
Abstract
Introduction
An abundance of recent experimental evidence suggests that H2S plays a prominent role in normal physiology and pathophysiology, and many therapeutic targets exist for H2S therapy (Fig. 1). The molecular targets of H2S include proteins, enzymes, transcription factors, and membrane ion channels. Cysteine is the major source of H2S in mammals, catalyzed by the enzymes: cystathionine beta-synthase (CBS), cystathionine gamma-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST) (Fig. 2). Whereas 3-MST is mainly localized in mitochondria, CBS and CSE exist in the cytosol.


CBS and CSE generate H2S by using many different substrates (71). 3-MST catalyzes only sulfur transfer reactions from 3-mercaptopyruvate (3-MP) to various donors, for example:

The enzymatic sulfur transfer yields a hydropersulfide, not H2S (10). Release of H2S requires a further redox reaction between RSSH and a biological thiol such as glutathione (GSH):

Recently Kimura et al. demonstrated that 3-MST depends on a biological dithiol-thioredoxin (Trx) or dihydrolipoic acid- for the production of H2S from 3-MP (128).
H2S is enzymatically generated in the vasculature, heart, liver, kidney, brain, nervous system, lung, airway tissues, upper and lower GI tract, reproductive organs, skeletal muscle, pancreas, synovial joints, connective tissues, cochlea, and adipose tissues (105, 112). The key role of H2S in health and disease is clearly borne out by the correlations found to exist between low levels of plasma/tissue endogenous H2S/sulfane sulfur and/or H2S-generating enzymes on the one hand, and on the other the presence and progression of adiposity, marked endothelial dysfunction/insulin resistance, hypertension, hyperhomocysteinemia, diabetes, exacerbated cardiac injury following ischemia-reperfusion injury, Alzheimer disease, cirrhosis, chronic kidney disease, GI tract irritation, asthma, wound healing, and cancer (19, 46, 57, 58, 62, 85, 109, 156, 159, 210, 211, 219, 221, 239).
Physiological Actions of Hydrogen Sulfide
Nutrition, metabolism, and homeostasis
The main dietary sources of sulfur compounds in human nutrition are inorganic sulfates in drinking water and proteins derived from plants and animals. Only two of the twenty amino acids normally present in proteins are sulfur-containing amino acids (SAAs), namely methionine and cysteine. Methionine cannot be synthesized by the human body and must be supplied by the diet, whereas cysteine requirements can, in principle, be met by an excess of dietary methionine. However, cysteine is known as a semi-essential amino acid because humans can synthesize it from methionine to a limited extent (51, 143). Furthermore, the enzymes required for conversion of methionine to cysteine decline with age (17, 21). Dietary excess of cysteine and methionine is stored as GSH (17) (a thiolic antioxidant tripeptide) or, once the GSH pool has been replenished, converted to taurine or oxidized to sulfate (169). In fact, the availability of cysteine appears to be the rate-limiting factor for GSH biosynthesis from glutamate, glycine, and cysteine (9).
The “sulfane sulfur” pool (Fig. 7) performs an essential function in the brain, upon neuron excitation the bound sulfane sulfur releases H2S (80, 194). It is highly likely that H2S formation from sulfane sulfur requires reduced GSH as both hydrogen and electron donor. In the brain, H2S is produced mainly in astrocytes, which contain larger amounts of GSH than neurons.
There is compelling evidence that the reversible formation of mixed disulfides between GSH and low-pKa cysteinyl residues of proteins (e.g., S-glutathionylation) is an important mechanism for dynamic, post-translational regulation of a significant number of regulatory, structural, and metabolic proteins, and signaling pathways (9, 53, 97, 114). Mitochondrial GSH has been shown to act as a “sulfide buffer” when H2S starts to build up in the cell (208). In mice and humans, ethylmalonic encephalopathy (EE) responds well to treatment with high doses of N-acetylcysteine (NAC, a cysteine/GSH prodrug) (208). This disorder is caused by mutations in ETHE1, a mitochondrial matrix sulfur dioxygenase involved in oxidative sulfide catabolism. Mitochondrial GSH can accept the sulfur atom of H2S through the action of sulfide-CoQ reductase, yielding GSH persulfide (GSSH), which is better tolerated by the cell than H2S (70, 71). Importantly, thiosulfate is excreted in massive amounts in the urine of mice and humans presenting with EE, with high thiosulfate and H2S concentrations present in mouse tissues (200). In the five children treated by Viscomi et al, serum thiosulfate concentrations consistently decreased during treatment (208).
In spite of the critical role of sulfur in our diet, and especially of an adequate cysteine intake, dietary consumption of cysteine is generally suboptimal (51, 143). On the other hand, homeostatic regulation of cysteine and GSH pools declines with age, with the onset appearing in men at a younger age than in women (17). Since high dietary intakes of methionine have been shown to raise plasma levels of homocysteine (190), despite adequate intake of B vitamins, and since free cysteine can be a prooxidant (8, 75, 167), cysteine supplementation is nowadays achieved by oral administration of NAC, L-2-oxothiazolidine-4-carboxylate (OTC, another cysteine-GSH prodrug) or IMMUNOCAL (an undenatured protein concentrate rich in SAAs) (8). High-dose oral NAC has been shown to counter the intertwined redox and inflammatory imbalances in cystic fibrosis (201), and in several clinical trials, cysteine supplementation improved skeletal muscle function, decreased the body fat/lean body mass ratio, decreased plasma levels of pro-inflammatory cytokines NF-κB and TNF-α, improved immune function, and increased albumin levels (9, 20, 53, 103, 122, 217). However, Palmer et al. found that oral administration of NAC to mice (10 mg/ml in drinking water) daily for 3 weeks led to development of pulmonary arterial hypertension that mimicked the effects of chronic hypoxia (150). These findings raise the concern that chronic NAC therapy might have similar consequences in patients (124).
The H2S-cysteine-GSH connection has been documented often in the biomedical literature (26, 59, 93, 94, 141, 147, 158). Five factors are currently considered to contribute to the H2S–stimulated increase in intracellular GSH levels: (i) enhancement of cellular glutamate uptake (194), (ii) a H2S–induced increase in the level of gamma-glutamylcysteine synthetase and cystine transporter activity in the cell (94) (iii), reduction of cystine into cysteine by H2S in the extracellular space, and transport of cysteine into cells by the cysteine transporter (93), (iv) H2S stimulation of nuclear transcription factor Nrf2, which in turn upregulates GSH synthesis and transport (9, 26), and (v) a decrease in the activity of GSH-catabolizing enzymes (184). We believe the H2S-cysteine-GSH connection to be strongly dependent on the fact that H2S and L-serine act as co-substrates of cystathionine for CBS to yield L-cysteine (99, 153). This reaction is widely acknowledged to proceed in the opposite direction, producing H2S from cysteine, but its ready reversibility is firmly established (76, 153).
In summary, GSH is the most important intracellular thiolic antioxidant, a major determinant of the thiol/disulfide redox state, and a critical regulator of immune function, cell senescence, apoptosis, and vital redox-sensitive signaling pathways. Adequate levels of GSH are essential for effecting detoxification of xenobiotics and endogenously-generated toxins, for the biosynthesis of many essential biomolecules, and for protecting all cells from oxidative stress. Through the H2S-cysteine-GSH connection, an H2S prodrug may function not only as a source of H2S but also as precursor of L-cysteine and GSH.
Inflammation and immunity
H2S regulates inflammation and cell death, possibly exerting its beneficial effects through action on ATP-sensitive K+ channels (KATP) (196), inhibition of activation of NF-κB and p38 MAPK, scavenging of oxidants, upregulation of intracellular cAMP, and inhibition of caspase-3 cleavage (212). Chronic inflammation is involved in some of the most common human diseases such as rheumatoid arthritis, tuberculosis, asthma, inflammatory bowel disease, vasculitis, and Crohn's disease. Chronic inflammation is an influential factor in type II diabetes, cardiovascular disease, and tumor development (1, 107, 133, 245). Infiltration of macrophages into the cellular mass is a common characteristic of atherosclerotic lesions and tumors. Since Virchow first showed that the inflammatory process influences atherosclerosis and tumor development, a growing body of evidence supports the hypothesis that macrophages play an important role in initiating and promoting both pathologies. In both cases, the combined effects of reactive oxygen species (ROS), cytokines, chemokines, and angiogenic factors, produced by tumor-associated macrophages and other inflammatory cells, explain the abnormal growth of cells: once a cellular mass becomes infiltrated by macrophages, the ability of tumor and atherosclerotic tissue to survive the immune response increases exponentially (163).
Ischemia-reperfusion (I/R) injury is regarded as a form of acute inflammation in which leukocytes play a key role. Experimental studies carried out during the last 20 years contributed to develop the concept that oxidant-induced leukocyte–endothelial tissue interactions are largely responsible for the microvascular dysfunction induced by reperfusion. Recognition of the vital role of the inflammatory process in I/R injury has provided the impetus for an intensive research effort aimed at preventing leukocyte infiltration into post-ischemic tissue (110, 242).
In atherosclerosis, monocyte adhesion to endothelial cells is stimulated by an oxidized cysteine/cystine redox status. The specific mechanism involves intracellular generation of hydrogen peroxide, activation of NF-κB, and transcriptional activation and increased cell surface expression of cell adhesion molecules (CAM's) (83). H2S is an extremely potent inhibitor of leukocyte adherence to the vascular endothelium (243). H2S might interfere with inflammatory processes by diminishing the tissue injury induced by neutrophils via induction of apoptosis and/or scavenging of neutrophil-derived HOCl (220). Importantly, H2S exerts opposite effects on the viability of lymphocytes and granulocytes, which is probably the reason for the potentiation of the acute inflammatory and bactericidal responses and the depotentiation of the chronic inflammatory cellular response (243).
ROS/reactive nitrogen species (RNS) are mediators of NF-κB activation and this process can be blocked by antioxidants, in particular, cysteine and GSH (83). H2S has been shown to downregulate several pro-inflammatory cytokines including NF-κB, TNF-α, IL-1β, IL-6, and IL-8 (55, 98, 144, 151), to modulate leukocyte adhesion and leukocyte-mediated inflammation (55,181), to mediate the cardioprotection induced by ischemic postconditioning (241) and to protect from NF-κB and TNF-α mediated endotoxic shock (113). The powerful reducing/antioxidant/free radical scavenging properties of H2S can explain its wide-ranging anti-inflammatory and cytoprotective effects, including protection against: ischemia-reperfusion injury in heart, brain, retina, liver, and intestine; endothelial dysfunction; hydrogen peroxide-induced damage in rat gastric epithelial cells; hyperhomocysteinemia in rats; methionine- and homocysteine-induced oxidative stress; and hemin-mediated oxidation of low-density lipoprotein (112).
Cytoprotection and pharmacological conditioning
Cardiovascular system
H2S strongly influences the body's redox status through various mechanisms, such as increasing GSH levels in the cytosol, mitochondria, and nucleus of cells, increasing the GSH/GSSG ratio, activating the reperfusion injury salvage kinase (RISK) pathway with upregulation of protective heat-shock proteins, and acting as “master switch” of Nrf2 nuclear translocation, resulting in persistent activation of the antioxidant responsive elements (AREs) of antioxidant genes and concomitant overexpression of antioxidant and phase II enzymes (151, 165). H2S not only exerts anti-apoptotic and anti-inflammatory effects but also anti-nociceptive and blood pressure-lowering effects by activating KATP channels (196). The cardioprotective effect of H2S also involves activation of cardiac extracellular signal-regulated-kinase and/or Akt pathways (196).
Evidence on the cardioprotective effects of H2S has been obtained by many researchers. It has been shown that H2S has profound protective effects on the heart in murine models and that genetic overexpression of CSE in the heart is highly protective from I/R injury (26, 55). Exogenous administration of H2S and its donors in the settings of atherosclerosis, myocardial I/R injury, chronic heart failure, and cardiopulmonary resuscitation shows significantly improved outcomes in small animal models (25, 26, 119, 131, 156). These results are being translated into large animal models (155, 179, 181). The observed protection is associated with improved heart mechanics, reduced myocardial inflammation, preserved mitochondrial function, Nrf2 activation, and reduced cardiomyocyte apoptosis (25, 26, 55, 131, 156).
Yusof et al. reported the first evidence that preconditioning by exposing the small bowel of rats to NaHS induces an anti-inflammatory phenotype, such that postcapillary venules fail to support leukocyte rolling and adhesion when subjected to I/R injury 24 hours later (242). I/R injury is a major source of morbidity and mortality, not only in myocardial infarction, but also in many other clinical settings, including solid organ transplantation and ischemic cerebral and retinal vascular episodes. It is also a cause of irreversible damage to skeletal muscle made ischemic either as the result of pathologic hypoperfusion or of a planned surgical intervention. On the basis of results of both in vitro and in vivo experiments, it was recently concluded that the preischemic or postischemic delivery of NaHS limits I/R-induced cellular damage and confers significant long-term protection, that intravenous or even intra-arterial delivery of an H2S donor would provide more focused treatment of target tissue and, when administered in appropriate doses and within the proper time frame, H2S holds significant promise as a cytoprotective agent (65).
Peripheral arterial disease (PAD) affects over 5% of the older population (>60 years). PAD is considered a marker for systemic atherosclerosis and is frequently complicated by coronary and cerebral events (116). In PAD, oxidative stress is implicated in the correlation of a reduction in flow-mediated dilation (FMD) with a higher risk of developing CV complications. Therefore, treatment with antioxidants, aimed at improving peripheral arterial dilatation, is being investigated (116). In a rat unilateral hind limb ischemic model, treatment with NaHS (50 μmol kg−1day−1) promoted significant angiogenesis and improved regional blood flow. These effects are associated with an increase in vascular endothelial growth factor (VEGF) expression in skeletal muscle and VEGF receptor 2 (VEGFR2) phosphorylation in neighboring vascular endothelial cells. In addition, Akt phosphorylation is increased in ischemic muscles following NaHS treatment. However, treatment with 200 μmol kg−1 day−1 has no angiogenic effect (215).
Angiogenesis is triggered when the effects of pro-angiogenic factors, such as hypoxia inducible factor (HIF) and tumor growth factor (TGF), present in the tissue overcome those of the anti-angiogenic factors. It is possible that, at the higher dose, H2S/HS- inhibits nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (27) and/or binds to multiple cellular targets evoking mechanisms that counteract the pro-angiogenic effects (24, 215). In 2007 Isenberg et al. presented the results of a study on modulation of angiogenesis by certain simple dithiolethiones (DTTs), certain dithiolethione-modified nonsteroidal anti-inflammatory drugs (S-NSAIDs) and valproic acid, and H2S (79). Simple DTTs, S-NSAIDs and S-valproate demonstrated significant anti-angiogenic activities, inhibiting endothelial cell proliferation and vascular cell outgrowth and invasion of extracellular matrix. H2S, on the other hand, dose-dependently inhibited vascular cell outgrowth (at concentrations between 0.1 and 1000 μM) while stimulating endothelial cell proliferation in a dose-dependent manner within the same concentration range. Importantly, vascular outgrowth from muscle tissue was completely abrogated by H2S at a concentration of 0.01 μM, whereas endothelial cell proliferation increased by a factor of less than two between 0.1 and 1000 μM (79). According to Sparatore et al., H2S-donating hybrids-containing a DTT moiety inhibit angiogenesis and cell proliferation, these effects being related to their ability to slowly and gradually release H2S (184).
Nervous system
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a neurotoxin that can induce Parkinson's disease (PD)-like symptoms and biochemical changes in animals and humans. Inhaled H2S has been shown by Kida et al. to prevent MPTP-induced movement disorder, neuron degeneration, and neuron apoptosis and gliosis in mice (91). These effects were attributed to upregulation of genes encoding anti-inflammatory and antioxidant proteins, including heme oxygenase-1 (HO-1) and glutamate-cysteine ligase. Levodopa (L-DOPA) is widely used in PD therapy, but it does not prevent loss of substantia nigral dopaminergic neurons. The main factors responsible for this loss are oxidative stress and inflammation, which can be controlled by L-3,4-dihydroxyphenylalanine (L-DOPA) derivatives capable of being converted in vivo into L-DOPA and H2S by chemical and/or enzymatic means such as ACS83, ACS84, ACS85, and ACS86 (Fig. 3).
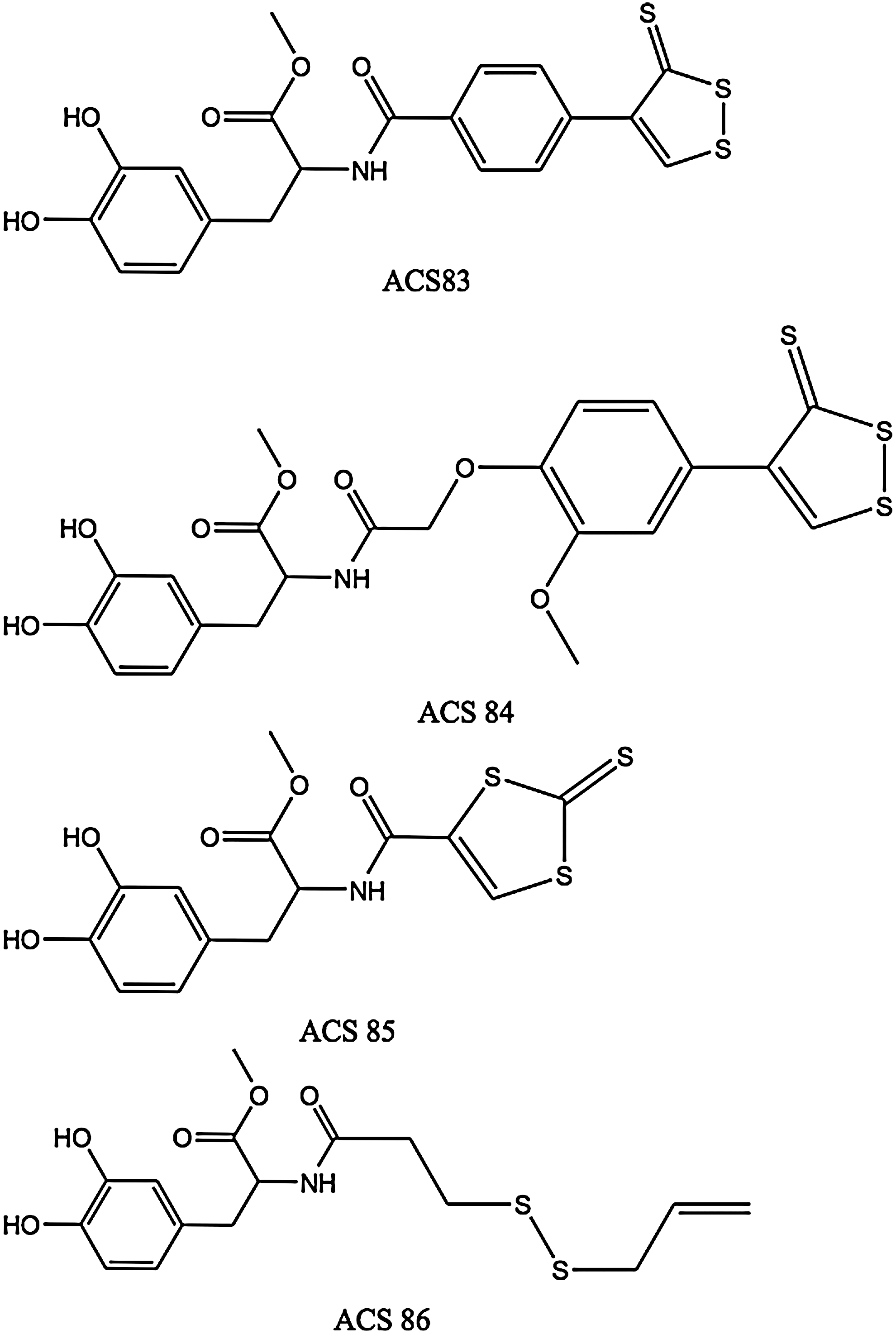
The four molecules in Figure 3 were synthesized and studied by Sparatore et al. (106). ACS83 and ACS84 are [1,2]-dithiole-3-thione derivatives, ACS85 is a [1,3]-dithiole-2-thione derivative, and ACS86 is a disulfide containing an allylmercapto moiety, which is expected to release H2S upon nonenzymatic reduction by GSH. ACS84 was converted by isolated mitochondria into H2S. This conversion was also observed in vivo, with a large increase in intracerebral dopamine (30% more than with L-DOPA) and GSH after intravenous administration to rats. The four L-DOPA hybrids reduce release of TNF-α, IL-6, and NO from stimulated microglia and astrocytes. They proved superior to L-DOPA itself as neuroprotectants.
Emerging evidence suggests that H2S may have therapeutic potential in Alzheimer's patients since it reduces mRNA levels and protein levels of beta-site amyloid precursor protein-cleaving enzyme 1 in nerve growth factor differentiated PC12 cells (105, 248). The depletion of H2S in the brains of Alzheimer's patients may be due to high levels of myeloperoxidase. Abnormally low brain levels of endogenous H2S and inflammatory stress are hallmarks of Alzheimer's (219).
Total GSH was shown to be substantially lowered in mitochondria from severely ischemic rat brain tissue (3). Hideo Kimura and colleagues recently showed that exogenous H2S increases GSH production and suppresses oxidative stress in isolated rat mitochondria (93). These in vitro findings were mirrored by in vivo observations that H2S protects ischemic brain by reinstating GSH levels decreased by oxidative stress. H2S has been shown to protect neurons against hypoxic injury by upregulating expression of heat shock protein (HSP) 90 (127, 199). HSP90 is a ubiquitous molecule that contributes to cell survival by regulating the folding of various cellular proteins, including survival factors, and by binding to apoptotic protease activating factor-1 (Apaf-1), thereby preventing apoptosis.
The slow-releasing H2S prodrug ACS67 (a latanoprost-dithiolethione conjugate) was shown to attenuate retinal ischemic damage following experimental elevation of retinal pressure in rats, with ACS67 being more potent than latanoprost (147). The same authors found that ACS67 significantly attenuated hydrogen peroxide-induced damage to transformed neural precursor cells known to exhibit a number of characteristics associated with retinal ganglion cells (RGC). It is pertinent to point out that, according to Osborne et al. (147), the neuroprotective effect of ACS67 probably involves several mechanisms, prominently including stimulation of GSH formation. Biermann et al. (14) recently demonstrated in rats that preconditioning with inhaled H2S (80 ppm in air) significantly attenuates apoptosis of RGCs after retinal ischemia/reperfusion injury. Their results revealed that H2S is able to attenuate caspase-3 cleavage and caspase-3 activity and significantly upregulated induction of cytoprotective chaperone HSP90, and strongly suggest that NF-κB downregulation is one component of this neuroprotective action. Furthermore, Mikami et al. demonstrated that H2S protects the retina from light-induced damage by regulation of intracellular calcium via activation of vacuolar type H+-ATPase (129).
Elevated levels of homocysteine (Hcy) in the blood (hyperhomocysteinemia), may cause mental retardation, seizures, and Alzheimer disease (205) via Hcy-induced oxidative stress and increased cerebrovascular permeability. Tyagi et al. (205) suggest that H2S functions as a type of armor in the brain, and could be a beneficial therapeutic candidate for the treatment of hyperhomocysteinemia-associated pathologies, such as stroke and neurologic disorders.
Digestive system
Exogenous administration of H2S prevents ethanol-induced gastric damage in mice and has a protective role against oxidative stress in rat gastric mucosal epithelium (126, 240), and is effective at preventing damage to the gastric mucosa induced by nonsteroidal anti-inflammatory drugs (NSAIDs) (58) and at promoting resolution of colitis in rats (213). Fiorucci et al. point out that, in addition to the firmly established contribution of exogenous H2S to gastric mucosal protection, its role in accelerating repair of mucosal injury might soon emerge. The therapeutic dose range of NaSH/Na2S was found to be very narrow (240). Takeuchi et al. present evidence supporting the assumption that endogenous H2S is involved in regulation of acid-induced bicarbonate ion secretion and mucosal protection in the duodenum (193).
H2S, as well as precursors containing a dithiolethione moiety, are potent inducers of the antioxidant and cytoprotective enzyme HO-1. This is clinically significant because HO-1 promotes ulcer healing (183). Other mechanisms believed to contribute to the GI-protective effect of H2S are increased epithelial secretion and mucosal blood flow, activation of KATP channels and of capsaicin-sensitive afferent nerves, reduction of leukocyte adhesion/infiltration, downregulation of TNF-α/IL-1β/IFN-gamma expression and scavenging of oxidants (212).
Liver and kidneys
Mounting evidence suggests that H2S regulates intrahepatic blood flow (microcirculation) in the normal and cirrhotic liver (58), with insufficient production of H2S in the cirrhotic liver and downregulation of H2S-producing enzymes in kidney and liver of patients with chronic kidney disease (2). Administration of H2S donors has been found to protect the liver and kidneys from ischemia-reperfusion damage (82, 115). In kidneys, H2S has been found to be beneficial to the prevention or treatment of diabetic kidney disease via alleviating renal glycative injury (115), increasing renal blood flow, glomerular filtration rate, and urinary sodium excretion (230), and ameliorating hyperhomocysteinemia-associated chronic renal failure (173). In the liver, H2S effectively attenuates stress-mediated liver injury and hepatic mitochondrial dysfunction in acutely ethanol-exposed mice (244), and markedly alleviates acetaminophen-induced hepatotoxicity in mice (135). The hepatoprotective and nephroprotective effects of H2S are mostly mediated by the “Nrf2 regulon”, (i.e., by activation of the many cytoprotective and lipogenesis-regulating genes controlled by the Nrf2-ARE pathway) (95,96,174). Additionally, H2S is also of benefit in hyperlipidemic and/or hypercholesterolemic prevention and therapy (75) via both enzymatic and nonenzymatic activities.
Diabetes and metabolism
Diabetes mellitus and its CV complications have been associated with increased production of ROS and perturbations of thiol redox homeostasis. Increased oxidative stress and oxidative damage are considered mediators of vascular injury in CV pathologies, including hypertension and atherosclerosis. In fact, CV disease is the major cause of morbidity and mortality for diabetic individuals. In order to reduce these risks, it is necessary to develop therapies aimed simultaneously at improving energy metabolism, insulin resistance, vascular function, blood pressure, and inflammatory/procoagulant status (125, 161).
The rate of ROS production depends on the metabolic status of the cell, as hyperglycemia increases the steady-state superoxide concentration. The rate of enzymatic reduction of glucose to sorbitol increases as well, with concomitant decreases in NADPH and GSH concentration. This depletion of reducing equivalents results in augmented sensitivity to oxidative stress (123). Thus, oxidative stress from excessive ROS and depleted mitochondrial GSH (mtGSH) can lead to cardiomyocyte apoptosis in the diabetic heart. Similarly, in diabetic retinopathy, superoxide levels in retinal mitochondria of diabetic mice are twice as high as those in nondiabetic controls, and mtGSH levels in the same retinas undergo a 40% decrease due to hyperglycemia (123).
According to Niki and his colleagues, endogenous H2S protects pancreatic β cells of mice from apoptosis induced by oxidative stress and/or glucotoxicity. They also found that NaHS was able to suppress ROS production induced by cytokines or hydrogen peroxide, via activation of Akt signaling (90, 197, 198). These findings are consistent with the effect of DATS (a hydrogen sulfide precursor, see below) on the level of blood sugar and oxidative stress markers in rats with type II diabetes mellitus (54).
It is now apparent that H2S biosynthesis declines as the severity of diabetes increases over time, and that therapies based on administration of different H2S donors to animals or patients in different stages of type I or type II diabetes may be highly successful (19, 46, 109, 221). Interestingly, it has been reported that plasma H2S levels are reduced in overweight individuals, with increasing adiposity being a major determinant of said levels (221). On the other hand, emerging evidence points to diminished Nrf2/ARE activity as a major contributor to increased oxidative stress, disrupted lipogenesis, mitochondrial dysfunction in the vasculature leading to endothelial dysregulation, insulin resistance, and the abnormal angiogenesis observed in diabetes (37, 95, 174, 192, 195, 209). Taken together, the aforementioned findings suggested that, in diabetes, blunted H2S biosynthesis is a major contributor to increased oxidative stress/mitochondrial and endothelial dysfunction and insulin resistance, the causal link being diminished Nrf2/ARE activity.
Last, recent evidence indicates that H2S (or its donors) exerts an anti-atherogenic effect by counteracting the oxidation of low-density lipoprotein (LDL) via HOCl scavenging, H2O2 scavenging, myeloperoxidase inhibition, and inhibiting foam cell formation by downregulating CD36, SR-A (scavenger receptor A) and ACAT1 (acyl-coenzyme A:cholesterol acyltransferase-1) expression via the KATP/ERK1/2 pathway in human monocyte-derived macrophages (100, 250). Lynn and Austin have reviewed experiments demonstrating that H2S supplementation ameliorates atherogenic processes, and therefore that such supplementation may be of therapeutic benefit in the prevention and treatment of atherosclerosis (119). For a full discussion of the relationship between H2S and the metabolic syndrome, please refer to the recent review by Desai et al. (48).
Benavides et al. proposed that endogenous H2S production from garlic-derived organic polysulfides provides the basis for the long-term beneficial effects obtained from the habitual consumption of garlic (13), in particular, the reduction in risk factors associated with the metabolic syndrome such as increased oxidative stress, obesity, hypertension, high blood glucose levels, hypercholesterolemia, hyperlipidemia, platelet aggregation, and blood coagulation, that together greatly increase the risk of developing CV disease and type II diabetes (148). Benavides et al. stressed endogenous H2S production from allyl, di-, and polysulfides derived from garlic (13), but did not mention the presence in garlic extracts of significant amounts of S-substituted L-cysteine derivatives (cysteine S-conjugates) which are also important H2S precursors, such as S-allyl-L-cysteine, S-allylmercapto-L-cysteine, S-propylmercapto-L-cysteine, and S-(penta-1,3-dienyl)mercapto-L-cysteine. These compounds are substrates of CBS (β-cystathionase, which also possesses beta-lyase activity). The mercapto-substituted derivatives are thereby converted into hydropersulfides (RSSH), which readily yield H2S upon reduction by GSH (13, 152). Taken together, these findings suggest that most of the organosulfur compounds in garlic preparations are potential H2S precursors in the body.
H2S as an antioxidant and free radical scavenger
At 37°C and physiological fluid pH (pH 7.4), about 80% of the H2S molecules dissociate to yield HS- (hydrosulfide anion), which is therefore the predominant sulfur-containing species in extracellular fluids and plasma (49), whereas within the cell (pH about 7.2) the amounts of H2S and HS- are nearly equal (145). Hydrosulfide anions are powerful one-electron chemical reductants capable of quenching free radicals by hydrogen atom transfer or by single electron transfer usually at or near diffusion-controlled rates. Their reaction with dioxygen is fast when catalyzed by divalent metal ions. They are also strong nucleophiles as evidenced by their reaction with S-nitrosothiols to release NO (202). The oxidation of hydrosulfide anions by biochemically relevant two-electron oxidants (e.g., hypochlorous acid and hydrogen peroxide) yields initially hydrogen disulfide (H2S2, also known as disulfane) which is also a highly reactive oxidizing agent (139, 160) capable of regenerating H2S by reaction with a thiol (13) or by disproportionation (118, 139). H2S will readily scavenge ROS and RNS, including hypochlorous acid, hydrogen peroxide, lipid hydroperoxides,
Under oxidative stress conditions, H2S may be converted to sulfite by activated neutrophils (132). Mitsuhashi et al. found that when NaHS was added in vitro to the supernatant of activated neutrophils, a significant amount of sulfite could be detected. Furthermore, a NADPH oxidase inhibitor markedly suppressed the production of sulfite. The chemical production of sulfite from H2S by neutrophil oxidative bursts is associated with inflammation, which might be responsible for the high levels of serum sulfite found in patients with pneumonia (132).
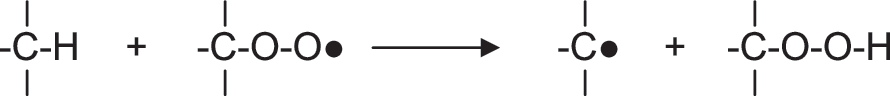
Although seldom acknowledged, simple species containing an SH group such as H2S, HS-, HS-SH, and HSS- excel at undoing the damage inflicted to biomolecules by free radicals through hydrogen atom donation to carbon-centered radicals (Fig. 4) (157, 191). Although hydrogen atom transfer to carbon-centered radicals is a diffusion-controlled reaction, the extremely low concentrations of H2S and H2Sx in blood and tissues limit their efficiency at repairing free radical damage to biomolecules (145).

Typically, carbon-centered free radicals react with oxygen to yield alkylperoxyl radicals:

which react further by abstracting hydrogen atoms from other biomolecules:
An inability to repair oxidized DNA, lipids, and proteins contributes to the damage induced by oxidative stress. Examples of macromolecular repair include DNA repair by base or nucleotide exclusion, protein repair by thioredoxin and glutaredoxin (162), and lipid repair by GSH peroxidase.
In order to appreciate the importance and uniqueness of the role of H2S as an antioxidant/free radical shield/cytoprotector, it is essential to recall that single antioxidants as pharmacologically active agents have not been found to exhibit extremely powerful therapeutic effects (177). This rather limited success might seem at first surprising in view of the decreased levels of selected major antioxidants consistently found in a number of disease states, but the limited success of this “single direct antioxidant approach” can be rationalized by recalling that mammals possess highly evolved and well-integrated antioxidant mechanisms that require the concerted and synergistic action of both antioxidant enzymes and low-molecular-weight antioxidants, with different antioxidants operating extracellularly and/or in specific cell compartments and having limited functional overlap: some destroy peroxidic species and/or peroxynitrite, others break free radical chains, and still others quench singlet oxygen (45). In addition, due to their short half-lives, direct antioxidants (vitamins C, E, etc.) must be administered frequently and at relatively high dosages to sustain their physiological efficacy (84). Furthermore, use of high-dose direct antioxidants may elicit pro-oxidant effects (45). However, H2S is not just another antioxidant to be added to the list of “direct antioxidants”, but it is also a powerful cytoprotective agent capable of activating nuclear transcription factor Nrf2 and consequently of inducing the expression of over 200 genes. These Nrf2-dependent genes encode proteins involved in lipid homeostasis, phase 2 detoxifying/antioxidant enzymes, directly acting antioxidant proteins, synthesis of low molecular weight antioxidants, and several P450 enzymes (84, 95, 174).
Cell signaling
The interaction of H2S with nuclear transcription factors has been intensively scrutinized. Many researchers have shown, using both cells in culture and whole animals that, in most cases, H2S inhibits NF-κB (112, 222). Slow-releasing H2S donors such as DATS, GYY4137, and S-diclofenac have also been shown to block NF-κB nuclear translocation in mouse macrophages and rat liver. Administration of GYY4137 to LPS-injected rats resulted in activation of signal transducer and activator of transcription-3 (STAT3), which is known to regulate the expression of many genes that mediate cell survival, proliferation, and angiogenesis (1, 113). H2S administration induces activation of transcription factor Nrf2 (26). In the nematode, Caenorhabditis elegans, H2S upregulates HIF-1 (23).
Many mechanisms of action of H2S may be mediated by protein S-sulfhydration (138,172). Sen et al. recently showed that S-sulfhydration of NF-κB by H2S is responsible for its anti-apoptotic actions (171). Mustafa et al. pioneered the concept of S-sulfhydration (SHY) as a signaling system (138). They define SHY as a physiological process wherein H2S attaches an additional sulfur atom to the thiol (-SH) groups of cysteine (Cys) residues within proteins, yielding a hydropersulfide group (-SSH). SHY usually activates enzymes (138). S-sulfhydration of GAPDH, for instance, results in a 7-fold increase in catalytic activity (138). Among the 49 proteins that were found to be basally S-sulfhydrated by liver-generated H2S are albumin, actin, β tubulin, CSE, CBS, several phosphatases, and catalase, and these authors estimate that from 10 to 25 percent of endogenous GAPDH, β tubulin. and actin are S-sulfhydrated in vivo (138).
Sulfane sulfur results following sulfhydration, and may also serve as a biological source of H2S. Operationally, sulfane sulfur was defined by Wood in 1987 (224) as sulfur that reacts, at pH 8.5–10, with cyanide to yield thiocyanate (Fig. 5) (88, 89, 225). From a structural viewpoint, a sulfane sulfur atom in an electrically neutral molecule is always attached to another sulfur atom and is either in an oxidation state of zero, or in an oxidation state of −1, and is attached to a hydrogen atom or to an “activating group” such as allyl, benzyl, phenacyl, etc. The “outer” sulfur atom of a hydropersulfide group is highly redox-labile, and is readily converted into H2S by reducing agents such as dithiothreitol, cysteine, or GSH:

“Activated organic disulfides” such as those shown in Figure 6 are organic sulfane sulfur compounds (12). The molecules of organic hydropersulfides, and hydropolysulfides contain the –S-S-H moiety, and therefore also contain sulfane sulfur. The most important hydropersulfides in biology are probably thiocysteine (Cys-SSH) and GSH hydropersulfide (G-SSH). “Bound sulfur” was defined by Ogasawara et al. as “divalent sulfur that is easily liberated as sulfide by reduction with dithiothreitol” Therefore, the “sulfane sulfur pool” constitutes a major portion of the labile sulfur pool (Fig. 7) present in tissues of plants and animals.

Acid-labile sulfur comprises various metalloproteins, which contain sulfide ions as part of metal/sulfur clusters (mainly Fe/S and Zn/S clusters) (81). Acidification may liberate the S−2 ions, which are released as SH- and H2S. The brain, heart, and liver contain significant amounts of acid-labile sulfur, whereas lung and muscle contain less (87). The labile sulfur pool (206) comprises both inorganic and organic chemical species, the simplest being disulfane (HS-SH), which is present as
The conversion of a thiol into a hydropersulfide by H2S requires one equivalent of an oxidant (81, 86, 139):
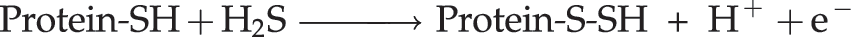
Although Nagy and Winterbourn recently proposed hypochlorous acid as a candidate (139), we believe hydrogen peroxide and the superoxide radical anion to be much more widely available oxidants in living tissues. Three likely mechanisms for S-sulfhydration are shown in Figure 8.

At physiological pH, most cysteine thiol groups in proteins are protonated (-SH) and hence display low reactivity towards H2O2. However, in some proteins where the cysteine residue is flanked by basic amino acids, the cysteine-SH group exists as the highly oxidizable thiolate anion (-S-). This introduces an element of specificity in H2O2-mediated signaling, suggesting that mainly proteins containing low pKa cysteine residues undergo S-sulfhydration. Hydrogen peroxide is the physiological oxidant of choice because it is constitutively produced inside most cells at various loci such as mitochondria, peroxisomes, and the cytosol mainly via enzymatic processes mediated by SOD, NADPH oxidases, xanthine oxidases, sulfhydryl oxidases, thiol oxidases, and monoamine oxidases (27, 83), and the reactivity of hydrogen peroxide toward thiols and H2S is high (140). Hydrogen peroxide generation in mammals is probably in the vicinity of 50 μmol kg−1min−1 (83).
S-sulfhydration of an enzyme may be accomplished through interaction with the proper substrate and does not require a discrete oxidation step involving a thiol group at the active site (as in mechanisms A, B or C, Fig. 8). Thus, 3-mercaptopyruvate has been reported to react with Trx, yielding pyruvate and Trx hydropersulfide (218). Therefore, H2S might S-sulfhydrate Trx through the following enzymatic pathway:

As efficient mitochondrial pathways for H2S oxidation are available (71), steady state tissue concentration can be held at very low levels and it is possible for H2S to function as an oxygen sensor (146). Thus, under hypoxic conditions, H2S catabolism would be blocked, leading to increased H2S levels with activation of specific responses (146). This hypothesis is consistent with similarities between the effects of hypoxia and H2S, enhancement of hypoxic signaling by H2S precursors, and abolishment of hypoxic signaling by H2S synthesis inhibitors.
Sexual function
Moore and his co-workers (185 —187) have described some pioneering studies that provide evidence for the endogenous formation of H2S and its pro-erectile relaxant effect on the corpus cavernosum of mammals, as well as on the effects of H2S in female sexual function. The first set of results were corroborated in a recently published article (40). There is also evidence that oxidative stress is implicated in erectile dysfunction (ED) in diabetic rodents (15,) and that interventions based on administration of tetrahydrobiopterin (182) and upregulation of antioxidant enzymes may be useful (44). For a discussion of the roles of endogenous and exogenous H2S in the endocrine and reproductive systems and the possibility of developing new therapies for ED that target this pathway, please see the recent articles by D´Emanuelle Di Villa Bianca et al. (41) and Zhu et al. (252).
Sparatore et al. have developed an H2S-donating derivative of sildenafil (ACS6) with possible clinical indications in ED, benign prostatic hypertrophy, and low urinary tract symptoms (184). ACS6 is a hybrid obtained by esterification between a phenolic dithiolethione and a carboxylic acid derived from sildenafil by attachment of a carboxyl moiety (CO2H) to the N-methyl group joined to the piperazine ring. The H2S released by S-sildenafil (ACS6) inhibits both PDE5 and NOX expression and activity. Furthermore, H2S applied ex vivo or overexpression of CSE has been shown to increase cGMP levels by phosphodiesterase inhibition in aortic ring preparations (22). Hence, this mechanism may constitute the basis of a new and effective approach to the treatment of patients suffering from ED, benign prostatic hypertrophy, and lower urinary tract symptoms.
In fact, ACS6 and sildenafil citrate relaxed cavernosal smooth muscle equipotently and ACS6 inhibited superoxide formation more than sildenafil citrate (175). Shukla et al. concluded that ACS6 not only promotes erection, but also affords effective protection from oxidative stress through upregulation of GSH synthesis. Additionally, in an investigation of the effect of NaHS on pregnant rat uterine contractility in vitro, Sidhu et al. found that this “hydrogen sulfide donor” produced significant dose-dependent decreases in uterine spontaneous contractility (176).
Life span modulation
Many lines of evidence suggest that oxidative stress plays an important role in aging. In C. elegans and Drosophila melanogaster, mutations resulting in resistance to toxic stresses, oxidative or not, tend to result in increases in longevity. In C. elegans, recent studies have shown that the Nrf2 homologue, SKN-1 (121), is necessary for the life span extension seen with dietary restriction, and overexpression of SKN-1 can increase life span. In D. melanogaster, increased Nrf2 activity correlates with oxidative stress resistance and increased life span of male flies (192). In mice, decreased Nrf2 signaling with age, and increased Nrf2 signaling with caloric restriction have been observed (111). H2S augments the life span of C. elegans through a sirtuin, a process that may involve protein S-sulfhydration (130). Since sirtuins are also found in vertebrates and since H2S signaling pathways are highly conserved, it is possible that this effect/mechanism might be found in mammals as well. According to Powolny et al, treatment of the worm C. elegans with DATS increases its mean lifespan, even if the treatment is initiated during young adulthood (154). Since DATS readily yields H2S in vivo, we consider it likely that this effect of DATS is mediated by H2S.
Leiser and Miller describe a series of studies that lend support to the hypothesis that augmented Nrf2 activity contributes to several forms of stress resistance observed in long-lived Snell dwarf mice that live about 40% longer than littermate controls and show delays in the onset of many aging-related pathologies (111). Importantly, Dwarf-derived fibroblasts exhibit many of the traits associated with enhanced Nrf2/ARE activity, including higher levels of GSH and higher GSH/GSSG ratios. In a related development, Guayerbas et al. concluded that a 4-week treatment of mice with NAC and thioproline protected all animals against early age-associated behavioral impairment, but the improvement was more evident in prematurely aging mice (61). On the other hand, Brown-Borg and collaborators found that in long-lived Ames dwarf mice the flux of methionine through the transsulfuration pathway is enhanced (in part because of upregulation of CBS and CSE), leading to an increased reduced GSH pool, mainly in the liver (136), with heightened resistance to toxic/oxidative challenges, and 50%–64% longer lives than their wild counterparts (males and females, respectively) (207). Importantly, Ames dwarf mice have a delayed occurrence and reduced incidence of presumably fatal neoplastic disease compared with their normal siblings (78).
Protection from NSAID toxicity
Nonsteroidal anti-inflammatory drugs (NSAIDs) also possess analgesic and anti-pyretic effects. The main adverse drug reactions associated with use of NSAIDs are gastrointestinal tract irritation, inhibition of cyclooxygenase (COX)-1 and COX-2 (211), inhibition of enzymatic H2S synthesis (211, 212), development of cardio- and cerebrovascular pathologies, and development of altered renal function. In fact, in the USA, an estimated 5% of all visits to a doctor are related to prescription of NSAIDs, and NSAID-related upper gastrointestinal adverse drug reactions are believed to result in over 100,000 hospitalizations and around 16,500 deaths yearly (204). Recent studies have shown that over 50% of patients taking NSAIDs have suffered mucosal damage to their small intestine (69). In a very recent and comprehensive meta-analysis, Sven Trelle et al. concluded that significantly increased CV risks are associated with taking naproxen, ibuprofen, diclofenac, celecoxib, etoricoxib, lumiracoxib, and rofecoxib (204). Since millions of persons with chronic musculoskeletal symptoms are long-term users of NSAIDs, their doubled risk of heart failure and increased risks of myocardial infarction and stroke are of the utmost concern.
Administration of NSAIDs results in a significant decrease in endogenous H2S enzymatic production. This effect was most profound with indomethacin, but was also observed with aspirin, diclofenac, and ketoprofen (58). Since endogenous H2S contributes significantly to mucosal defense (212), it is reasonable to expect that exogenous administration of this mediator would be effective at preventing NSAID-induced mucosal damage. Indeed, H2S donors such as NaHS (58) and diallyl disulfide (DADS) (212) were shown to confer mucosal protection from NSAIDs, preventing gastric damage in rodents. Furthermore, DADS prevented naproxen-induced decreases in gastric blood flow and increases in leukocyte adherence. Based on these findings, several research groups have developed NSAID derivatives that release H2S in vivo. These are obtained by conjugating a molecule of an NSAID with one of an H2S releasing compound. Typically, these H2S–releasing NSAID derivatives are carboxylic acid esters with general formula RCOOR´, obtainable (at least in principle) by condensing the NSAID molecule, which bears the carboxyl moiety with a sulfur-containing phenolic molecule (Fig. 9). The sulfur-bearing /H2S releasing phenols that have been used are shown in Figure 10.


One such H2S-releasing NSAID, S-diclofenac (ACS15, see below), showed greater anti-inflammatory activity than diclofenac at equimolar doses in several experimental models (184). Treatment with S-diclofenac, but not diclofenac, resulted in a marked reduction in severity of pancreatitis-associated lung injury. Moreover, S-diclofenac has much lower gastrointestinal toxicity than diclofenac and provides marked cardioprotection in a well-characterized experimental model of ischemia-reperfusion injury in the rabbit (164). Furthermore, S-diclofenac effects were accompanied by a significant increase in GSH, inhibition of angiogenesis and cell proliferation, and inhibition of NF-κB and TNF-α.
Hibernation and protection against hemorrhage
In 2005, Blackstone et al. revealed that H2S induces a hypometabolic state in naturally nonhibernating mice (16). When exposed to nontoxic H2S concentrations, mice rapidly and reversibly entered a hibernation-like state, which Blackstone et al. designated as “suspended animation-like” (16). An 80 ppm H2S treatment induced, within minutes, a 60% reduction in CO2 production and oxygen consumption, which can be lowered to over 90%. Additionally, core body temperature decreases to near-ambient, and heart rate and breathing frequency are significantly lowered. Oxygen demand is so drastically diminished that H2S-treated mice survive for over 6 hours in an atmosphere containing 5% oxygen, whereas untreated controls die within 15 min. Upon cessation of H2S exposure, the mice awoke without displaying neurological or behavioral abnormalities.
Following up on these highly newsworthy studies, Morrisson et al. showed that inhaled H2S or intravenous-administered Na2S can protect rats from lethal hemorrhage, with surviving rats free from functional or behavioral deficits (134). In the introduction to this article, the authors state that “clinicians and investigators have long hypothesized that reducing metabolic demand could buy time for patients suffering from insufficient blood supply until they can receive definitive treatment”. In effect, this goal is still being actively pursued in many quarters (5, 18, 47, 50), but it is proving extremely difficult to translate the protective effects displayed by H2S treatment of rats to larger mammals.
In fact, attempts to protect piglets and pigs from hemorrhagic shock failed (50), as well as administration of gaseous H2S-via extracorporeal membrane lung ventilation to sheep, in an attempt to avoid the potential pulmonary toxicity of H2S (47). It seems that these failures are related to the fact that the higher doses of H2S required to depress metabolism in larger mammals elicit toxic effects, systemic and/or pulmonary, and the possibility that the ability of H2S to abate metabolism depends on the specific metabolic rate of animals. H2S may reduce metabolism when the baseline metabolic rate is high (i.e., in awake mice), but not when metabolic rate is already depressed, for instance, in anesthetized mice or sheep (47).
Cancer prevention and treatment
Consideration of the many differential anti-cancer effects of H2S (collected in Table 1), shows that H2S and H2S prodrugs seem to be capable of inhibiting all stages in cancer development. These studies will be further expanded upon below. However, we will first discuss briefly the events that lead to the development of cancer to better understand how H2S may be used to intervene.
1, Initiation; 2, Promotion; 3, Progression; 4, Metastasis.
Normal cellular homeostasis is maintained by a balance between the processes of cell proliferation and cell death (apoptosis). An imbalance may lead to uncontrolled cell proliferation and cancer. The causal role played by ROS/RNS in carcinogenesis is now firmly established (203) and two mechanisms are thought to operate: (i) modulation of gene expression, with numerous oncogenes and tumor-suppressor genes operating through redox mechanisms that may be amenable to pharmacological intervention (223), and (ii) induction of genetic modifications. Redox dysregulation contributes to mutations and malignant transformation/progression through mitogenic signaling and modulation of apoptotic and survival pathways. Usually, pro-oxidant deviations from redox homeostasis relate to many aspects of the cancerous phenotype including alterations in metabolism, modulation of the cell cycle, upregulation of anti-apoptotic survival signaling, and upregulation of pro-angiogenic signaling.
According to the “differential redox set points” hypothesis, pro-oxidant-induced upregulation of intracellular ROS/disulfide stress specifically targets cancer cells, the therapeutic index being determined by the redox differential between the set points of normal and malignant cells. Wondrak (223) uses a highly descriptive analogy of this process with the operation of a car engine, where the red bar displayed on car tachometers denotes the maximum speed at which the car's engine is designed to operate without being damaged. In cancer cells, with a high set point of oxidative stress, pro-oxidant manipulation induces a redox shift that “redlines” and ruins the cancer cell's proliferative machinery. In contrast, normal cells tolerate the same pro-oxidant shift. It is important to note that many redox-targeted cancer drugs (including H2S donors) have been shown to potentiate the effect of other anticancer agents and radiation, which is consistent with preferential sensitization of cancer cells to the cytotoxicity of the nonredox-directed agent.
H2S is a Janus-faced molecule that can also behave as a pro-oxidant (6, 7, 189) via its interaction with dioxygen and/or the superoxide ion to generate sulfur-centered and oxygen-centered free radicals as well as higher sulfides H2Sn (1<n<8). A likely mechanism for H2S2 formation from NaHS and O2 in aqueous solution at pH close to 7 is:

Please note that Ka for HS• is greater than Ka1 for H2S by a factor of about 1000 (118).
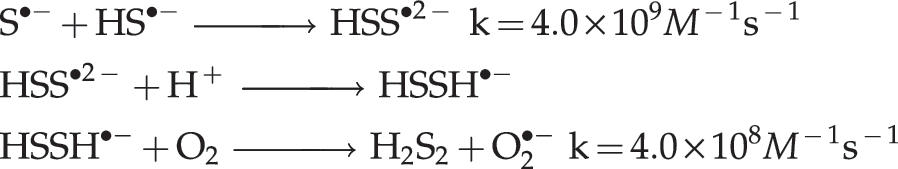
The first step would be the slowest in the sequence, but it is efficiently catalyzed by transition metal ions (117).
In turn, inorganic polysulfides (H2Sn) and organic hydropolysulfides (RSnH, n>1) are known to possess a high tendency to undergo homolysis and generate perthiyl radicals,
There is evidence in favor of pro-oxidant redlining of cancer cells by treatment with H2S or its prodrugs. Treatment of human neuroblastoma SH-SY5Y cells with NAC and ribose-cysteine resulted in elevation of sulfane sulfur level and inhibition of their proliferation (85). Diallyl disulfide (which releases H2S in vivo), was found by Filomeni et al. to induce neuroblastoma cell death (56). These researchers presented evidence that supports mediation of cytotoxicity by a ROS-dependent c-Jun NH2-terminal kinase/c-Jun signaling cascade (56). An extensive series of publications by Singh et al. (4, 66 –68, 92, 154, 231 –237), Seki et al. (72, 73, 170), Das et al. (42), and Lee et al. (104) demonstrate that DATS selectively targets DU145 and PC-3 cells in prostate cancer models, amazingly without damaging a normal prostate cancer cell line (237), kills cells of human gastric cancer cell lines, arrests the cell cycle in human cancer cell lines, and is cytotoxic towards a human breast cancer cell line (104), lung adenocarcinoma (228), prostate (231), colon (72), and human glioblastoma cells. This differential effect of DATS has been attributed to induction of intracellular oxidative stress through ROS generation. In a recent report, Lee et al. postulate that mitochondria are the main source of ROS generation and that DATS-induced oxidative stress is detected through glutaredoxin (GRX) (104).
DATS contains sulfane sulfur and is an excellent source of H2S in vivo (see section Diabetes and Metabolism), as evidenced by its ability to increase the intracellular GSH level and enhance the antioxidant and detoxification capabilities of rat primary hepatocytes (226). While we consider it likely that the effect of DATS on cancer cells is mediated by H2S, it has yet to be definitively demonstrated. Although the anticancer effects have been attributed in some cases to reversible covalent modification of specific proteins (73), it is estimated that this may be just an epiphenomenon (81, 137). In fact, Hosono's demonstration that cysteine residues Cys-12 and Cys-354 of beta tubulin are oxidized by DATS to S-allylmercaptocysteine residues constitutes indirect proof of H2S formation in this system (73):
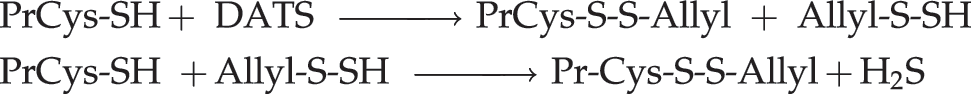
DATS has been intensively studied in China during the last 25 years. However, most research results were published in obscure Chinese periodicals. Validation of the anticancer and cancer chemopreventative activities of DATS is found in that body of literature, of which the following studies are worth mentioning: • DATS inhibits mouse colon tumors in mouse CT-26 cells allograft model in vivo (227). The authors conclude that DATS may represent a colon cancer-preventing agent. • A double blind intervention study was performed on 2526 experimental subjects and 2507 persons in the control group, with those in the first group taking doses of 200 mg synthetic DATS daily plus 100 μg of selenium every other day for one month of each year from November 1989 to December 1991. After a 5-year follow-up, it was concluded that the DATS+Se treatment had the effect of decreasing the incidence of digestive cancer by over 50% (64, 251). • DATS enhances the antitumor function of macrophages either by priming macrophages alone or by synergic action, meanwhile increasing the susceptibility of some tumor cells to macrophage cytotoxicity (247). • DATS augments the activation of T lymphocytes. This effect is related to inhibition of NO production by macrophages. In addition, DATS can antagonize the inhibition of tumor-derived immunosuppressive factors produced by S180 cells and Ehrlich ascitic cancer cells on the activation of T lymphocytes and reduce the inhibitory rate significantly. The authors state that DATS is potentially useful in tumor therapy (246). • Apoptosis of human cholangiocarcinoma FRH-0201 cells can be induced by DATS in vitro in a dose-dependent manner (38). • DATS can induce mitotic arrest in human gastric cell line SGC-7901 (35). • DATS can cause gastric cancer cell (MGC803 and SGC7901 cell lines) arrest in the M-phase, and this may be one of the mechanisms for inhibiting cell proliferation (63). • DATS induces apoptosis in human gastric cancer cell line BGC-823 through downregulation of Bcl-2 and increased caspase-3 expression and activity (101, 102).
There are many studies from several laboratories on the cytotoxic effects of organic isothiocyanates (e.g., sulforaphane and benzyl isothiocyanate) derived from cruciferous vegetables (36, 168, 180, 188, 203, 229, 249). These authors report that organic isothiocyanates selectively kill cancer cells (human prostate, human pancreatic, etc.) in culture through ROS-mediated mechanisms. Since hydrolysis of organic isothiocyanates under physiologic conditions may generate H2S, it is likely that these selective cytotoxic effects are H2S-mediated (238).
Allyl sodium thiosulfate, also known as 2-propenyl thiosulphate (2-PTS) and also found in garlic, has been shown by two research groups to behave similarly to DATS in many respects (28 –30, 166). These authors found that 2-PTS reacts with GSH, under physiologically relevant conditions, generating H2S. In vitro, 2-PTS inhibits proliferation of human tumor cell lines WiDR, 293, HL-60, and HuT78 (human T-lymphoblastoid cell line) in a dose-dependent manner, and caused oxidative damage and apoptosis to HL-60 and HuT 78 cells. Cytotoxicity of 2-PTS is related to a blockage in the G2/M phase of the cell cycle, which was linked to an early increment in ROS flux, and to inactivation of rhodanese, with concomitant thiolation to yield a protein disulfide highly sensitive to proteolytic degradation.
Moore, Deng and co-workers provided further evidence on the anticancer effects of H2S (108). They studied the interaction of two H2S donors (NaHS and GYY4137) with cancer cells in vitro and the effect on mice tumors of intraperitoneal injection of 100–300 mg/kg/day of GYY4137, a slow-releasing H2S donor that persists in the culture medium for up to 7 days, versus only 2 hours for NaHS. GYY4137 (but not NaHS) is cytotoxic to human cancer cells in a concentration-dependent manner. The two H2S donors studied did not affect the survival of normal human lung fibroblasts (IMR-90 and WI-38), and GYY4137 promoted cancer cell (MCF-7), but not normal cell (IMR-90) apoptosis. It also induced cell cycle arrest of cancer cells in the G2/M phase. Daily administration of GYY4137 to immunodeficient mice for 14 days caused a dose-dependent reduction in the growth of tumors induced by prior injection of a human leukemia cell line (HL-60 or MB4-11).
Chattopadhyay et al. show, in a recent series of articles, that H2S-releasing-NSAIDS are effective at inhibiting the growth of a variety of cancer cells (32 –34). H2S-releasing-NSAIDS inhibited cell proliferation, promoted apoptosis, and caused G0/G1 cell cycle block of eleven different cancer cell lines (33). The H2S-releasing-NSAIDS had potencies of 28- over 3,000-fold compared to their NSAID counterparts in these effects, and H2S-releasing-aspirin (HS-ASA) was consistently more potent than the other H2S-releasing-NSAIDS tested (33). HS-ASA not only inhibits the growth of HT-29 human colon and Hepa 1c1c7 mouse liver adenocarcinoma cells in culture, it also induces Nrf2 expression and Phase-II detoxifying enzymes in vivo (34). HS-ASA also shows promise as a therapeutic agent in estrogen receptor negative breast cancer (32).
Conclusion
H2S has come to the forefront of some very exciting and promising research to treat a variety of diseases, and several H2S-releasing prodrugs are currently under development by the pharmaceutical industry. H2S possesses a very diverse biological profile that includes: potent antioxidant, anti-apoptotic, anti-inflammatory, metabolic, vasoactive, and cytoprotective actions on normal cells that could potentially be harnessed to treat a number of pathological states. The robust antioxidant actions of H2S involving direct scavenging of toxic reactive oxygen species combined with the effects on antioxidant enzyme expression and function are highly attractive features of this gaseous signaling molecule. It is possible that H2S prodrugs and novel agents that modulate H2S bioavailability might be efficacious for acute myocardial infarction, stroke, diabetes, arthritis, peripheral artery disease (PAD), metabolic syndrome, organ transplantation, erectile dysfunction, diabetes, inflammatory bowel disease and pulmonary hypertension. Contrastingly, H2S appears to exert powerful prooxidant and proapoptotic actions on cancer cells of different origins, which suggests that H2S prodrugs might be developed into effective anticancer agents capable of achieving high specificity and broad efficacy across different cancer types. However, it is important to fully consider the highly toxic actions of supraphysiological levels of hydrogen sulfide, and great care must be taken during the development of H2S-based therapeutic agents. This is true of any agent that exerts potent actions on the redox status in both normal cells and cells undergoing oxidative stress during pathological states.
Footnotes
Acknowledgments
DJL and BLP are supported by grants from National Institutes of Health (NIH) National Heart Lung and Blood Institute (NHLBI) 5R01HL-092141-03, and 1R01HL-093579-03 to DJL, and by the Carlyle Fraser Heart Center at Emory University Hospital Midtown. GG would like to express his gratitude to Marcela del Rocio Rosas del Real for her invaluable assistance in preparing the manuscript.
Disclosure Statement
DJL is a co-founder and consultant for Sulfagenix. GG is a founder and consultant for Sulfagenix. GG is an inventor of several United States patents for the use of hydrogen sulfide-based therapeutics for a number of disease states.