
Abstract
Introduction
In this article, the ability of H2S to modulate inflammation is reviewed, with a particular focus on the role of the mediator in resolution of inflammation. The use of H2S as a therapeutic modality is also reviewed, along with the potential effects of bacteria-derived H2S in modulating inflammation and mucosal integrity in the digestive tract and possibly in other organs.
H2S and Inflammation
One of the first physiological effects of H2S that was identified was its ability to relax vascular smooth muscle (73, 75), resulting in vasodilation, a hallmark of inflammation. Several studies have subsequently highlighted the importance of H2S in inflammation (27, 30, 54, 56). As was the case for nitric oxide, the literature on H2S in inflammation was initially contradictory, but in recent years a general pattern has emerged consistent with this mediator exerting anti-inflammatory effects, except at high concentrations (56). Moreover, there are now substantial data supportive of a role of H2S in promoting resolution of inflammation and healing of injured tissue. Figure 1 summarizes some of the key effects of H2S with respect to inflammation and injury. These effects include the ability of H2S to inhibit leukocyte adherence to the vascular endothelium and the subsequent extravasation of leukocytes (72). The impact of this effect of H2S can be seen in various models of inflammation, in which sulfide salts or H2S donors are able to reduce infiltration of neutrophils and lymphocytes (17, 72). These effects are likely due, at least in part, to reduced expression of endothelial and/or leukocyte adhesion molecule expression following exposure to H2S (18). H2S acts as a tonic down-regulator of leukocyte adherence; thus, inhibition of H2S synthesis leads to leukocyte adherence (72). Treatment of rats with inhibitors of H2S synthesis resulted in a marked increase in mucosal inflammation (elevated granulocyte levels) and an increase in susceptibility to injury (18, 58, 59) in the gastrointestinal tract. This may have been in part due to reduced basal levels of cyclooxygenase-2 (COX-2) expression and a parallel reduction of mucosal prostaglandin E2 (PGE2) synthesis (61). COX-2 and PGE2 play important roles in the maintenance of mucosal defense in the digestive tract, as well as in modulating mucosal inflammation (3, 49, 61, 70, 71).
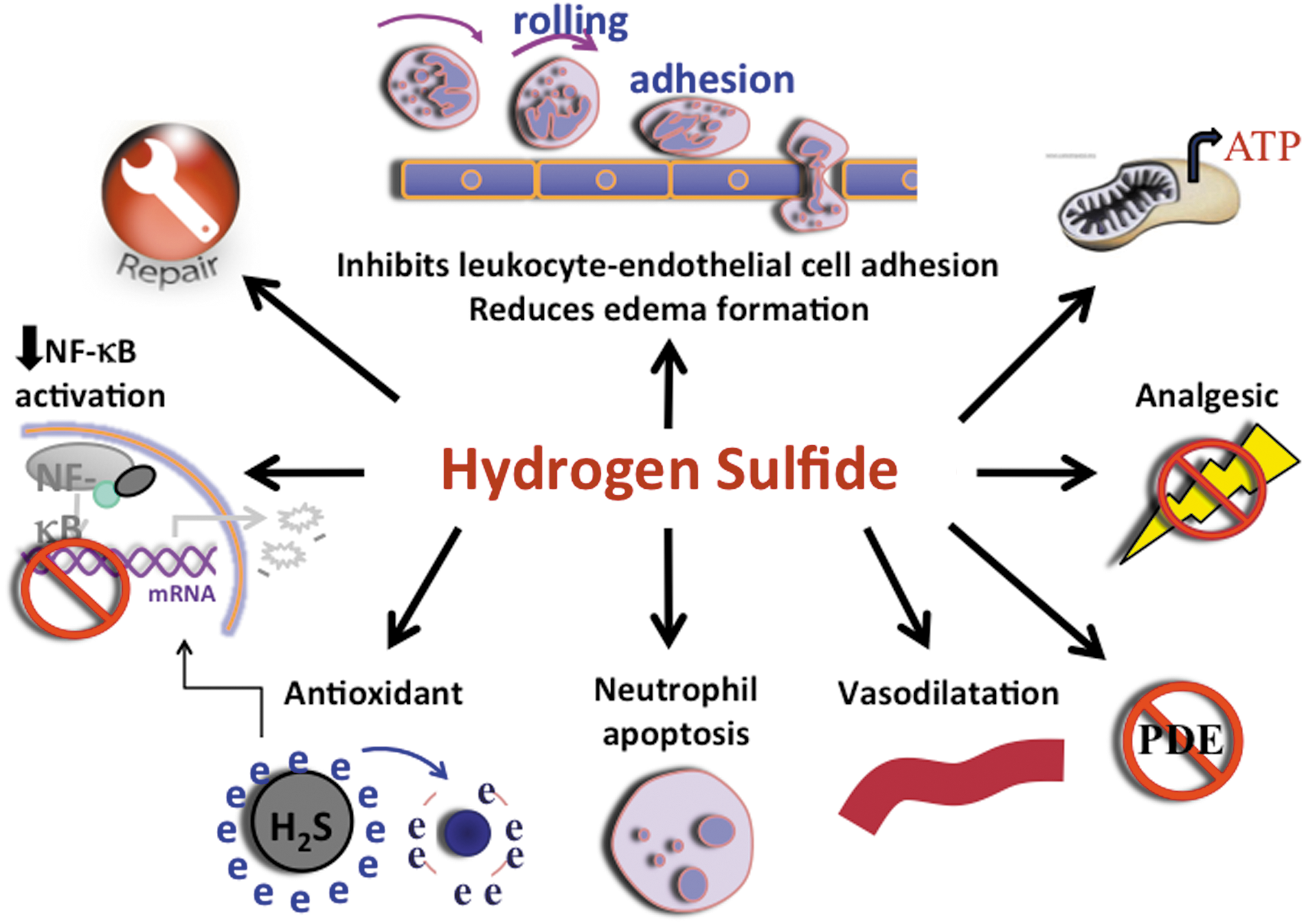
In addition to modulating leukocyte adhesion and recruitment, H2S can reduce plasma exudation (edema formation), while inhibitors of H2S augment edema formation triggered by an inflammatory agent (59). These effects may contribute to the enhanced edema-reducing effects of H2S-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) in rat models of acute (carrageenan) and chronic (Freund's adjuvant) paw swelling (58, 59).
The ability of H2S to reduce inflammation has been demonstrated in a variety of animal models, including kaolin/carrageenan-induced monoarthritis in rats (2), tobacco-smoke induced lung inflammation in mice (12, 23), synovitis in rats (17), ischemia–reperfusion injury in mice (76), and in rat and mouse models of colitis (19, 56, 61). Whiteman et al. (66) demonstrated that H2S is present in synovial fluid of patients with rheumatoid arthritis and osteoarthritis, with levels correlating to disease activity, but the role of H2S in those conditions remains unclear.
H2S was recently shown to inhibit phosphodiesterase activity, and this may contribute to anti-inflammatory actions in some circumstances (via elevation of intracellular cyclic AMP and/or cyclic GMP) (9). Scavenging of oxidants and peroxynitrite may also contribute to the anti-inflammatory activity of H2S (64, 65). Inhibition of nuclear transcription factor-κB (NF-κB) has been reported in several models (27, 28, 38), and consistent with this, H2S reduces pro-inflammatory cytokine, chemokine, and enzyme (e.g., inducible nitric oxide synthase [iNOS]) expression (17, 19, 51, 67). In addition, the antioxidant activity of H2S can be mediated by up-regulation of enzymes such as superoxide dismutase, glutathione peroxase dismutase, and thioredoxin as assessed in rats subjected to intestinal ischemia-reperfusion injury (31) or in brain endothelial cells under methionine-induced oxidative stress in vitro (53) or even by inhibition of NADPH oxidase activity, as in the case of osteoblasts exposed to hydrogen peroxide in vitro (69). With many of these putative mechanisms of action of H2S, it is unclear if the concentrations required for such actions are achieved in a physiological setting. Difficulties in measuring H2S levels in vivo further complicate the assessment of the relative significance of these mechanisms in various scenarios.
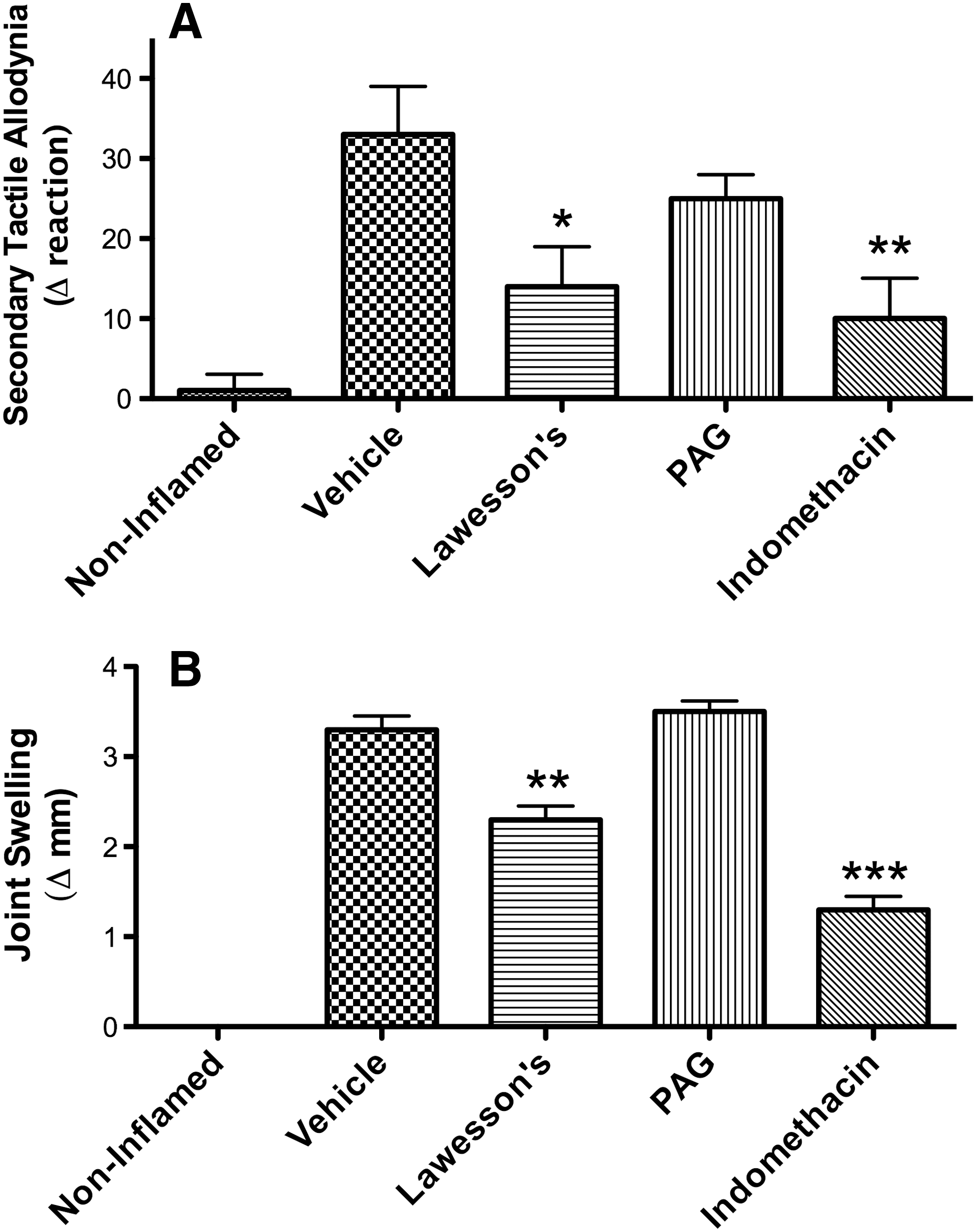
H2S has been shown to exert anti-nociceptive effects in some (15, 18, 19, 62), but not all (37) visceral pain studies. The discrepancies may be related to the different methods for measuring pain or to different doses or routes of administration of the H2S-releasing agents. In a recent study performed in our laboratory, we found that two H2S-generating substances (NaHS and Lawesson's reagent) dose-dependently reduced gastric distention–induced visceral pain (measured through cardio-autonomic responses) in the rat in an ATP-sensitive potassium channel–dependent manner (62). The ability of H2S to exert peripheral anti-nociceptive effects has also been demonstrated in some, but not all studies. While anti-nociceptive effects of H2S were reported by Cunha et al. (13) in a model of peripheral pain induced by bacterial endotoxin and by Ekundi-Valentim et al. (17) in rats with carrageenan-induced knee joint synovitis (see Fig. 2), Andruski et al. (2) observed a significant inhibition of leukocyte recruitment by an H2S donor in a rat model of monoarthritis, but no significant effect on pain sensitivity.
H2S and Resolution of Inflammation/Injury
With the ability to inhibit so many elements of acute inflammation, it is not surprising that H2S contributes significantly to the resolution of inflammation and injury. Inflammatory reactions are driven largely by soluble, pro-inflammatory mediators, such as leukotrienes, histamine, bradykinin, platelet-activating factor, and interleukin (IL)-1, to name just a few (49). Counteracting the effects of these pro-inflammatory mediators are a variety of soluble mediators that down-regulate inflammation, including lipoxins, certain prostaglandins, annexin-1 (AnxA-1), and IL-10 (49). An over-production of pro-inflammatory mediators or an under-production of anti-inflammatory mediators can lead to progression from acute to chronic inflammation. Resolution of inflammation occurs through removal of the triggers of the inflammatory response (e.g., a foreign body or organism), inhibition of the recruitment of neutrophils to the site of injury, and induction of apoptosis of the infiltrated neutrophils and their subsequent clearance by macrophages (Fig. 3). Macrophages undergo a phenotype shift from pro-inflammatory to anti-inflammatory during this process.
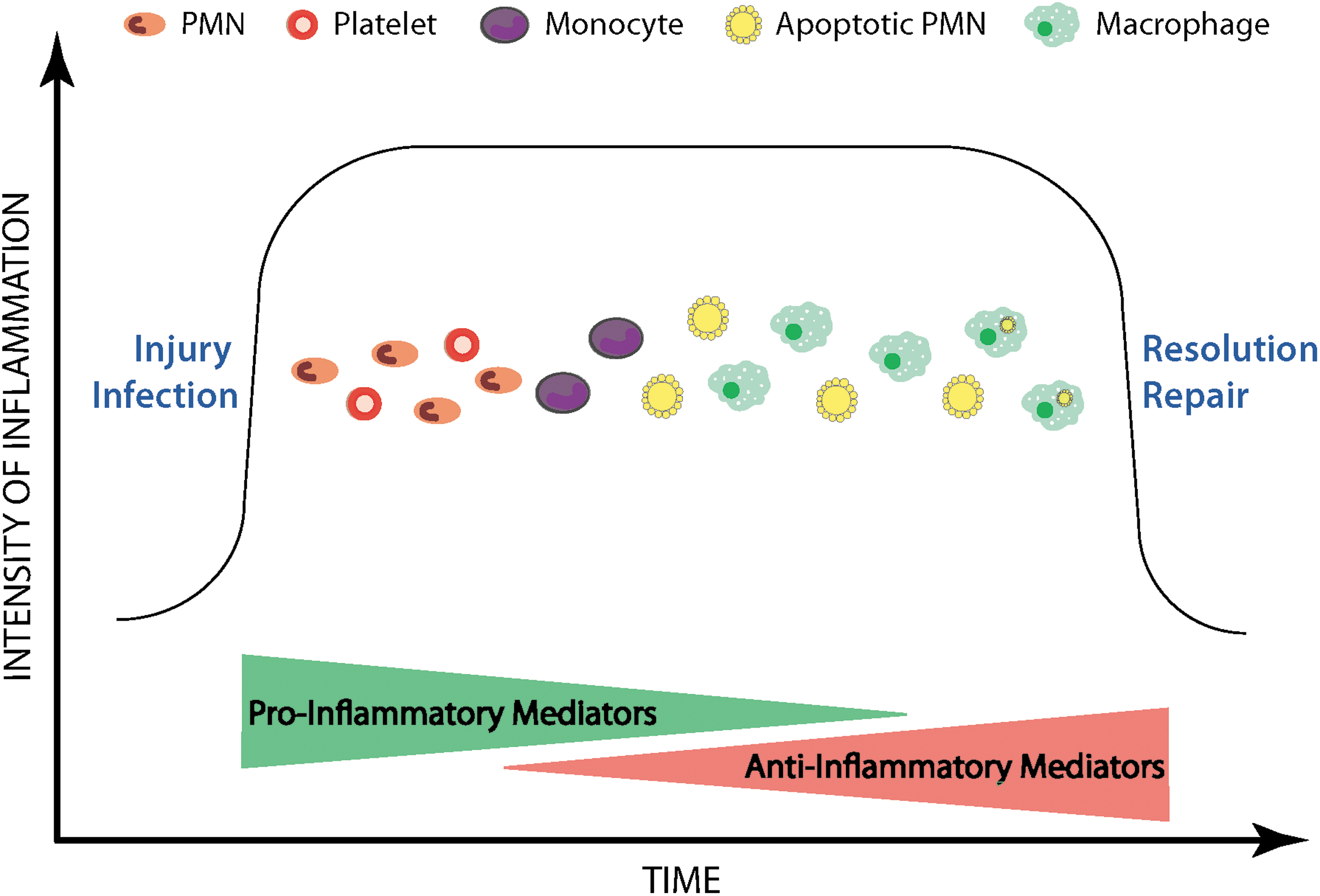
There is emerging evidence that H2S may participate in several stages of the process of resolution of inflammation. As already mentioned, H2S can reduce leukocyte adherence to the vascular endothelium and leukocyte migration to sites of injury (72). H2S can also induce neutrophil apoptosis (34), and there is recent evidence that it can trigger significant changes in macrophage function consistent with a shift to a pro-resolution phenotype (16). Specifically, exposure of murine bone marrow–derived macrophages to H2S resulted in a significant enhancement of phagocytosis of bacteria. H2S also suppressed endotoxin-induced tumor necrosis factor α (TNFα) production by macrophages, while enhancing chemotaxis. In vivo, in a mouse peritonitis model, H2S significantly reduced granulocyte infiltration while maintaining macrophage numbers (16).
H2S also appears to interact with other pro-resolution mediators. H2S modulates COX-2 expression in the gastrointestinal tract, which plays a crucial role in resolution of inflammation and injury (3, 49, 56, 61, 70, 71). Recently reported work from Brancaleone et al. (8) demonstrates an important interaction between H2S and AnxA-1. AnxA-1 is a well-characterized anti-inflammatory and pro-resolution mediator that has also been shown to contribute to gastrointestinal integrity and repair (36, 41). Brancaleone et al. (8) observed that an H2S donor (NaHS, 10–100 μM) elicited intense mobilization of AnxA-1 from the cytosol to the membrane of human neutrophils. It also markedly suppressed IL-1–induced leukocyte adhesion and emigration in mesenteric venules of wild type, but not AnxA1-deficient mice. There were also effects of endogenous AnxA1 on H2S synthesis. Thus, mice deficient of AnxA1 displayed marked up-regulation of cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) in a variety of tissues as compared with wild-type mice. Moreover, H2S could down-regulate other inflammatory pathways in macrophages from wild-type mice (i.e., significant suppression of iNOS and COX-2 expression in lipopolysaccharide-stimulated bone marrow-derived macrophages) but not in macrophages obtained from AnxA1-deficient mice. The authors concluded that these data demonstrate interlinks between the H2S and AnxA1 pathways that are likely very important in the resolution of inflammation.
The importance of H2S in promoting resolution of inflammation and repair of injury has been clearly demonstrated in animal models of gastrointestinal inflammation and ulceration. In rat and mouse models of gastric ulcer, administration of H2S-generating agents has been shown to significantly accelerate ulcer healing, while inhibition of endogenous H2S synthesis was associated with impaired ulcer healing (58, 60). Administration of the precursor for H2S synthesis, L-cysteine, promoted ulcer healing in the rat at doses that did not affect tissue glutathione levels (60). One of the significant limitations to the use of NSAIDs for treatment of inflammatory conditions is their ability to retard the healing of gastrointestinal ulcers. As shown in Fig. 3, this can be shown in a mouse model of gastric ulceration in which NSAIDs such as naproxen and celecoxib significantly impaired ulcer healing. However, an H2S-releasing derivative of naproxen (ATB-346) not only did not impair ulcer healing, it significantly accelerated the healing process (58); thus, enhanced repair occurred despite marked suppression of COX-2 activity in the damaged tissue. Similar effects have been demonstrated previously in the rat with an H2S-releasing derivative of mesalamine (19, 56). The mechanism underlying the enhancement of ulcer healing by H2S is not clear, but may be related, at least in part, to its vasodilatory activity, its ability to enhance COX-2 expression, and its reported ability to promote angiogenesis (40).
Further evidence for an important role of H2S in resolution of inflammation and repair of injury comes from studies of experimental colitis in rodents. When the colonic mucosa is inflamed, there is a marked increase in the capacity of the tissue to produce H2S (20, 58). Most of the H2S in this context is produced via CBS, as is the case in non-inflamed colon (35), but synthesis via CSE is also significant (61). Preliminary data suggested that there is also a marked elevation of colonic H2S synthesis via other enzymatic pathways (i.e., non-CSE and non-CBS) when the colon is inflamed. Inhibition of H2S synthesis once colitis is established led to a marked worsening of the inflammation, with perforation of the bowel wall and death occurring in most animals within a few days (61). In the rats that survived a week-long treatment with an inhibitor of H2S synthesis, the colonic damage was significantly worse than that observed in vehicle-treated controls. Interestingly, there was a marked thickening of the smooth muscle in the colon in the rats treated with an inhibitor of H2S synthesis (61).
Additional evidence that H2S could promote resolution of colitis came from studies in which H2S donors were administered to rats or mice with colitis (19, 61, 68). Irrespective of the H2S donor used, a significant reduction of the severity of colitis was observed, with a marked inhibition of granulocyte infiltration into the colonic tissue (19, 61). The latter observation is important, since much of the tissue injury associated with colitis is likely produced by infiltrating granulocytes. In these studies, the H2S-generating agents significantly reduced colonic expression of TNFα (protein and mRNA); in other studies, they also reduced expression of interferon-γ, IL-1β, and iNOS (19, 61, 68). Consistent with these findings, allyl trisulfide, a garlic-derived substance that releases H2S (7), was shown to suppress TNFα expression and NF-κB activation in colonic biopsies from patients with ulcerative colitis (6).
H2S in Novel Therapeutics
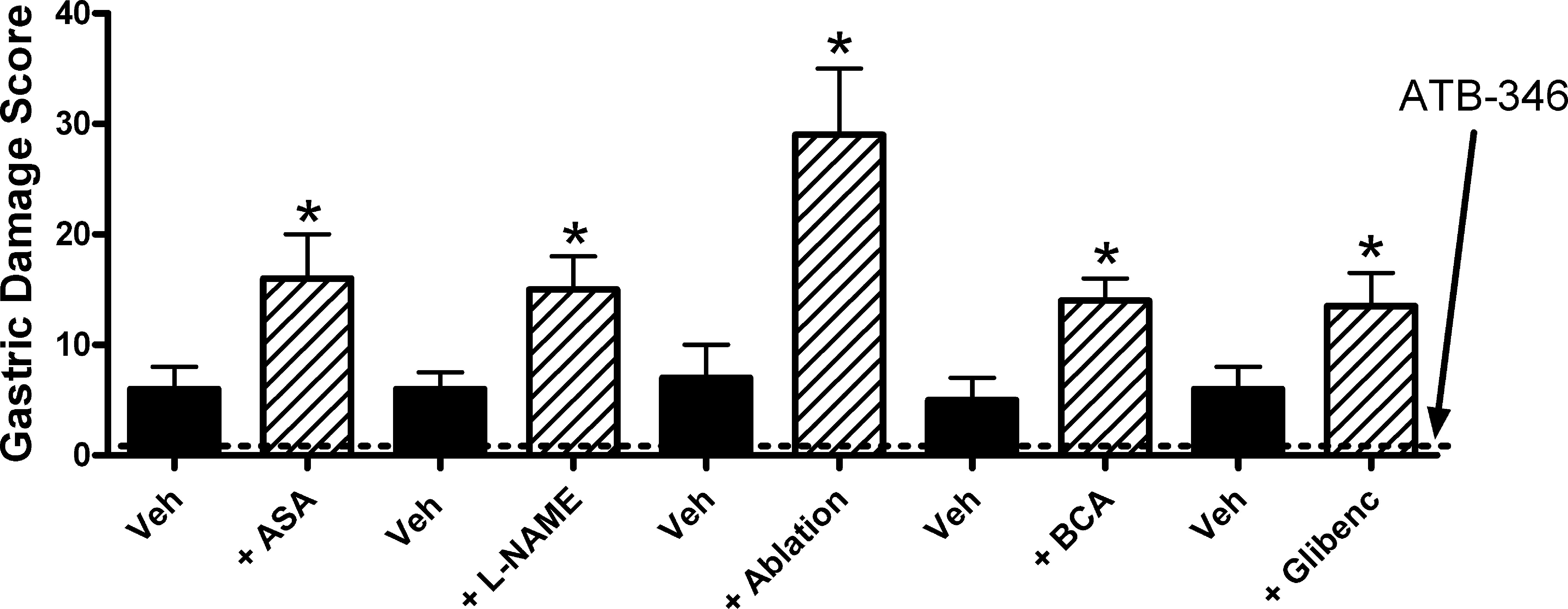
The ability of H2S to enhance gastrointestinal resistance to injury, to promote repair of damaged tissue, and to reduce mucosal inflammation make it an attractive substance to exploit in designing novel drugs for treatment of gastrointestinal injury and inflammation (10, 54). We have synthesized and characterized the effects of an H2S-releasing derivative of mesalamine in animal models of colitis (mesalamine is the first-line therapy for colitis) and have observed a significant enhancement of anti-inflammatory activity as compared to the parent drug (19, 56). H2S-releasing derivatives of several NSAIDs have been assessed in animal models, with the consistent finding of greatly reduced gastrointestinal toxicity and, in some cases, enhanced anti-inflammatory activity (30, 54, 58, 59). An H2S-releasing derivative of naproxen has been particularly well characterized. The anti-inflammatory activity of the compound, called ATB-346, is comparable to that of the parent drug (58). However, even at exceptionally high doses (100 times the human dose on a per kilogram basis), ATB-346 caused negligible gastric damage in healthy rats. While impressive, studies performed in healthy rodents may not serve as a good predictor of how an anti-inflammatory drug will behave in humans with diseases such as osteoarthritis and rheumatoid arthritis alongside other co-morbidities for gastrointestinal damage. To more rigorously assess the gastric safety of ATB-346, studies were performed in rats in which gastric mucosal defense was significantly compromised (58). This was achieved by interfering with the production of mediators known to contribute to mucosal defense (e.g., nitric oxide, hydrogen sulfide), co-administering another drug that can damage the stomach (e.g., low-dose aspirin, frequently co-administered with NSAIDs in a clinical setting), blocking receptors believed to contribute to mucosal defense (e.g., glibenclamide to block ATP-sensitive K+ channels), or ablating sensory afferent nerves (via neonatal administration of capsaicin), which play a crucial role in mucosal responses to luminal irritants (55). As illustrated in Fig. 4, naproxen produced a low level of damage in control rats at the dose tested (60 μmol/kg). However, with each approach to compromising mucosal defense, the extent of injury induced by naproxen was significantly increased (two- to sixfold). In contrast, ATB-346 at an equimolar dose did not produce significant damage in controls or in mucosal defense–compromised animals. Particularly interesting is the observation that ATB-346 was still gastric safe in rats pretreated with glibenclamide. Many actions of H2S have been attributed to activation of ATP-sensitive K+ channels (14, 62, 74). This observation suggests that the mechanism underlying the gastric tolerability of ATB-346 is unrelated to activation of ATP-sensitive K+ channels. It is also noteworthy that ATB-346 suppressed gastric prostaglandin synthesis as effectively as equimolar doses of naproxen (58). Thus, the compound can compensate for the reduced mucosal resistance to injury that occurs when prostaglandin synthesis is markedly reduced (presumably because H2S can exert many of the same effects, in terms of mucosal defense, as prostaglandins do).
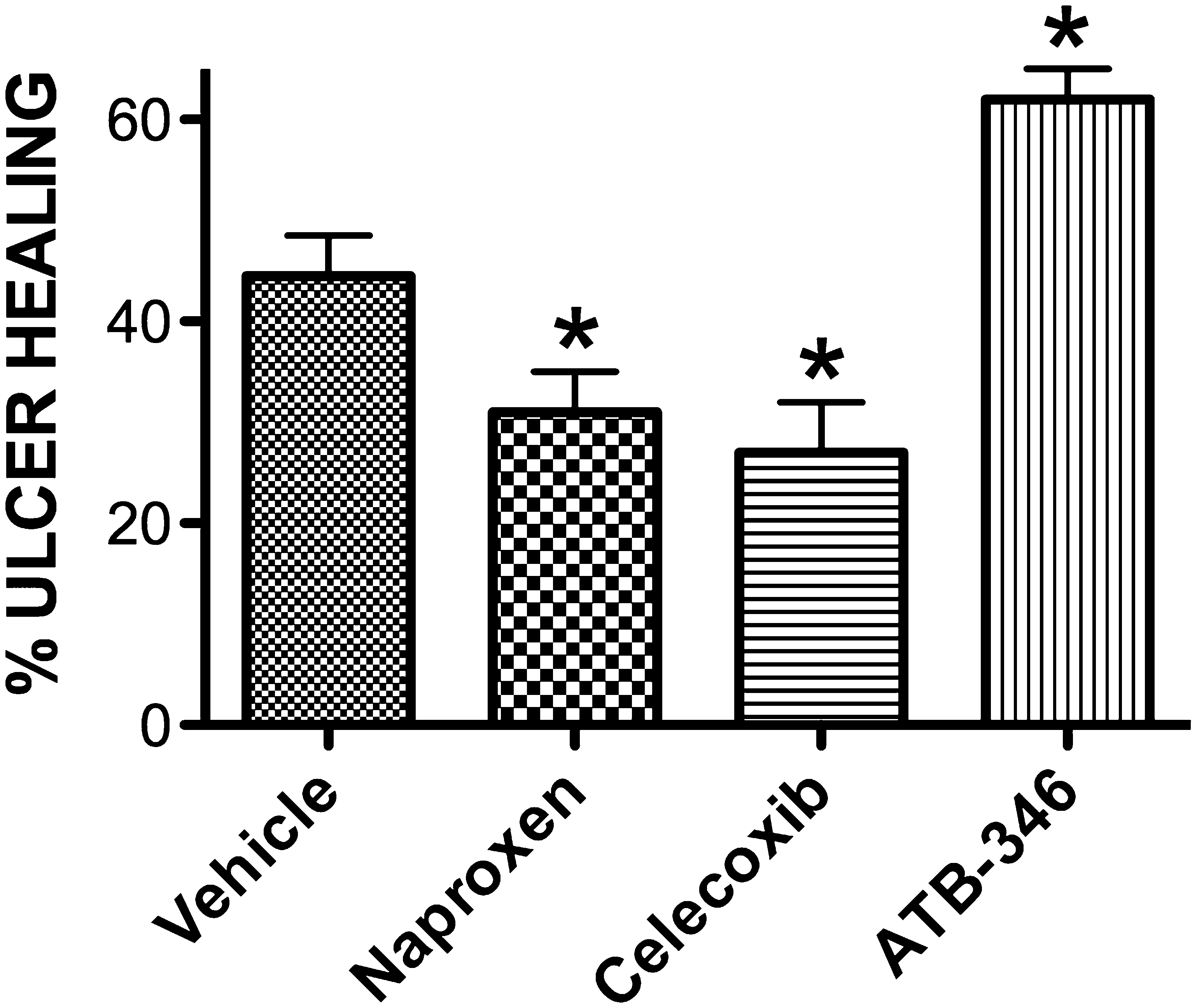
Another major clinical concern with respect to the use of NSAIDs is their ability to interfere with the healing of ulcers (55). This effect is likely related to suppression of COX-2 activity by the NSAIDs. COX-2–derived prostaglandin synthesis by cells at the ulcer margin is critically important for re-epithelialization and angiogenesis (55). As shown in Fig. 5, naproxen and celecoxib (nonselective COX inhibitor and selective COX-2 inhibitor, respectively) each significantly impaired the healing of gastric ulcers in mice when administered twice daily over a 4-day period. In sharp contrast, the H2S-releasing derivative of naproxen (ATB-346) significantly accelerated the healing of pre-existing gastric ulcers (58). This is consistent with previous reports that L-cysteine and H2S-generating agents could accelerate experimental ulcer healing, while inhibitors of H2S synthesis impaired ulcer healing (56, 60). The beneficial effects of H2S on ulcer healing may be attributable to the stimulatory effects of H2S on angiogenesis (40).
From a clinical perspective, the main focus in terms of the gastrointestinal toxicity of NSAIDs is the stomach and proximal duodenum. This is largely due to the relative ease of endoscopically viewing these regions. However, it is becoming increasingly clear that NSAIDs frequently produce significant injury in the small intestine. Indeed, the jejunum and ileum may be the major sites of NSAID-induced bleeding (55, 57). Damage to these regions induced by NSAIDs is determined by a number of factors, but most important is the enterohepatic circulation of the NSAIDs, leading to repeated exposure of the epithelium to the NSAIDs and bile, and also to marked changes in the numbers and type of bacteria in the small intestine (57). The H2S-releasing derivative of naproxen (ATB-346), when given twice daily to rats for 5 days, did not produce detectable small intestinal damage (58). This was in marked contrast to the parent drug, naproxen, which elicited widespread ulceration and bleeding in the small intestine.
Bacteria-Derived H2S: A Modulator of Inflammation or Mucosal Function?
Many of the species of bacteria residing in the human gastrointestinal tract are capable of producing H2S. Some early studies suggested that the concentrations of H2S in the lumen of the gut were extremely high relative to levels that occur in the body (32, 33). Largely based on these data, and observations of adverse effects of such concentrations of H2S on colonocyte function (5), roles for H2S in the pathogenesis of inflammatory bowel disease and colon cancer were suggested (4, 45, 46). However, this hypothesis has been challenged (25, 26, 38, 42), as has the notion that there are millimolar concentrations of free H2S in the lumen of the gut (27, 28, 38, 42, 43). Most of the H2S that is produced in the lumen of the intestine is bound to fecal material and therefore not available to diffuse through the epithelium. The H2S that is available, likely in micromolar concentrations, is absorbed and rapidly metabolized (51). Indeed, efficient detoxification occurs via a number of enzymes present in the mucosa, and no impairment of these detoxification systems has been detected in patients with ulcerative colitis or Crohn's disease (42). Based on studies utilizing a rat model of colitis (dextran sodium sulfate), Furne et al. (21) concluded that “excessive H2S production” did not contribute to tissue injury.
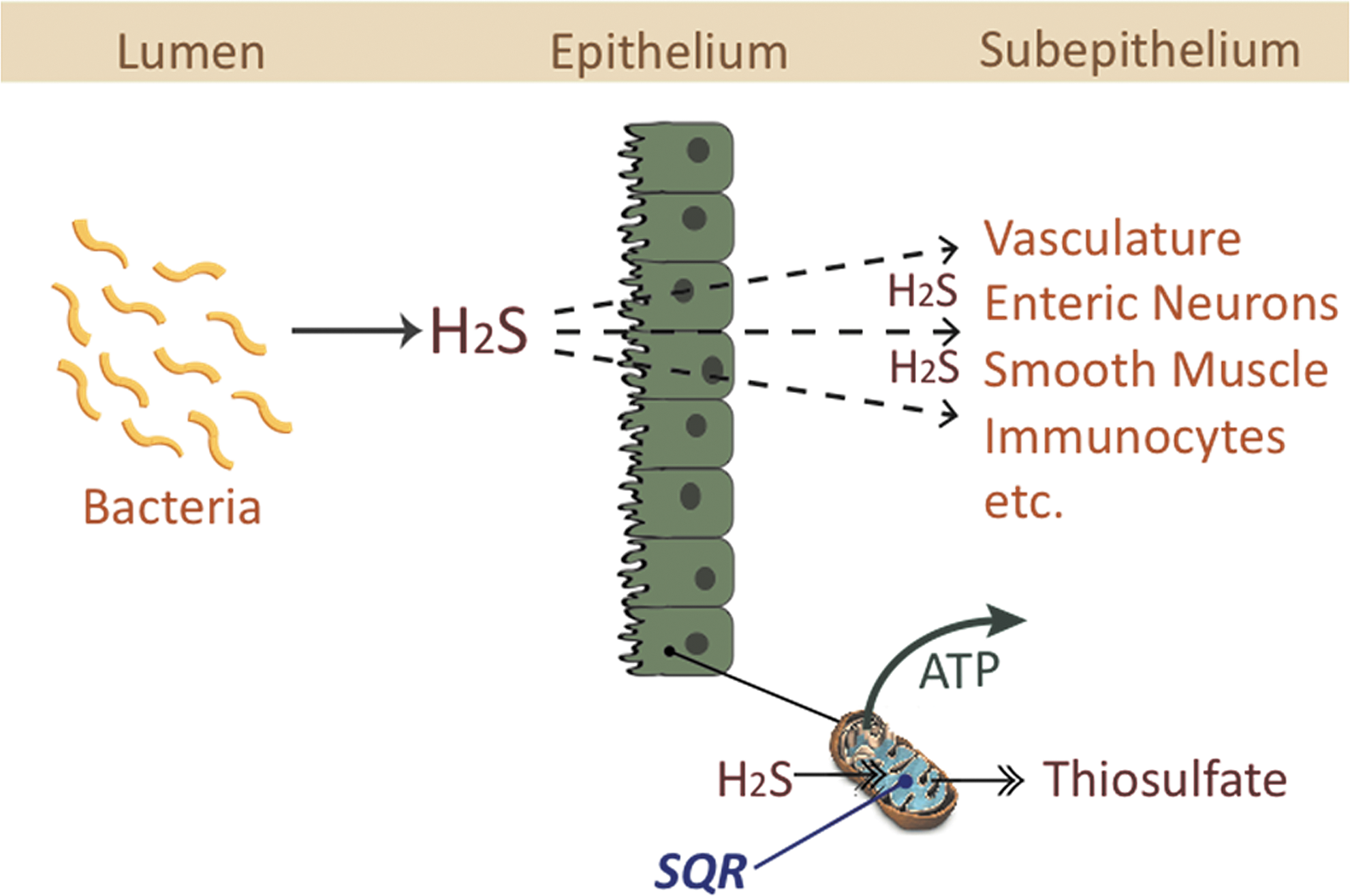
Even before reaching the mucosa, however, there is substantial metabolism of H2S in the mitochondria of enterocytes and colonocytes. Virtually all H2S that crosses the apical membrane of enterocytes and colonocytes is rapidly oxidized to thiosulfate (22, 28). This is accomplished mainly by a mitochondrial enzyme, sulfide quinone reductase, which rapidly consumes H2S, thereby providing energy to the cell and keeping H2S concentrations at nontoxic levels (26) (Fig. 6). This ancestral capacity, predating photosynthesis, is common to organisms living in low-light, low-oxygen conditions (52). As mentioned above, efficient detoxification also occurs via a number of mucosal enzymes (52). The epithelium could therefore be viewed not only as a physical barrier, protecting organisms from potentially harmful substances in the lumen of the digestive tract, but also a metabolic barrier providing additional protection. When there is an intact, healthy epithelium, luminally produced H2S likely has little, if any, effect on mucosal function. However, in cases in which the epithelium is dysfunctional or damaged, it remains possible that H2S produced by bacteria could exert significant effects on several aspects of mucosal function, including secretion (via effects on enteric neurons) (47), pain sensation (14), blood flow (18, 63), and smooth muscle contractility (29, 63). Indeed, an impaired colonic “barrier” to diffusion of H2S from the lumen may explain in part the marked beneficial effects of H2S donors, when administered by enema, in models of colitis (19, 61).
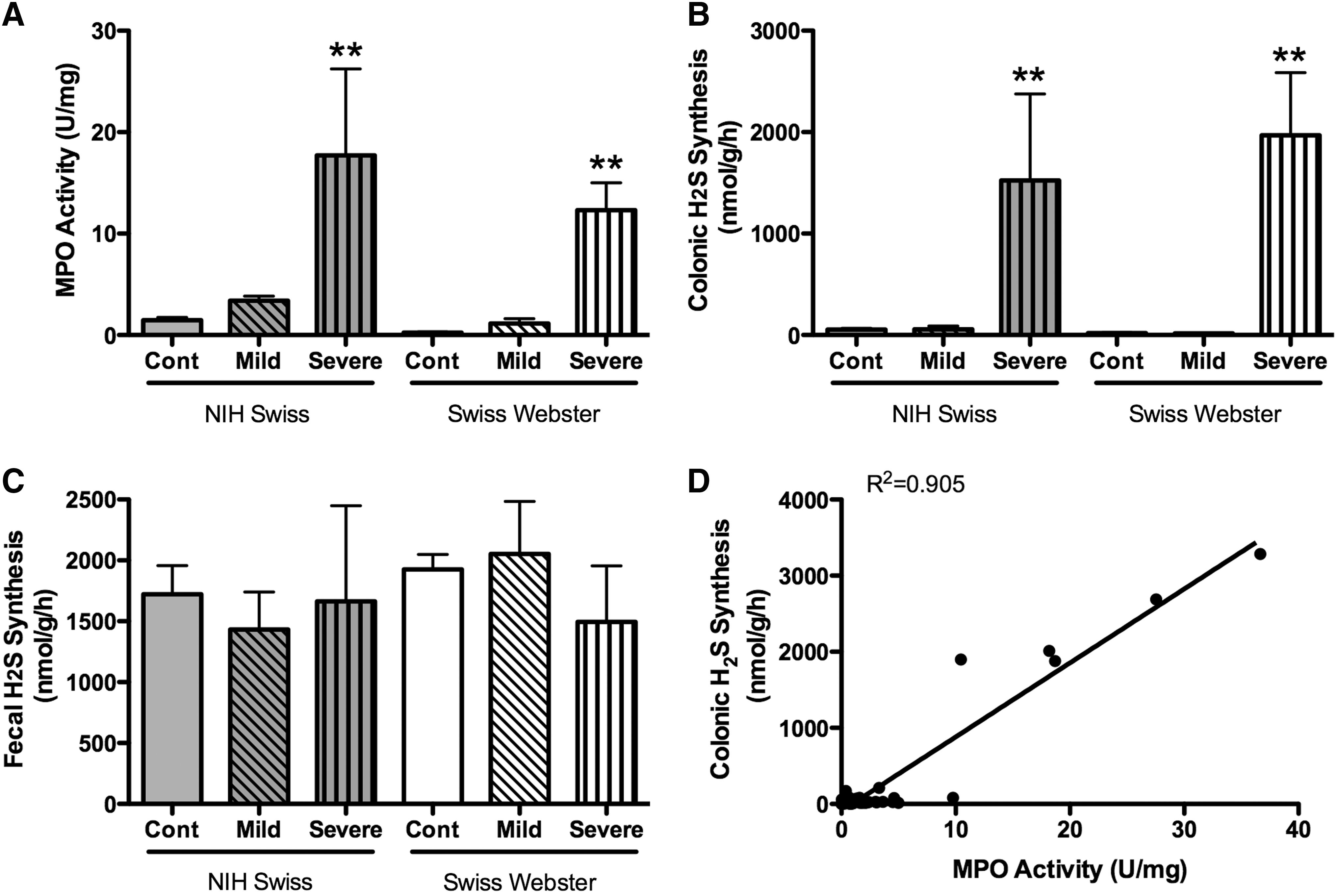
As already reviewed, colonic production of H2S is markedly increased when the mucosa is inflamed (20, 61). Because measurements of H2S in this context are generally performed in vitro, and because of the capacity of some colonic bacteria to produce H2S, we performed a series of studies to determine if some portion of what we measure as “colonic H2S synthesis” is actually bacterial H2S synthesis (20). These studies involved the use of germ-free mice and mice colonized with Altered Schaedler flora. There was no difference in colonic H2S synthesis between these two groups of mice, indicating that any bacterial contribution (in the colonized mice) was negligible (Fig. 7). We also measured tissue and fecal H2S production from healthy mice and mice with colitis induced by trinitrobenzene sulfonic acid. Colonic H2S synthesis was markedly increased in parallel with the severity of colitis and the extent of granulocyte infiltration (Fig. 8), but neutrophils (the main infiltrating granulocytes) were not a significant source of H2S synthesis. Taken together, these studies clearly demonstrated that the H2S synthesis measured using the in vitro zinc-trapping method was derived from the colonic tissue itself, rather than from any bacteria adherent to the tissue samples. On the other hand, certain bacteria may be able to modulate colonic H2S synthesis. For example, butyrate, which is produced by Bifidobacteria and Faecalibacterium prausnitzii, has been shown to regulate H2S production, at least in transformed colonocytes (WiDr cells) (11). Butyrate increased expression of CBS and CSE, the major enzymatic sources of H2S in these cells.
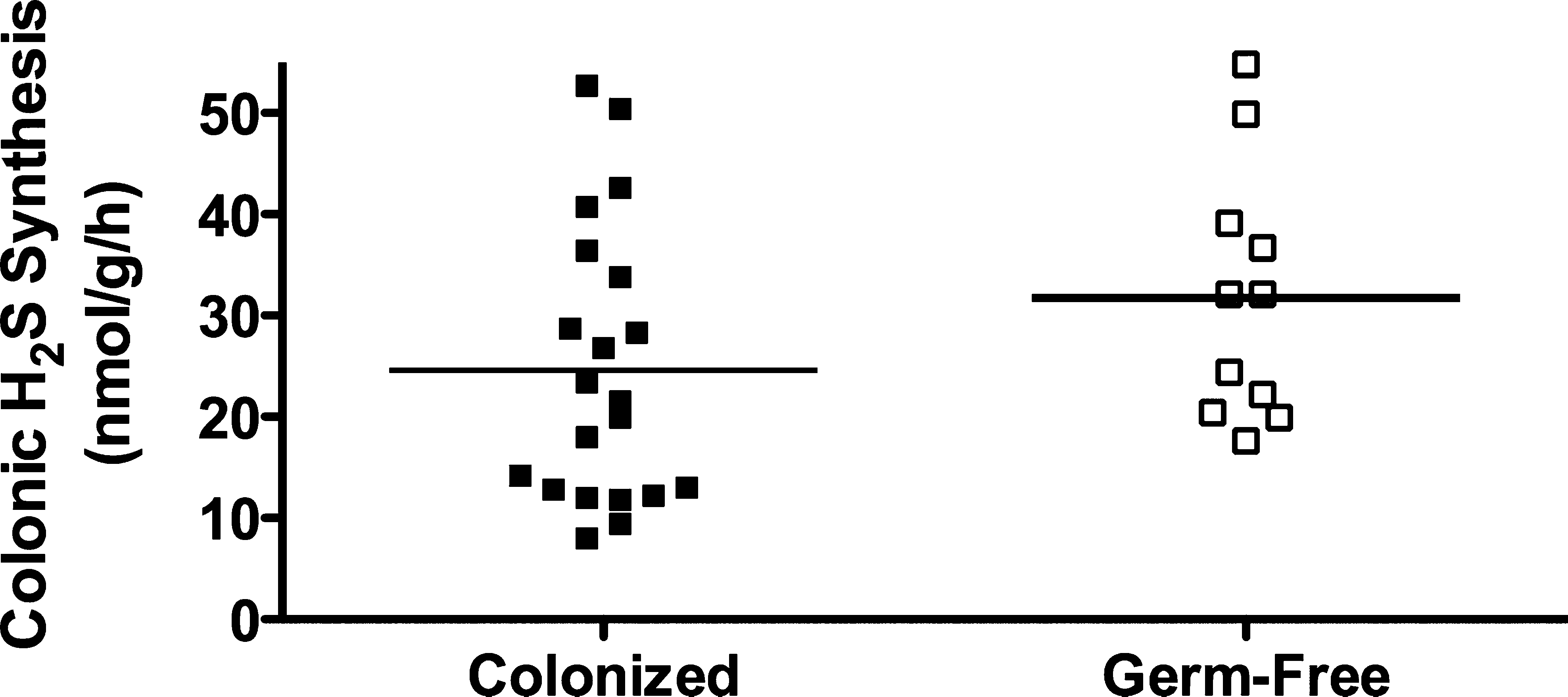
Conclusions and Future Directions
The contribution of H2S as a modulator of inflammation is becoming more clear, but studies of this gaseous mediator continue to be limited by the lack of highly selective inhibitors of its synthesis and simple methods for measuring its production in vivo. Pathways of synthesis of H2S other than via CSE and CBS are becoming evident and represent an important area for future research.
Novel therapeutic agents that release H2S and exhibit significant anti-inflammatory and/or gastrointestinal mucosal protective effects look promising in preclinical studies. The mechanisms through which H2S increases resistance to mucosal injury, promotes repair of injury, and accelerates resolution of inflammation remain incompletely understood. Evaluation of H2S-releasing drugs in a clinical setting will provide insight as to whether or not the exploitation of H2S as a therapeutic agent will live up to the promise.
The ability of many enteric bacteria to produce H2S and the possibility that bacterially derived H2S could affect mucosal function are intriguing. The intestinal epithelium appears to act as a metabolic barrier to diffusion of significant concentrations of H2S into the mucosa, at least when the epithelium is healthy. On the other hand, H2S is an important metabolic fuel for enterocytes. Given the widespread actions that H2S can exert on various cell types in the subepithelial compartment (e.g., blood vessels, enteric nerves, smooth muscle cells, resident and infiltrating immunocytes), it is important to determine the extent to which bacterial H2S can “escape” epithelial and mucosal inactivation in certain circumstances and disease conditions.
Footnotes
Acknowledgments
This work is supported by grants from the Canadian Institutes of Health Research and the Crohn's and Colitis Foundation of Canada.
Author Disclosure Statement
Dr. Wallace holds shares in Antibe Therapeutics, a company developing H2S-releasing anti-inflammatory drugs. No conflicts of interest exist for the other authors.