
Abstract
Introduction
Innovation
Until now, the role of PRDX4 in the development of atherosclerosis has not been investigated. We show for the first time that PRDX4 ameliorates atherosclerosis progression in apoE−/− mice fed a high-cholesterol diet by protecting the aorta from oxidative stress and reducing apoptosis of macrophages and SMCs. We also show that PRDX4 might induce a stable plaque phenotype by reducing endothelial dysfunction, suppressing inflammatory cell infiltration, and enhancing SMC migration together with a collagen-rich matrix. In conclusion, our findings provide novel insight into the protective role of PRDX4, a member of the antioxidant PRDX family, in the development of atherosclerosis.
Increased oxidative stress is a risk factor for the initiation and progression of atherosclerosis, and plays a crucial role in atherosclerosis that is manifested by the accumulation of lipid peroxidation products, such as oxidized low-density lipoprotein (oxLDL) and thiobarbituric acid reactive substances (TBARS), and the formation of ROS (5, 21, 29). Several studies have revealed that antioxidative markers, including other PRDXs, function as antiatherogenic factors in vivo. The size of atherosclerotic lesions in the aortic sinus decreases significantly after 20 weeks of high-fat feeding in mice lacking glutathione peroxidase-1 (GPX1) (GPX1−/−) as compared with control wild-type (WT) mice (2). Cu,Zn-superoxide dismutase 1 (SOD1) transgenic mice develop smaller atherosclerotic lesions than do WT mice (25). Overexpression of catalase reduces the severity of atherosclerosis in apolipoprotein E (apoE)-deficient mice (apoE−/−) (33). Notably, ApoE and PRDX1 double-knockout mice (PRDX1−/−/apoE−/−) develop larger, more macrophage-rich aortic sinus lesions than do control (PRDX1+/+/apoE−/−) mice in the early stages of atherosclerosis (10). PRDX2 knockout mice (PRDX2−/−) have a greater influence on the growth and migration of smooth muscle cells (SMCs) with increased neointimal thickening as compared with WT mice during vascular remodeling in wire-injured carotid arteries (1). PRDX6 knockout mice (PRDX6−/−) raised on C57BL/6 and 129/SvJ mixed backgrounds and fed a high-fat diet had more pronounced atherosclerotic lesions than did control mice (28). Taken together, these in vivo data demonstrate that PRDXs could play a pivotal role in antioxidative signaling and in the signaling cascades that protect against atherosclerosis-prone pathophysiological processes.
Recently, we generated hPRDX4 transgenic (Tg) mice and evaluated the role of PRDX4 in a type 1 diabetes mellitus model. We found that insulin-secreting β-cells in the islets of Tg mice were significantly protected against inflammation and apoptosis, unlike those of control WT mice (3). In the present study, we crossed Tg mice with apoE−/− mice and generated hPRDX4+/+/apoE−/− mice to investigate the effects and roles of this unique antioxidant enzyme in atherosclerosis. We used two experimental models: hPRDX4+/+/apoE−/− mice with high-cholesterol diet (HcD)-induced atherosclerosis and apoE−/− mice with HcD-induced atherosclerosis after bone marrow transplantation (BMT) from Tg mice.
Results
Expression of hPRDX4 in mice under basal and hypercholesterolemic conditions
The expression of apoE and hPRDX4 in animals fed the normal-cholesterol diet (NcD) was analyzed as shown in Supplementary Figure S1B, supplementary data are available online at
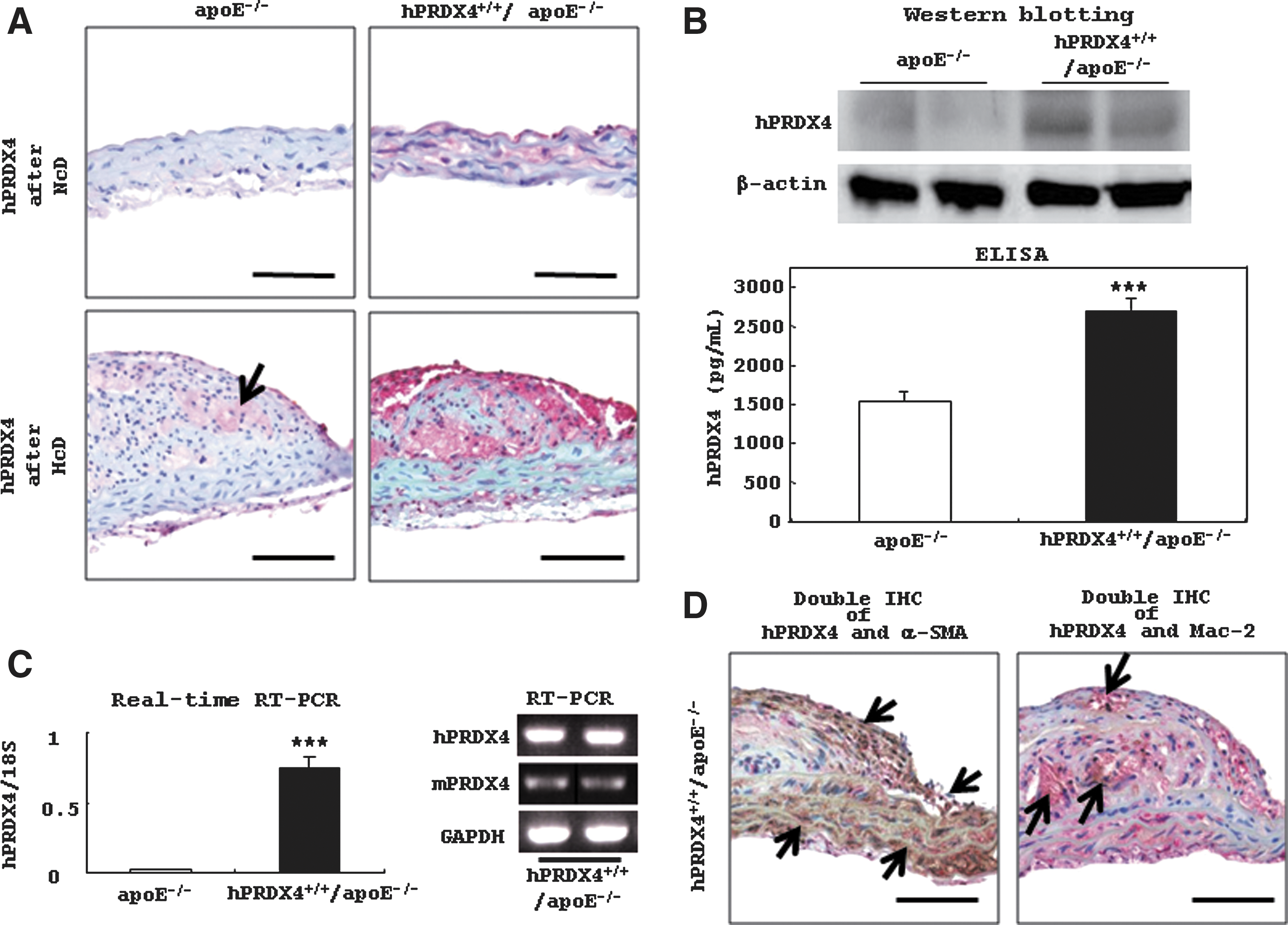
RT-PCR (Supplementary Fig. S2A) also revealed that hPRDX4+/+/apoE−/− mice expressed hPRDX4 mRNA in all tissues under basal conditions, with greater expression in the heart, kidney, brain, and testis. hPRDX4 mRNA was not detected in apoE−/− mice. Endogenous mPRDX4 was expressed in all tissues, except for the pancreas, in both groups of mice under basal conditions. Additionally, IHC (Supplementary Fig. S2B) showed that hPRDX4 was highly expressed in cardiac muscle, bronchioloalveolar cells, and glial cells in hPRDX4+/+/apoE−/− mice. A small number of hepatocytes, splenic white pulp lymphocytes, renal glomerular cells, pancreatic acinar cells, intestinal columnar epithelial cells, testicular spermatogenic cells, and thymic lymphocytes of hPRDX4+/+/apoE−/− mice also expressed hPRDX4.
In mice fed the HcD, hPRDX4 was highly expressed throughout the atherosclerotic aortas of hPRDX4+/+/apoE−/− mice, including macrophages (foam cells), SMCs, and endothelial cells (Fig. 1A–1C). To determine the level of hPRDX4 protein present in the circulatory system, we performed enzyme-linked immunosorbent assays (ELISA) using serum samples, which showed that only hPRDX4+/+/apoE−/− mice had markedly elevated PRDX4 levels (apoE−/− 1529.5±126.2 pg/mL vs. hPRDX4+/+/apoE−/− 2697.7±147.9 pg/mL; p<0.0001) (Fig. 1B), consistent with Western blotting (Fig. 1B) and RT-PCR (Fig. 1C) results. Macrophages in the atherosclerotic aortas of HcD-fed apoE−/− mice were also faintly positive for anti-hPRDX4 antibody, indicating cross-reactivity between hPRDX4 and mPRDX4 (Fig. 1A). Additionally, RT-PCR showed hPRDX4 expression in atheromatous aortas was significantly greater than that of mPRDX4 in hPRDX4+/+/apoE−/− mice (Fig. 1C). Double IHC staining showed relatively weak hPRDX4 expression in macrophages in the atherosclerotic lesions of apoE−/− mice but not in SMCs (data not shown), whereas it was highly expressed in both SMCs and macrophages of hPRDX4+/+/apoE−/− mice (Fig. 1D).
Comparison of metabolic parameters in hypercholesterolemia model mice
All mice developed mild obesity typical of hypercholesterolemia-induced atherosclerosis. However, body weight was not significantly different between the hPRDX4+/+/apoE−/− and apoE−/− mice (Supplementary Fig. S3A). Arterial blood pressure was significantly elevated in apoE−/− mice after 12 weeks of HcD feeding, as compared with that before starting HcD feeding and hPRDX4+/+/apoE−/− mice fed the HcD for 12 weeks. However, there was no significant difference in the blood pressure of hPRDX4+/+/apoE−/− mice between before and after 12 weeks of HcD feeding (Supplementary Fig. S3B). In terms of glucose metabolism, there were no significant differences in glucose levels between the two groups of mice measured before and at 15, 30, 60, and 120 minutes after intraperitoneal glucose injection (Supplementary Fig. S3C). We also determined the serum cholesterol profiles by high-performance liquid chromatography in mice fed the HcD for 12 weeks (22, 25). Both apoE−/− and hPRDX4+/+/apoE−/− mice developed hyperlipidemia with increased levels of total cholesterol, very low-density lipoprotein, and LDL, and decreased levels of high-density lipoprotein and triglycerides (TG). There were no significant differences in these levels, except for TG, between the two groups (Supplementary Fig. S3D).
En face and histological evaluation of the aortas of hypercholesterolemia model mice
En face quantitative analysis of oil red-O stained aortas (Fig. 2A) revealed significantly fewer atherosclerotic areas in hPRDX4+/+/apoE−/− mice after 12 weeks of HcD feeding compared with those of apoE−/− mice (apoE−/− 24.7%±1.5% vs. hPRDX4+/+/apoE−/− 14.3%±1.2%; p<0.0001).
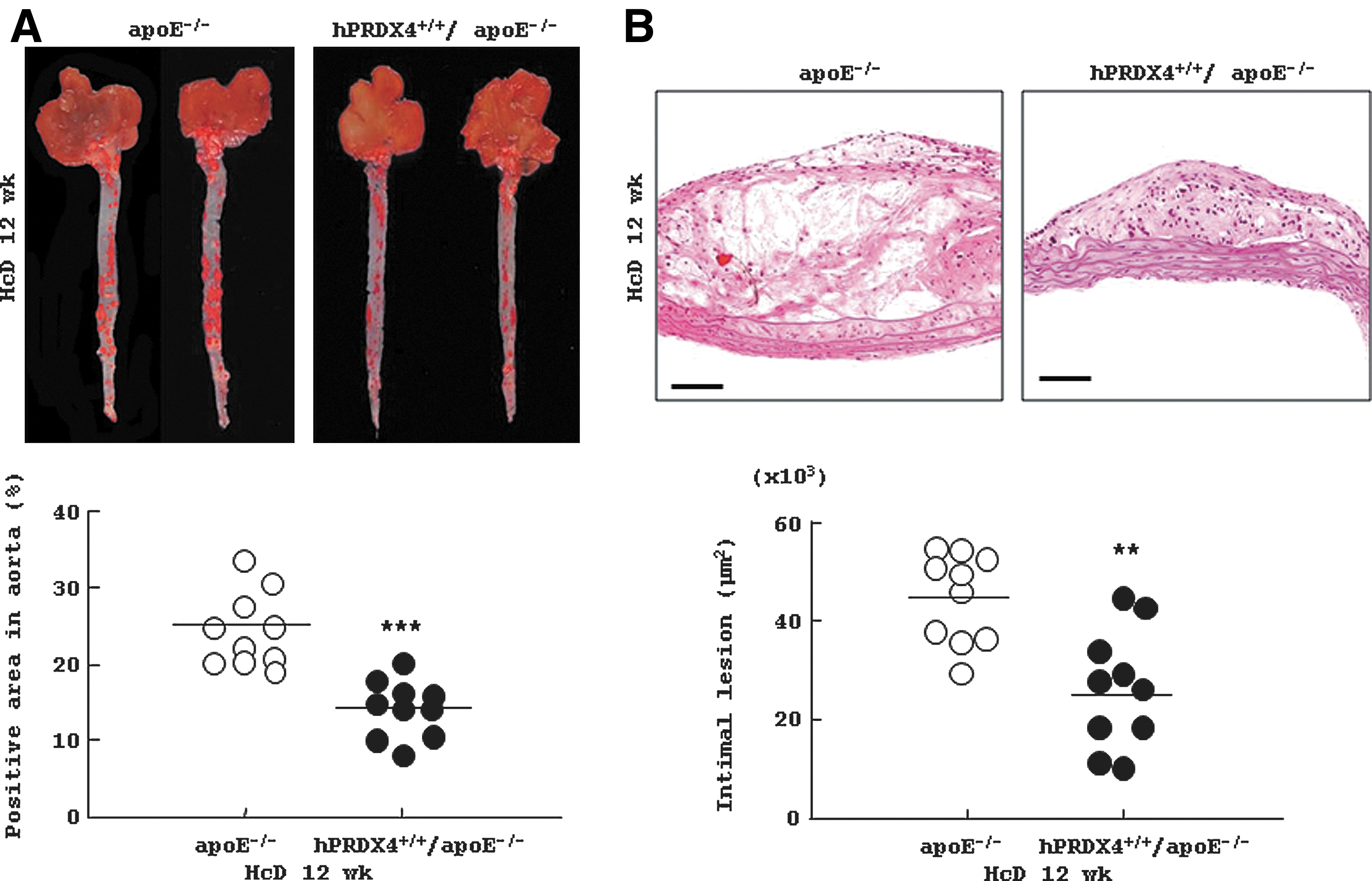
As shown in Figure 2B, quantitative analysis of sequential sections of the atherosclerotic aortas showed that the intimal lesion area was significantly smaller in hPRDX4+/+/apoE−/− mice than in apoE−/− mice after 12 weeks of HcD feeding (apoE−/− 44.5±2.9×103 μm2 vs. hPRDX4+/+/apoE−/− 22.6±4.4×103 μm2; p<0.001).
We also microscopically evaluated the thickened intimal areas, focusing on the three valves at the aortic root level. Consistent with the above data, quantitative analysis of the atherosclerotic aortic valves showed that hPRDX4+/+/apoE−/− mice had significantly smaller intimal lesions than did apoE−/− mice (apoE−/− 762.4±82.2×103 μm2 vs. hPRDX4+/+/apoE−/− 523.0±42.0×103 μm2; p<0.05) (Supplementary Fig. S4).
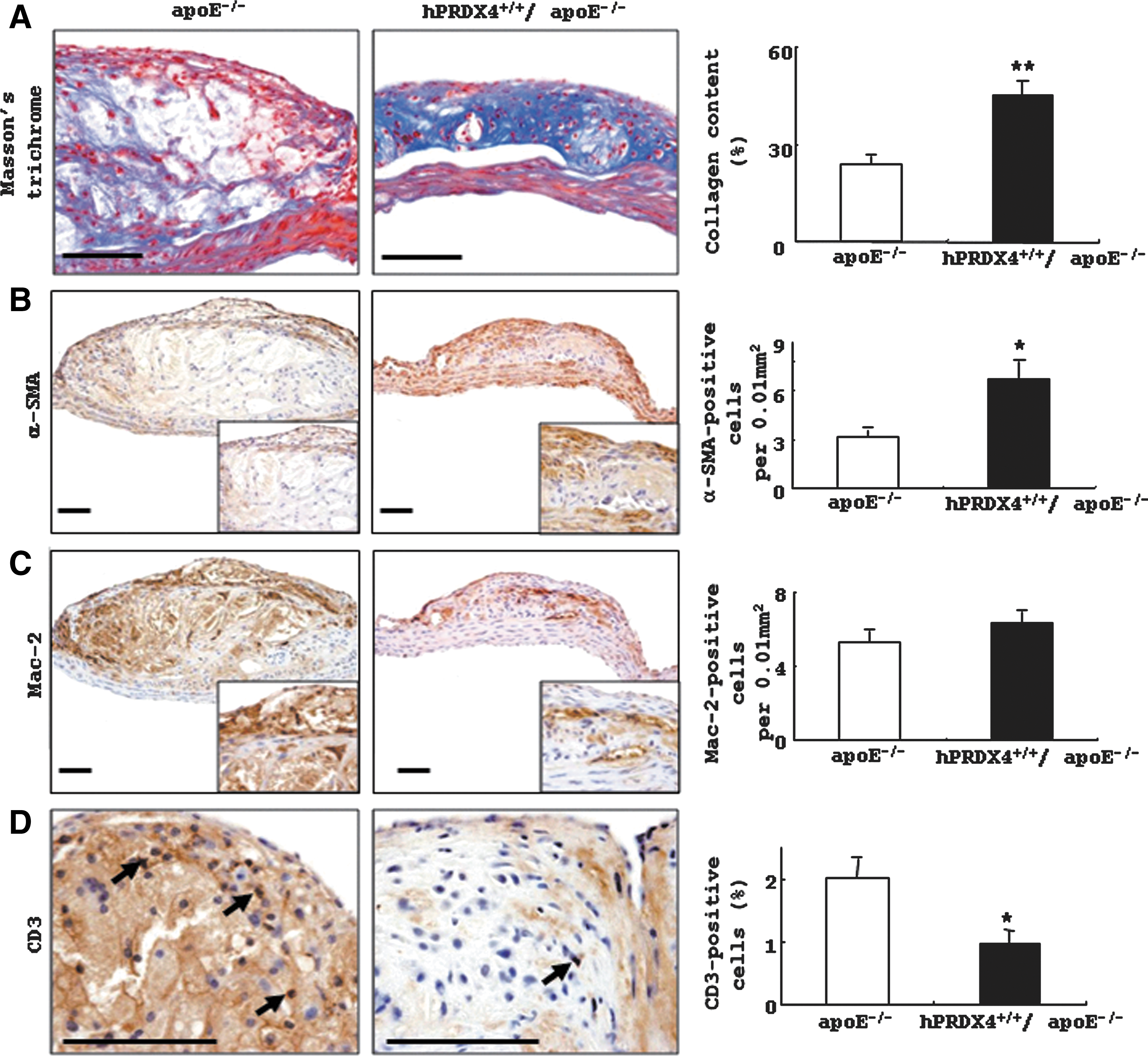
Staining of collagen, smooth muscle cells, macrophages, and T cells in atheromatous lesions in hypercholesterolemia model mice
Masson's trichrome staining (Fig. 3A) showed larger and thicker collagen deposits in aortic atherosclerotic lesions in hPRDX4+/+/apoE−/− mice compared with those in apoE−/− mice. IHC staining of whole intimal lesions revealed significantly more α-smooth muscle actin (SMA)-positive SMCs in hPRDX4+/+/apoE−/− mice (6.7±1.1 per 0.01 mm2) than in apoE−/− mice (3.2±0.4 per 0.01 mm2, p<0.05) (Fig. 3B), whereas the number of Mac-2-positive macrophages (foam cells) was not significantly different between these mice (apoE−/− 5.3±0.6 per 0.01 mm2 vs. hPRDX4+/+/apoE−/− 6.3±0.7 per 0.01 mm2) (Fig. 3C). IHC staining also demonstrated that the number and percentage of CD3-positive cells were significantly lower in the atheromatous plaques of hPRDX4+/+/apoE−/− mice than in those of apoE−/− mice (apoE−/− 2.0±0.3% vs. hPRDX4+/+/apoE−/− 1.0±0.2%, p<0.05) (Fig. 3D).
8-hydroxy-2′-deoxyguanosine and oxLDL levels in the aortas of hypercholesterolemia model mice
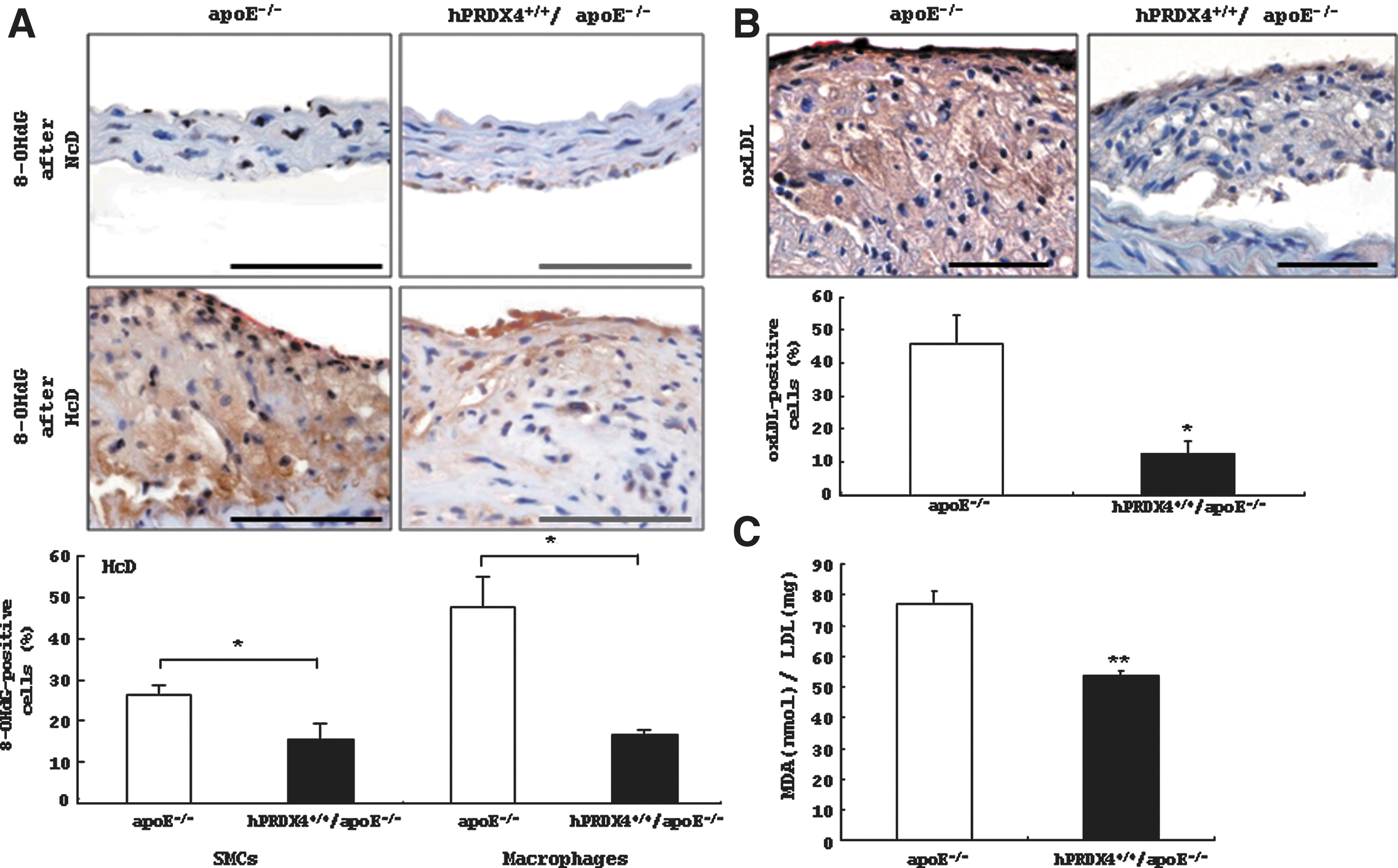
We determined the expression of 8-hydroxy-2′-deoxyguanosine (8-OHdG) and oxLDL, markers of oxidative stress, in the atheromatous plaques of HcD-fed mice; 8-OHdG (Fig. 4A) and oxLDL (data not shown) were not detected in the aortas of NcD-fed mice. The atherosclerotic lesions of hPRDX4+/+/apoE−/− mice contained significantly less 8-OHdG than did the apoE−/− mice after 12 weeks of HcD feeding, particularly in the intimal SMCs and macrophages that could be identified morphologically (31) (Fig. 4A). The percentages of 8-OHdG-positive SMCs (apoE−/− 26.5±1.9% vs. hPRDX4+/+/apoE−/− 15.7±3.5%, p<0.05) and macrophages (apoE−/− 47.4±7.4% vs. hPRDX4+/+/apoE−/− 16.8±1.1%, p<0.05) were significantly lower in the atheromatous plaques of hPRDX4+/+/apoE−/− mice than in apoE−/− mice (Fig. 4A). Moreover, the atherosclerotic lesions in HcD-fed hPRDX4+/+/apoE−/− mice showed significantly lower expression of oxLDL, as compared with lesions in apoE−/− mice (apoE−/− 45.8±8.4% vs. hPRDX4+/+/apoE−/− 12.4±3.9%, p<0.05; Fig. 4B); most of these oxLDL-positive cells could be identified as macrophages.
Serum thiobarbituric acid reactive substances levels in hypercholesterolemia model mice
We next measured the serum levels of another oxidative stress marker, thiobarbituric acid reactive substances (TBARS), which is closely associated with the development of atherosclerosis. Serum TBARS levels [expressed as nmol malondialdehyde (MDA)/mg LDL protein] were significantly lower in hPRDX4+/+/apoE−/− mice than in apoE−/− mice after 12 weeks of HcD feeding (apoE−/− 77.3±3.8 nmol MDA/mg LDL protein vs. hPRDX4+/+/apoE−/− 53.5±1.6 nmol MDA/mg LDL protein, p<0.0001) (Fig. 4C).
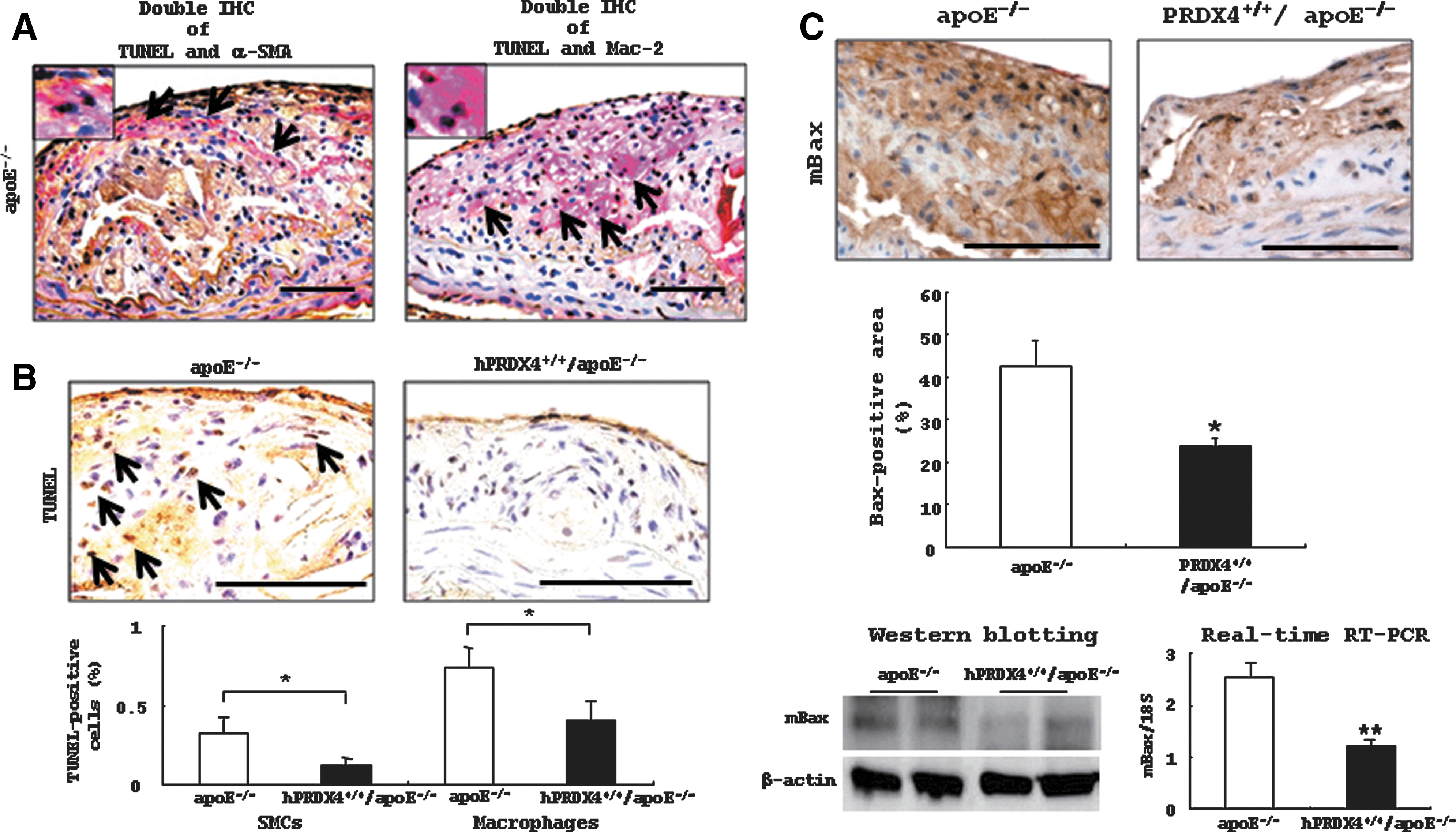
Analysis of apoptotic and proliferating activities in atheromatous plaques in hypercholesterolemia model mice
Double IHC staining confirmed that some macrophages and SMCs of apoE−/− mice were positive for terminal deoxynucleotidyl transferase end-labeling (TUNEL) (Fig. 5A). Although a small number of apoptotic cells were observed in the intimal lesions of both groups of mice, the percentages of TUNEL-positive SMCs (apoE−/− 0.3%±0.1% vs. hPRDX4+/+/apoE−/− 0.1%±0.04%; p<0.05) and macrophages (apoE−/− 0.7%±0.1% vs. hPRDX4+/+/apoE−/− 0.4%±0.1%; p<0.05) were significantly lower in hPRDX4+/+/apoE−/− mice than in apoE−/− mice (Fig. 5B). Western blotting and real-time RT-PCR showed that aortic Bax expression was lower in hPRDX4+/+/apoE−/− mice than in apoE−/− mice (Fig. 5C). On IHC analysis of pro-apoptotic Bax in the aortas from both groups of mice, the atherosclerotic lesions in hPRDX4+/+/apoE−/− mice showed significantly lower expression of Bax compared with lesions in apoE−/− mice (apoE−/− 42.8±5.8% vs. hPRDX4+/+/apoE−/− 24.0±2.1%, p<0.05; Fig. 5C); most of these Bax-positive cells could be identified as SMCs and macrophages. IHC staining also showed that the percentage of cleaved caspase-3-positive area was significantly lower in the atheromatous plaques of hPRDX4+/+/apoE−/− mice than in those of apoE−/− mice (apoE−/− 30.9±1.8% vs. hPRDX4+/+/apoE−/− 19.9±3.0%, p<0.05; Supplementary Fig. S5A). This was confirmed by Western blotting of activated caspase-3, which showed the expression of the large fragment of caspase-3 (17 kDa) (Supplementary Fig. S5B). In terms of proliferation activity, the percentage of MIB-1-positive cells in the atherosclerotic intimal lesions was not significantly different between the two groups of mice (apoE−/− 1.6±0.3% vs. hPRDX4+/+/apoE−/− 1.1±0.3%; data not shown).
Free cholesterol loading-induced apoptosis in peritoneal macrophages
The nuclear morphology of macrophages was observed following double-staining with propidium iodide (red) and Hoechst 33258 (blue); TUNEL-positive apoptotic cells (green) were also analyzed in the same fields (Supplementary Fig. S6). Only a few apoptotic cells were detected in untreated macrophages (apoE−/− 1.7±0.1% vs. hPRDX4+/+/apoE−/− 1.5±0.2%). Stimulation with acetyl-LDL and 58035 (free cholesterol [FC] loading) significantly increased the numbers of apoptotic macrophages. However, the number of apoptotic cells was significantly lower in hPRDX4+/+/apoE−/− mice than in apoE−/− mice after FC loading (apoE−/− 12.5±1.5% vs. hPRDX4+/+/apoE−/− 8.8±0.9%; p<0.05). The apoptotic macrophages occasionally showed condensed chromatin and fragmented nuclei (Supplementary Fig. S6, inset).
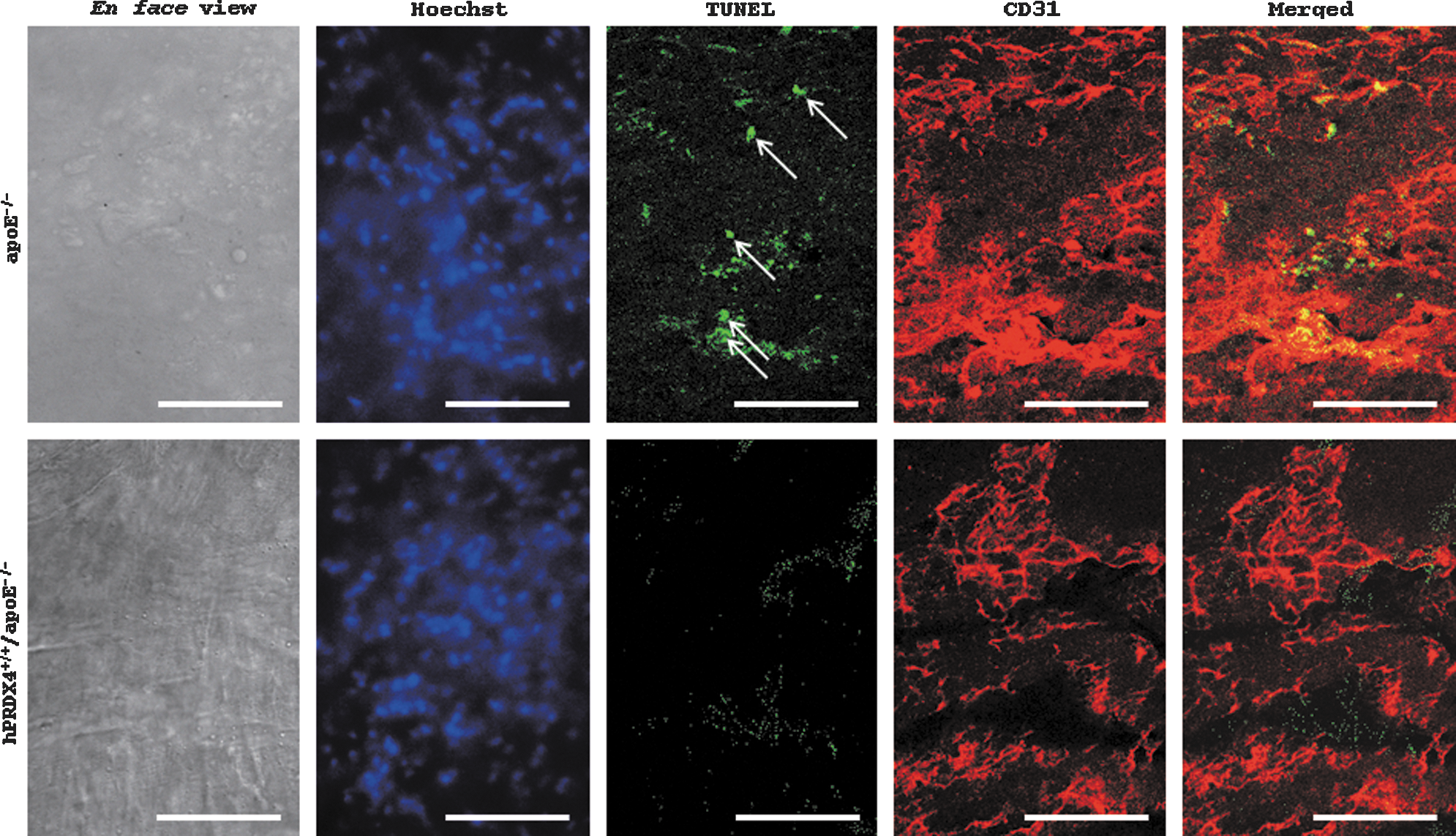
En face aortic double-immunofluorescence of hypercholesterolemia model mice
The percentage of TUNEL- and CD31-positive endothelial cells was significantly higher in apoE−/− mice than in hPRDX4+/+/apoE−/− mice (apoE−/− 4.3±1.2% vs. hPRDX4+/+/apoE−/− 0.7±0.3%; p<0.05) (Fig. 6).
Pathological parameters of atheromatous plaques in hypercholesterolemia model mice
As shown in Figure 2B and Supplementary Table S1, the fibrous cap thickness of atherosclerotic plaques in hPRDX4+/+/apoE−/− mice was significantly greater than that in apoE−/− mice (apoE−/− 34.2±3.8 μm vs. hPRDX4+/+/apoE−/− 46.7±3.6 μm; p<0.05). The cellularity of intimal SMCs in the atheromatous plaques was significantly increased in hPRDX4+/+/apoE−/− mice (apoE−/− 36.8±4.6% vs. hPRDX4+/+/apoE−/− 51.0±8.4%; p<0.05), whereas that of intimal macrophages was decreased (apoE−/− 61.2±7.0% vs. hPRDX4+/+/apoE−/− 48.0±5.3%; p<0.05), compared with those in apoE−/− mice. By contrast, oil red-O staining confirmed that the necrotic lipid core area was significantly smaller in hPRDX4+/+/apoE−/− mice than in apoE−/− mice (apoE−/− 13.4±1.3% vs. hPRDX4+/+/apoE−/− 8.8±0.7%; p<0.05). All of these features (Figs. 2B and 3A–3C) indicate that the vulnerability index was markedly lower in hPRDX4+/+/apoE−/− mice than in apoE−/− mice (apoE−/− 1.23±0.14 vs. hPRDX4+/+/apoE−/− 0.59±0.06; p<0.001) (Supplementary Table S1).
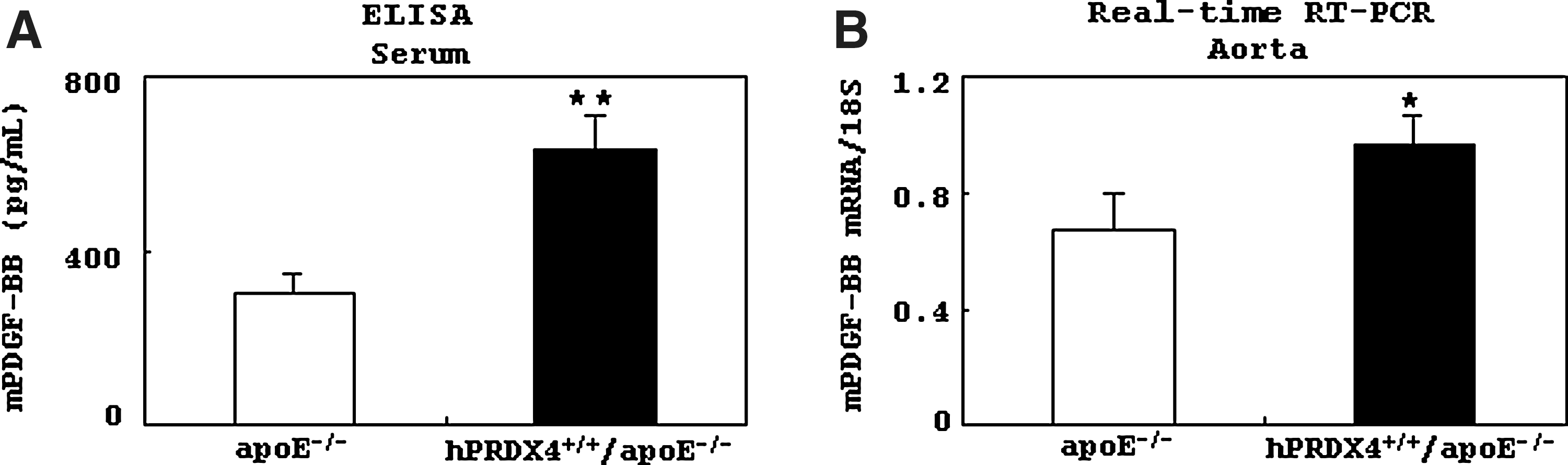
Analysis of murine platelet-derived growth factor BB expression in hypercholesterolemia model mice
Results of ELISA showed that serum murine platelet derived growth factor-BB (mPDGF-BB) levels were significantly higher in hPRDX4+/+/apoE−/− mice than in apoE−/− mice after 12 weeks of HcD feeding (p<0.001) (Fig. 7A). Real-time RT-PCR also showed significantly greater expression of mPDGF-BB in the atherosclerotic aortas of hPRDX4+/+/apoE−/− mice than in apoE−/− mice (p<0.05) (Fig. 7B).
Analysis of gene expression of antioxidative enzymes and inflammatory signaling factors in the aortas of hypercholesterolemia model mice
Real-time RT-PCR showed that the gene expression levels of mPRDXs (except for mPRDX4), GPX1, GPX2, catalase, and nuclear factor-erythroid 2-related factor 2 were not significantly different between the two groups (Supplementary Figs. S7 and S8). GPX3 and glutathione synthetase (GSS) were significantly upregulated, whereas SOD1, SOD2, and glutathione reductase (GSR) were significantly downregulated in the atherosclerotic aortas of hPRDX4+/+/apoE−/− mice compared with those in apoE−/− mice (Supplementary Fig. S8). There were no marked changes in the expression of most of the inflammatory signaling factors examined, although the expression levels of toll-like receptor 2 (TLR2), TLR3, nuclear factor κ-B1a, signal transduction and activator of transcription 1, and inducible nitric oxide synthase were significantly downregulated in the atherosclerotic aortas of hPRDX4+/+/apoE−/− mice compared with those in apoE−/− mice (Supplementary Fig. S9).
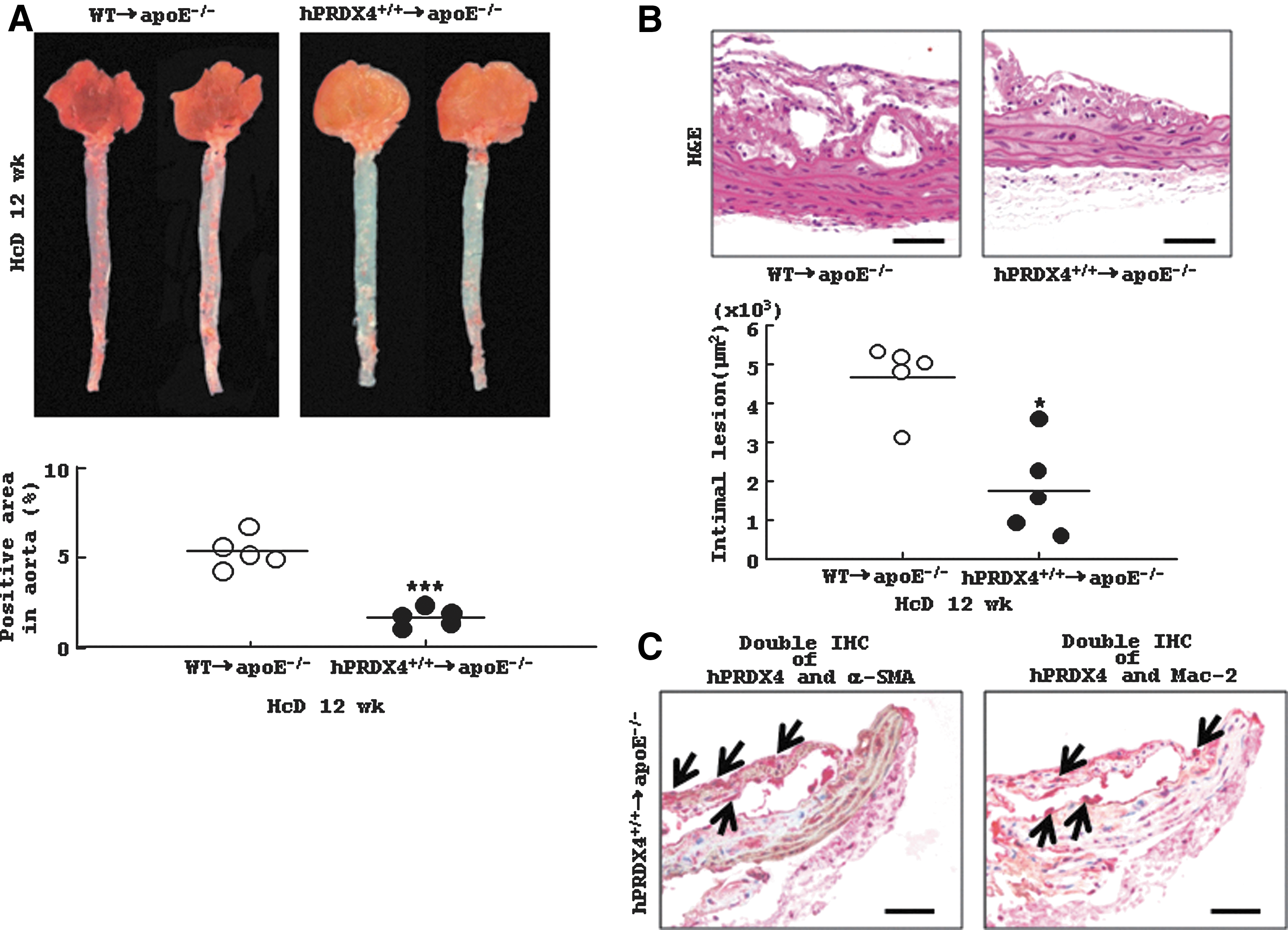
En face, histological, and IHC analysis of the aortas of BMT hypercholesterolemia model mice
Oil red-O staining demonstrated that atherosclerotic area was significantly smaller in apoE−/− mice after BMT from hPRDX4+/+ (Tg) donors as compared with BMT from WT donors (WT donors 5.2±0.4% vs. hPRDX4+/+ donors 1.6±0.2%; p<0.0001) (Fig. 8A). Similarly, BMT from hPRDX4+/+ donors significantly reduced intimal lesion areas as compared with BMT from WT donors (WT donors 4.7±0.4×103 μm2 vs. hPRDX4+/+ donors 1.8±0.5×103 μm2; p<0.05) (Fig. 8B). In IHC analysis, the percentages of α-SMA-positive SMCs (WT donors 6.5±2.0 per 0.01 mm2 vs. hPRDX4+/+ donors 6.9±1.9 per 0.01 mm2) and Mac-2-positive macrophages (WT donors 10.3±2.8 per 0.01 mm2 vs. hPRDX4+/+ donors 10.0±3.6 per 0.01 mm2) in intimal lesions were not significantly different between the two groups of mice (data not shown). The atherosclerotic intimal areas were significantly smaller and less advanced in the BMT model mice than in non-BMT model mice (31). Additionally, double IHC revealed that these intimal SMCs and macrophages were positive for hPRDX4 in BMT mice from hPRDX4+/+ donors, but not from WT donors (Fig. 8C).
Discussion
Consistent with a previous study (3), we detected the expression of hPRDX4 in aortic medial SMCs, although little was apparent on IHC staining (Fig. 1A). Interestingly, after checking the specific expression of hPRDX4 in vitro in aortic vascular SMCs and peritoneal macrophages harvested from hPRDX4+/+/apoE−/− mice (Supplementary Fig. S1 C and D), we found marked expression of hPRDX4 in SMCs and macrophages in vivo (Fig. 1 A and D). We also demonstrated that the expression of the hPRDX4 transgene could overcome that of the endogenous mPRDX4 gene (Fig. 1C) in the atherosclerotic plaques of hPRDX4+/+/apoE−/− mice. In addition, the aortas of HcD-fed apoE−/− mice revealed faint hPRDX4 expression, particularly in macrophages, indicating cross-reactivity of the antibody against hPRDX4 to endogenous mPRDX4 (Fig. 1A), even while the mPRDX4 gene is normally (but weakly) expressed in macrophages and SMCs (Fig. 1C and Supplementary Figs. S1C and S7). Moreover, it is presumable that the circulating serum levels of hPRDX4 protein as an additive effect were particularly elevated in hPRDX4+/+/apoE−/− mice (Fig. 1B), in addition with those of mPRDX4. These findings indicate that the establishment of hyperlipidemia in the current model increases oxidation by generating ROS in atheromatous plaques, as shown in Figure 4. This could lead to increased expression of activated antioxidative proteins of PRDX4, in addition to other markers, such as GPX3 and GSS (Supplementary Figs. S7 and S8), reminiscent of a PRDX4-induced antioxidative cascade. In fact, the CMV enhancer/promoter is notorious for its cross-talk with other promoters and is prone to be stimulated by ROS and inflammation during the progression of atherosclerosis in this HcD model (14), and therefore significantly upregulates transgenic hPRDX4 expression. Since it remains unclear why we also observed an increase in endogenous mPRDX4 expression of hPRDX4+/+/apoE−/− mice on the HcD model, as compared to that of apoE−/− mice (Supplementary Fig. S7), further studies are needed to understand these findings. By contrast, the expression of SOD1, SOD2, and GSR was significantly reduced in the atherosclerotic aortas of hPRDX4+/+/apoE−/− mice (Supplementary Fig. S8), suggesting complementary mechanisms are responsible for the overexpression of hPRDX4 (2).
There were no marked differences in the gene expression of the other antioxidant markers, including the other mPRDXs, or inflammatory signaling molecules between the two types of mice (Supplementary Figs. S7, S8, and S9). Analyses of body weight, serum cholesterol lipoprotein profiles, and glucose metabolism also revealed no significant differences between these groups of mice, although blood pressure was significantly lower in hPRDX4+/+/apoE−/− mice than in apoE−/− mice (Supplementary Fig. S3). One possibility is that the slightly, but significantly lower blood pressure, might be due to presumably systemic additive effects of the elevated circulating antioxidant hPRDX4 (Fig. 1B) and the reduced circulating oxidant marker, TBARS (Fig. 4C), and that the subsequent suppression of endothelial dysfunction or wall tension and decreased turbulence or increased shear stress could have protective effects on atherosclerosis and plaque morphology in hPRDX4+/+/apoE−/− mice. However, of the lipoproteins assessed, only TG levels in hPRDX4+/+/apoE−/− mice were reduced, but they were still much higher than those in apoE−/− mice (Supplementary Fig. S3D). In these hypercholesterolemia model mice, serum TG levels might not significantly affect the progression of atherosclerosis, as compared with other cholesterol parameters, similar to that reported by other research groups (4, 13). In fact, we only measured lipoproteins present in blood samples, and it is unclear how the serum concentrations reflect the local levels in the aortic wall, where atherosclerosis was analyzed (2). Therefore, the correlation between TG and PRDX4 overexpression also remains to be elucidated. Nevertheless, these data demonstrate that hPRDX4+/+/apoE−/− mice provide a good model for determining the local (i.e., intracellular) and presumably systemic (i.e., circulating) additive effects of hPRDX4 in addition with endogenous mPRDX4 on hypercholesterolemia-induced atherosclerosis and possibly on metabolic syndrome.
Our experiments clearly showed that hPRDX4 attenuates advanced atherosclerosis in hPRDX4+/+/apoE−/− mice with HcD-induced hypercholesterolemia. The pathological parameters of atheromatous plaques in HcD-fed mice are summarized in Supplementary Table S1. Morphological observation showed that hypercholesterolemia-induced macrophage-rich atherosclerosis was suppressed although more SMC-rich plaques were observed in hPRDX4+/+/apoE−/− mice (Figs. 2 and 3). These plaques were characterized by a thicker fibrous cap, a greater amount of collagen, and fewer central necrotic lipid cores, as compared with those in apoE−/− mice. The presence of SMCs, underlying macrophage accumulation, and central cores was reminiscent of the structure of human atherosclerotic plaques. Plaque instability is the most critical factor in cardiac attack. These data indicate that the vulnerability index (34) during advanced atherosclerotic formation is significantly reduced in hPRDX4+/+/apoE−/− mice. Conversely, it is possible that hPRDX4+/+/apoE−/− mice might show suppressed levels of a marker of vulnerable plaque formation under hypercholesterolemic conditions (Supplementary Table S1), suggesting a potentially important role for PRDX4 in plaque stability. BMT from Tg or WT mice to apoE−/− mice confirmed these observations, particularly the antiatherogenic properties of PRDX4, demonstrating that recipients of hPRDX4+/+ donors had fewer HcD-induced atheromatous plaques than did recipients of WT donors (Fig. 8A and B). Intimal SMCs and macrophages also revealed the expression of hPRDX4 in BMT mice from Tg (hPRDX4+/+) donors, but not from WT donors, which was confirmed by double IHC (Fig. 8C). Based on these features, the greater suppression and potentially less vulnerable plaque composition should be locally elicited by PRDX4. These effects might, at least in part, be dependent on the expression of PRDX4 by bone marrow-derived cells, particularly circulating monocytes and SMC precursor cells, or adventitial SMC progenitor cells.
It has been reported that apoE−/− mice on long-term HcD and high-fat diet-feeding is a useful and reproducible model of atherosclerotic plaque rupture (7). The sites of ruptured vulnerable plaques typically contain large lipid cores and are covered by thin fibrous caps that harbor activated T cells (12, 26). Several studies have proposed that activated T cell-driven inflammation attenuates collagen maturation in atherosclerotic plaques by inhibiting differentiation and collagen gene expression in SMCs. This, in turn, leads to reduced SMC proliferation; thus, T cells modulate plaque stability (12, 16, 18). In fact, the number of CD3-positive T cells was significantly decreased, especially in the fibrous caps of hPRDX4+/+/apoE−/− mice, but the number of Mac-2-positive macrophages was not, indicating that hPRDX4 specifically suppresses the infiltration of T-lymphocytes (Fig. 3C and D).
In terms of SMC migration, among many cytokines or growth factors, only the expression of PDGF was significantly different between the groups of mice (Fig. 7). PDGF-BB, a strong activator of SMC migration or replication, and an important proatherogenic factor, is produced by platelets, macrophages, and vascular cells at sites of inflammation and/or damage to the arterial wall (12, 18, 24). Considering our finding that the SMCs in the advanced atherosclerotic lesions of hPRDX4+/+/apoE−/− mice showed very low proliferative activity based on MIB-1 staining, the activated PDGF expression and secretion would enhance SMC migration, particularly in the thickened fibrous caps of atheromatous plaques, and may therefore reduce plaque vulnerability in hPRDX4+/+/apoE−/− mice. Accordingly, overexpression of hPRDX4 might be closely related to PDGF-induced migratory signaling in neointimal SMCs, the possible origins of which could include PRDX4-expressed medial SMCs, circulating SMC precursor cells, or adventitial SMC progenitor cells (19, 20, 32).
In our recent study using Tg mice, which showed high hPRDX4 expression, particularly in pancreatic islets, the β cells of Tg mice were significantly protected against insulitis and apoptosis, as compared with those in WT mice. This protection was accomplished by enhanced scavenging of ROS/oxidative stress and suppressed inflammatory signaling (3). Atherosclerosis is a complex inflammatory disease; disturbed blood flow, inflammatory cytokines, and the subsequent generation of ROS derived from macrophages, SMCs, and endothelial cells all contribute significantly to its pathogenesis (5, 29). As hPRDX4 was specifically expressed in the medial SMCs and in the SMCs that migrated to the intima, and macrophages in the atheromatous plaques of hPRDX4+/+/apoE−/− mice (Fig. 1), it seems very likely that overexpression of hPRDX4 in the atherosclerotic aorta protects against chronic inflammation and subsequent disease progression by preventing inflammatory cell-derived ROS generation and scavenging the generated extracellular pool of ROS. Indeed, we detected lower levels of 8-OHdG, oxLDL, and circulating TBARS as markers of oxidative stress (3, 21, 23) (Fig. 4), and reduced cellularity of macrophages as secretory cells in the fibrous caps of the atheromas (Supplementary Table S1) in HcD-fed hPRDX4+/+/apoE−/− mice as compared with apoE−/− mice.
Oxidative stress is closely associated with apoptosis. The expression of Bax and caspase-3, key apoptotic factors, was suppressed in hPRDX4+/+/apoE−/− mice (Fig. 5C and Supplementary Fig. S5). We also provide the first in vitro evidence showing that apoptotic cell death is partly processed by the overexpression of hPRDX4 (Supplementary Fig. S6). Generally, most apoptotic cells in the early to advanced stages of atherosclerosis are macrophages. Therefore, macrophage apoptosis may be an important feature of vulnerable plaques, with an increased size of the necrotic lipid core as a result of weakened or defective phagocytic clearance (11, 22, 31). In the current study, significantly fewer TUNEL-positive macrophages and SMCs were found in fewer atherosclerotic intimal lesions in hPRDX4+/+/apoE−/− mice than in apoE−/− mice (Fig. 5A and B). Apoptosis of SMCs may also lead to a loss of interstitial collagen fibers, resulting in unstable plaques that are prone to rupture (11). Furthermore, en face aortic double immunofluorescence staining (Fig. 6) indicated that hPRDX4 can protect CD31-positive endothelial cells against apoptosis. Apoptosis of endothelial cells is also important because the initial step in ROS-induced atherosclerosis involves endothelial damage, increased expression of adhesion molecules, and inflammatory cell migration (5, 12, 27). In fact, we found that the cellularity of intimal macrophages and T cells in the atheromatous plaques of hPRDX4+/+/apoE−/− mice was significantly lower than that in apoE−/− mice (Fig. 3D and Supplementary Table S1). Therefore, our present data suggest that overexpression of hPRDX4 plays a significant role in (i) ameliorating atherosclerotic progression by reducing the size of the central necrotic core by suppressing macrophage apoptosis, and reducing inflammatory cell migration by suppressing endothelial cell apoptosis, and (ii) reducing possible markers of plaque instability by thickening the fibrous caps and increasing the collagen-rich matrix by suppressing SMC apoptosis.
These observations indicate that specific induction of PRDX4 could be a potential therapeutic strategy to protect against atherosclerosis progression possibly supporting plaque stability by suppressing oxidative damage and apoptosis of macrophages, SMCs, and endothelial cells.
Materials and Methods
Detailed Materials and Methods are given in the on-line supplement.
Animals and quantitative assessment of atherosclerotic lesions in hypercholesterolemia model mice
The experiments were performed using young male apoE−/− mice and hPRDX4 transgenic (Tg or hPRDX4+/+) and apoE−/− (hPRDX4+/+/apoE−/−) mice, weighing 20–25 g (Supplementary Fig. 1A) (3, 30). These mice were analyzed using RT-PCR, Western blotting, real-time RT-PCR, hematoxylin and eosin (H&E) staining, and IHC staining. At 8 weeks of age, apoE−/− and hPRDX4+/+/apoE−/− mice were fed the HcD containing 1.25% cholesterol, 15.0% lard, and 0.5% sodium cholate (Oriental Yeast, Tokyo, Japan) or the NcD. They were killed 12 weeks later by intraperitoneal anesthesia with an overdose of ketamine and medetomidine, as described elsewhere (19, 27, 31).
We excised the aortas and stained them with oil red-O after fixation in 10% neutral-buffered formalin for 24 h. To measure the atherosclerotic lesions, we captured en face aortic images with an Olympus Camedia E-10 digital camera (Olympus, Tokyo, Japan). The red area relative to the whole aorta surface area was measured using NIH imaging software (27, 31, 32). Next, the aortas were divided into four equally sized pieces and embedded in paraffin for histological examination. The specimen from the aortic root to the abdominal aorta was cut into 2,500 sequential 4-μm-thick cross-sections and subjected to H&E, IHC, Masson's trichrome, and oil red-O staining. Of each set of 20 serial sections, at least one section was stained with H&E (i.e., 80-μm intervals). For quantitative analysis, images were captured of approximately 125 sequential sections, and the thickened intimal or medial areas were evaluated by a NanoZoomer Digital Pathology Virtual Slide Viewer (Hamamatsu Photonics Corp., Hamamatsu, Japan). These indices were quantified by averaging all measurements for an individual mouse, and we decided one representative section with values similar to the mean for each mouse. All of the representative sections were taken from the aortic root to the upper part of the thoracic aorta. To calculate the relative collagen content and necrotic lipid core to intima ratio, we measured the intimal lesion areas, Masson's trichrome-positive blue area and necrotic areas, including acellular foci (e.g., cholesterol crystals) in at least three sequential sections of each set, including the representative section, as previously described (31, 32). Oil red-O staining confirmed that these central necrotic areas were lipid cores. We also measured the fibrous cap thickness of advanced atherosclerotic lesions.
The Ethics Committee of Animal Care and Experimentation, University of Occupational and Environmental Health (Japan) approved the study protocols. All animal procedures were performed according to the Institutional Guidelines for Animal Experiments and the Law (no. 105) and Notification (no. 6) of the Japanese government.
Immunohistochemistry
We studied 5–10 mice per experimental group, and one representative section per mouse was prepared for IHC staining. Images of at least three sequential sections of each 20-section set (i.e., 80-μm intervals), including the representative section, were captured and evaluated using NanoZoomer Digital Pathology Virtual Slide Viewer (Hamamatsu Photonics Corp.) to avoid potential bias (3, 27, 31, 32). To detect hPRDX4, we used a rabbit anti-human polyclonal antibody (1:100; Affinity BioReagents, Golden, CO) and a goat polyalkaline phosphatase secondary antibody (Vulcan Fast Red; Biocare Medical, Concord, CA). To our knowledge, no specific antibody for endogenous mPRDX4 is available. To detect the number of SMCs and macrophages in intimal lesions, we used a monoclonal mouse anti-human α-SMA antibody (1:1000; Dako Cytomation, Kyoto, Japan) and a rat anti-mouse Mac-2 monoclonal antibody (1:500; Cedarlane Laboratories, Burlington, Ontario, Canada), respectively. For the monoclonal mouse α-SMA antibody, we treated sections with the HistoMouse™–Plus Kit (Invitrogen Corporation, Camarillo, CA) to block endogenous IgG. Double IHC was also performed to detect the distribution of both hPRDX4- and α-SMA-positive SMCs or both hPRDX4- and Mac-2-positive macrophages in atherosclerotic lesions (hPRDX4 with Vulcan Fast Red and α-SMA or Mac-2 with DAB as a substrate, respectively) using the MACH 2 Double Stain system (Biocare Medical) (3, 23).
To analyze the apoptotic and proliferative activities of infiltrating cells in atherosclerotic lesions, we used a Bax rabbit polyclonal antibody (1:50; Santa Cruz Biotechnology, Santa Cruz, CA) to assess apoptotic activity and a Ki67 (MIB-1; 1:2000; Epitomics) rabbit monoclonal antibody to assess proliferation (3, 31).
To evaluate T-lymphocyte infiltration, especially in the fibrous caps of atherosclerotic lesions, sections were stained with a polyclonal rabbit anti-human CD3 antibody (1:1; Dako Cytomation), and the number of CD3-positive T cells was counted (3). To analyze ROS/oxidative stress levels in aortas, we used anti-8-OHdG monoclonal antibody (1:200; Japan Institute for the Control of Aging, Fukuroi, Japan) and anti-Cu2+-oxLDL polyclonal rabbit antibody (1:200; Merck, Ltd., Darmstadt, Germany). For these tests, we did not block endogenous peroxidase activity using hydrogen peroxide (3, 9).
Measurement of TBARS
The measurement of TBARS is a well-established method to evaluate lipid peroxidation (21). Therefore, to assess a second and more quantitative marker of oxidative stress that is closely associated with the development of atherosclerosis, we measured the TBARS levels using a TBARS Assay Kit (Cayman Chemical Company, Ann Arbor, MI) in serum collected from HcD-fed mice. Results are expressed as nmol MDA/mg LDL protein.
TUNEL staining
To determine the number of apoptotic cells in atherosclerotic lesions, we conducted TUNEL assays using the In Situ Cell Death Detection Kit, POD (Roche Applied Science, Lewes, UK) (3, 31). Double IHC was also performed to determine the distribution of TUNEL-positive/α-SMA-positive SMCs or TUNEL-positive/Mac-2-positive macrophages in atherosclerotic lesions (TUNEL with DAB and α-SMA or Mac-2 with Vulcan Fast Red as a substrate, respectively) as described above (Biocare Medical).
En face aortic double-immunofluorescence staining
Aortas of HcD-fed hPRDX4+/+/apoE−/− and apoE−/− mice were prepared en face, opened longitudinally to remove adventitial adipose tissue, fixed in cold acetone for 10 min, and air-dried for 10 min. En face vessels were labeled with TUNEL reaction mixture (Roche Applied Science, Mannheim, Germany) and rat monoclonal antibodies against mouse CD31 (1:20; Dianova, Hamburg, Germany), incubated with 0.5 μg/ml Hoechst 33258 (Dojindo, Kumamoto, Japan), and visualized with anti-fluorescein-antibodies (TUNEL POD; Roche Applied Science) or Alexa Fluor-conjugated goat anti-rat IgG and IgM antibodies (Invitrogen, Carlsbad, CA). The stained tissues were placed on a glass slide intimal side up, coverslipped by surface tension, and then viewed by confocal laser scanning microscopy (LSM5 PASCAL EXCITER; Carl Zeiss, Oberkochen, Germany) with a 40X UPlanApo oil immersion objective lens. Three to six representative high-power fields (∼100 cells) were counted to determine the number of TUNEL-positive (green)/CD31-positive (red) cells, and Hoechst-positive (blue)/CD31-positive (red) cells (31). The number of apoptotic endothelial cells was expressed as a percentage of the total number of cells. We performed three independent experiments per group of mice.
RT-PCR and real-time RT-PCR
RT-PCR and real-time RT-PCR were used to analyze gene expression in the aorta, as previously described (3, 27, 31, 32). The pairs of specific primers and probes for all of the genes tested are listed in Supplementary Table S2. The relative expression level of each gene was normalized for that of GAPDH and 18S ribosomal RNA (rRNA) using random primers (31).
Western blot analysis
Rabbit anti-human polyclonal hPRDX4 (1:100; Affinity BioReagents), rabbit anti-mouse polyclonal Bax (1:100; Santa Cruz Biotechnology), and mouse anti-chicken monoclonal β-actin (1:1000; Santa Cruz Biotechnology) antibodies were used for Western blotting (3).
Vulnerability index
The vulnerability index was calculated as previously described (34), as Index=(% Macrophages + % lipid cores / (% SMCs + % collagen content).
ELISAs for hPRDX4 and PDGF-BB
The serum levels of hPRDX4 and PDGF-BB in HcD-fed mice were measured using ELISA kits according to the manufacturers' instructions (hPRDX4 ELISA Kit; Abnova, Taipei, Taiwan and PDGF-BB ELISA Kit; R&D Systems, Minneapolis, MN).
Bone marrow transplantation
Bone marrow (5×106 cells) obtained from the femurs of WT (C57BL/6) or hPRDX4+/+ (WT background) donor mice was transplanted into whole-body-irradiated (9 Gy from a cesium γ source) recipients (8-week-old male apoE−/− mice). Three weeks after BMT, all recipient mice were fed the HcD for 12 weeks (19, 31).
Statistical analysis
Results are expressed as means±standard error. Significant differences were analyzed using Student's or Welch's t-test, or analysis of variance where appropriate. Values of p<0.05 were considered statistically significant.
Footnotes
Acknowledgments
We would like to thank Hiroko Isagai, Hana Nishimura, and Naoko Une for their expert technical assistance.
This work was supported in part by Grants-in-Aid for Scientific Research (19590413 and 20590416) from the Ministry of Education, Culture, Sports, Science, and Technology, Tokyo, Japan (to Y.S., K.K., and A.T.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.