
Abstract
Introduction
E
Certain cellular systems can produce ROS via enzymes such as NADPH oxidases (Nox) (41) and xanthine oxidase (4). In mitochondria, ROS can form during oxidative metabolism through one-electron reduction of molecular oxygen, leading to superoxide anion formation, and subsequent dismutation would lead to hydrogen peroxide formation. Electron transport chain complexes I, II, and III contain sites where electrons are uncoupled to generate ROS (26, 44). Data suggest cross-talk regulation of ROS production and redox signaling between Nox and mitochondria, as recently reviewed (6). Nox-derived ROS activate mitochondrial ATP-sensitive potassium channel opening, which leads to depolarization of mitochondrial membrane potential, followed by mitochondrial ROS formation and respiratory dysfunction. Mitochondrial ROS subsequently enter the cytosol through mitochondrial permeability-transition pores (mPTPs) (6). The major constituents of mPTPs are voltage-dependent anion channels (VDACs), adenine nucleotide translocase (ANT), and cyclophilin D (CypD) (6).
Modification of protein thiols by ROS and electrophiles is an important process in redox signal transduction. Although mitochondrial proteins undergo redox-based thiol modifications, the specificity and biological impacts of such modifications caused by diverse ROS/electrophiles remain largely elusive. The current study discovered that, by utilizing a newly developed proteomic method, the endogenous electrophile 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) clearly induced redox-based modification of mitochondrial heat-shock proteins (HSPs) via S-guanylation, which may be involved in mitochondrial permeability-transition pore (mPTP) opening. Our data thus suggest that mitochondrial HSPs may be novel targets for regulation of mitochondrial redox signaling via mPTP opening.
Modification of protein thiols by ROS and/or electrophiles is an important part of redox signal transduction (20). Proteins possessing redox-sensitive thiols include kinases/phosphatases, transcription factors and their regulators, and channels, which participate in regulating ROS-/electrophile-dependent redox signaling (20). Protein S-guanylation is a unique post-translational modification (PTM) of protein thiols by the electrophilic nitrated cyclic nucleotide 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) (12, 36). 8-Nitro-cGMP is mainly synthesized via soluble guanylate cyclase from 8-nitroguanosine 5′-triphosphate (8-nitro-GTP), derived via GTP nitration by nitric oxide and ROS, with involvement of mitochondrial ROS production stimulated by Nox2-derived ROS (2, 12). Protein S-guanylation by 8-nitro-cGMP occurs on Keap1, a negative regulator of transcription factor Nrf2, which leads to an antioxidant adaptive response against oxidative stress (12, 36). Thus, 8-nitro-cGMP may play an important role in regulating antioxidant redox signaling downstream of mitochondrial ROS production.
Mitochondrial protein thiols are susceptible to modifications by ROS and electrophiles because of the relatively alkaline pH in the mitochondrial compartment (17, 22), which promotes deprotonation of thiols to produce reactive thiolate anions. Because 8-nitro-cGMP is colocalized with mitochondria under certain circumstances (36, 42), 8-nitro-cGMP may participate in regulating mitochondrial redox signaling, possibly by induction of protein S-guanylation. In this study, we developed a proteomic method—S-guanylation proteomics—to identify protein targets for S-guanylation in mitochondria. We identified mitochondrial proteins, including mitochondrial stress-70 protein (mortalin) and 60-kDa heat-shock protein (HSP60), which are S-guanylated endogenously during lipopolysaccharide (LPS)/cytokine stimulation. Mortalin (34) and HSP60 (13) reportedly regulate mPTP opening, at least partly, by interacting with CypD (13), a component of mPTP (14). We found that mPTPs opened in cells after stimulation with LPS/cytokines in a cyclosporine A (Cs)-sensitive manner, which suggests CypD-dependent mPTP opening. Furthermore, 8-nitro-cGMP induced mPTP opening, which was inhibited by Cs. These observations suggest that mitochondrial heat-shock proteins (HSPs) may be novel targets for redox modification via protein S-guanylation that participates in mPTP regulation and mitochondrial redox signaling.
Results
Development of S-guanylation proteomics
S-Guanylation proteomics comprised two approaches (Fig. 1): (i) direct protein digestion, followed by immunoaffinity capture of S-guanylated peptides that were subjected to liquid chromatography–tandem mass spectrometry (LC-MS/MS) (immunoaffinity capture and LC-MS/MS), and (ii) 2D polyacrylamide gel electrophoresis (PAGE) separation of S-guanylated proteins that were extracted and subjected to in-gel digestion, followed by LC-MS/MS (2D-PAGE and LC-MS/MS).
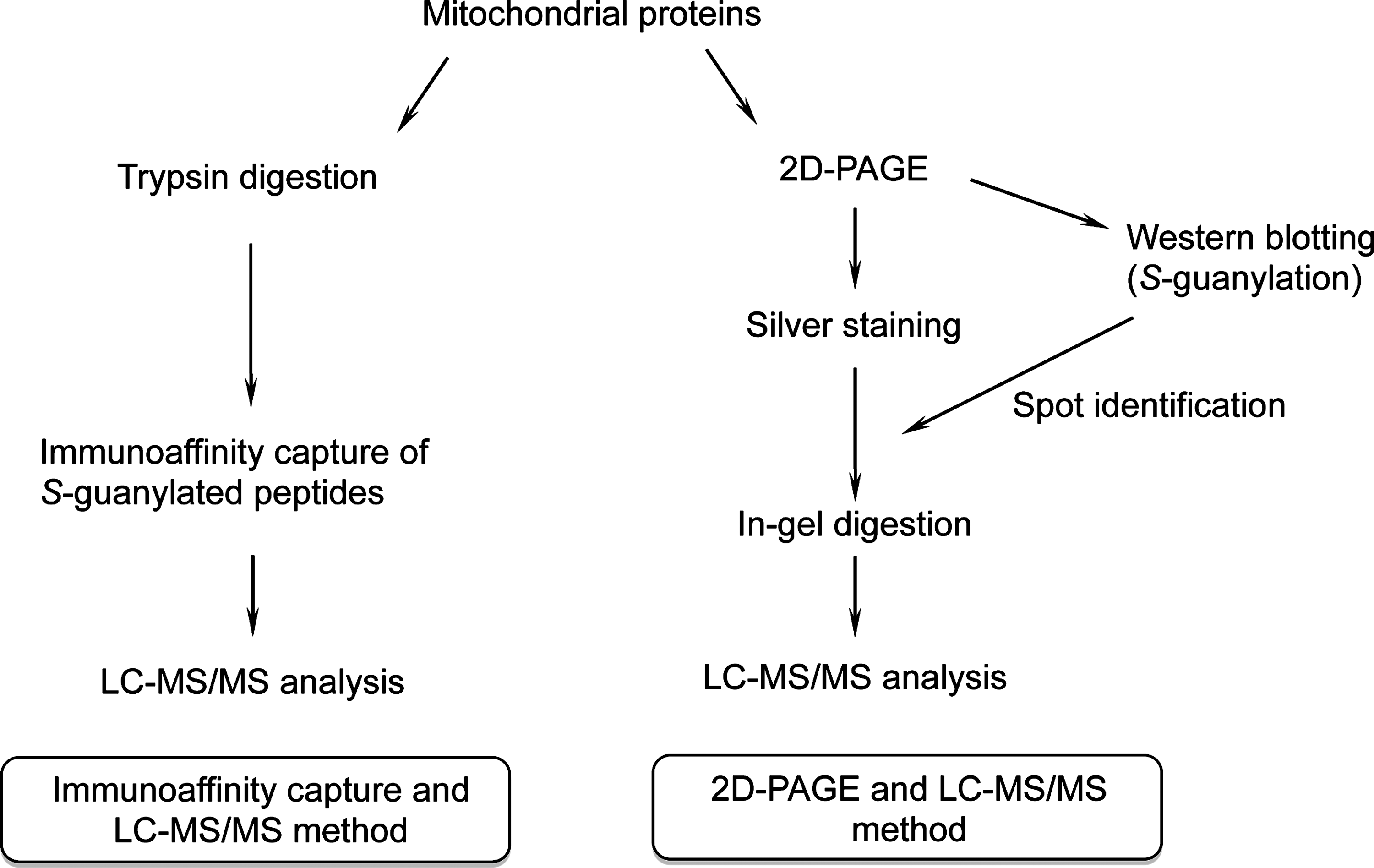
Immunoaffinity capture and LC-MS/MS
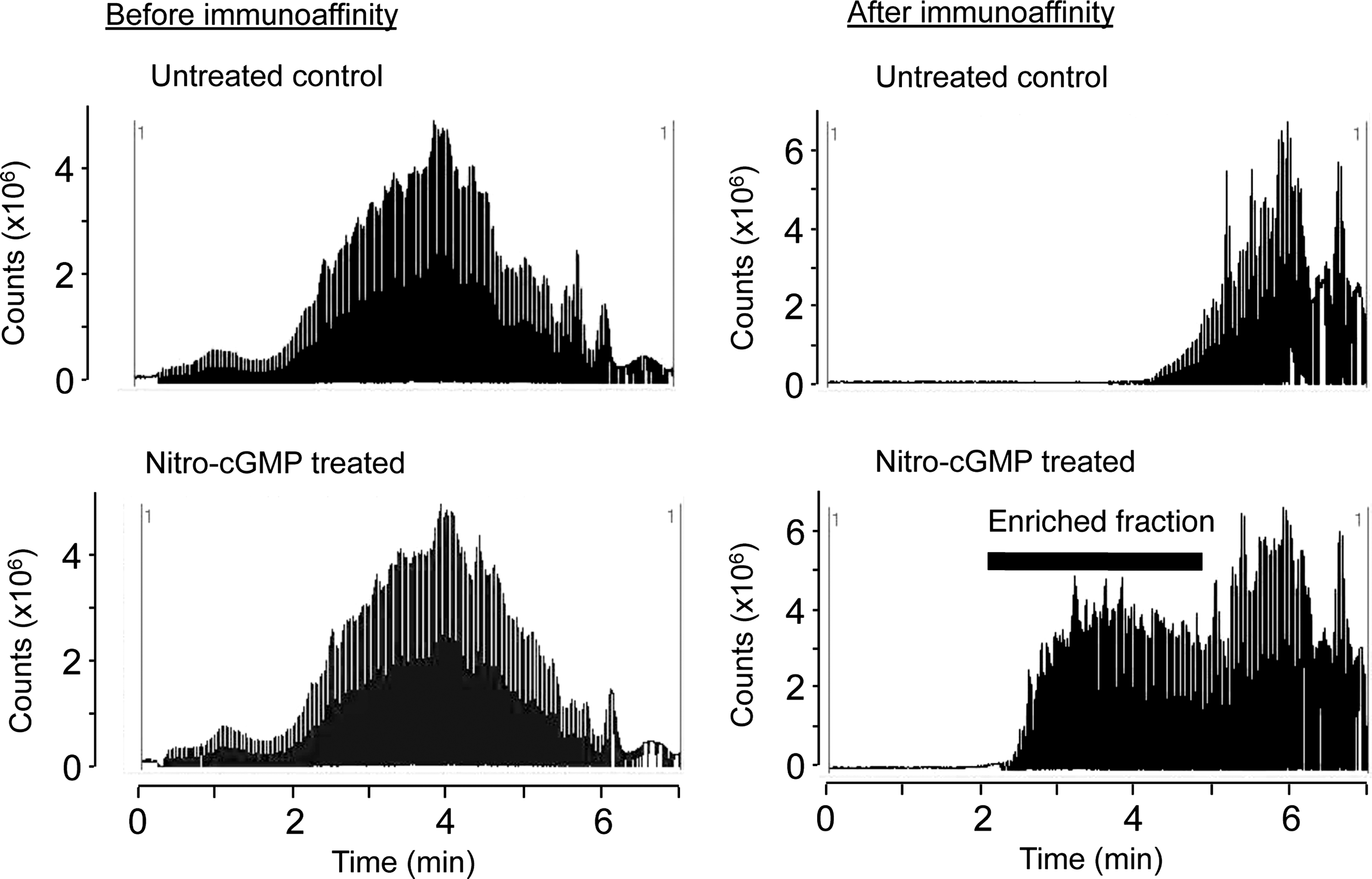
To test the efficacy and selectivity of immunoaffinity capture and LC-MS/MS, we first analyzed mitochondrial proteins treated with authentic 8-nitro-cGMP. Mitochondrial proteins solubilized in an RIPA buffer were reacted with 8-nitro-cGMP or were untreated, followed by direct trypsin digestion and immunoaffinity capture, as Figure 1 shows. Figure 2 shows total-ion chromatograms (TICs) of trypsin-digested peptides before and after immunoaffinity capture. Before immunoaffinity capture, TIC profiles were similar for peptides obtained from mitochondria with or without 8-nitro-cGMP treatment. After immunoaffinity capture, however, TIC intensity markedly increased for peptides obtained from mitochondria treated with 8-nitro-cGMP compared with controls. This result suggests that our immunoaffinity capture effectively and selectively enriched S-guanylated peptides from the mixture of S-guanylated and non-S-guanylated peptides.
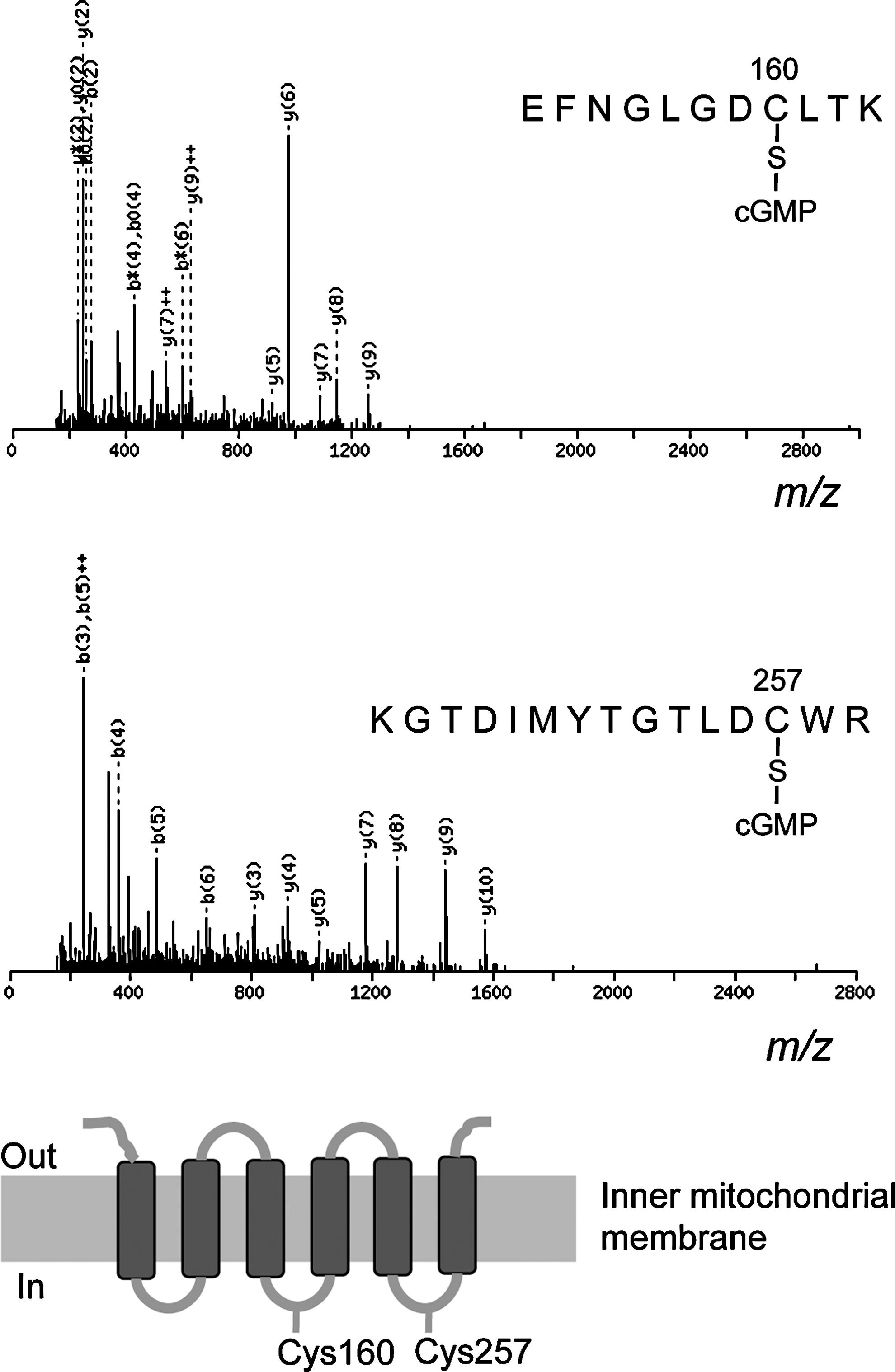
We also evaluated mitochondria treated with 8-nitro-cGMP for S-guanylation by using MS/MS data, which we analyzed via a Mascot search of valuable modifications at cysteine residues with carbamidomethyl and cGMP moieties. Figure 3 provides MS/MS spectra for S-guanylated peptides of ANT. Amino acid sequences of S-guanylated peptide ions were established by MS/MS fragmentation patterns. By this method, we identified 107 putative S-guanylated tryptic peptides from 89 proteins reacted with 8-nitro-cGMP in vitro in the RIPA buffer (Supplementary Table S1; Supplementary Data are available online at
2D-PAGE and LC-MS/MS
We next examined the identification of S-guanylated proteins by using 2D-PAGE and LC-MS/MS. As Figure 4 illustrates, multiple Western blot spots proved with the anti-S-guanylated protein antibodies were detected in both solubilized and intact mitochondria treated with 8-nitro-cGMP. Of 56 immunoreactive spots (Fig. 4A), 2D-PAGE and LC-MS/MS successfully identified 39 as S-guanylated proteins in solubilized mitochondria (Supplementary Table S3). Similarly, of 34 immunoreactive spots (Fig. 4B), 34 proteins were identified in intact mitochondria (Supplementary Table S4).
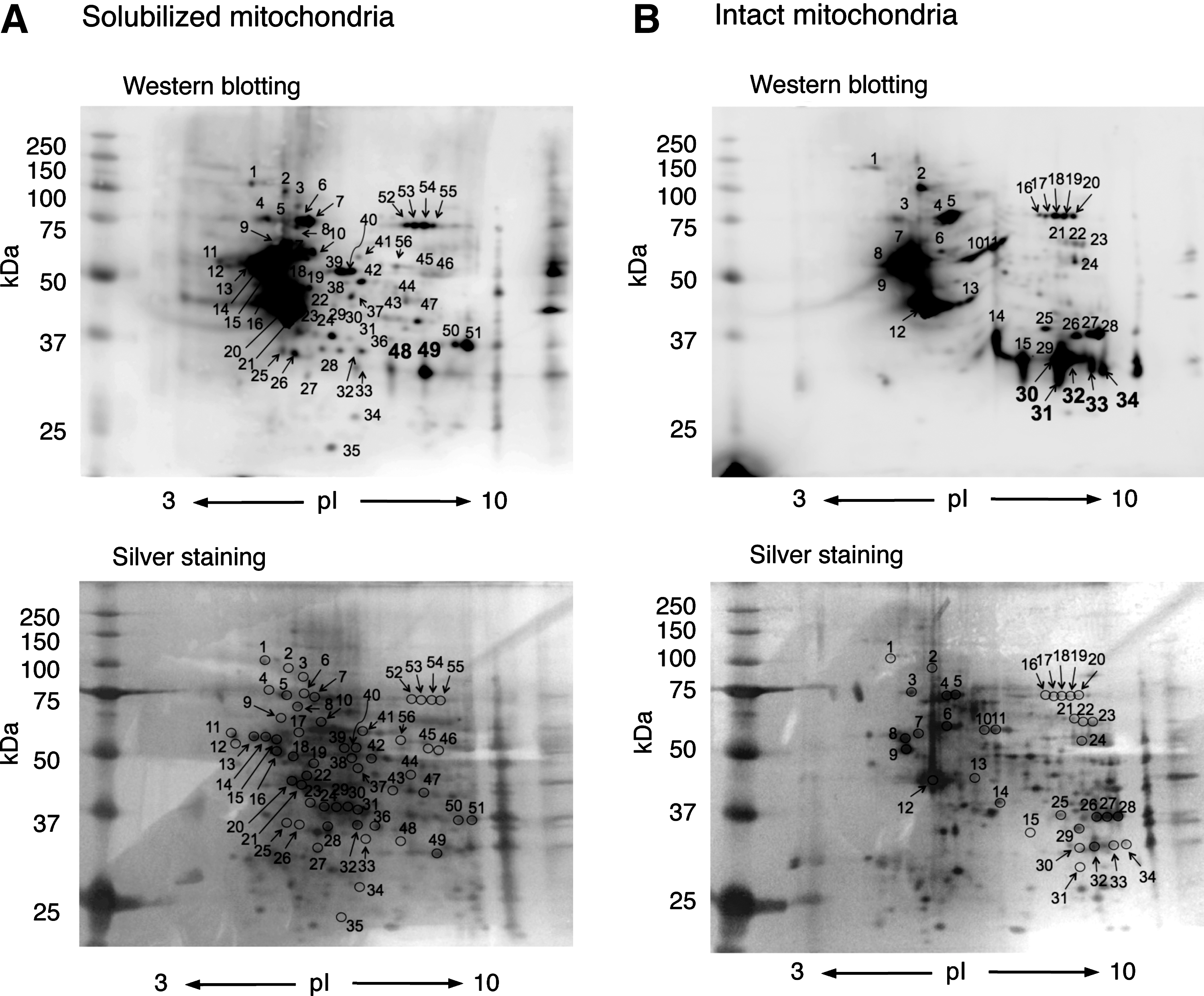
One advantage of 2D-PAGE and LC-MS/MS is semiquantitative comparison of the degree of S-guanylation of Western blot spots. Spots 30–34 in intact mitochondria (Fig. 4B) showed intense staining compared with spots 48 and 49 in the solubilized mitochondria (Fig. 4A). These spots were identified as VDACs (Supplementary Tables S3 and S4). 2D-PAGE analyses suggest that VDAC S-guanylation may be more effectively induced when VDACs are localized in the mitochondria than when they exist homogeneously in RIPA buffer.
The analyses of S-guanylated proteins by two different methods showed that some proteins were commonly detected by both methods, but others were not overlapped each other. This may be due to apparently distinct principles of these methods used. In general, the 2D-SDS-PAGE method has limitation to detect hydrophobic or membrane proteins, or small and highly basic (pI>8) proteins. In fact, smaller proteins such as ribosomal proteins were detected in an immunocapture LC-MS/MS method, but not in the 2D-SDS-PAGE method under current conditions (e.g., Supplementary Tables S1 and S3). These limitations can be solved by employing narrower pH-gradient 2D gels or higher-polyacrylamide-percentage gels. On the other hand, the strength of the current 2D-SDS-PAGE method is that much smaller amounts of proteins suffice for their detection (0.1 mg protein/analysis) compared with that for the immunocapture and LC-MS/MS method (∼1 mg protein/analysis). Thus, the 2D-SDS-PAGE method is suitable for initial screening of protein samples of limited availability.
Several nonmitochondrial proteins were identified as S-guanylated targets in mitochondrial preparations (e.g., Supplementary Tables S1 and S3). This may be due, at least in part, to two possibilities as suggested by Wong and Liebler (46); protein contaminations from other subcellular organelles during sample preparation even with use of a widely accepted method for isolation of mitochondria, and some of the proteins may have multiple subcellular localizations.
Identification of endogenously S-guanylated mitochondrial proteins
We successfully identified, for the first time, endogenously S-guanylated mitochondrial proteins. S-Guanylation patterns differed for untreated control and LPS/cytokine-stimulated cells (Fig. 5). As summarized in Table 1, HSP60, mortalin, prelamin-A/C, and vimentin were detected only in the treated condition. In contrast, S-guanylation of protein disulfide isomerase A6 was only observed in mitochondria obtained from untreated cells. Solomon et al. reported that the expression of protein disulfide isomerase A6 was strongly suppressed by tumor necrosis factor-α (40). Further study is needed to clarify whether similar suppression of protein disulfide isomerase A6 expression may be induced by LPS/cytokine treatment in C6 cells. Other proteins such as heterogeneous nuclear ribonucleoprotein K, tubulin, ATP synthase subunit-beta mitochondrial, and actin were detected in both untreated and LPS/cytokine-treated conditions.
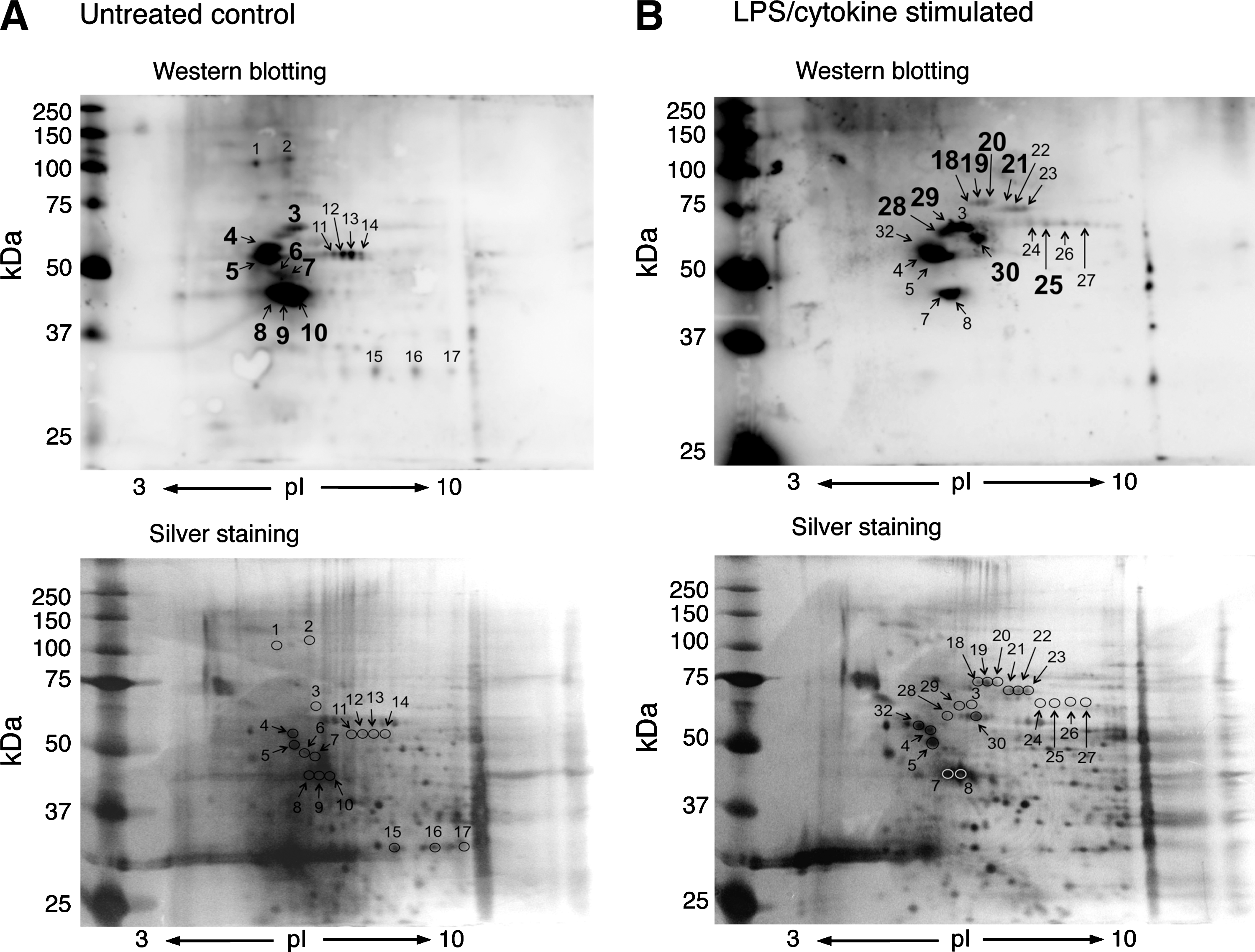
Supplementary Tables S5 and S6 provide detailed information on protein identification.
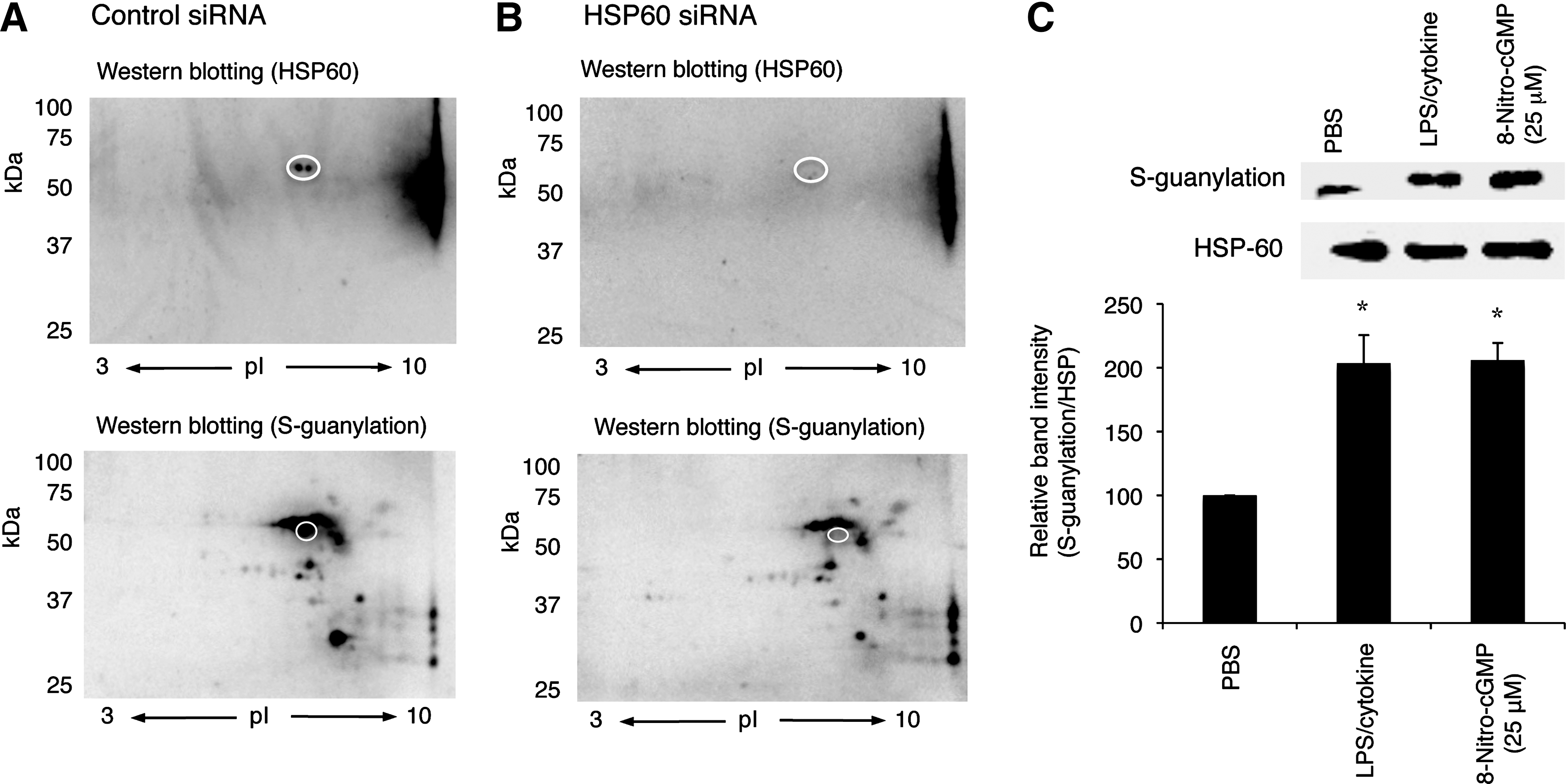
Mortalin and HSP60 are mitochondrial HSPs that were recently reported to be involved in regulation of mPTP opening (13, 34). Our data clearly indicated that these proteins were significantly S-guanylated after LPS/cytokine stimulation when endogenous 8-nitro-cGMP formation is upregulated. This observation was verified by the means of alternative approaches: siRNA knockdown and immunoprecipitation. As shown in Figure 6A and B, downregulation of HSP60 by siRNA resulted in the remarkable reduction of the spot intensity corresponding to that for HSP60 S-guanylation Western blotting. This was further supported by immunoprecipitation assay. HSP60 was first immunoprecipitated with anti-HSP60 antibody, followed by analyses with anti-S-guanylation Western blotting. As shown in Figure 6C, LPS/cytokine stimulation significantly increased the content of S-guanylated HSP60 than untreated control. It is also important to mention that 8-nitro-cGMP treatment (25 μM, 24 h) resulted in the similar extent of HSP60 S-guanylation as that induced by LPS/cytokine stimulation. In the following study, we therefore performed experiments to examine whether 8-nitro-cGMP can modulate mPTP activity in cells.
Heterogeneous nuclear ribonucleoprotein K, vimentin, and prelamin-A/C are other possible targets for endogenous S-guanylation under immunological stimulation. The first protein regulates mitochondrial transcription (31) and affects mitochondrial responses to insulin (9). Vimentin modulates mitochondrial motility (28). Although S-guanylation of these proteins may affect mitochondrial transcription and motility, further study will be needed to investigate the roles of 8-nitro-cGMP in those processes.
Induction of mPTP opening by 8-nitro-cGMP
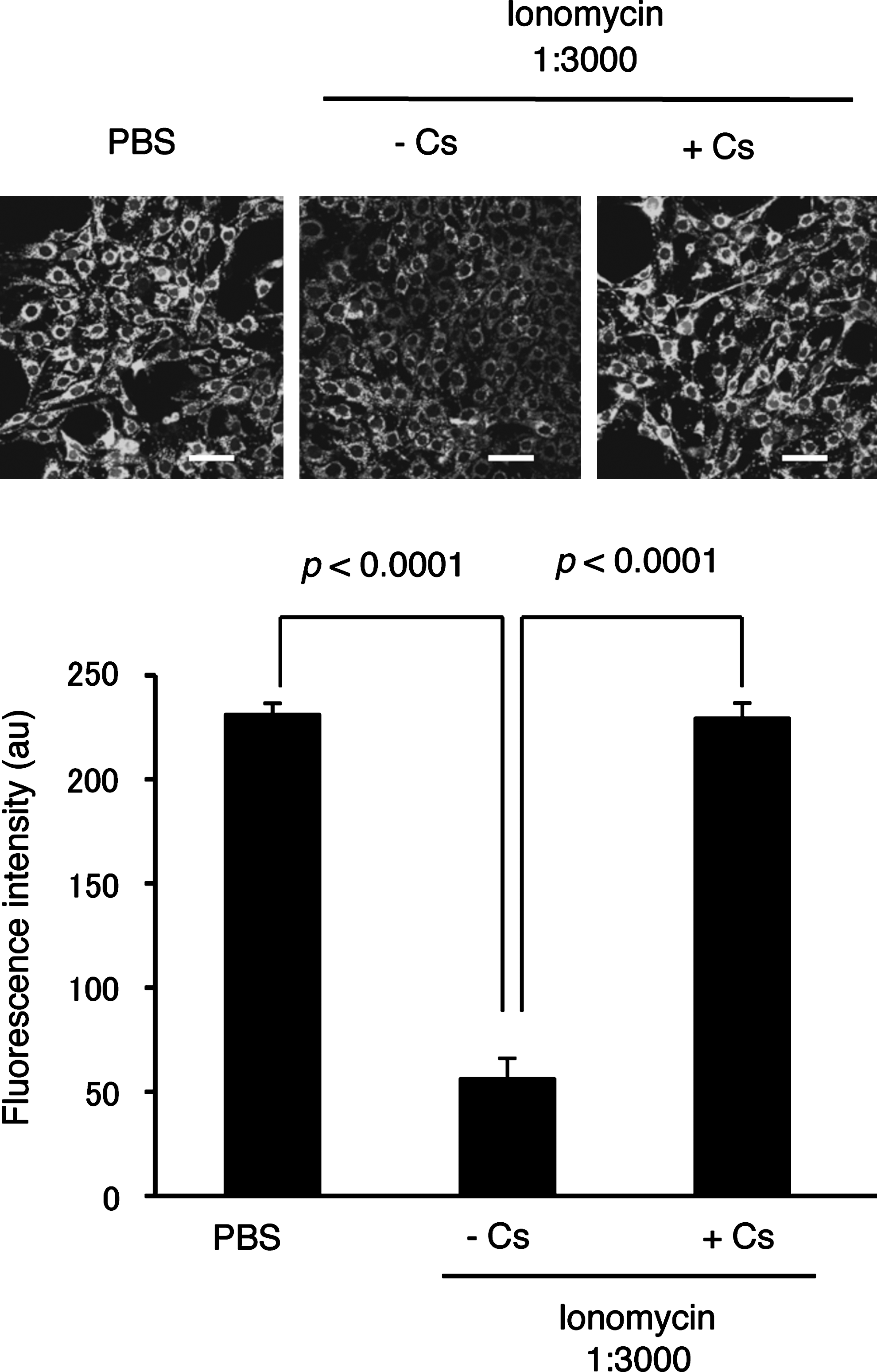
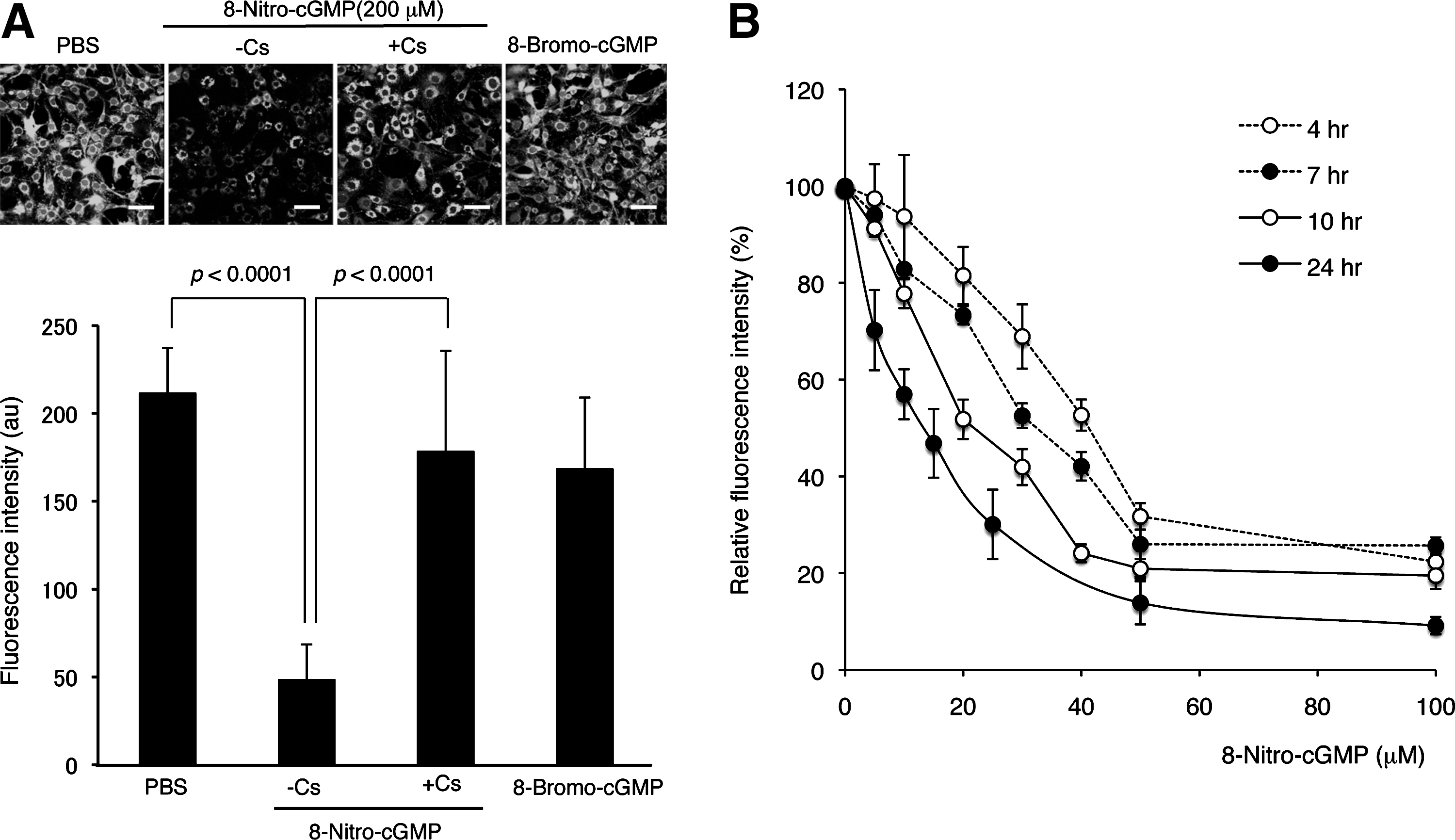
To test whether LPS/cytokine stimulation and/or 8-nitro-cGMP treatment induced mPTP opening in rat C6 glioma cells, we employed a calcein-quenching assay according to the literature (33). Calcein incorporated in cells distributes both in the cytosolic compartment as well as in mitochondria. Fluorescence of cytosolic calcein can be quenched by Co2+ treatment, whereas mitochondrial calcein is protected from Co2+-mediated quenching. The mPTP opening results in calcein release from mitochondria to cytosol, leading to reduced fluorescence. Figure 7 shows strong fluorescence in untreated C6 cells; ionomycin treatment, which induces mPTP opening via increased cellular calcium, significantly reduced calcein-derived fluorescence. Cs treatment abolished this ionomycin-induced reduction of fluorescence, which suggests CypD-dependent mPTP opening.
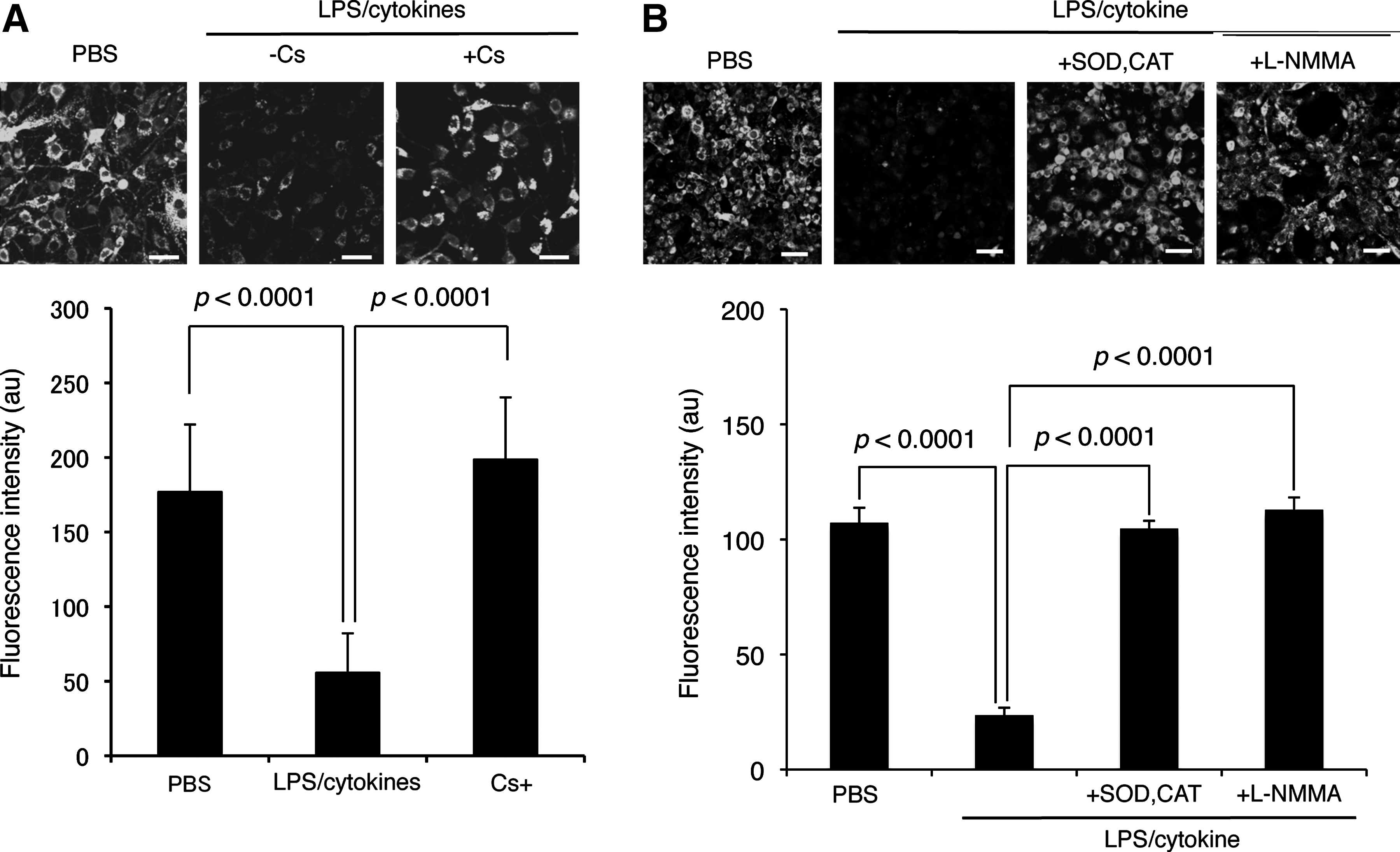
By using this calcein-quenching assay, we demonstrated that LPS/cytokine stimulation induced mPTP opening in C6 cells (Fig. 8A). This mPTP opening was almost completely reversed by Cs, which suggests involvement of CypD activation in mPTP opening. The mPTP opening triggered by LPS/cytokine stimulation was inhibited by the treatment with ROS scavengers (pegylated superoxide dismutase [SOD] and catalase) or an NO synthase inhibitor (N
ω-monomethyl-
Discussion
8-Nitro-cGMP is the first known endogenously formed electrophilic nucleotide (3, 12, 18, 36, 48). 8-Nitro-cGMP reacts with protein thiols to induce a unique PTM, protein S-guanylation (3, 12, 36). We previously demonstrated that Keap1 is susceptible to S-guanylation in cells (12, 36). Keap1 S-guanylation thus induced is clearly involved in Nrf2-mediated induction of adaptive responses to oxidative stress. To understand how 8-nitro-cGMP mediates redox signal transduction via S-guanylation, identifying the S-guanylation proteome is necessary. In this study, we developed an MS-based proteomic method combined with immunoaffinity capture or 2D-gel electrophoresis to investigate S-guanylation. We found that immunoaffinity capture and 2D-gel electrophoresis combined with LC-MS/MS effectively identified target mitochondrial proteins for S-guanylation.
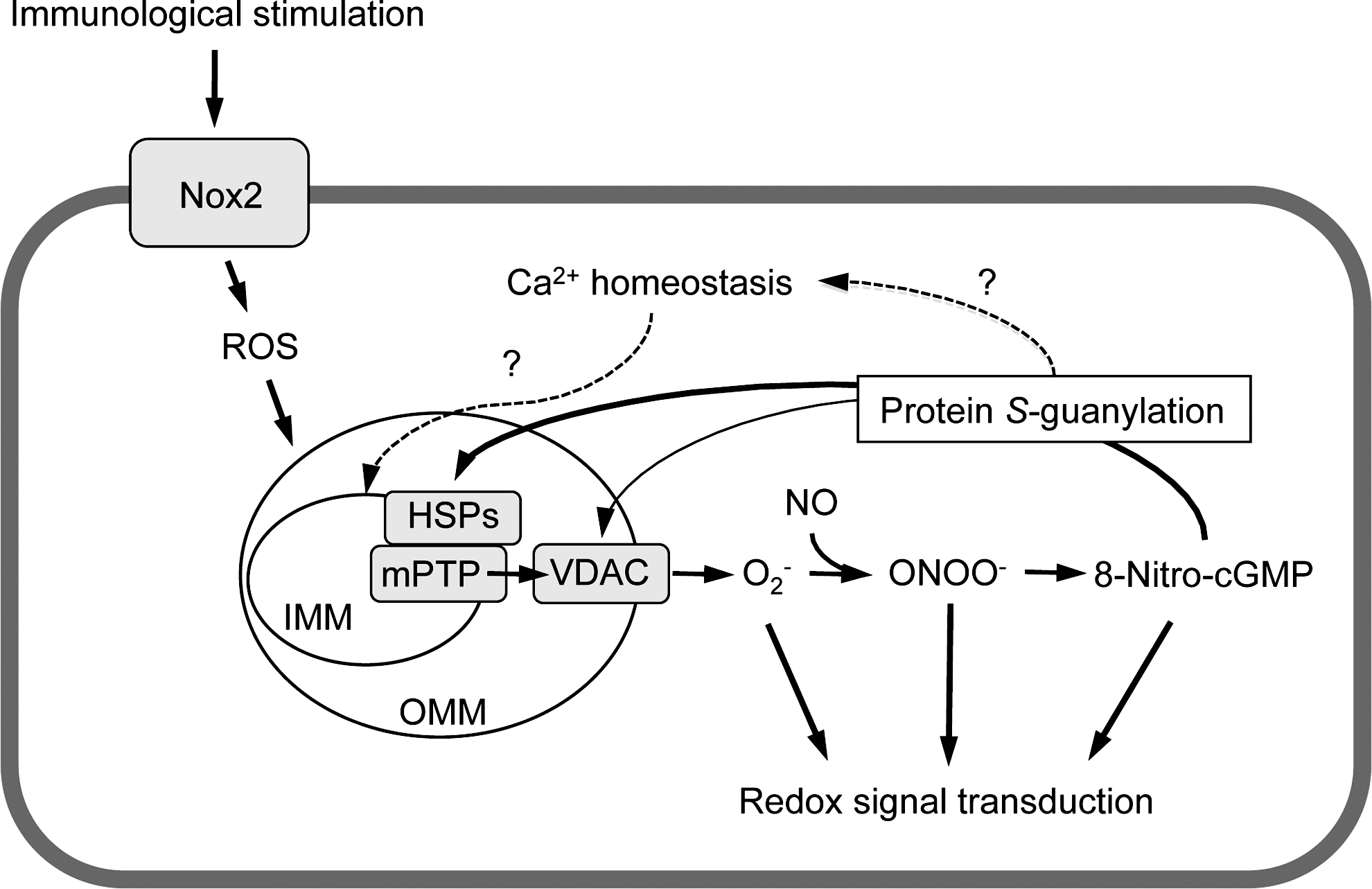
mPTP is a nonselective pore spanning the inner mitochondrial membrane (IMM) and the OMM and consists of several proteins, including ANT in the IMM, VDAC in the OMM, and CypD in the matrix (15). These proteins are redox sensitive, and ROS and thiol-reactive electrophiles induced mPTP opening through redox-based modifications of thiols of those proteins (25, 29). The effects of electrophiles on mPTP opening have been reported to be varied dependent on the types of electrophiles examined. Landar et al. reported that the lipid-derived electrophile 15-deoxy-Δ12,14-prostaglandin J2 induced mPTP opening in isolated mitochondria (21). On the other hand, 4-hydroxynonenal, a naturally occurring electrophile derived via lipid oxidation, inhibited mPTP opening in isolated mitochondria (19). Our study revealed for the first time that 8-nitro-cGMP, as an endogenous electrophile, induced mPTP opening in cells (Fig. 9). Proteomic analyses showed no S-guanylation of ANT, VDAC, and CypD after LPS/cytokine stimulation to promote endogenous formation of 8-nitro-cGMP (Fig. 5 and Table 1). We found however that several mitochondrial proteins, including HSPs, underwent endogenous S-guanylation after LPS/cytokine stimulation. Recent studies suggested that mitochondrial HSPs, including HSP60 and mortalin, regulate mPTP activity. Ghosh et al. showed that a multichaperone complex comprising HSP60, HSP90, and tumor necrosis factor receptor-associated protein-1 interacts with CypD to suppress mPTP opening (13). siRNA-mediated knockdown of HSP60 triggers CypD-dependent mPTP opening, which suggests a critical role of HSP60 in multichaperone complex formation (13). Our immunoaffinity capture and LC-MS/MS analyses indicated that Cys442 may be redox sensitive and hence a possible target for S-guanylation in cells (Supplementary Tables S1 and S2). Cys442 of HSP60 is located near the ATP-binding site. Chemical modification of this residue disrupted HSP60 oligomerization and suppressed chaperone activity of HSP60 (27). Thus, S-guanylation at Cys442 of HSP60 may influence the multichaperone complex stability. In addition to HSP60, mortalin was determined to be a negative regulator of mPTP opening (34). Our study identified Cys317 as a putative S-guanylation site for mortalin (Supplementary Table S2). No information is available however about the role of Cys317 in chaperon activity or other biological functions of mortalin.
We observed that several S-guanylated proteins were detected not only in LPS/cytokine-stimulated cells but also in untreated cells (Fig. 5A, B, and Table 1). It has been reported that the mitochondrial compartment is maintained as slightly alkaline pH and more reducing than other cellular compartment (17, 20, 22), and hence mitochondrial proteins may be susceptible for thiol modifications via S-guanylation. This may result in the background formation of protein S-guanylation in untreated cells via baseline levels of 8-nitro-cGMP (12). Some S-guanylated proteins undergo rapid turnover, for example, proteasomal and autophagic degradation, and metabolisms of S-guanylated proteins may be regulated by formation and degradation (unpublished observation).
In this study, mitochondrial pellets were obtained by centrifugation. During this process, mitochondrial matrix proteins may be released to the extramitochondrial buffer if the mPTP opening was induced by 8-nitro-cGMP in vitro. We analyzed the supernatants obtained during mitochondrial preparation treated with 8-nitro-cGMP in vitro. We observed that several proteins were detected as S-guanylated proteins in the supernatants, including HSP60 and mortalin. However, we could not differentiate whether these proteins were S-guanylated in mitochondria before release to extramitochondrially or were S-guanylated in the supernatant after centrifugal separation. Further, careful examination is needed to clarify the process of S-guanylation for extramitochondrially released proteins.
Although this study focused on the identification of protein S-guanylation in mitochondria, proteins in other compartments such as the cytosol, plasma membrane, endoplasmic reticulum (ER), and nucleus may be modified by 8-nitro-cGMP. As mentioned above, Keap1 was identified as a susceptible target for S-guanylation in response to oxidative stress conditions (12, 36). We recently found that H-Ras, a membrane-bound small G-protein, is another highly susceptible target for S-guanylation in cells and in tissue (30). Importantly, S-guanylation of H-Ras induced H-Ras activation, leading to the activation of the downstream signaling cascade. In relation to mPTP opening, regulation of calcium homeostasis plays an important role. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) is a protein that is involved in the regulation of Ca2+ release from ER. The present study showed that SERCA may be a potential target for S-guanylation (Supplementary Table S1). Interestingly, some protein targets for S-guanylation, including Keap1, HSP60, H-Ras, actin, and tubulin, have also been reported to be the targets for protein S-glutathionylation, a reversible attachment of glutathione moiety to protein thiols via disulfide formation (8). These similarities may be suggestive of that certain redox-reactive thiols are prone for both S-guanylation and S-glutathionylation, and hence protein thiol modifications via S-guanylation and S-glutathionylation may play a role in determination of subsequent redox signal transduction. Further study is warranted to identify protein targets for S-guanylation in different cellular compartments and to elucidate its impact on redox-based signal transduction.
mPTP opening plays a role in ROS release from mitochondria. Although many reports suggest the pathological impact of mPTP-mediated ROS release, physiological and cytoprotective functions of mPTP-dependent ROS release have also been documented, as exemplified by the role of ROS in ischemic preconditioning (24). Cardiac ischemic preconditioning is the phenomenon in which short repeated bouts of ischemia and reperfusion protect the heart from damage associated with prolonged ischemia and reperfusion (32). During ischemic preconditioning, ROS, likely produced by the mitochondria, seem to be required (32). It is therefore of interest to investigate whether S-guanylation-mediated mPTP opening has a role in ischemic preconditioning and its related cellular events such as hypoxic response.
In summary, we developed an effective proteomic method called S-guanylation proteomics that enabled identification of susceptible targets for protein S-guanylation in cells. We identified several mitochondrial proteins, including HSP60, mortalin, vimentin, heterogeneous nuclear ribonucleoprotein K, and prelamin-A/C, as putative targets for endogenous S-guanylation. Our data also revealed that 8-nitro-cGMP affects mPTP opening, most probably via a redox-based protein modification. These data thus suggest that mitochondrial HSPs may be novel targets for redox modification via protein S-guanylation that functions in mPTP regulation and mitochondrial redox signaling.
Materials and Methods
Materials
8-Nitro-cGMP, pegylated SOD, and pegylated catalase were prepared according to the method reported previously (2, 12, 36). 8-Bromo-cGMP and L-NMMA were obtained from Sigma-Aldrich Corporation. Calcein-AM was purchased from Invitrogen Corporation, Molecular Probes. Cs was obtained from Santa Cruz Biotechnology. All other chemicals were of analytical grade and were used without further purification.
Cell culture
Rat C6 glioma cells obtained from the Japan Health Sciences Foundation were cultured at 37°C in the Dulbecco's modified Eagle's medium (Wako Pure Chemical Industries) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Cells were plated at a density of 2×106 cells per 100-mm dish for mitochondrial isolation, and at a density of 1×105 cells per chamber in BD Falcon Culture Slides (BD Biosciences) for mPTP-opening assays as described below.
Mitochondrial isolation
C6 cells were plated at a density of 2×106 cells per 100-mm dish. After overnight incubation, cells were washed twice with ice-cold phosphate-buffered saline (PBS). The mitochondria were isolated by homogenization and differential centrifugation according to the literature (23). Briefly, to the dishes were added 1 ml of the ice-cold isolation buffer consisting of 10 mM HEPES (pH 7.4), 250 mM sucrose, 1 mM ethylene glycol tetraacetic acid, and protease inhibitor cocktail (cOmplete Mini; Roche). Cells were collected with a cell scraper (BD Biosciences) into 15-ml polypropylene tubes, followed by sonicating with a probe-type sonicator. The resulting cell suspensions were centrifuged at 700 g at 4°C for 10 min. The supernatant was collected and centrifuged at 10,000 g at 4°C for 10 min to obtain the mitochondrial pellet. The mitochondrial pellet thus obtained was resuspended in 1 ml of the isolation buffer. After centrifugation at 10,000 g at 4°C for 10 min, the resultant pellet was resuspended again in 0.1 ml of the isolation buffer.
S-Guanylation of mitochondrial proteins in vitro
Mitochondrial proteins were reacted with 8-nitro-cGMP under different conditions to induce protein S-guanylation. First, proteins were S-guanylated under solubilized conditions in a detergent-containing buffer. The isolated mitochondria were lysed in an RIPA buffer (10 mM Tris–HCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 150 mM NaCl, pH 7.4) to achieve a protein concentration of 1 mg/ml, followed by 200 μM 8-nitro-cGMP treatment at 37°C for 3 h. Second, proteins in intact mitochondria were treated with 8-nitro-cGMP. The isolated mitochondria were suspended in an isolation buffer to obtain a protein concentration of 1 mg/ml, followed by 200 μM 8-nitro-cGMP treatment at 37°C for 3 h. After the reaction, the mitochondrial suspension was centrifuged at 10,000 g at 4°C for 10 min to obtain the mitochondrial pellet. The pellet was subjected to further analyses as described below.
S-Guanylation of mitochondrial proteins in C6 cells by endogenously formed 8-nitro-cGMP
Cells were untreated or stimulated with a mixture of 10 μg/ml LPS (from Escherichia coli; L8274; Sigma-Aldrich Corporation) and 200 U/ml interferon-γ, 500 U/ml tumor necrosis factor α, and 10 ng/ml interleukin-1β for 36 h (all cytokines from R&D Systems) (2, 12, 18). After stimulation, mitochondria were isolated from cells as mentioned above and were subjected to S-guanylation proteomics by using 2D-PAGE as described below.
Immunoaffinity capture of S-guanylated peptides
Mitochondrial proteins were subjected to trypsin digestion according to the method described by Wisniewski et al. (45) with slight modifications. In brief, to a Microcon Centrifugal Filter (Ultracel YM-30, MWCO 30,000; Millipore) was added a mitochondrial protein sample (∼0.2 mg of proteins per filter unit, total 1 mg of protein), followed by centrifugation at 12,000 g for 15 min at room temperature. The filter unit was washed with 200 μl of 0.1 M ammonium bicarbonate (Ambic), followed by incubation with 0.1 M Ambic containing 6 M urea and 5 mM Tris(2-carboxyethyl)phosphine at 37°C for 30 min. After incubation, 20 mM iodoacetamide was added to the filter unit and incubation continued for 15 min at room temperature in the dark. Excess Tris(2-carboxyethyl)phosphine and iodoacetamide were removed by centrifugation at 12,000 g for 15 min at room temperature. The filter unit was then washed twice with 200 μl of 0.05 M Ambic. Mitochondrial proteins remaining on the filter were digested with trypsin (Sequencing Grade Modified Trypsin; Promega Corporation; enzyme-to-protein ratio, 1:100) in 0.05 M Ambic at 37°C overnight. Peptides thus formed were collected by centrifugation at 12,000 g for 15 min at room temperature.
Peptide mixtures containing S-guanylated peptides were extracted by using anti-S-guanylated protein affinity gel as previously reported (12).
2D-gel electrophoresis
Mitochondrial protein samples (0.1 mg of protein) were treated with the 2-D Clean-Up Kit (GE Healthcare UK Ltd.), according to the manufacturer's instruction. After clean-up, protein samples were dissolved in a rehydration buffer consisting of 2 M urea, 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 0.5% dithiothreitol (DTT), 0.002% bromophenol blue, and 0.5% immobilized pH gradient (IPG) Buffer (pH 3–10 NL; GE Healthcare). Protein S-guanylation is stable in the presence of DTT and other reducing agents such as 2-mercaptoethanol. IPG strips (7 cm, pH 3–10 NL; GE Healthcare) were rehydrated with protein samples for 10 h at 20°C by using the Ettan IPGphor 3 apparatus (GE Healthcare). Isoelectric focusing was performed as follows: 300 V for 4 h, 1000 V (gradient) for 30 min, 5000 V (gradient) for 1.5 h, and then at 5000 V for another 3 h. The IPG strips were then incubated in an equilibration buffer consisting of 50 mM Tris–HCl (pH 8.8), 6 M urea, 30% glycerol, 1% SDS, and 0.25% DTT for 15 min, followed by incubation again in an equilibration buffer containing 4.5% iodoacetamide for 15 min. IPG strips were then placed on SDS-PAGE gel (10% acrylamide). Proteins separated were stained with silver (Silver Stain MS Kit; Wako Pure Chemical Industries) for visualization of protein spots or underwent to Western blotting to show S-guanylated proteins. A protein size marker (Precision Plus Protein standards; Bio-Rad Laboratories) was S-guanylated by incubation with 1 mM 8-nitro-cGMP at 37°C for 1 h for Western blotting visualization.
Western blotting
Proteins separated via SDS-PAGE were subjected to Western blotting with self-made rabbit polyclonal anti-S-guanylated protein antibodies (12, 36) used with 2000 dilution as reported previously (12, 36). The immunoreactive spots were detected by using a chemiluminescence reagent (ECL Plus Western Blotting Reagent; GE Healthcare) with a luminescent image analyzer (LAS-1000 UV mini; Fujifilm).
Spot excision
The silver-stained gel image was covered with the S-guanylation Western blotting image to identify protein spots containing S-guanylated proteins. Overlap images of silver staining and S-guanylation Western blotting were prepared by using Microsoft Power Point. The silver-stained images were to be 50% transparent, and lay over on the Western blotting images to identify protein spots containing S-guanylated proteins. S-Guanylated protein marker, which is positive for both silver staining and S-guanylation Western blotting, was used to adjust the position of the images. These spots were subjected to in-gel digestion according to the literature (39). These peptide samples were subjected to LC-MS/MS.
LC-MS/MS
Peptide samples obtained via immunoaffinity capture and 2D-PAGE were analyzed in an electrospray ionization quadrupole time-of-flight (ESI-Q-TOF) tandem mass spectrometer (6510; Agilent Technologies, Inc.) A microfluidic reversed-phase HPLC chip (5-μm particle size, 75-mm i.d., 43-mm length; Zorbax 300SB-C18; Agilent) was used for peptide separation, with nanoflow at 600 nl/min of the mobile phase of 0.1% formic acid in MS-grade water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). The gradient was 5%–75% B in 9 min. A capillary pump was used for loading samples with solvent A at 4 μl/min. The Agilent ESI-Q-TOF instrument was operated at 300°C in the positive ionization mode (ESI+) with an ionization voltage of 1850 V and a fragmentor voltage of 175 V. Protonated molecular ions were fragmented in the auto-MS/MS mode starting with a collision energy voltage of 3 V that was increased by 3.7 V per 100 Da. The m/z ranges were 300–2400 Da in the MS mode and 59–3000 Da in the MS/MS mode.
Mass lists in the form of Mascot generic files were created and used as input for Mascot MS/MS ion searches of the National Center for Biotechnology Information-nonredundant (NCBI nr) database via the Matrix Science Web server Mascot version 2.2. Default search parameters were the following: enzyme, trypsin; maximum missed cleavage, 1; variable modifications, carbamidomethyl (Cys) and cGMP (Cys); peptide tolerance,±1.2 Da; MS/MS tolerance,±1.2 Da; peptide charge, 2+ and 3+; instrument, ESI-Q-TOF. For positive identification, the score of the result of [−10×log(P)] had to exceed the significance threshold (p<0.05).
Immunoprecipitaton of HSP60
The isolated mitochondria were lysed in a modified RIPA buffer (10 mM Tris–HCl, 0.5% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, pH 7.4). To the mitochondrial solution (1 mg protein) was added 50 μl of Protein A/G plus Agarose (Santa Cruz Biotechnology, Inc.) and incubated for 2 h at 4°C to remove any protein that was nonspecifically bound to the agarose beads. Supernatant was then collected. Monoclonal anti-HSP60 antibody LK1 (Enzo Life Sciences, Inc.) was added (1:100) to the supernatant and incubated for overnight at 4°C. Protein A/G plus agarose (50 μl) was then added and further incubated for 1 h at 4°C. Agarose beads were washed twice with PBS, and incubated with an SDS buffer (100 μl) at 95°C for 5 min to liberate bound HSP60. The supernatant thus obtained was subjected for Western blotting with anti-S-guanylated protein antibodies, or rabbit polyclonal anti-HSP60 antibody (3000 dilution, HSP 60, H-300; Santa Cruz Biotechnology, Inc.).
Transfection of HSP60 siRNA
A pair of 21-nucleotide HSP60 siRNA (manufactured by Sigma, Oligonucleotide # 11102001 and 11102002) named Rn_Hspd1_1430_s with the sequence 5′rGrCUrCUUrArGrCrArCrArCUrGrGUUUTT and Rn_Hspd1_1430_as with the sequence 5′rArArArCrCrArGUrGUrGrCUrArArGrArGrCTT was used for transfection using Lipofectamine™ RNAiMAX transfection reagent (Invitrogen). Briefly, C6 cells were seeded in 10-cm dishes at a density of 1×106 cells/dish. Cells were transfected with HSP60 siRNA (600 pmol/10 cm plate) using Lipofectamine™ RNAiMAX transfection reagent. At 72 h after transfection, cells were harvested, but just before the harvest, they were treated with LPS/cytokines for 36 h. Stealth RNAi (RNA interference) negative control (high GC; Invitrogen) was used as a negative control siRNA.
Calcein/Co2+ staining assay for mPTP opening
mPTP opening in C6 cells was examined via calcein/Co2+ staining as described by Petronilli et al. (33). Antagonists for CypD such as Cs and sanglifehrin A were reported to inhibit mPTP opening via inhibition of peptidyl-prolyl cis–trans isomerase activity of CypD (5). Cs was used to confirm whether calcein fluorescent changes depended on mPTP opening. Cells were treated with 50 μM Cs before 8-nitro-cGMP treatment or LPS/cytokine stimulation. In some experiments, C6 cells were treated with pegylated SOD (200 U/ml) plus pegylated catalase (200 U/ml) or 10 mM L-NMMA 1 h before LPS/cytokine stimulation. At 20 min before the end of the above-mentioned treatment or stimulation, cells were loaded with calcein to a final concentration of 1 μM in the presence of 1 mM Co2+ and 0.1% Pluronic F-127, in Hank's buffer. After 20 min of incubation, calcein was washed out with ice-cold Hank's buffer (5 min), and cells were examined with a Nikon EZ-C1 confocal laser microscope. Excitation was at 488 nm (the green photomultiplier channel of the confocal microscope was used for image acquisition).
Statistical analysis
All data are given as means±SEM. Data for each experimental condition were acquired from at least three separate experiments. Student's t-test was used for statistical analyses.
Footnotes
Acknowledgments
We thank Judith B. Gandy for her editing of the manuscript. Thanks to K. Ono and J. Yoshitake (Kumamoto University) for their technical assistance. This work was supported in part by Grants-in-Aid for Scientific Research ([B], [C]); Grants-in-Aid for Scientific Research on Innovative Areas (Research in a Proposed Area) from the Ministry of Education, Sciences, Sports, Technology (MEXT), Japan; and a grant from the JST PRESTO program.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.