH will now allow fuller analyses of the part played by the protein in other CQR lines.
Materials and Methods
Materials
P. falciparum GC03, C2GC03, C3Dd2, C67G8, T76K-1Dd2, and C-1Dd2 were a gift from Professor D. Fidock, (New York, USA) (18, 42) (Table 1). The plasmids PfHSP86 5′-pENTR4/1, GFP-pENTR2/3p, CHD-Hsp86, and pCHD-3/4 were a gift from Professor G. I. McFadden (Melbourne). Anti-P. falciparum glutathione S-transferase (PfGST) antiserum was obtained from Professor E. Liebau (University of Münster, Germany). Antiserum against P. falciparum casein kinase 2α (PfCK2a) was provided by Professor C. Doerig (Monash University, Australia) (16). RPMI 1640 medium and Albumax II were from Invitrogen. All chemicals, unless otherwise stated, were from Sigma.
Parasite culture and determination of IC50s
Parasites were cultured according to Trager and Jensen (46), synchronized using sorbitol (19), and freed from erythrocytes using saponin (47). Parasite drug susceptibility was determined by measuring the incorporation of [3H]-hypoxanthine (20 Ci/mmol; ARC) in the presence of increasing drug concentrations (10) and modifiers of intracellular GSH concentrations as detailed in the figure legends.
Localization studies
Expression constructs pCHD- Hsp86-γGCS-GFP and pCHD- Hsp86-GS-GFP contained the γgcs or gs genes of P. falciparum cloned in frame with a 3′gfp gene to generate C-terminal-tagged γ-GCS or GS fusion proteins. The chimeric genes were expressed under the control of the Pfhsp86 promoter and were generated using the Invitrogen MultiSite Gateway system in combination with the pCHD-3/4 destination vector (48). To generate gene entry clones, the full-length genes for γgcs and gs were amplified using the primer pairs, defined in Table 2, using Pfx SuperMix (Invitrogen) and subcloned according to manufacturer's instructions. The Multisite Gateway LR recombination reaction was performed according to the manufacturer's instructions (Invitrogen).
RT-PCR, real-time quantitative-polymerase chain reaction.
Determination of total GSH levels
Intracellular GSH levels of saponin-isolated parasites were determined by HPLC (51). Cells were washed once with Earle's balanced salt solution (EBSS) (6.8 g/l NaCl, 0.4 g/l KCl, 0.2 g/l MgSO4×7 H2O, 0.158 g/l NaH2PO4×2 H2O, 0.264 g/l CaCl2×2 H2O, 2.2 g/l NaHCO3, and 1 g/l
CQ uptake and equilibrium binding assays
CQ uptake by P. falciparum trophozoites was measured as described previously (5, 18). Steady-state CQ uptake was determined over 20 min using 5 nM of [3H]-CQ (specific activity 4.7 Ci/mmol; from ARC) in the presence or absence of various concentrations of CDNB. Equilibrium binding studies were performed as described previously, using 2 nM [3H]-CQ and a range of concentrations of unlabelled CQ (18). After correcting for nonspecific uptake (5), the resulting binding isotherms were fitted using nonlinear regression, and binding parameters calculated using the Grafit single-site ligand-binding model (Erithacus).
Determination of parasite hemozoin/heme concentration
P. falciparum-infected RBCs were saponin-lysed, and hemozoin was purified as previously described (18). The protein concentration of parasite lysates was determined using Bradford reagent following the manufacturer's instructions (BioRad). Purified hemozoin was converted into heme by dissolving in 0.5 N NaOH, and heme concentration was determined using a QuantiChrom Heme Assay kit DIHM-250 according to the manufacturer's instructions (Universal Biologicals Ltd). Hemozoin concentration was normalized to mM/mg of protein.
Expression of pfcrt in X. laevis oocytes
pfcrt cDNA products were synthesized with Thermoscript (Invitrogen) from total RNA of P. falciparum and amplified by PCR (primers see Table 2) with Taq High-Fidelity polymerase (Invitrogen) following manufacturer's recommendations. Amplified pfcrt (MAL7P1.27) genes from P. falciparum HB3 and Dd2 and a third gene only harboring the K76T mutation in the HB3 background constructed using the Quikchange site-directed mutagenesis kit (Stratagene) were directionally cloned into the pSP64T vector (using XhoI-NcoI sites) for expression in X. laevis. The Atclt1 (Arabidopsis thaliana
Capped complementary RNA (cRNA) was transcribed in vitro using the SP6 or T7 Message Machine kits (Ambion) using as templates EcoRI-linearized recombinant pSP64T or SalI-linearized pT7TS plasmids, respectively. Oocytes were obtained from X. laevis mature females purchased from Xenopus Express (Vernassal). X. laevis were immersed in euthanasic concentrations (0.5% w/v) of ethyl 3-aminobenzoate methanesulfonate, and 5 mM Tris–HCl, pH 7.4. Ovary lobes were removed and divided into smaller sacs. Individual oocytes were isolated manually with a platinum loop (43). After two washes, oocytes were left to recover at 18°C for 2 h in oocyte Ringer's solution (115 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgSO4, 1 mM Na2HPO4, 1.18 mM KH2PO4, and 5 mM HEPES, pH 7.4; or 5 mM MES for pH 6.0; adding 1 ml/l of 10,000 U penicillin/10,000 U streptomycin solution). Stage V–VI oocytes were selected and injected the following day with ∼50 ml of DEPC-treated water or cRNA solutions at 1 μg/μl using a semiautomatic injector (Drummond, Nanoject).
GSH uptake assays into X. laevis oocytes
Enzymatic defolliculation of oocytes was completed by 30-min incubation in 1 mg/ml collagenase in Ringer's solution (pH 7.4). Radiotracer uptake studies (12 oocytes per condition) were carried out at room temperature in 1 ml Ringer's solution, pH 7.4, with 2 μCi/ml of [3H]-GSH (38.6 Ci/mmol; ARC) and 5% dithiothreitol; the final concentration of GSH was 52 nM. [3H]-CQ (4.7 Ci/mmol; ARC) was used at 2 μCi/ml in Ringer's solution, pH 6.0; the final concentration was 425 nM. Uptake studies were terminated by washing the oocytes in Ringer's solution, pH 7.4, at 4°C. Individual oocytes were collected and immersed in 1 ml of scintillation liquid. After overnight incubation, radioactivity was determined using a Wallac 1450 Microbeta scintillation counter.
Indirect Immunofluorescence of X. laevis oocytes expressing PfCRT
Individual oocytes were immersed in 1 ml of an optimal cutting temperature medium (RA Lamb Ltd), and snap-frozen in 2-methylbutane in liquid N2 before sections of 10 μm were prepared on glass slides coated with Chrome Alum gelatin solution. Sections were fixed with cold acetone for 10 min, followed by blocking with 4% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 1 h, and incubation with primary antibody (1:500 in 4% BSA) for 1 h. After three washes with PBS, secondary anti-rabbit antibody (1:500 in 4% BSA) was applied for 1 h followed by washes as before. Samples were mounted using a VectaShield HardSet mounting medium (Burlingame) and examined by confocal microscopy (Zeiss Axiovert 200 M; L5M5 Pascal laser module).
Isolation of RNA and real-time PCR
RNA was extracted from synchronized trophozoites using Trizol according to the manufacturer's instructions (45). RNA was treated with TURBO-DNA-free before synthesis of cDNA using the RETROscript kit (both Ambion). Real-time quantitative PCR was performed using QuantiTect SYBR Green master mix (Qiagen) and primers at a final concentration of 0.3 μM in a 7500 Real-Time PCR system (Applied Biosystems). Transcription levels of γgcs, gs, and pfcrt were examined using specific primers (Table 2). Seryl-t-RNA synthase was used as an endogenous control (primers see Table 2), as it is transcribed uniformly throughout the parasite life cycle (25, 38). PCR cycling conditions were 50°C for 2 min, 95°C for 15 min, followed by 40 cycles of 95°C for 15 s, 54 for 30 s, and 68°C for 35 s. Relative expression levels were calculated by the ΔΔCT method (User Bulletin 2, Applied Biosystems,
Western blotting
About 5 μg of protein from four different parasite lines were separated on a 15% SDS-PAGE and blotted onto nitrocellulose. The membrane was probed with the primary antibodies raised against PfGST (1:5000), P. falciparum GR (PfGR) (at 1:15,000 dilution), and an antibody raised against PfCK2a (1:200) as a loading control. The secondary anti-rabbit antibody (Promega) was used at 1:10,000 dilution, and the signals were visualized using the Immobilon Western kit (Millipore). Relative expression was analyzed using LabImage 1D software (Kapelan Bio-Imaging Solutions).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 3.0. Parametric data were analyzed by one-way analysis of variance followed by Newman-Keuls post-test if differences were significant. Nonparametric data were analyzed by Mann–Whitney U-test. The use of the term significant in the text means a statistically significant difference p<0.05.
Footnotes
Acknowledgments
This work was supported by the Wellcome Trust [WT061173MA-SM]. The research was also supported by the European Community's Seventh Framework Programme [FP7/2007–2013] under grant agreement N° 242095. The Medical Research Council supported PGB and JES-S with the career-developing grant G0400173-69712. The Biotechnology and Biological Sciences Research Council supported SCM and JAHM under the grant BB/C515047/1. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would like to thank Professors Hagai Ginsburg and Graham H. Coombs for critical reading of the manuscript and many helpful comments.
Ethical Statement
This work involving malaria parasites was carried out using in vitro culture of parasites with human RBCs obtained from the West of Scotland Blood Transfusion Service. Ethics approval to use donated blood for this purpose has been obtained from the Scottish National Blood Transfusion Service Review of Nontherapeutic Issues of Blood & Components. All work on genetically modified Plasmodium falciparum has been approved by the Health and Safety Executive (GM37/K.09/008, “Genetic manipulation of human malaria parasites for functional genetic analyses”; HSE reference: GM37/02.5). P. falciparum wild-type and mutant parasites are contained in Category 3 facilities for all the experimental work and not released into the environment.
Author Disclosure Statement
The authors declare that no competing financial interests exist.