
Abstract
Respiratory syncytial virus (RSV) is one of the most important causes of upper and lower respiratory tract infections in infants and young children, for which no effective treatment is currently available. Although the mechanisms of RSV-induced airway disease remain incompletely defined, the lung inflammatory response is thought to play a central pathogenetic role. In the past few years, we and others have provided increasing evidence of a role of reactive oxygen species (ROS) as important regulators of RSV-induced cellular signaling leading to the expression of key proinflammatory mediators, such as cytokines and chemokines. In addition, RSV-induced oxidative stress, which results from an imbalance between ROS production and airway antioxidant defenses, due to a widespread inhibition of antioxidant enzyme expression, is likely to play a fundamental role in the pathogenesis of RSV-associated lung inflammatory disease, as demonstrated by a significant increase in markers of oxidative injury, which correlate with the severity of clinical illness, in children with RSV infection. Modulation of ROS production and oxidative stress therefore represents a potential novel pharmacological approach to ameliorate RSV-induced lung inflammation and its long-term consequences. Antioxid. Redox Signal. 18, 186–217.
I. Introduction
The treatment of RSV infection is primarily symptomatic, except possibly in immunocompromised individuals (248). The administration of supplemental oxygen to maintain oxygen saturations of ≥93% and the replacement of fluid deficits are sufficiently effective, so that the majority of infants hospitalized with RSV infection may be discharged within 72 h. Bronchodilators, corticosteroids, and other modalities are ineffective in reducing the rate of hospitalization when administered to outpatients with RSV infection and in shortening the duration of hospitalization among inpatients. As far as specific antiviral agents, ribavirin is the only licensed antiviral drug for treating RSV infection, but its use is restricted to high-risk or severely ill infants, and its utility has been limited by cost, variable efficacy, and tendency to generate resistant viruses (195). Postinfection (p.i.) treatment of RSV with anti-RSV immunoglobulin shows no benefit (187), and the current need for additional effective anti-RSV agents is well acknowledged (234).
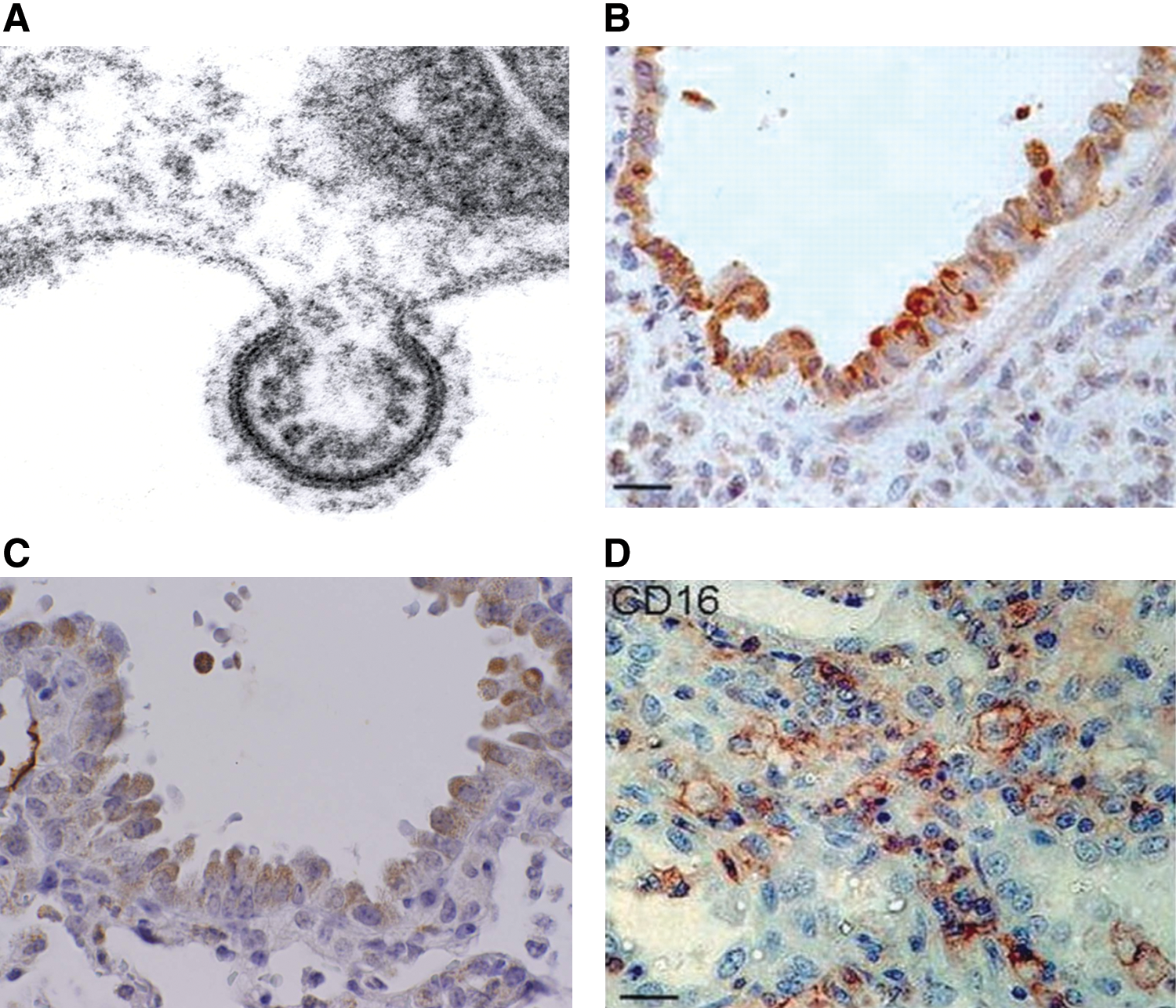
RSV vaccines with immunogenic, protective, and nonreactogenic properties are currently under investigation, but a significant obstacle to the development of vaccines for RSV is our still limited understanding of the pathogenetic mechanisms that determine the severity of ALRI caused by the virus. Paradoxically, intense research efforts boosted by the unfortunate experience with the first RSV formalin-inactivated vaccine (136) has significantly contributed to our understanding of the immune-mediated mechanisms responsible for the enhanced disease that occurred in a subset of vaccinated infants, but has incompletely identified the immunopathogenic components in naturally acquired infections (81). In this regard, although infants with certain risk factors (prematurity, chronic lung disease, congenital heart disease, or immunodeficiencies) have an increased risk for more severe RSV disease, the large majority of infants with RSV infections that require hospitalization were previously healthy (246). Therefore, the spectrum of RSV disease severity in otherwise previously healthy infants points to both host determinants and viral-specific factors that can influence the outcome of infection, and while different circulating RSV strains may in part explain differences in disease severity, these differences are relatively minor and do not at the moment provide convincing proof of the large range of disease manifestations (68, 170). On the other hand, more than 50 years of research on RSV-mediated disease has favored the role of the host response and the immunopathogenesis hypothesis, either as causing excessive/enhanced immune/inflammatory responses in the lower airways or as failing to restrict and terminate viral replication as a result of impaired innate immune response. These only apparently contrasting pathways to disease, likely to be genetically determined, may coexist and/or represent temporally distinct aspects of the host response against RSV infections, which are triggered by the initial infection of respiratory epithelium. Indeed, after entering the respiratory tract primarily through fomite or hand-to-nasal transmission after contact with infectious secretions, RSV infects the local respiratory epithelium. At that point, infection may be self-limited or may spread to the lower respiratory tract in part by a cell-to-cell transfer of the virus along intracytoplasmic bridges (96), although the mechanism by which RSV reaches the lower airway is not clearly defined, as the distribution of infected cells in LRTI is patchy. Thus, the epithelium of the respiratory mucosa, the main function of which is to provide a protective physical barrier against injurious inhaled stimuli, is clearly the main target of RSV replication, as shown by studies of infected patients (178, 250), experimental murine models (224), and by a variety of different in vitro culture systems (75, 185, 203, 258) (Fig. 1). Release of viral particle progeny occurs by a budding process, which takes place from the apical surface when assessed in cultures of polarized epithelial cells (258). As the infection progresses, necrosis and disorganized proliferation of the bronchiolar epithelium and destruction of ciliated epithelial cells become prominent features of RSV ALRI (mainly bronchiolitis) (4), with a significant amount of sloughed epithelial cell debris accumulated in the airway spaces. Influx of polymorphonuclear cells and eosinophil degranulation within the airways cells are also well-recognized features of RSV bronchiolitis (69, 137, 250), along with the peribronchial infiltrate of mononuclear cells. The exact type of such mononuclear cells has not been fully characterized, but paucity of T cells, either CD4 or CD8, in postmortem samples of RSV-infected lung suggests that lymphocytes may be absent at the time when infants with bronchiolitis are experiencing their most severe symptoms (250). Another important pathological feature of RSV bronchiolitis is submucosal edema and excess mucus secretion, which combined with cell debris and inflammatory cells cause bronchiolar obstruction, air trapping, and emphysema (4, 178). Gas exchange becomes compromised, resulting in hypoxemia, and in more severe cases the need for supportive respiratory therapy. Overall, severe RSV infections are associated with recruitment of inflammatory cells to the airway mucosa and release of potent inflammatory mediators, processes that are initiated by viral replication in epithelial cells.
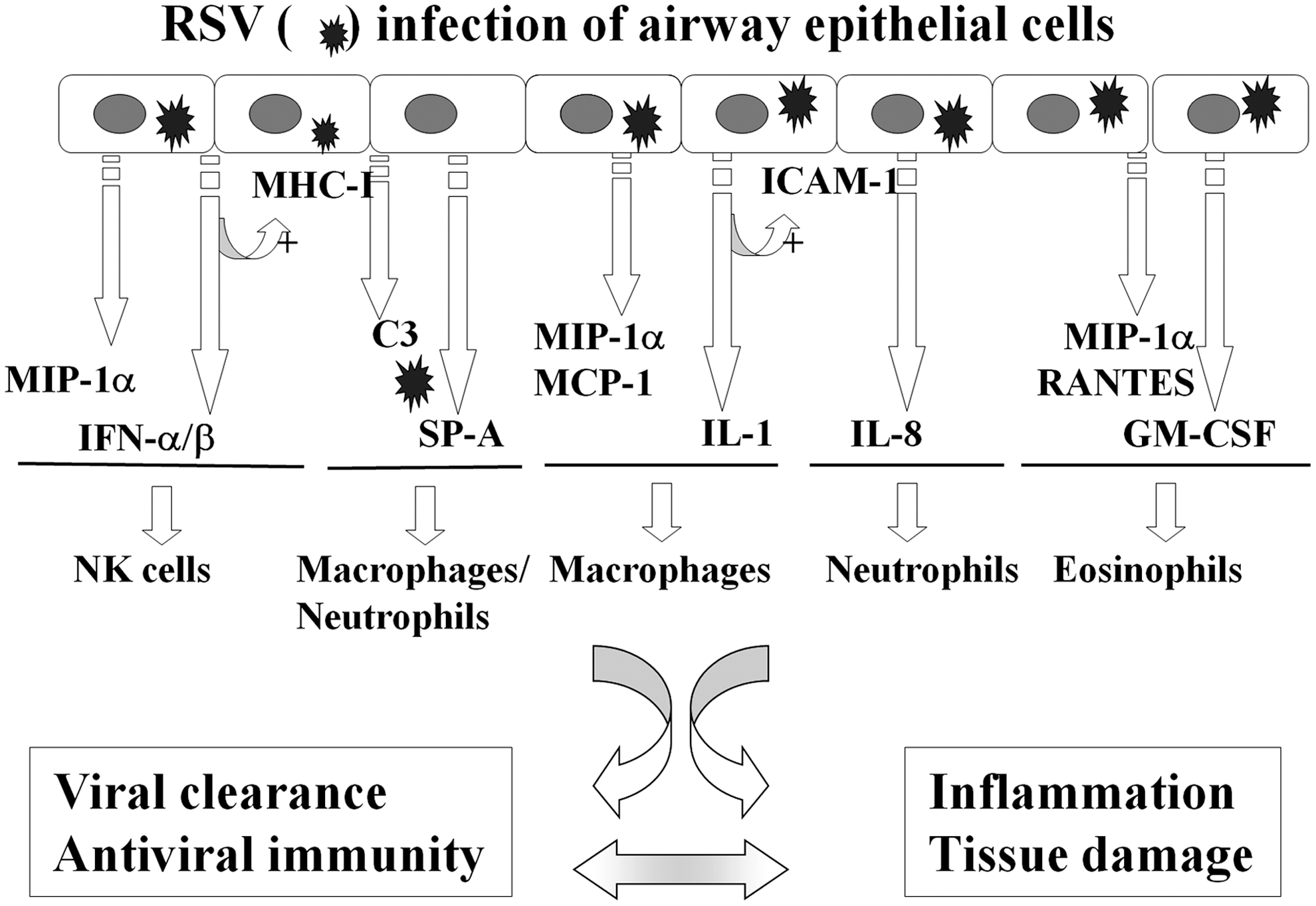
Although the mechanisms responsible for recruitment of circulating leukocytes into the lung as a consequence of RSV infection are largely unknown, chemokines are recognized as critical molecules in the recruitment and activation of leukocytes in a variety of inflammatory conditions of the lung [reviewed in (14)]. Chemokines are a superfamily of proteins divided into functionally distinct groups: three groups of small basic (heparin-binding) proteins, termed the C, CC, and CXC chemokines (based on the number and spacing of highly conserved NH2-terminal cysteine residues), and a fourth, distantly related group, the CX3C chemokines, composed of large, membrane-bound glycoproteins attached through a COOH-mucin-like domain. Chemokine receptors are expressed in a cell type-restricted fashion, allowing specificity of chemokine activity; for example, members of the C group primarily activate lymphocyte chemotaxis; members of the CXC group induce neutrophil chemotaxis, and the CC group stimulates monocyte, lymphocyte, and eosinophil chemotaxis. These data suggest that pulmonary inflammation associated with neutrophilic and monocytic infiltration is the result of coordinate expression of diverse chemokines with distinct cellular specificities. To identify chemokines produced in the course of RSV infection, a number of clinical studies have been conducted in children with RSV bronchiolitis. These studies have shown that RSV-infected infants have increased production of chemokines, including CXCL8/IL-8, CXCL10/interferon gamma (IFN-γ)-induced protein 10 (IP-10), chemokine (C-C motif) ligand 5 (CCL5)/RANTES, CCL3/macrophage inflammatory protein (MIP)-1α, and CCL2/monocyte chemotactic protein 1 (MCP-1), as measured in nasopharyngeal secretions (NPS) and tracheal aspirates or bronchoalveolar lavage (BAL) of mechanically ventilated patients (82, 167, 184, 211, 232). In some studies, the concentration of chemokines (MIP-1α and MCP-1) has been shown to significantly and inversely correlate to the degree of oxygen saturation, an objective measure of disease severity in RSV-infected infants (82). In some studies, plasma samples of RSV-infected infants with bronchiolitis have been shown to contain higher levels of IL-8 compared to infants with the milder form of disease (26). Interestingly and in contrast with all other studies, analysis of RSV-infected children at the time of visit in the emergency room has shown that those with increased levels of the cytokines IL-6, IL-10, IFN-γ, and the chemokines IL-8 and MIP-1β required less-extended supplemental oxygen therapy, suggesting that a robust immune response may play a protective role at the early phases of infection (23).
Functional studies addressing the role of certain chemokines in RSV infection have been possible only in animal models using mice genetically deficient for chemokines or chemokine receptors, or by neutralizing antibodies. These studies have identified a critical role of chemokines and chemokine receptors, including CCL5/RANTES (231), KC and its receptor CXCR2 (169), CCR1 (the receptor that binds CCL5), and CCL3/MIP-1α (95), in mediating different aspects of RSV-mediated immunopathology and airway pathophysiology. Studies of genetic polymorphism (mostly single-nucleotide polymorphism [SNP]) for single chemokine and chemokine receptor genes, including IL-8, IL-8 receptor, RANTES, CCR5 receptor (for RANTES and MIP-1α), and their association with RSV disease severity have been so far inconclusive, based on the fact that these studies have not been confirmed in more than one population or have reported conflicting results [reviewed in (170)]. Greater scientific consensus on the other hand exists in the recognition of respiratory epithelial cells as a major initiator and a modifier of host innate responses and inflammation by secreting chemokines in response to RSV infection (75, 81, 95, 183, 203). Most of these studies have been performed in cell culture, but some studies have shown RSV-mediated expression of chemokines in airway epithelial cells in vivo (95). Some chemokines appear to be selectively expressed only in epithelial cells from the lower respiratory tract, after RSV infection (185). In the most comprehensive study so far conducted to examine chemokine production, the kinetics and patterns of chemokine expression in RSV-infected lower-airway epithelial cells (A549 and normal human small-airway epithelial cells [SAECs]) have been investigated by membrane-based cDNA microarrays and high-density oligonucleotide probe-based microarrays (257). In A549 cells, RSV induced expression of a complex network of CC (I-309, Exodus-1, thymus and activation regulated chemokine (TARC), RANTES, MCP-1, macrophage-derived chemokine (MDC), and MIP-1α and -1β), CXC (GRO-α, β, and γ, ENA-78, IL-8, and I-TAC), and CX3C (Fractalkine) chemokines. Some chemokines were independently confirmed by multiprobe RNase protection assay, Northern blotting, and reverse transcription–PCR. High-density microarrays performed on SAECs confirmed a similar pattern of RSV-inducible expression of CC chemokines (Exodus-1, RANTES, and MIP-1α and −1β), CXC chemokines (I-TAC, GRO-α, β, and γ, and IL-8), and Fractalkine. In contrast, TARC, MCP-1, and MDC were not induced, suggesting the existence of distinct genetic responses for different types of airway-derived epithelial cells.
In summary, infection of respiratory epithelial cells is the first event occurring after RSV inhalation or inoculation into the nasal mucosa. This is rapidly followed by the induction of a network of epithelial cell cytokines and chemokines that have profound immune and inflammatory regulatory functions. These early elements of the host response to RSV are major determinants of the elimination or the progression of the infection, significantly affect airway mucosa inflammation, and ultimately may dictate the nature of the specific adaptive immune response to the virus (Fig. 2). Thus, understanding the mechanisms that control viral-induced gene transcription in airway epithelium is critically important to identify new therapeutic opportunities to treat ALRI caused by RSV and other respiratory viral pathogens.
II. Redox-Sensitive Transcription Factors in RSV Infection
Starting with the discovery of the complex network of cytokines and chemokines produced in response to RSV infection, a number of studies have characterized in great detail the transcriptional mechanisms that control gene expression in respiratory epithelial cells, the major target of RSV infection. RSV replication in these cells results in the activation of multiple cellular signaling pathways involved in the expression of early response genes, such as cytokines, chemokines, and type I IFN, which is coordinated by a small subset of transcription factors. Most of the studies investigating the transcriptional regulation of RSV-induced gene networks have utilized A549 cells, an alveolar type II-like cell line, as a prototype of lower airway respiratory cell, with confirmation of some of the major findings in primary human airway epithelial cells, and many have focused on gene promoter analysis of proinflammatory molecules, such as the C-X-C chemokine IL-8 and the C-C chemokine RANTES. In the following sections, we will discuss the major transcription factors that have been shown to regulate RSV-induced gene expression in epithelial cells and are known to be redox sensitive, together with some of the major signaling pathways leading to their activation. Figure 3 illustrates a part of the signaling network induced by RSV infection in airway epithelial cells.
A. Nuclear factor-IL6
The CCAAT/enhancer-binding proteins (C/EBP) are basic domain/leucine zipper-containing transcription factors involved in the inducible expression of cytokine and adhesion molecule genes, as well as in cell differentiation (8). C/EBPα, C/EPBβ/NF-IL6, and C/EPBδ are the three major members of this family. RSV infection of A549 cells induces the rapid synthesis of a single 45.7-kDa isoform of nuclear factor (NF)-IL6 in a time-dependent manner (119). NF-IL6 is first detectable after 3 h of infection and continues to accumulate until 48 h. NF-IL6 production could not be induced by ultraviolet (UV)-inactivated virus, demonstrating the requirement of viral replication for NF-IL6 synthesis. Immunoprecipitation studies after [35S]-methionine metabolic cell labeling were performed to investigate the mechanism for NF-IL6 production. In that study, there was robust NF-IL6 protein synthesis within RSV-infected cells. Protein synthesis occurred without detectable changes in the abundance or size of the single 1.8-kb NF-IL6 mRNA. RNase protection assay of transfected chloramphenicol acetyltransferase reporter genes driven by either wild-type or mutated NF-IL6 binding sites showed a virus-induced increase in NF-IL6-dependent transcription. The mechanism for enhanced translation of preformed mRNA used by RSV appears to be distinct from that used by influenza virus (241). Influenza virus infection increased rapidly and transiently the DNA-binding activity of preformed NF-IL6 protein without changing its steady-state levels. Thus, the NF-IL6 activity appears to be inducible by several distinct post-translational mechanisms in viral infections. In studies of RANTES promoter activation by RSV infection, deletions/site mutations of the NF-IL6-binding site decreased RSV-induced reporter (luciferase) activity by ∼50% (38). Similar results have been reported in studies of the intercellular adhesion molecule (ICAM-1) promoter in RSV-infected epithelial cells (45). C/EBP-β and C/EBP-δ appear to be the major components of the NF-IL6 nucleoprotein complex formed on the RANTES promoter (38).
B. Nuclear factor-kappa B
Nuclear factor kappa B (NF-κB) is a family of inducible transcription factors related by a common NH2-terminal Rel homology domain (RHD). This family includes the proteolytically processed DNA-binding subunits NF-κB1 and NF-κB2, also known as p50 and p52, and the transcriptional activators p65/RelA, cRel, and RelB [reviewed in (126)]. NF-κB is complexed and inactivated in the cellular cytoplasm by binding inhibitors of NF-κBs IκBs, which are ankyrin repeat domain-containing proteins that bind the RHD and block nuclear translocation and DNA-binding activity. The major IκBs include the isoforms α/β/ɛ and the 100-kDa NF-κB2 precursor.
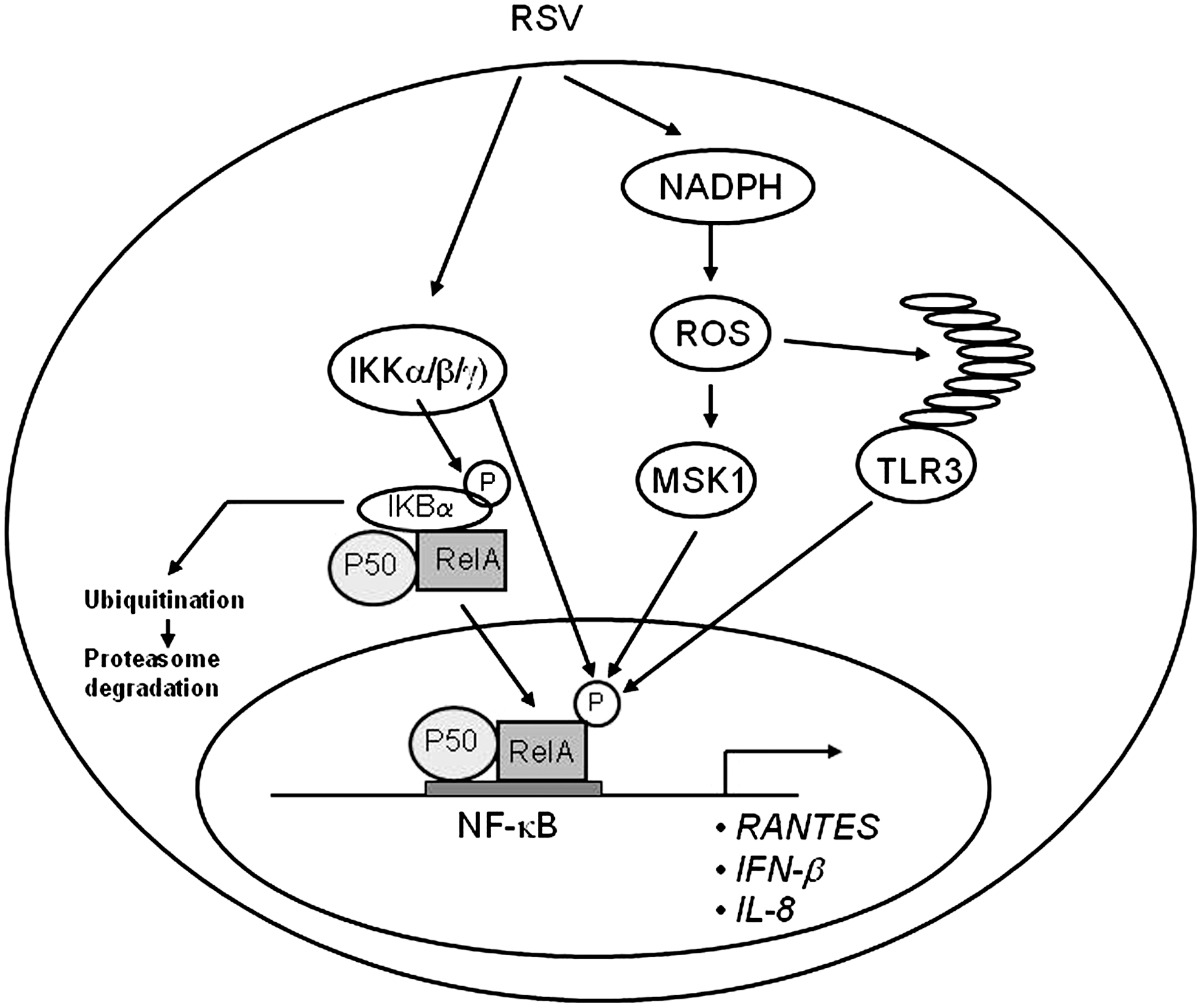
RSV is a potent activator of NF-κB in airway epithelial cells (27, 38, 83, 118). A number of RSV-inducible inflammatory and immunoregulatory genes require NF-κB for their transcription and/or are dependent on an intact NF-κB-signaling pathway. Transient transfection of the human IL-8 promoter mutated in the binding site for NF-κB demonstrated that this sequence was essential for RSV-activated transcription (27, 83, 161). Gel mobility shift assays demonstrated RSV induction of sequence-specific binding complexes, and these complexes were supershifted by antibodies directed to the NF-κB subunit RelA. Both by Western immunoblot and indirect immunofluorescence assays, it was shown that cytoplasmic RelA in uninfected cells became localized to the nucleus after RSV infection. Moreover, RelA activation requires replicating RSV, because neither a conditioned medium nor UV-inactivated RSV was able to stimulate its translocation. In addition to IL-8, NF-κB activation is essential for RSV-induced expression of RANTES (38), as well as other chemokines, cytokines, secreted proteins, and signaling molecules (27, 233). In vivo, in a mouse model of infection, RSV activates NF-κB early in the course of infection (92), and inhibition of NF-κB activation reduces cytokine production and clinical disease, without decreasing viral replication (93). Together, these findings suggest that activation of the host inflammatory response via NF-κB is a central step in the immunopathogenesis of RSV infection. As a result, the mechanisms by which RSV activates RelA have been intensively studied, particularly in the context of chemokine gene transcription.
In response to RSV infection, NF-κB translocation is mediated by two distinct pathways termed the canonical and the cross-talk pathways (118, 153). The canonical pathway is induced by cytokines, Toll-like receptor (TLR) ligands, and viral double-stranded RNA (dsRNA), and it requires a multiprotein signaling complex composed of two highly homologous serine/threonine kinases, IκB kinase (IKK)-α and β, and a regulatory subunit, IKK-γ, which exists in a precise stoichiometric relationship of two catalytic subunits to two regulatory subunits. Gene knockout studies have shown that IKK-β is the major IκB-α kinase. IKK-γ, also known as the NF-κB essential modulator (NEMO), is essential for inducible IKK activity, as IKK-γ-deficient cells are unable to activate IKK in response to all stimuli tested. IKK-β phosphorylation in its activation loop induces IκB-α NH2-terminal phosphorylation, ubiquitination, and degradation, releasing RelA to translocate into the nucleus [reviewed in (29)]. Infection of airway epithelial cells with RSV induces a progressive degradation of IκB-α and IκB-β, paralleled by the increase in RelA DNA-binding activity, through a mechanism that is partially independent of the proteasome pathway (118). RSV infection also induces IKK-β activation, demonstrated by kinase assays, starting at 3 h p.i. and peaking between 6 and 12 h p.i. with a gradual decrease at later timepoints of infection. IKK-β is necessary for RSV-induced NF-κB activation, as demonstrated using IKK-β dominant-negative (DN) mutants in reporter gene assays, or the NEMO-binding domain peptide (a known inhibitor of IKK-β catalytic activity) (59).
Upstream of the IKK complex, the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase kinase 1 (MEKK1) and transforming growth factor-beta-activated kinase 1 (TAK1), which are serine/threonine kinases in the MAP kinase kinase kinase (MAPKKK) family, have been shown to activate intracellular kinases such as p38 MAPK, JNK, and IKKs, leading to NF-κB and AP-1 induction (182). Inhibition of TAK1 activation, by overexpression of kinase inactive TAK1 or using cells lacking TAK1 expression, significantly reduces RSV-induced NF-κB nuclear translocation and DNA-binding activity, as well as NF-κB-dependent gene expression, identifying TAK1 as an important upstream signaling molecule regulating RSV-induced NF-κB activation (59).
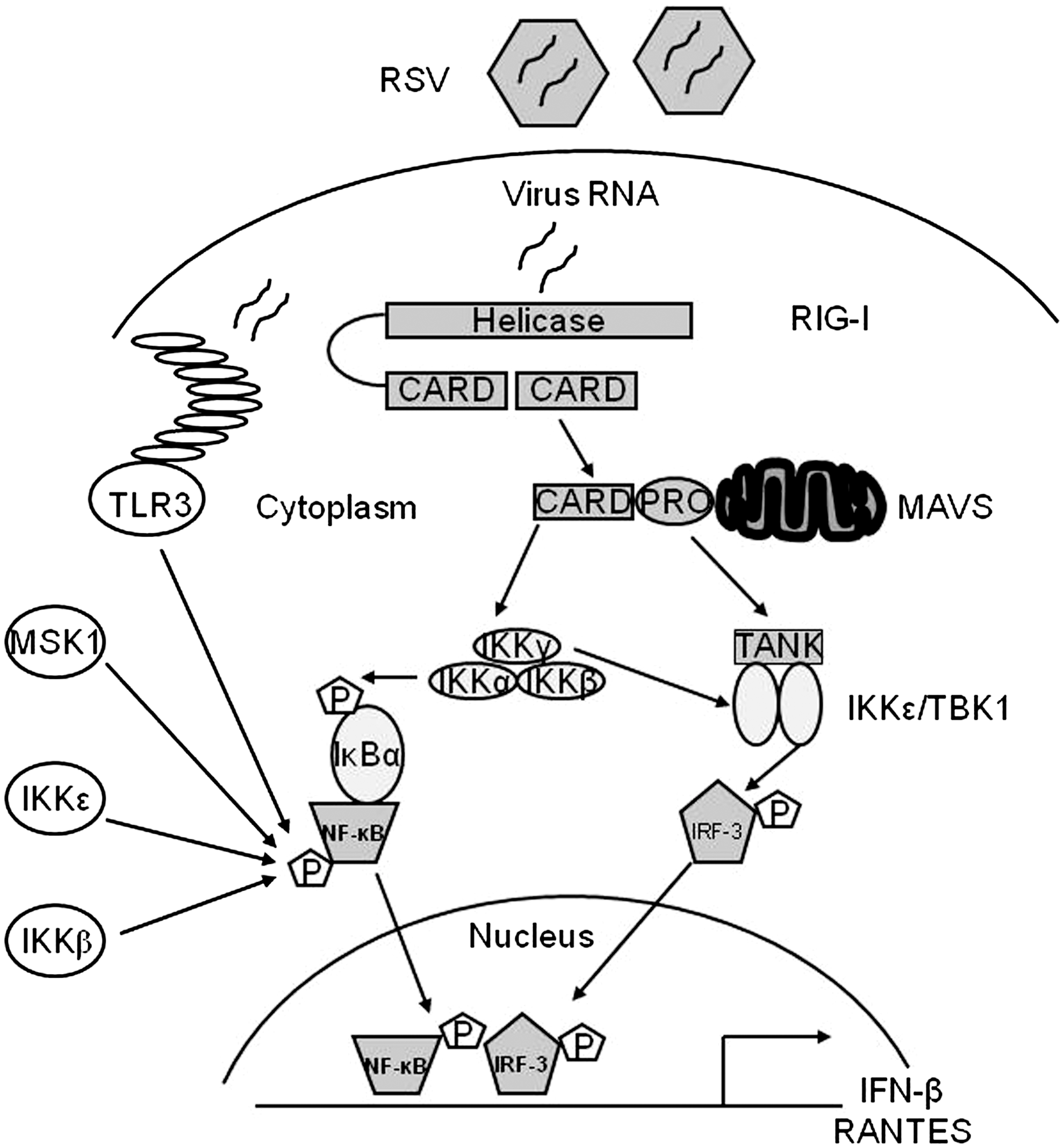
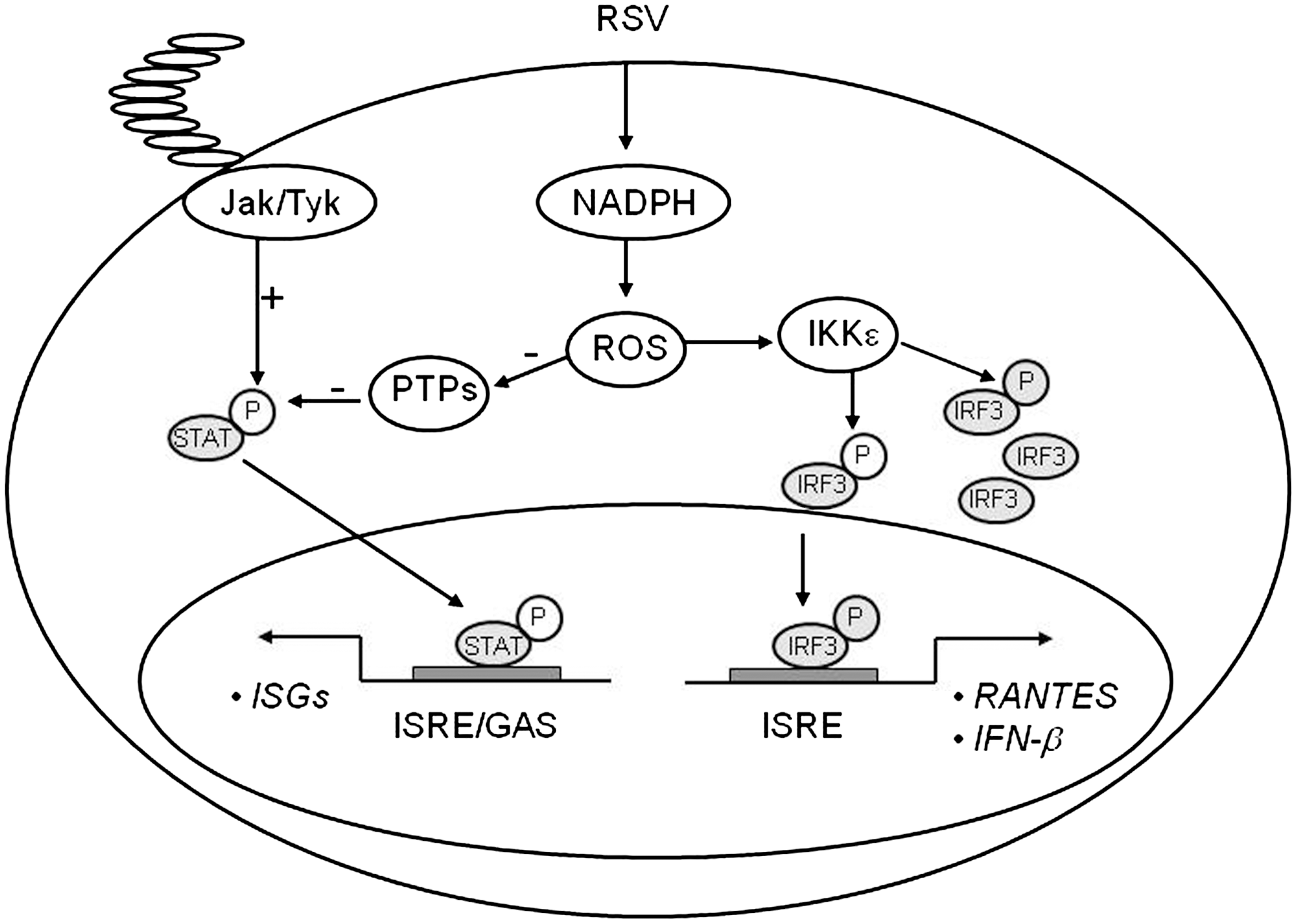
Infected host cells detect and respond to RNA viruses using different mechanisms in a cell-type-specific manner, including the retinoic acid-inducible gene I (RIG-I)-dependent and TLR-dependent pathways. In studies addressing the upstream pathways of canonical NF-κB activation, we have shown that RIG-I helicase binds RSV transcripts within 12 h of infection (152). Short interfering RNA (siRNA)-mediated RIG-I knockdown significantly inhibited NF-κB nuclear translocation and DNA binding. Consistent with this finding, RSV-induced IFN-β, IP-10, CCL5, and IFN-stimulated gene 15 (ISG15) expression levels were decreased in RIG-I-silenced cells, indicating that the RIG-I-mitochondrial antiviral signaling protein (MAVS) complex is upstream of the NF-κB canonical pathway. In addition, TLR3 (which binds dsRNA) also seems to play a role in RSV-induced NF-κB activation in airway epithelial cells, as siRNA-dependent knockdown of its expression results in the inhibition of RSV-induced chemokines and antiviral cytokines, including IP-10 and IFN-β (152, 202).
The noncanonical pathway induces processing of the 100-kDa NF-κB2 precursor into its mature 52-kDa DNA-binding form (NF-κB2) and liberation of sequestered p100-associated complexes [reviewed in (29)]. In the noncanonical pathway, p100 processing is initiated by IKK-α-mediated serine phosphorylation of the p100 COOH-terminus. IKK-α activation in the noncanonical pathway is controlled by NF-κB-inducing kinase (NIK). NIK serves as a rate-limiting upstream activator that phosphorylates IKK-α and also serves as a docking protein to recruit both p100 NF-κB2 and IKK-α into a complex. Although the focus of the noncanonical pathway has been on the RelB-NF-κB2 complexes, recent work has shown the existence of a p100-sequestered RelA-NF-κBl complex whose liberation can be induced by lymphotoxin-beta. The latter represents a cross-talk pathway where prototypical ReIA-NF-κBl DNA-binding complex is released as a consequence of activating the noncanonical NIK-IKK-α kinases. A part of the RelA activation and RelA-dependent chemokine response to RSV infection also appears to be induced through a noncanonical/cross-talk pathway involving the NIK-IKK-α complex downstream of RIG-I-MAVS activation (49, 152). In airway epithelial cells, RSV infection induces NIK kinase activity, processing of p100 to p52, and nuclear translocation of a ternary complex with IKK-α and processed p52, leading to the expression of a subset of NF-κB-dependent genes (49). Upon RSV infection, NIK associates with the RIG-I-MAVS complex via the RIG-I NH2-terminal caspase activation and recruitment domain (CARD) and the COOH-terminus of NIK, and RIG-I silencing in RSV-infected epithelial cells inhibits p100 processing to p52, suggesting that RIG-I is functionally upstream of the noncanonical regulatory kinase complex composed of NIK-IKK-α subunits (153).
Optimal NF-κB activity also requires signal-induced phosphorylation. Several NF-κB family members, in particular RelA, have been shown to be phosphorylated on specific serine residues, either constitutively or in an inducible manner, by stimuli such as lipopolysaccharide (LPS), IL-1, and tumor nuclear factor (TNF) (244, 245, 259, 260). This event is associated with an increase in p65 transcriptional activity, without modification of nuclear translocation or DNA-binding affinity (260). Among the inducible serine phosphoacceptor sites that regulate NF-κB transcriptional activity, serines 276 (Ser276) and 536 (Ser536) have been shown to be potential targets for protein kinase A (PKA) and mitogen- and stress-activated protein kinase 1 (MSK1) (239, 260) or casein kinase II and IKK-β kinases, respectively (204, 245). In addition, the IKK-like molecule IKKɛ, a critical component of the virus-activated kinase complex responsible for interferon regulatory factor (IRF)-3 activation (73, 209), has also been shown to modulate NF-κB activation in response to LPS and PMA stimulation (191, 212). IKKɛ mediates p65 phosphorylation in response to IL-1 and TNF stimulation (33, 251), and regulates constitutive NF-κB activity in cancer cells through a similar mechanism (2).
In airway epithelial cells, RSV induces a time-dependent RelA phosphorylation on both the residues Ser-276 and Ser-536 (16, 120). RelA Ser-276 phosphorylation in infected cells depends on activation of MSK1 (120), a serine/threonine kinase downstream of the extracellular signal-regulated kinases 1 and 2 (ERK1/2) and p38 MAPKs that play key roles in the immune response, cell proliferation, and apoptosis (63). Inhibition of MSK1 using H89 and siRNA knockdown reduces both RSV-induced phospho-Ser276 RelA formation and expression of a subset of NF-κB-dependent genes (120). In addition, siRNA-mediated knockdown of TLR3 (which binds dsRNA) in infected epithelial cells also results in the reduction of RSV-induced phospho-Ser276 RelA formation and inhibition of RSV-induced chemokines and antiviral cytokines, including IP-10 and IFN-β (152, 202).
The mechanism by which RSV-induced RelA Ser276 phosphorylation induces transcriptional activation is not completely understood. In other contexts, activated NF-κB mediates promoter-specific recruitment of coactivators to produce chromatin remodeling, transcription factor acetylation, and stable enhanceosome formation on inflammatory gene promoters (121). In recent investigations, we found that RSV-induced RelA Ser276 phosphorylation is required for acetylation at lysine 310, an event required for transcriptional activity and stable association of RelA with the activated positive transcriptional elongation factor-b complex proteins, bromodomain-4 (Brd4) and CDK9 (29).
The mechanisms leading to RSV-induced RelA Ser536 phosphorylation have also been investigated. We have recently shown that IKKɛ, a kinase known to regulate IRF-3 activation in response to viral infections (73, 114), also controls NF-κB transcriptional activity in response to RSV infection, by inducing RelA Ser536 phosphorylation (16). Expression of catalytically inactive IKKɛ significantly inhibits RSV-induced IL-8 secretion, promoter activation, and NF-κB-driven gene transcription, indicating a fundamental role of this kinase in the pathway leading to RSV-induced NF-κB activation. Lack of IKKɛ does not affect RelA nuclear translocation and DNA binding, but it greatly reduces RelA Ser536 phosphorylation, a post-translational modification important for RSV-induced NF-κB-dependent gene expression, as indicated by reconstitution experiments of RelA−/− MEFs (using a wild-type or Ser536Ala RelA mutant). In these cells, expression of wild-type RelA, but not Ser536Ala RelA mutant, results in enhanced expression of both basal and RSV-inducible expression of growth-regulated protein beta (Gro-β), an RSV-inducible NF-κB-dependent gene, indicating that phosphorylation of Ser536 plays an important role in RSV-induced NF-κB transcriptional activity (16). Similar results have been reported by a different group who showed that this RelA phosphorylation site is important in modulating NF-κB-dependent gene transcription in A549 cells infected with RSV (72). The presence of residual phospho-Ser536 p65 levels in cells lacking IKKɛ expression suggests the presence of potential additional pathways involved in viral-induced Ser536 p65 phosphorylation. Recent work from Yoboua et al. has shown that in airway epithelial cells, initial RSV-induced p65 Ser536 phosphorylation is dependent on RIG-I, through a pathway involving MAVS, TNF receptor-associated factor 6 (TRAF6), and the IKK-β kinase (254).
C. Activator protein-1
Studies addressing the mechanisms of inducible IL-8 and RANTES gene expression by RSV infection in epithelial cells have identified other critical transcription factors that contribute to the expression of these chemokines. Using transfection studies with reporter plasmids containing mutations in the binding sites for different transcription factors in the 5′-flanking region of the IL-8 gene promoter, it was first shown that mutation in the region of the activator protein-1 (AP-1)-binding site resulted in a diminished response to RSV, even in the presence of intact binding sites for NF-κB and NF-IL6 (163). Interestingly, when the AP-1 site is removed, but not when it is present, mutation of the NF-IL6-binding site significantly decreases responsiveness to RSV. Increased binding of AP-1 after RSV infection of A549 cells was also shown by electrophoretic mobility shift assay in the same studies. Other studies of the IL-8 promoter have shown that the presence of an AP-1-binding site is necessary for RSV inducibility, whereas TNF inducibility of the promoter stimulation mainly requires an intact NF-κB/NF-IL6 binding (39). Indeed, site mutations of the AP-1 site affect both the basal and RSV inducibility of the promoter, the latter being reduced to ∼50%. The AP-1 family of transcription factors consists of homodimers and heterodimers of Jun (c-Jun, JunB, and JunD), Fos (c-Fos, FosB, Fra-1, and Fra-2), or the activating transcription factor (ATF-2 and ATF-3) proteins (127). RSV infection induces the activation of several members of the AP-1 family, including c-Jun and ATF-2, and overexpression of c-Jun DN significantly inhibits IL-8 gene transcription (Casola A, personal communication), as well as RANTES gene transcription (38), indicating an important role of this family of transcription factors in RSV-induced gene expression.
D. Interferon regulatory factor
Other detailed studies have subsequently compared the mechanisms for induction of IL-8 in A549 cells by RSV infection and by stimulation with TNF (39). Promoter deletion and mutagenesis experiments indicated that although the region from −99 to −54 nt is sufficient for TNF-induced IL-8 transcription, this region alone is not sufficient for RSV-induced IL-8 transcription. Instead, RSV requires participation of the previously characterized element at −132 to −99 nt, containing an AP-1-binding site and of a previously unrecognized element, spanning from −162 to −132 nt, which was termed the RSV-response element (RSVRE). Analysis of the RSVRE sequence identified two potential transcription factor-binding sites: a GATA site between −151 and −147 nt, and an IFN-stimulated responsive element (ISRE)-like site between −144 and −132 nt. Mutation of the GATA site did not affect RSVRE binding, and competition assays, using oligonucleotides corresponding to consensus sequences of GATA, could not identify the RSVRE as one of them. On the other hand, Western blot analysis performed on cytoplasmic and sucrose cushion-purified nuclear extracts of A549 cells control and infected for various lengths of time demonstrated that IRF-1 protein, which modulates transcription of ISRE-containing genes, was highly inducible after RSV infection, starting between 3 and 6 h p.i. By a two-step microaffinity isolation/Western blot assay, we confirmed that RSV-induced IRF-1 was indeed binding to the IL-8 RSVRE (39).
IRF-1 belongs to a growing family of transcription factors, the IRFs [reviewed in (102)]. Nine human IRFs have been identified (IRF-1–IRF-9). Each member shares extensive homology in the N-terminal DNA-binding domain (DBD), characterized by five tryptophan-repeat elements, located within the first 150aa of the protein. The IRF DBD mediates specific binding to GAAANN and AANNNGAA sequences, termed ISRE, present in IFN-stimulated genes (ISGs). Each IRF contains a unique C-terminal domain, termed the IRF-association domain (IAD); the unique function of a particular IRF is accounted for by the ability of the IAD to interact with other members of the IRF family and other factors, its intrinsic transactivation potential, and cell-type-specific expression of the IRFs. IRF-1 is of particular interest, as this virus-inducible protein activates IFN-β, a gene highly expressed in RSV-infected epithelium. Interestingly, on the IFN-β gene, IRF-1 activates transcription only when NF-κB is coexpressed (85), indicating a common mechanism of activation with our studies on IL-8. Several studies have investigated the promoter elements involved in the regulation of IRF-1 gene expression and have identified both the NF-κB site and the gamma-IFN-activated sequence (GAS), which bind transcription factors belonging to the signal transducers and activators of transcription (STAT) family, as promoter regulatory elements necessary for inducible IRF-1 gene transcription (99). IRF-3, a critical player in the induction of type I IFNs after virus infection, is a constitutively expressed phosphoprotein. Transcriptional activity of IRF-3 is controlled by carboxyl-terminal phosphorylation events on serines 385 and 386, as well as the serine/threonine cluster between aa 396 and 405 mediated by the IKK-related kinases TBK-1 and IKKɛ. These events induce a conformational change in IRF-3 that allows homo- and heterodimerization, nuclear localization, and association with the coactivator CREB-binding protein (CBP)/p300, which retains IRF-3 in the nucleus and induces transcription of IFN-β and other genes. IRF-7 is a multifunctional protein with transcriptional activity that, like IRF-3, depends on C-terminal phosphorylation. While constitutive IRF-7 expression is restricted to B cells and dendritic cells, in the majority of the other cell types IRF-7 is virus- and IFN-inducible. IRF-7 gene transcription is controlled mainly through activation of the promoter ISRE site, which binds transcription factors of the STAT and IRF families (157). In addition, cytokine stimulation and viral infections lead to IRF-7 phosphorylation of the C-terminal region between aa 471 and 487. Transient transfection studies of the RANTES promoter using a series of 5′-deletion show that a deletion of the RANTES promoter to nt −120 completely abolishes RSV-induced RANTES transcription (38). The promoter region spanning nt −138 to −117 contains a functional ISRE site, and site-directed mutagenesis experiments clearly show that this site plays a fundamental role in promoter activation after RSV infection, since the ISRE mutant is no longer RSV inducible. Supershift assays and microaffinity isolation experiments have shown that IRF-1, 3, and 7 are components of the RSV-inducible complex formed on the RANTES ISRE site. Among the IRF proteins, IRF-3 seems to be essential for RSV-induced RANTES transcription, since overexpression of a transcription-inactive protein completely abolished RANTES promoter activation. Furthermore, Western blot analysis of cytoplasmic and nuclear proteins extracted from A549 cells infected with RSV for various lengths of time have shown that RSV infection induced de novo synthesis of IRF-7 and its nuclear translocation starting around 12 h p.i. By contrast, IRF-3 was constitutively expressed, and RSV infection induced its nuclear translocation starting around 6 h p.i. (37).
E. Signal transducers and activators of transcription
STAT proteins are constitutively expressed and, in unstimulated cells, are located in the cytoplasm. Seven STAT proteins have been identified in mammalian cells: STAT1, 2, 3, 4, 5a, 5b, and 6. Upon activation, they are phosphorylated on specific tyrosine residues, a post-translational modification necessary for dimerization and nuclear translocation, both of which are required for DNA binding [reviewed in (113)]. Upon IFN-α/β stimulation, STAT1/STAT2 heterodimers bind to the ISRE promoter sites, in the presence of an additional DNA-binding protein, p48/IRF-9, to form the ISGF3 complex. In response to IFN-γ or other cytokines, STAT dimers bind to GAS motifs on target genes (113). A number of receptor-associated and nonreceptor-associated tyrosine kinases have been shown to phosphorylate STAT proteins. Among them, the best-characterized ones are the Janus-activated kinases (JAKs). JAKs are a family of tyrosine kinases that includes JAK1, 2, 3, and Tyk2. JAK1, JAK2, and Tyk2 are ubiquitously expressed, whereas JAK3 is tissue specific. JAK1 can form heterodimers with JAK2, JAK3, or Tyk2, depending on the activating stimulus (149). In one of the first reports, RSV infection was shown to induce a rapid (within 30 min) phosphorylation and nuclear translocation of STAT1 in A549 cells and in normal human bronchial cells after RSV infection (142). Treatment of cells with heparin or heparinase blocked RSV cell attachment/infection and RSV-induced activation of STAT1. When A549 cells were preincubated with AG490, an inhibitor of JAK, decreased RSV-induced STAT1 phosphorylation was observed. In the same study, RSV also activated STAT3 via an IL-6-dependent pathway. We have also shown that RSV infection of A549 cells leads to a time-dependent activation of STAT1, STAT2, and STAT3 and their binding to the IRF-1 GAS site and to the IRF-7 ISRE site (155). STAT1 and STAT3 bind IRF-1 GAS, whereas STAT1, STAT2, IRF-1, and IRF-9 bind IRF-7 ISRE, leading to induction of gene expression. In our study, we were not able to consistently show a role of JAK in RSV-induced STAT activation; however, we found that RSV infection inhibits intracellular tyrosine phosphatase activity, which is critical for STAT activation, suggesting that modulation of phosphatases could be an important mechanism of virus-induced STAT activation (155).
F. Hypoxia-inducible factor
Hypoxia-inducible factor 1 (HIF)-1α is a transcription factor that functions as a master regulator of mammalian oxygen homeostasis. HIF-1 is composed of two subunits: constitutively expressed HIF-1β and oxygen-regulated HIF-1α. Under normoxic conditions, HIF-1α is subjected to hydroxylation on proline residues. The modification is required for the binding of the von Hippel-Lindau (VHL) tumor suppressor protein, the recognition component of an E3 ubiquitin protein ligase that targets HIF-1α for proteasomal degradation. Under hypoxic conditions, hydroxylation is inhibited, and the VHL protein does not bind to HIF-1, eventually leading to stabilization of the alpha-subunit, heterodimerization, nuclear translocation, and transcription of HIF-dependent genes [reviewed in (207)]. Although the erythropoietin (EPO) gene has been identified as a primary target of HIF-1 transcription, binding of HIF-1 to the EPO enhancer promoter region results in the transcriptional induction of a number of genes that are involved in inflammation or infection processes (216). HIF-1α has also been identified as a key regulator of NF-κB (243), and a recent study suggests that NF-κB is a critical transcriptional activator of HIF-1α, and that basal NF-κB activity is required for HIF-1 protein accumulation under hypoxic conditions (199). RSV infection of respiratory cells leads to HIF-1α activation via nitric oxide (NO)-dependent protein stabilization (94). Activation is replication-dependent, as addition of UV-inactivated virus to epithelial cells is unable to increase HIF-1α protein levels. HIF-1α activation in viral-infected cells regulates the expression of several genes, including vascular endothelial growth factor (VEGF), CD73, and cyclo-oxygenase-2 (COX-2), whose induction is abolished in pulmonary epithelia after siRNA-mediated repression of HIF-1α. Hypoxia does not seem to be the major trigger of RSV-induced HIF-1α activation, as measurements of the partial pressure of oxygen in the supernatants of RSV-infected epithelia or controls revealed no apparent differences in oxygen content. Finally, in vivo studies of RSV infection have confirmed HIF-1α activation in the lung of experimentally infected mice (94).
III. ROS in RSV-Induced Cellular Signaling and Oxidative Stress
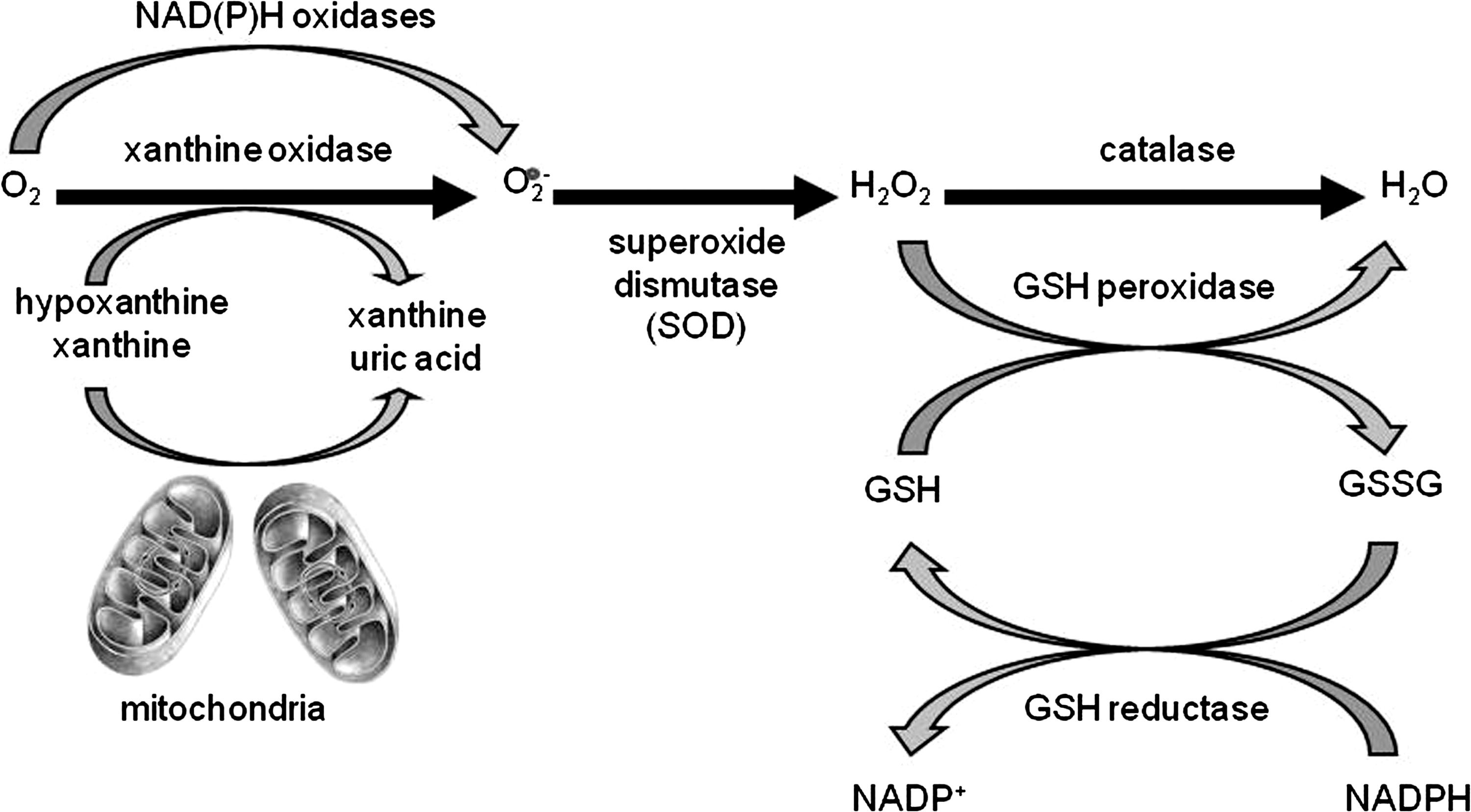
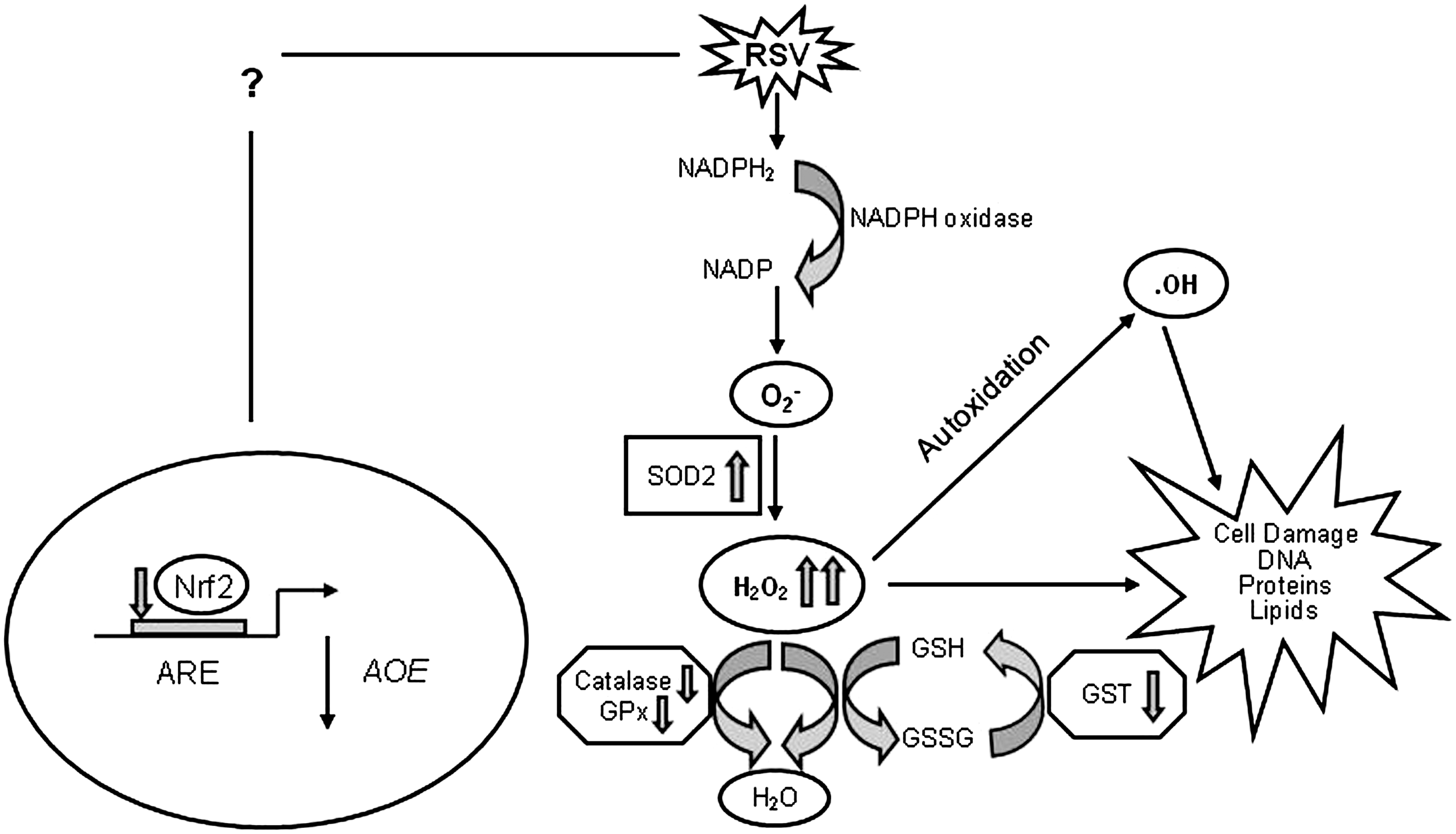
Reactive oxygen species (ROS) are ubiquitous, highly diffusible, and reactive molecules produced as a result of reduction of molecular oxygen, including species such as superoxide anion radical (O2 −), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), and they have been implicated in damaging cellular components like lipids, proteins, and DNA. The toxicity of ROS depends on the presence of a Fenton catalyst, such as iron ions and peroxidases, which in the presence of O2 − and H2O2 give rise to the extremely reactive •OH radical. ROS and in particular •OH can interact with a variety of molecules, such as membrane lipids, leading to lipid peroxidation, which impairs membrane functions, inactivates receptors, and increases tissue permeability [reviewed in (66)]. Aldehydes, generated during the process of lipid peroxidation, such as malonaldehyde (MDA) and 4-hydrynonenal (4-HNE), can diffuse within and escape from cells and attack targets far from the site of the original free-radical generation. ROS formation takes place constantly in every cell during metabolic processes. Cellular sites for ROS generation include the mitochondria, microsomes, and various enzymes like COX, lipoxygenase, xanthine oxidase, and membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Fig. 4). Excessive levels of ROS can be generated by increased stimulation of the otherwise tightly regulated NADPH oxidase (mitochondrial- and cell membrane-associated) or by other mechanisms that produce ROS accidentally, which is often due to mitochondrial dysfunction or increased activity of the above-mentioned enzymes. Endogenous antioxidants comprise a number of enzyme systems, such as superoxide dismutase (SOD) (three isoforms of SOD have been identified in mammals: the cytoplasmic Cu/ZnSOD or SOD1, the mitochondrial MnSOD or SOD2, and the extracellular ECSOD or SOD3), catalase, and glutathione peroxidase (GPx), as well as nonenzymatic molecules such as reduced glutathione (GSH) [reviewed in (66)] (Fig. 4). Depending on the source and type of ROS generation, each system plays a major role in limiting intracellular and extracellular oxidative stress. In the lungs, neutrophils, eosinophils, monocytes, and macrophages recruited at sites of infection/inflammation are a major source of supraphysiological levels of O2 − production, most of which undergoes dismutation resulting in H2O2 formation. The presence of eosinophil- and neutrophil-derived peroxidases, EPO and myeloperoxidase (MPO), respectively, then greatly enhances the oxidizing potential of H2O2 by leading to the formation of •OH.
In addition to being responsible for cellular oxidative stress, there has been increased recognition of the role of ROS as central regulators of cellular signaling [reviewed in (10, 66, 194)]. Modulation of gene expression in response to ROS formation is a complex phenomenon, as a variety of signal transduction molecules have been shown to be redox sensitive, from transcription factors to kinases and phosphatases, to chromatin-remodeling proteins. In the following sections, we will describe the suggested mechanisms of RSV-induced ROS production and their role in cellular signaling and oxidative cellular damage in response to RSV infection.
A. ROS generation in RSV infection
Several viruses, including human immunodeficiency virus (HIV), Hepatitis B and C, rhinovirus, and influenza, have been shown to induce ROS in a variety of cell types [reviewed in (189, 190)]. In the past few years, we have shown that infection of airway epithelial cells with RSV induces ROS formation, measured by oxidation of the cell membrane permeable compound 2′,7′ dichlorodihydrofluorescein diacetate (DCFDA), which is trapped intracellularly after cleavage by cellular esterases and becomes fluorescent once oxidized (144, 159). RSV-inducible ROS production was detectable as early as 2 h p.i., with a progressive increase observed up to 24 h p.i., resulting in several-fold induction in DCFDA fluorescence in infected cells compared to uninfected (37, 120). In addition to epithelial cells, RSV interaction with professional phagocytes, such as monocytes and polymorphonuclear cells, in particular neutrophils and eosinophils, has been shown to lead to superoxide production (70, 137). H2O2 generated in these two types of cells becomes the substrate of the eosinophil and neutrophil MPO, leading to the release of potent pro-oxidative mediators in the extracellular environment.
As mentioned before, cellular sites for ROS generation include the mitochondria, microsomes, and various enzymes such as COX, lipoxygenase, xanthine oxidase, and membrane-bound NADPH oxidase. The latter one is an important source of inducible intracellular ROS, generated in response to a variety of stimuli [reviewed in (13)]. Initially thought to be restricted to phagocytic cells and used to kill invading microorganisms, superoxide production by the NADPH oxidase system has been reported in a variety of nonphagocytic cells, including epithelial cells. This system consists of the catalytic subunit gp91phox (known as NOX2), together with the regulatory subunits p22phox, p47phox, p40phox, p67phox, and the small GTPase RAC. The enzyme activity of gp91phox is regulated by the assembly of these regulatory subunits with gp91phox to form an active complex. In addition to NOX2, six functional oxidase homologs, NOX1, NOX3, NOX4, NOX5, Duox1 and 2, and two homologs of the regulatory units, NOXO1 and NOXA1, have been identified in the past several years, with different tissue expression [reviewed in (147)]. Although the role of NADPH oxidases in innate responses to bacterial infection is well documented, little is known regarding their role in ROS generation and modulation of cellular responses after viral infections, in particular in cells other than phagocytes, such as airway epithelial cells, the primary target of RSV infection.
The first demonstration that RSV induces superoxide production and chemokine secretion through a NADPH oxidase-dependent pathway came from the observation that skin fibroblasts derived from a patient with congenital granulomatous disease, due to lack of p47phox expression, showed a significant reduction in RSV-induced ROS production and IL-8 secretion, compared to skin fibroblasts of normal donors (130). Using a variety of inhibitors, including diphenylene iodonium chloride (DPI), apocynin, and 4-(2-aminoethyl) benzene sulfonyl fluoride (AEBSF), we have shown that NADPH oxidases play an important role in modulating RSV-induced RANTES expression, as well as activation of the transcription factors IRF-3 and STAT1 and of the upstream kinase IKKɛ in airway epithelial cells (114, 154). Inhibition of NADPH oxidases also resulted in a modest decrease of viral replication (114). The importance of the NADPH oxidase system in regulating redox-dependent cellular signaling in response to RSV infection was confirmed by the inhibition of NF-κB serine phosphorylation and transcriptional activity, as well as RIG-I/MAVS-dependent IRF-3 activation in airway epithelial cells in which the expression of NOX2 was significantly downregulated by siRNA treatment (72, 218).
In regard to other respiratory viruses, rhinovirus infection of bronchial epithelial cells has been shown to induce ROS formation (24) through NOX1 (53), whereas NOX2 seems to play an important role in inflammatory cell superoxide production in response to influenza infection (240).
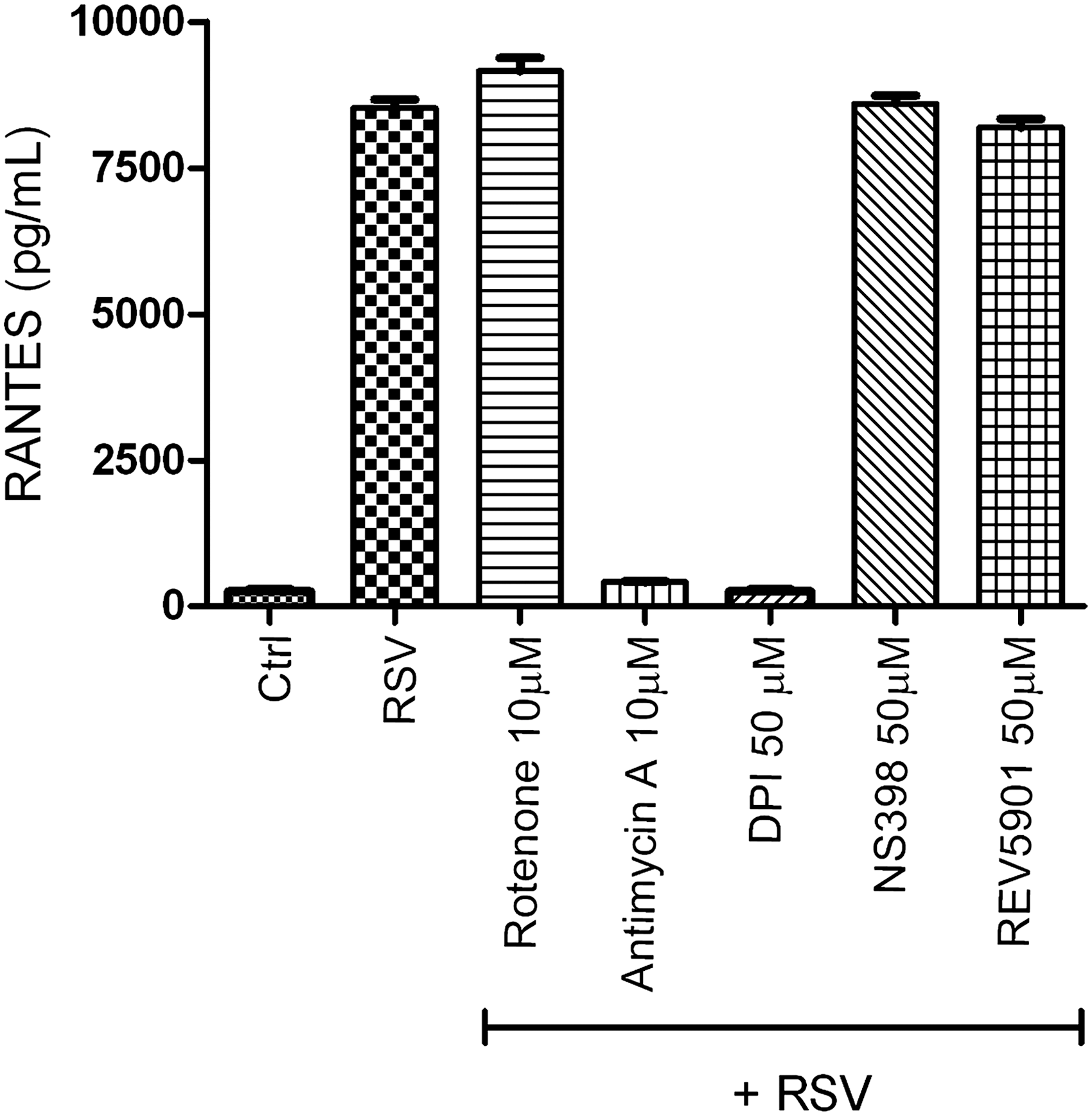
In an attempt to identify the other sources of RSV-induced ROS generation, we tested the effect of inhibiting different sources of intracellular ROS on RSV-induced chemokine secretion (Casola A, unpublished observation). We infected A549 cells with RSV, MOI of 3, in the presence or absence of DPI, rotenone (an inhibitor of mitochondrial complex 1), antimycin A (a mitochondrial complex 3 inhibitor), NS 398 (a COX inhibitor), and REV 5901 (a lipoxygenase inhibitor). Cell supernatants were harvested 24 h after infection, and the RANTES concentration was determined by ELISA. As shown in Figure 5, RSV-induced RANTES production was significantly inhibited by DPI and antimycin A treatment, suggesting that the mitochondria could represent also an important site of ROS production in viral-infected epithelial cells.
B. ROS as mediators of cellular signaling in RSV infection
As described before, RSV infection of airway epithelial cells results in activation of a subset of transcription factors, including NF-κB, AP-1, IRF, and STAT proteins, which control the expression of a variety of proinflammatory/immunological mediators, such as cytokine, chemokines, and type I IFNs. While the role of ROS in activation of some of these transcription factors has been extensively investigated, as in the case of NF-κB, AP-1, and STAT proteins (35, 205, 213), little is known on the potential redox regulation of others, such as IRF proteins. We summarize the evidence in support of ROS-dependent regulation of RSV-induced cellular signaling in the following sections.
1. NF-κB/NF-IL6/AP-1 activation
The first evidence that changes in oxidant tone regulate RSV-induced cellular signaling came from a study of Mastronarde et al., in which RSV-infected epithelial cells treated with the antioxidant dimethyl sulfoxide (DMSO) showed a dose-dependent decrease in IL-8 secretion and IL-8 mRNA, without significant changes in viral replication (162). The same group demonstrated that DMSO, as well as the other antioxidants dimethyl pyrroline N-oxide and N-acetylcysteine (NAC), significantly reduced RSV-induced nuclear translocation and DNA-binding activity of AP-1 and NF-IL6 to their respective binding sites of the IL-8 promoter, whereas there was no effect on NF-κB binding (163). That study however did not examine the effect of antioxidants on NF-κB transcriptional activity, which is regulated by a variety of post-translational modifications, including phosphorylation and acetylation (42, 43). In particular, inducible phosphorylation on distinct serine residues, including Ser276 and Ser536, has been shown to regulate NF-κB transcriptional activity without modification of nuclear translocation or DNA-binding affinity (260). We have recently shown that treatment of airway epithelial cells with NAC or DMSO significantly reduced RSV-dependent NF-κB serine phosphorylation, resulting in the inhibition of RSV-induced expression of several NF-κB-dependent genes, without affecting nuclear translocation (120). Inhibition of RSV-induced NF-κB serine phosphorylation was also shown in epithelial cells in which the expression of NOX2 was significantly downregulated by siRNA treatment (72), as previously mentioned, indicating that in airway epithelial cells, ROS regulation of RSV-induced NF-κB activation occurs at a step subsequent to activation through the canonical pathway. Cellular treatment with either NAC or DMSO had no significant effect on RSV gene transcription, as N-protein mRNA expression was the same in treated versus untreated infected cells, although overall viral replication, assessed in terms of formation of a fully assembled virus, was not assessed. As mentioned earlier in this review, MSK1 is one of the upstream kinases regulating RSV-induced NF-κB serine phosphorylation (120), and its kinase activity has been reported to be redox sensitive (3). Airway epithelial cell exposure to either NAC or DMSO strongly inhibited viral-induced MSK1 activity to nearly that of uninfected cells, suggesting that this is an important mechanism of redox control of viral-induced NF-κB activation (120) (Fig. 6). The molecular basis of ROS-dependent activation of MSK1 in RSV-infected cells is currently unknown.
In addition, RSV-induced NF-κB phosphorylation is also controlled through activation of TLR3 (90, 152), and TLR3 activation in airway epithelial cells has recently been shown to be enhanced by oxidative stress (140). Treatment of macrophage with melatonin, a pineal gland-secreted hormone with antioxidant properties, significantly reduced TLR3-dependent NF-κB activation and gene expression in RSV-infected cells (109), suggesting a possible additional ROS-dependent mechanism controlling NF-κB induction in response to RSV infection (Fig. 6).
A role of ROS in IL-8 and NF-κB activation has been demonstrated for other respiratory viruses as well, such as rhinovirus (24) and influenza (48, 139), suggesting a common regulatory mechanism of this pathway in response to viral infections of airway epithelial cells.
2. IRF/STAT activation
IRF transcription factors have been shown to play a fundamental role in the induction of several genes involved in the immune/inflammatory response to viral infections, including type I IFN, cytokines like IL-15, adhesion molecules, major histocompatibility complex-I (MHC-I) molecules, and inducible nitric oxide synthase (iNOS) [reviewed in (180)]. As mentioned before, IRF protein binding to the ISRE of the RANTES promoter is necessary for viral induction of RANTES transcription and gene expression (151). Pretreatment of RSV-infected airway epithelial cells with a panel of chemically unrelated antioxidants can effectively inhibit RANTES secretion, mRNA induction, and gene transcription, indicating the involvement of ROS in RANTES gene expression (37). Treatment of airway epithelial cells with the antioxidant butylated hydroxyanisole (BHA) blocks RSV-induced de novo IRF-1 and 7 gene expression and protein synthesis, and inhibits IRF-3 nuclear translocation and DNA binding to the RANTES ISRE (37), an event required for RSV-induced RANTES gene transcription. The mechanism by which BHA affects RSV-induced IRF-3 activation involves inhibiting its serine phosphorylation, a fundamental mechanism of IRF protein activation, necessary for nuclear translocation, dimerization, binding to DNA, and activation of transcription [reviewed in (208)].
Treatment of airway epithelial cells with BHA or a panel of NADPH oxidase inhibitors, including DPI, apocyanin, and AEBSF, inhibited gene expression and protein synthesis of IKKɛ, a kinase that plays a major role in RSV-induced IRF-3 activation (114), indicating for the first time that ROS formation plays a major role in the signaling pathway leading to viral-induced IRF-3 activation. A confirmation of the redox sensitivity of this pathway, in the context of viral infections, was provided by the observation that interference with NOX2 expression inhibited RSV- and Sendai virus-induced activation of the mitochondrial-associated adaptor MAVS, as well as IRF-3 serine phosphorylation and dimerization, with subsequent inhibition of IRF-3-dependent gene expression (218) (Fig. 7).
One additional study has shown that induction of IFN-stimulated genes (ISGs) by LPS, which occurs in an IRF-3-dependent manner, requires the generation of ROS by the NADPH-dependent oxidase system. In that study, activation of ASK1 kinase linked LPS-induced ROS production to the activation of MKK6 and p38, two kinases that had previously identified as components of the LPS-induced IRF-3 activation cascade (44).
As mentioned above, BHA treatment of airway epithelial cells blocks RSV-induced IRF-1 and 7 gene expression, and this inhibition occurs at the level of gene transcription. IRF-1 gene expression is induced by IFN-γ and cytokines through the activation of STAT and NF-κB transcription factors (99). Similarly, IFN-γ activates IRF-7 gene transcription through an ISRE site that binds members of the STAT family (157). RSV infection induces STAT protein phosphorylation, nuclear translocation, and binding to the IRF-1 and 7 promoters (155), an event necessary for IRF-inducible transcription, as seen with IFN stimulation (99, 157). Antioxidant treatment completely abolishes RSV-induced STAT activation, by blocking tyrosine phosphorylation, therefore preventing STAT translocation and DNA binding to both the IRF-1 and IRF-7 ISRE promoter sequences (155). Recent studies have shown that activation of the JAK-STAT pathway is redox-sensitive. Simon et al. demonstrated that STAT1 and STAT3 are activated in response to H2O2 in fibroblasts (215), and JAK-STAT activation after stimulation with angiotensinogen II and oxidized low-density lipoprotein is inhibited by antioxidant treatment (165, 166). Little is known about the role of ROS in viral-induced STAT activation. Very recently, Gong et al. have shown that the human hepatitis C virus NS5A protein alters intracellular calcium levels, triggering ROS formation and nuclear translocation of NF-κB and STAT3, which are completely inhibited by the use of antioxidants (89). Similarly, the hepatitis B virus X protein induces ROS formation, through association with an outer mitochondrial anion channel, an event that leads to NF-κB and STAT3 activation, which is sensitive to antioxidant treatment, as well as SOD2 overexpression (247). The mechanism involved in ROS formation and STAT activation, after RSV infection, is not fully understood. In airway epithelial cells, we found that RSV infection significantly inhibited protein tyrosine phosphatase (PTP) activity, which was restored by antioxidant treatment (155). Very little information is available regarding viral-induced regulation of PTPs activity and their role in signaling pathways activated by viral infections. Exogenous H2O2 treatment has been shown to induce an increase in the overall intracellular levels of protein tyrosine phosphorylation, triggering activation of multiple signaling molecules and transcription factors, ultimately leading to gene expression (100), and it has been suggested that one of the mechanisms mediating the biological effects of H2O2 treatment is inhibition of protein phosphatases (100). More specifically, two studies have shown that H2O2 inhibits specific PTPs, whereas antioxidants can increase PTPs activity (58, 168, 200). It is possible that viral-induced ROS activate STAT through an imbalance between cellular tyrosine kinases and phosphatases, resulting in increased net phosphorylation and therefore activation of this transcription factor (Fig. 7).
C. RSV and oxidative stress
Oxidative stress has been implicated in the pathogenesis of several acute and chronic airway diseases, such as asthma and chronic obstructive pulmonary disease (COPD) [reviewed in (74)], as well as in influenza-induced lung disease (7, 226). Oxidant species have been shown to mimic in vivo of the pathophysiological features of airway diseases such as bronchoconstriction, airway hyper reactivity, mucous hypersecretion, enhanced arachidonic acid cascade, increased synthesis of chemoattractants, epithelial damage, and microvascular leakage [reviewed in (51)].
In the past few years, we have shown that RSV infection induces significant oxidative stress, defined as a disruption of the pro-oxidant–antioxidant balance in favor of the former, in vitro as well as in vivo, due to an impairment of the antioxidant defense system. Our findings of a progressive increase in lipid peroxidation products, such as 8-isoprostanes, MDA, and 4-HNE, and a progressive reduction of the GSH/glutathione disulfide (GSSG) ratio provide strong evidence of increased oxidative stress in RSV-infected airway epithelial cells (106). When lipid peroxidation products were measured in the BAL of RSV-infected mice, there was a significant increase of MDA and 4-HNE levels in BAL samples at all timepoints tested, when compared to uninfected mice, indicating that lung oxidative stress damage indeed occurs in RSV infection (40). In addition, we measured the levels of F2 8-isoprostane and MDA in NPS of children with RSV-proven infections of increasing clinical severity, that is, from milder upper respiratory tract infections (URTI), to bronchiolitis with or without hypoxia (the group of children with hypoxic bronchiolitis included also subjects who required intubation and ventilatory support [VS] because of respiratory failure) (105). Concentration of F2 8-isoprostane in NPS was slightly increased in subjects with mild (nonhypoxic) bronchiolitis compared to those with URTI, but the difference was not statistically significant. However, subjects with hypoxic bronchiolitis had significantly more F2 8-isoprostane in NPS than did subjects with URTI alone (p<0.01) or with nonhypoxic bronchiolitis (p<0.001). A similar trend was observed for MDA concentrations in a smaller number of NPS samples that were tested (105).
As mentioned before, cells are protected against oxidative damage by well-developed enzymatic and nonenzymatic antioxidant systems, including SOD, catalase, GSH-dependent enzymes, thioredoxin (TRX), and peroxiredoxins (Prdxs), which protect cells against ROS and cytotoxic products of lipid peroxidation. Antioxidant enzymes (AOEs) can either directly decompose ROS (e.g., SOD and catalase) or facilitate these antioxidant reactions (e.g., peroxidase using GSH as a reducing agent). In the case of RSV infection, the protective mechanism of upregulating antioxidant defenses after increased ROS formation occurred only at the very beginning of infection, with an increase in SOD1, SOD2, catalase, and glutathione S-transferase (GST) expression and GPx activity in airway epithelial cells at 6 h p.i. While SOD2 expression and total SOD activity continued to increase during the timecourse of RSV infection, there was a progressive decrease in the expression and activity of all the other tested AOEs. SOD enzymes convert superoxide anion to H2O2, and catalase and GPx convert H2O2 to water and oxygen (106). The increase in total SOD activity together with the progressive decrease in catalase, GPx, and GST expression and activity suggests that RSV infection likely results in enhanced intracellular H2O2 production, which is not detoxified by AOEs, leading to the generation of free radicals, such as the hydroxyl radical (•OH), which reacts with lipids, proteins, and DNA, causing structural cellular damage.
Using a proteomic approach to investigate changes in abundance/function of nuclear proteins in response to RSV infection, we also found that several family members of the Prdx family, such as Prdx-1, Prdx-3, and Prdx-4, were oxidized in response to RSV without changes in their total abundance (122). Cells lacking Prdx-1, Prdx-4, or both showed increased levels of ROS formation and a higher level of protein carbonylation in response to RSV infection. Using a saturation fluorescence labeling and 2-dimensional electrophoresis gel analysis, we showed that 15 unique proteins had enhanced oxidative modifications of at least >1.2-fold in the Prdx knockdowns in response to RSV, including annexin A2 and desmoplakin, suggesting that Prdx-1 and Prdx-4 are essential for preventing RSV-induced oxidative damage in a subset of nuclear intermediate filament and actin-binding proteins in epithelial cells. Prdxs are a family of AOEs that catalyze reduction of H2O2 and alkyl hydroperoxides in the presence of TRX, TRX reductase, and NADPH [reviewed in (193)]. There are six family members, all expressed in the lung, and they have been shown to play an important role not only in peroxide scavenging but also in peroxide-dependent cellular signaling (206). Enhanced TRX expression in a mouse model of influenza infection was protective against severe disease, with significant reduction in lung inflammation and pneumonia in TRX-expressing mice (177), indicating that this system plays an important role in regulating inflammatory process during host defense against respiratory viral infections.
Decreased expression/activity of antioxidant proteins was also observed in vivo, both in a mouse model of RSV infection, as well as in children with severe bronchiolitis (105). Using a proteomic approach to identify proteins whose expression in BAL changed during the course of RSV infection, we found that levels of several AOEs, from Prdx enzyme to catalase, SOD1, GPx 1, and various forms of GST, were significantly decreased in the lungs of infected animals compared to uninfected (Table 1 summarizes all antioxidant proteins whose expression in BAL changes in response to RSV infection). Most of the AOE levels were significantly reduced as early as day 1 p.i. (with the exception of Prdx-2), and they were significantly lower throughout the acute phase of RSV infection, compared to control mice, to return to basal or slightly above basal levels by day 25 p.i., indicating that RSV significantly diminishes antioxidant defenses in the lung. To confirm the data obtained in the mouse model, we measured levels of SOD1, 2, and 3, catalase, and GST-mu in NPS of infected children. For this analysis, infants on ventilator support (VS), that is, those with most severe illness, were analyzed as a separate group. SOD1 levels were lower in infants with bronchiolitis, hypoxic bronchiolitis, and on ventilatory support (VS), compared to those with URTI alone. The VS group also showed significantly lower levels of SOD3, catalase, and GST-mu compared to the other illness groups, suggesting a correlation between levels of AOE expression and severity of RSV infection.
Shown are high probability antioxidant protein identifications and their expression (in terms of fold changes in RSV BAL compared to control mice) at different days of postinfection from peptide mass fingerprinting in MALDI-TOF/MS.
Reprinted with permission of the American Thoracic Society. Copyright © 2011 American Thoracic Society (81).
BAL, bronchoalveolar lavage; —, not determined.
In support of the concept that oxidative stress could be linked to the pathogenesis of paramyxovirus-induced lung diseases, we have recently shown that human metapneumovirus (hMPV), a pneumovirus belonging to the Paramyxoviridae family responsible for a significant portion of lower respiratory tract infections in children (31), significantly affects AOE expression in vitro and in vivo. hMPV infection of airway epithelial cells induces a progressive decrease of SOD3, catalase, GST, and Prdx gene expression and protein levels, with a concomitant increase in SOD2 and no change in SOD1 expression (17), similar to what we have observed with RSV. Similar findings were also observed in a mouse model of hMPV infection (105).
In regard to other respiratory viruses, rhinovirus infection of bronchial epithelial cells has been shown to induce ROS formation (24) and to increase SOD1 expression and total SOD activity at early timepoints of infection, with no changes in SOD2, catalase, and GPx (129). However, AOE expression/activity was investigated only at 6 h p.i., but not at later timepoints. Similar to RSV, influenza virus induces SOD2 gene expression in airway epithelial cells, with a concomitant decrease in catalase gene expression (116). Similar findings were reported in a mouse model of influenza infection, with increased expression of SOD2 (47); however, total lung SOD and catalase activity, as well as the ratio of GSH/GSSG, have been shown to be reduced in mice after influenza infection (146). Taken together, these data clearly suggest that airway oxidant–antioxidant imbalance occurring during RSV infection, and possibly viral respiratory infections in general, could play a very important role in the pathogenesis of RSV-induced lung disease.
D. Potential regulatory mechanisms of AOE gene expression in RSV infection
The only AOE gene that showed significant induction after RSV infection of airway epithelial cells is SOD2. The human SOD2 gene promoter contains binding sites for several transcription factors such as NF-κB and AP-1 [reviewed in (138)], with NF-κB being necessary for SOD2 gene expression, in response to cytokine stimulation (54, 55). RSV being a potent activator of NF-κB in airway epithelial cells (83), it is likely that its induction is responsible for the observed increase in the SOD2 expression and activity level in the course of RSV infection. The mechanism leading to decreased expression/activity of the majority of AOEs after RSV infection is currently unknown. SOD3 and catalase expression has been shown to be negatively regulated in response to cytokine stimulation such as IL-1, tumor necrosis factor-α, and IFN-γ (50). Transcription of many oxidative stress-inducible genes is regulated in part through cis-acting antioxidant-responsive element (ARE) sequences. This element has been identified in the regulatory regions of many genes encoding phase-2 detoxification enzymes and various other cytoprotective proteins such as NAD(P)H:quinone oxidoreductase (NQO1), which catalyzes the reduction of a variety of quinones and quinoid compounds and many AOEs, including SOD1, catalase, heme oxygenase 1 (HO-1), GST, and GSH-generating enzymes such as glutamate cysteine ligase [reviewed in (117)]. NF-E2-related factor 2 (Nrf2) is a protein that has been well established as an important redox-responsive protein that helps protect the cells from oxidative stress and injury [reviewed in (117)]. It is a basic leucine-zipper transcription factor that is normally bound in the cytosol to an inhibitor called Kelch-like-ECH-associated protein 1 (Keap1). This association normally renders Nrf2 inactive by shuttling it toward degradation by the ubiquitin–proteasome pathway. However, when ROS are generated, Keap1 undergoes conformational change and releases Nrf2, which associates with small MAF proteins and translocates to the nucleus to bind to ARE or MAF recognition elements (MARE) and promote gene transcription (117).
Several transcription factors can antagonize Nrf2-dependent gene transcription by (i) competing for binding to the ARE, (ii) inhibiting Nrf2 activation through direct physical association, or (iii) interfering with recruitment of coactivators (CBP) to the ARE site [reviewed in (242)]. Bach1 is also a basic leucine-zipper protein that, similar to Nrf2, binds to small MAF proteins and then to ARE/MARE, acting as a transcriptional repressor (60). Transcription factors belonging to the AP-1 family, such as the immediate early proteins c-Fos and FRA1, can also compete with Nrf2 for binding to the ARE and inhibit gene transcription (242). On the other hand, ATF-3, as well as retinoic acid receptor α, can form inhibitory complexes with Nrf2, leading to displacement of the coactivator CBP from AREs and inhibition of Nrf2-dependent gene transcription (242). Finally, activation of NF-κB can lead to decreased Nrf2-dependent gene transcription by decreasing the availability of coactivators (CBP) and promoting the recruitment of corepressors (histone deacetylases) (242).
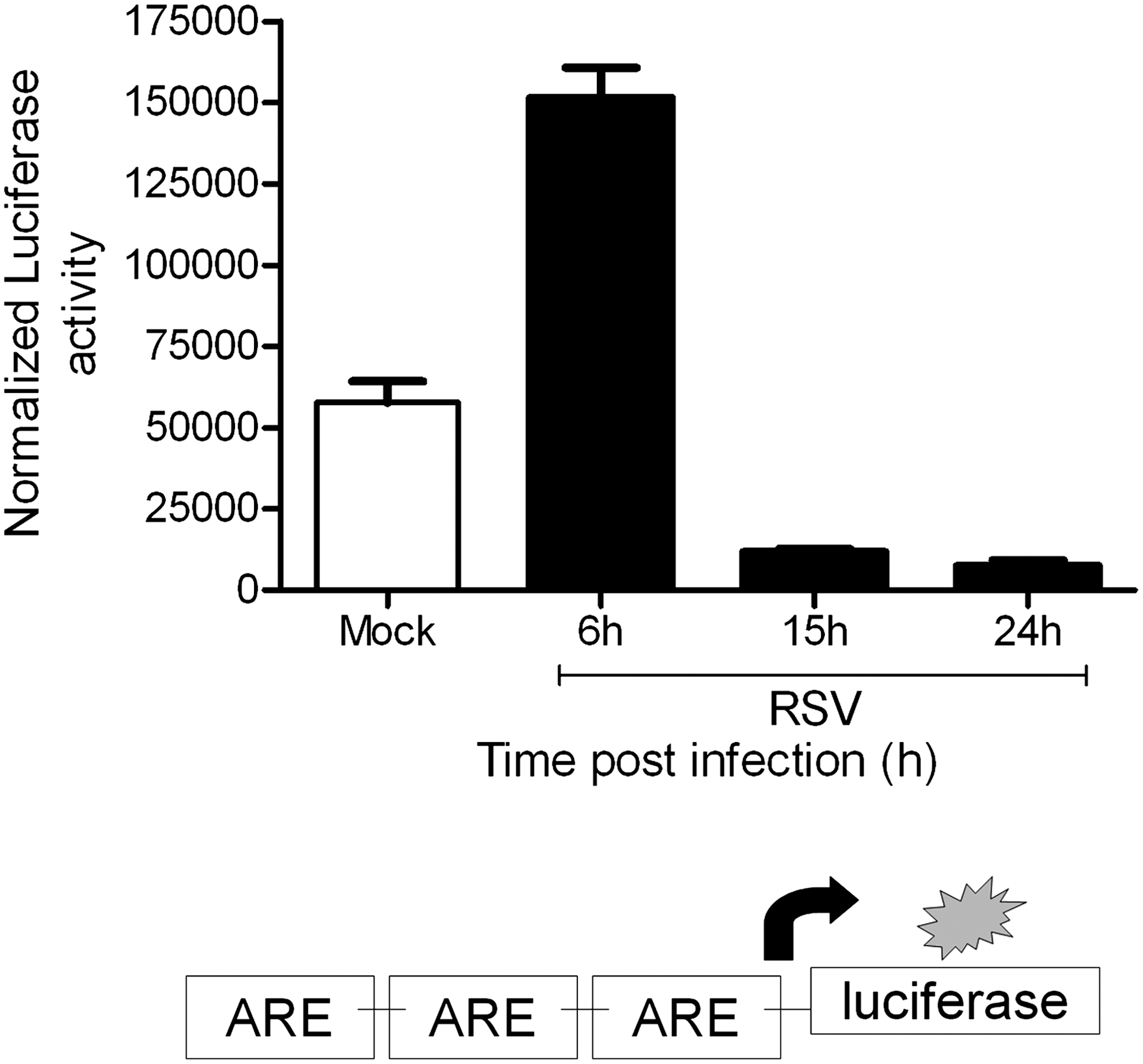
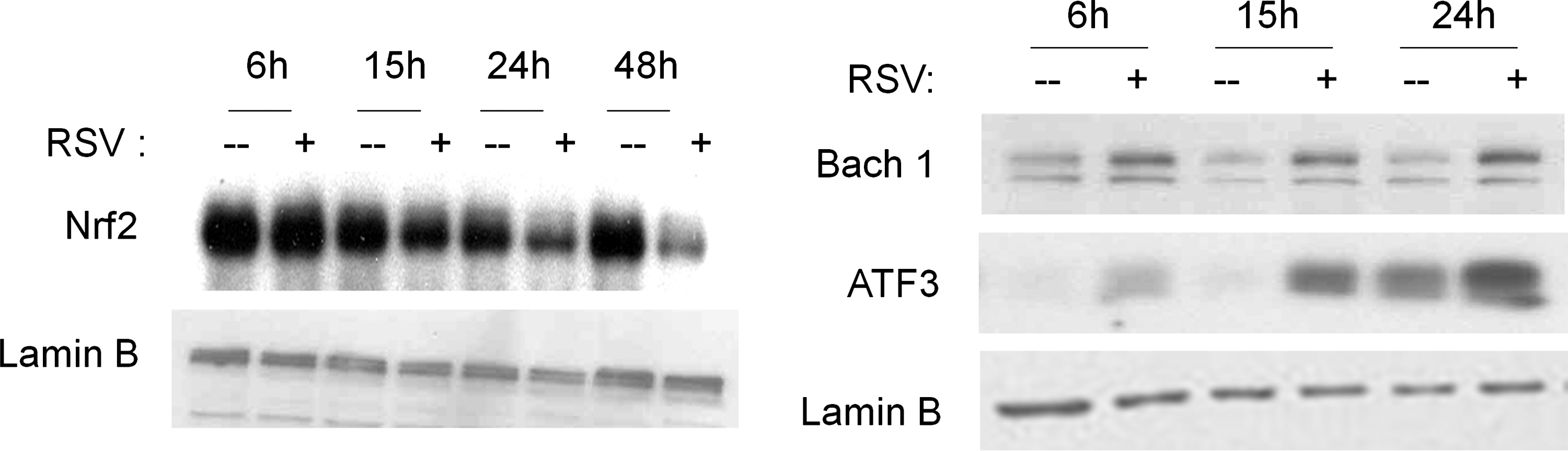
A recent study has shown that the Nrf2-ARE pathway plays a protective role in the murine airways against RSV-induced injury and oxidative stress (46). More severe RSV disease, including higher viral titers, augmented inflammation, and enhanced mucus production, and epithelial injury were found in Nrf2−/− mice compared to Nrf2+/+ mice. Preliminary studies indicate that RSV infection indeed induces a time-dependent decrease in ARE-dependent gene transcription, investigated using luciferase reporter assays (Casola A, unpublished observation) (Fig. 8). RSV leads to a decrease in nuclear levels of Nrf2 in A549 and in SAECs (106), while increasing nuclear levels of known ARE transcriptional repressors such as Bach1 and ATF-3 (Casola A, unpublished observation) (Fig. 9), suggesting a potential mechanism for viral-induced downregulation of AOE gene expression. Reduced nuclear levels of Nrf2 can occur as a result of various mechanisms, including decreased expression, increased degradation, or through increased nuclear export (128). As Nrf2 positively regulates its own gene transcription, there are reduced Nrf2 mRNA levels in airway epithelial cells at a late timepoint of RSV infection, as we recently published (106). In Figure 10, we summarized possible mechanisms leading to RSV-induced oxidative stress in airway epithelial cells.
IV. Role of Reactive Nitrogen Species in RSV Infection
Reactive nitrogen species (RNS) are a group of chemically reactive unstable metabolites generated by NO metabolism. NO, a gaseous nitrogen-centered inorganic radical, is an endogenously synthesized free radical first characterized as a noneicosanoid component of endothelial-derived relaxation factor (78). NO is produced by a variety of mammalian cells, including vascular endothelium, neurons, smooth muscle cells, macrophages, neutrophils, platelets, and pulmonary epithelium. NO is synthesized in biological tissues by the oxidative deamidation of
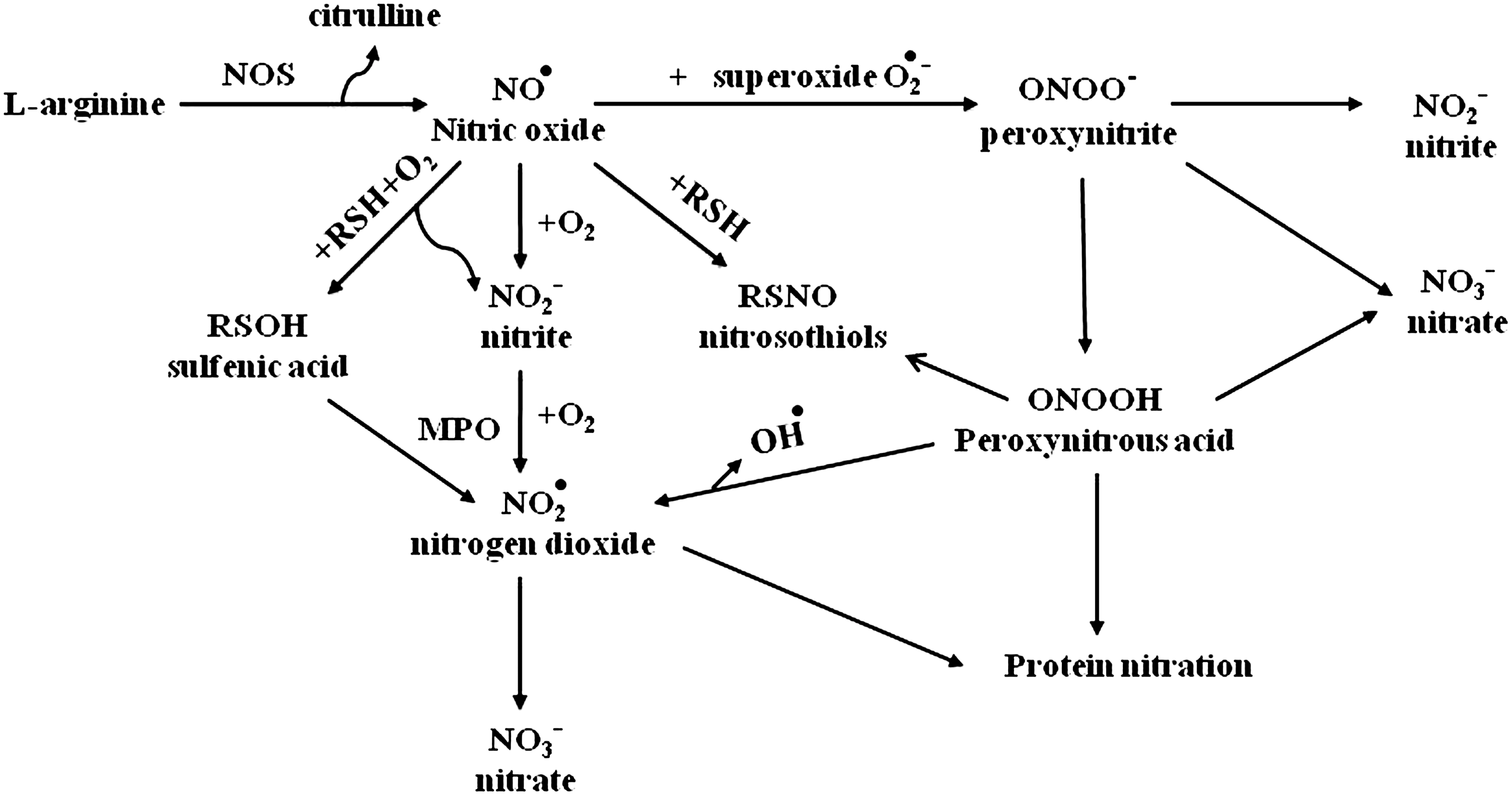
A variety of lung cells have been shown to produce NO. Human airway epithelial cells can express eNOS and iNOS, with the latter one highly induced after exposure to proinflammatory cytokines and oxidants (12). iNOS is also expressed in monocytes/macrophages and other types of antigen-presenting cells (52). In the respiratory system, NO derived from constitutive NOS (cNOS) has homeostatic effects, including dilation of the pulmonary blood vessels and relaxation of airway smooth muscle. In contrast, NO derived from iNOS, which usually produces larger amounts (100–1000 times more) of NO compared to cNOS enzymes, is involved in RNS formation and nitrosative stress, and has been implicated in the pathogenesis of a number of inflammatory lung diseases (22). In the following sections, we will briefly review the available information regarding the role of NO/NRS in RSV infection.
A. NO production and iNOS expression in RSV infection
NO production has been demonstrated in a variety of viral infection models (5, 6, 255), and iNOS induction is now being considered as a possible universal event in all viral infections. Induction of iNOS in response to RSV has been shown in A549 cells and primary SAECs (125, 235), Hep-2 cells (9), bronchial epithelial cells, and Clara cells (217). Tsutsumi et al. were the first to demonstrate induction of the iNOS gene by RSV infection in A549 cells (235). Although in several viral infections, including RSV, iNOS gene expression can be regulated through the secretion of cytokines such as IL-1β, TNF-α, and IFN-γ (6, 91), in case of RSV infection, initial iNOS induction appears to be independent of cytokine stimulation (235), similar to what has been reported with hepatitis B and polio virus (156, 158). Increase in iNOS protein, with a concomitant increase in nitrite levels, was also reported in SAECs after RSV infection (125). UV-inactivated RSV failed to increase either NO release or iNOS protein in these studies, suggesting again that iNOS expression in airway epithelial cells depends on viral replication. Pretreatment with IL-4 reduced the production of NO in response to RSV infection in A549 cells, without any effect on iNOS expression, suggesting that the Th1/Th2 balance affects the ability of epithelial cells to produce NO in response to RSV infection (125). In contrast to these observations in epithelial cells, no increase in NO production was reported in human monocytes infected with RSV (197).
Induction of iNOS occurs at the transcriptional level. The promoter of the iNOS gene has two NF-κB-binding sites and an ISRE site, to which IFN regulatory factors bind (230). Although the mechanism of RSV-induced iNOS expression has not been investigated, we and others have shown that RSV infection of airway epithelial cells strongly induces transcription factors belonging to both the NF-κB and IRF families (38, 83, 235).
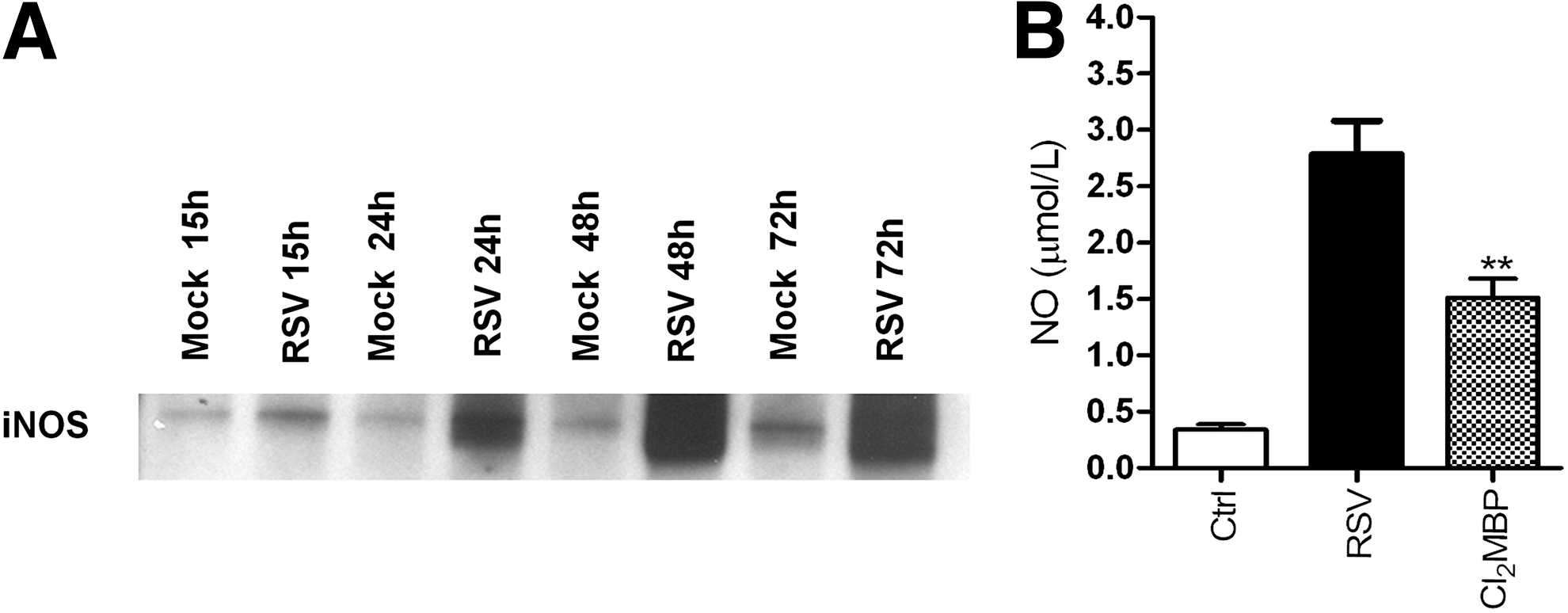
RSV induction of NO production and iNOS expression has also been investigated in vivo. Significantly increased production of NO, NOS activity, and expression of iNOS mRNA has been demonstrated in the lung of RSV-infected mice (222). Immunohistochemical analysis identified iNOS in the respiratory epithelium as the major NOS enzyme expressed during acute RSV infection. In agreement to this study, mouse lung epithelial cell extracts from the lungs of RSV-infected mice show a significant induction of iNOS protein (Kolli D, unpublished data) (Fig. 12A). In addition, using a macrophage-depletion model, we found that the NO/NRS production in mice infected with RSV also depends upon resident lung macrophages (Kolli D, unpublished data) (Fig. 12B). A similar role of macrophages in RSV-induced NO production was recently confirmed in a lamb model of RSV infection (219). Compared to the full term, the lungs of preterm lambs showed decreased nitrite levels and a trend toward increased arginase activity, suggesting that macrophages are either immature or differentially activated in preterm animals infected with RSV.
In contrast to the in vitro and animal studies, adult volunteers experimentally infected with RSV (but not showing clinical signs of lower respiratory tract infection) did not demonstrate a change in nasal and oral NO production compared to control subjects (86). In addition, a reduction of exhaled NO has been reported in infants during the acute phase of RSV bronchiolitis, compared with healthy controls, which returned to normal levels during the convalescence phase (79). High levels of NO metabolites (nitrite/nitrate) have been reported in the spinal fluid of children infected with RSV with central nervous system symptoms (173).
B. Effect of NO on RSV replication, cellular signaling, and lung disease
NO production in the course of a viral infection has been shown to have multiple effects; it can inhibit viral replication, cause viral mutation, and play a role in disease pathology. Using a series of HEp-2 cell clones that express iNOS constitutively and generate varying amounts of NO, Ali-Ahmad et al. demonstrated that iNOS and NO production results in a reversible, dose-dependent inhibition of RSV replication (9). Furthermore, when the HEp-2 parent cell line was treated with increasing concentrations of the chemical NO donor, S-nitroso-N-acetyl-penicillamine (SNAP), no inhibition of viral replication was observed except at high concentration of SNAP, suggesting that the NO derived from endogenous iNOS expression was much more efficient at inhibiting RSV replication than was the addition of the exogenous NO via the chemical donor SNAP (9). In a model of persistently infected dendritic cells, endogenous NO production seemed to be important in controlling RSV replication, as shown by increased viral titers in cells treated with the NOS inhibitor nitro-
NO production also seems to be important in modulating RSV replication in vivo. Using a mouse model of infection, Stark et al. demonstrated that inhibition of iNOS results in increased RSV lung titers (222). Similarly, in a mouse model of hypereosinophilia, due to overexpression of IL-5, which shows increased viral clearance, increased iNOS expression in the lungs correlated with decreased RSV replication (192). Treatment of mice with iNOS inhibitors before and during RSV infection significantly delayed RSV clearance in wild-type mice, and completely reversed the advantage conferred by the hypereosinophilic status of the IL-5 transgenic mice, suggesting that the eosinophil-derived NO contributes to innate protection against RSV (192).
Various intracellular signaling molecules are regulated by NO, from kinases to transcription factors such as NF-κB and AP-1, as they contain critical cysteine residues that undergo nitrosylation (221). Airway epithelial cell treatment with the iNOS inhibitors L-NAME, L-NG-monomethyl arginine (L-NMMA), and aminoguanidine did not have a significant effect on RSV-induced IL-8 or RANTES production (37, 162), suggesting that NO does not affect the intracellular signaling pathways leading to RSV-induced NF-κB, AP-1, and IRF activation. However, treatment of a human bronchial epithelial cell line (BEAS-2B) with free 3-nitrotyrosine (NO2Tyr) induced both a decrease in viral replication and a reduction of chemokine secretion via formation of nitrated α-tubulin, which resulted in alteration of cellular microtubule properties (110).
An important effect of increased NO production in response to RSV infection is the modulation of ion channel activity (217). Viral-infected cells show a significant reduction in activity of amiloride-sensitive epithelial Na+ channels, the main pathways through which Na+ ions enter lung epithelial cells (41). This inhibition appears to be mediated by NO, through upregulation of iNOS as a result of activation of the NF-κB pathway, as incubation of cells with the specific iNOS inhibitor 1400W significantly reverses viral-induced Na+ channel inhibition (217). This could represent an important mechanism by which RSV decreases alveolar fluid clearance, as demonstrated in BALB/c infected mice, resulting in increased levels of lung water and hypoxemia (56).
In addition, RSV-induced NO production causes stabilization of the transcription factor HIF-1α in a time-dependent manner (135). VEGF is one of the many gene targets of HIF-1α (76). Treatment of human primary bronchial epithelial cells infected with RSV with a NO inhibitor (Carboxy-PTIO) significantly reduced viral-induced HIF-1α expression and VEGF secretion (135). As VEGF is a cytokine known to alter vascular permeability, increased NO production in vivo could play an important role in airway mucosal edema, which is a prominent feature of RSV bronchiolitis and pneumonia.
Although NO production is associated with significant antiviral activity in vitro and in vivo against RSV, excessive NO formation has also been linked to the pathophysiology of RSV lung disease in vivo. In a mouse model of infection, prevention of NO formation was associated with a significant decreased recruitment of inflammatory cells into the lungs and with reduced airway hyper responsiveness (AHR) to methacholine (222). In summary, NO possesses antiviral activity against RSV, but is also involved in the recruitment of inflammatory cells and viral-induced AHR (Fig. 13). Modulation of NO during the early events in the course of RSV lower respiratory tract infection that cause inflammation and AHR could potentially ameliorate the resulting lung disease in children.
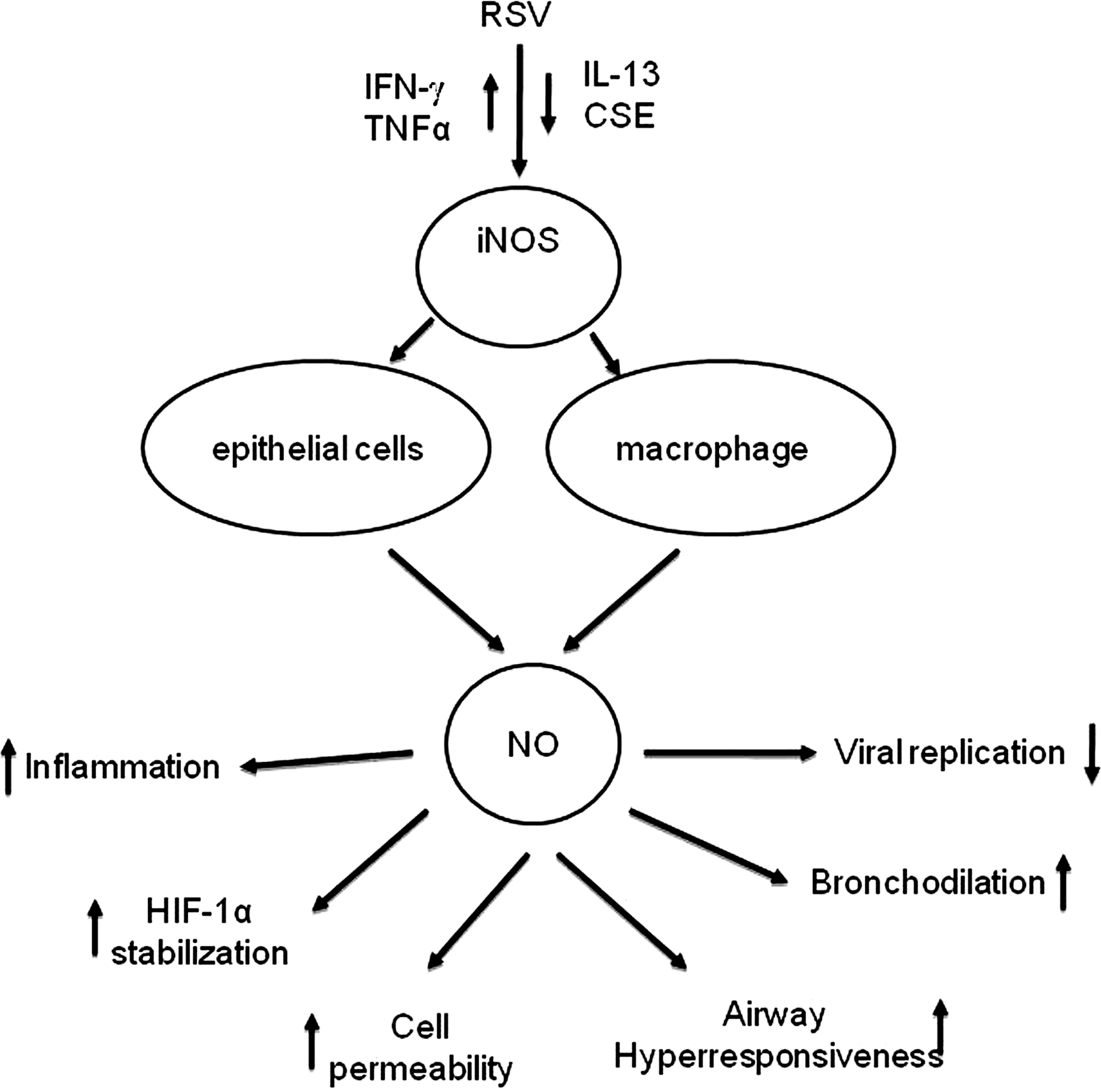
V. Potential Therapeutic Approaches
As there is strong supportive evidence that RSV-induced intracellular ROS formation regulates the expression of proinflammatory mediators and that oxidative stress, resulting from an imbalance between ROS production and airway antioxidant defenses, occurs in vitro and in vivo in response to RSV infection, antioxidant intervention would represent a rational approach for treatment of RSV lung disease. Two complementary approaches could be used to affect the outcome of RSV-associated lower respiratory tract infections; the first would be to increase airway antioxidant defenses by modulation of AOE expression/activity, and the second would be by enhancing nonenzymatic defenses through pharmacological intervention with molecules able to scavenge/detoxify ROS. In the following section, we will review several small-molecular-weight molecules with known antioxidant activity, from thiol to polyphenols, to antioxidant mimetics that have either been shown to affect RSV replication, viral-induced ROS production, and/or oxidative stress. We will also review compounds that stimulate Nrf2-dependent gene expression, with the potential to increase endogenous AOE levels in the context of RSV infection.
A. Vitamin A
There have been a few studies reporting low serum levels of vitamin A in severe cases of RSV bronchiolitis (179, 196), similar to what occurs in cases of severe infections with measles, another paramyxovirus for which vitamin A supplementation is currently recommended (1, 34). In two randomized clinical trials done in the United States, vitamin A administration in RSV infection did not show any clinical benefits (30, 196); however, there was some beneficial effect in other two studies performed in Chile and in Japan (64, 132), which showed a faster resolution of some symptoms, such as tachypnea, retractions, and wheezing, as well as the duration of hospitalization, in children treated with vitamin A, suggesting that possible underlying deficiencies can be a risk factor for developing more severe bronchiolitis.
B. Vitamin D
Although vitamin D does not have a direct effect on ROS formation, in the past few years, there has been increased recognition of its role in modulating pulmonary diseases, from asthma, COPD, and infections to cancer [reviewed in (101)], by affecting responses in a variety of cell types, from epithelial cells to antigen-presenting cells, such as monocytes/macrophages and dendritic cells, to lymphocytes, therefore shaping both innate and adaptive immune responses. In relationship to respiratory tract infections, there have been several studies in adults showing an inverse relationship between the levels of vitamin D3 and the number and severity of respiratory infections (101). In in-vitro studies, treatment of airway epithelial cells with vitamin D decreases RSV-induced NF-κB activation and proinflammatory mediator production. In addition, an SNP in the vitamin D receptor (123, 145) as well as low levels of vitamin D in the cord blood of neonates (21) have been both associated with increased risk of developing lower respiratory tract infection in response to RSV, suggesting a potential benefit of vitamin D treatment in the treatment/prophylaxis of RSV-associated pulmonary disease.
C. Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) is a secretory product of the pineal gland involved in synchronizing circadian and seasonal timing of several physiological and behavioral processes. It has also been shown to be a powerful free-radical scavenger with a broad-spectrum antioxidant capacity, as it interacts with both ROS and RNS, therefore providing protection against cellular oxidative damage [reviewed in (229)]. In addition, melatonin has been shown to play an important role in the immune function under both physiological and physiopathological conditions [reviewed in (36)]. Several immune functions appear to be modulated by melatonin, including the antitumor defenses of the host and cytotoxicity of natural killer cells, as well as antibody production. Melatonin has also been shown to have a protective effect in the pathophysiology of various viral infections [reviewed in (28)]. Melatonin was shown to prevent paralysis and death in mice infected with encephalomyocarditis virus, to reduce viremia and significantly postpone the onset of the disease and death in mice infected with the lethal Semliki Forest virus, and to attenuate noninvasive West Nile virus-induced disease. A similar protective effect was demonstrated in mice infected with Venezuelan equine encephalomyelitis virus, where melatonin treatment delayed the onset of the disease and reduced mortality. There is one report on the administration of melatonin in RSV infection in vivo. Huang et al. reported that mice intranasally inoculated with RSV showed oxidative stress changes demonstrated by increased NO, MDA, and •OH levels and decreasing GSH and SOD activities, whereas administration of melatonin significantly reversed all these effects. Furthermore, melatonin inhibited RSV-induced production of proinflammatory cytokines such as TNF-α (108). These results suggest that melatonin ameliorates RSV-induced lung inflammatory injury in mice via inhibition of oxidative stress and proinflammatory cytokine production.
D. Thiols
NAC is a thiol-containing compound that is used to reduce viscosity and elasticity of mucus. Moreover, it is able to scavenge H2O2, hydroxyl radicals, and hypochlorous acid (11). Pretreatment of human alveolar and bronchial epithelial cells with NAC protects both cell types against injurious effects of H2O2 (174). It has also been shown that NAC protects against hypochlorous acid-induced contraction of guinea pig tracheal smooth muscles (19) and inhibits LPS-induced leukocyte accumulation in rat lungs. Furthermore, NAC can be deacetylated to cysteine, an important precursor of cellular GSH synthesis, and thus stimulating the cellular GSH system (87). NAC has been used in clinical practice for the treatment of chronic bronchitis, cystic fibrosis, pulmonary oxygen toxicity, and ARDS (87), with mixed results, although a recent meta-analysis of clinical studies using NAC has shown positive results in reducing exacerbation rates in COPD (223). Administration of NAC significantly decreases mortality in mice infected with influenza virus, and two placebo-controlled trials of NAC supplementation suggest that it could be effective in improving influenza-induced severity of respiratory symptoms in humans [reviewed in (236)]. Treatment of airway epithelial cells with NAC greatly diminishes RSV-induced NF-κB activation, by affecting its serine phosphorylation, and NF-κB-dependent gene expression and protein secretion, including IL-8, RANTES, IL-6, and TNF-α (37, 120, 162, 164), as well as it reduces RSV-induced mucin synthesis (164). In one of the studies, but not the others, NAC inhibits viral replication in A549 cells (164).
E. Polyphenols
Natural and synthetic polyphenols are molecules known for their antioxidant properties. Flavonoids are a ubiquitous group of polyphenolic substances present in seeds, fruit skin or peel, and flowers of most plants. Among them, quercetin and catechins, such as epigallocatechin 3-gallate (EGCG), are the ones best investigated for their antioxidant properties. Quercetin and the related molecule isoquercetin have been shown in a recent study to inhibit the replication of both influenza A and B viruses [reviewed in (236)]. In a double treatment of isoquercetin and amantadine, synergistic effects were observed on the reduction of viral replication in vitro. The serial passages of virus in the presence of isoquercetin did not lead to the emergence of resistant virus, and the addition of isoquercetin to amantadine or oseltamivir treatment suppressed the emergence of amantadine- or oseltamivir-resistant virus. In a mouse model of influenza virus infection, isoquercetin administered intraperitoneally to mice inoculated with human influenza A virus significantly decreased the viral titers and pathological changes in the lung, suggesting that isoquercetin may have the potential to be developed as a therapeutic agent for the treatment of influenza virus infection and for the suppression of resistance in combination therapy with existing drugs. Similarly, EGCG and related compounds from green tea have been shown to reduce influenza replication in vitro and to possibly affect influenza-induced respiratory symptoms in the elderly [reviewed in (236)]. There is only one study investigating the effect of dietary flavonoids, including quercetin and catechins, on the infectivity and replication of RSV. In this study, preincubation of HEp2 cells with quercetin did not affect the ability of the virus to infect the cells or replicate, whereas catechins inhibited the infectivity, but not the replication of the virus (131).
Resveratrol, a nonflavonoid polyphenolic compound contained in grapes, has been shown to have important antioxidant and anti-inflammatory properties. It has been shown to improve outcomes in a variety of chronic diseases such as cardiovascular disease and diabetes, as well as cancer. There is experimental evidence that resveratrol could also be useful in the treatment of acute and chronic respiratory diseases, such as asthma, COPD, and possibly viral infections [reviewed in (252)]. A study of influenza virus has shown that resveratrol reduces influenza virus replication in MDCK cells by inhibiting nuclear–cytoplasmic translocation of viral ribonucleoproteins, therefore reducing expression of late viral proteins, and by inhibiting protein kinase C activity and its dependent pathways (186). The same study also shows that resveratrol treatment significantly improves survival and decreases pulmonary viral titers in a mouse model of influenza infection. There are only two reports on the use of resveratrol in models of RSV infection. The first in vitro study investigated its antiviral activity against RSV, showing that resveratrol is effective in inhibiting RSV replication in HEp2 cells at a concentration of 2 mg/ml (65). In the second study, administration of resveratrol in a mouse model of RSV infection reduced viral lung titers, viral-induced AHR, and lung inflammation (256), suggesting a potential benefit in treating RSV-associated pulmonary disease.
BHA and the related compound butylated hydroxytoluene (BHT) are phenolic compounds that are often added to foods to preserve fats. Oxygen reacts preferentially with BHA or BHT rather than oxidizing fats or oils. BHA can undergo several metabolic pathways in the body, such as dimerization, conjugation, and O-demethylation (238). In the context of RSV infection, treatment of airway epithelial cells with BHA greatly reduces RSV-induced chemokine production and modulates signaling pathways leading to IRF and STAT activation, with a slight reduction in viral replication (37, 114, 154). In vivo, treatment with BHA significantly decreases the content of MDA and 4-HNE in BAL of RSV-infected mice and ameliorates both body weight loss and clinical illness, indicating that modulation of oxidative stress in the context of RSV infection can lead to improvement in lung disease (40). These findings were reproduced by the use of another antioxidant agent, DMSO. Oral administration of DMSO at the time of infection significantly reduces RSV-induced body weight loss and clinical disease. Although it is difficult to determine the precise mechanisms by which BHA provides protection in the context of RSV infection, attenuation of inflammation is likely a key one. BHA treatment significantly reduces pulmonary cytokine and chemokine production, after RSV infection, resulting in inhibition of lung recruitment of inflammatory cells, especially neutrophils, which are the major cell type responsible for oxidative burst in response to infectious stimuli. BHA treatment is effective if given before or at the moment of RSV infection; however, it does not result in significant improvement of clinical disease administered 1 day after infection, when clinical disease and lung inflammation are already present. This finding suggests that early therapeutic intervention may be necessary to improve the clinical outcome in RSV infection. In fact, treatment of children with RSV-induced lower respiratory tract infection with anti-inflammatory drugs, such as steroids, has been shown to be mostly ineffective in improving clinical disease, as they are usually administered when children are hospitalized, days after manifestation of initial symptoms [reviewed in (32)].
In addition to reducing body weight loss and pulmonary inflammation, BHA treatment also attenuates RSV-induced AHR, possibly due to a reduction in cysteinyl-leukotriene (LT) production. LTs are potent bronchoconstrictors, as well as proinflammatory mediators, and their role in the pathogenesis of airway inflammation and obstruction has been recently recognized as a new target for therapeutic intervention [reviewed in (124)]. They have been shown to increase in the respiratory secretion of children infected with RSV (62, 84) and are associated with RSV-induced wheezing (237). In a mouse model of RSV infection, increased levels of cysteinyl-LTs correlate with increased airway responsiveness (249), and either inhibition of LT production (249) or treatment with LT receptor antagonist (77) inhibits AHR. In children, treatment with a cysteinyl-LT receptor antagonist decreases exacerbations of reactive airway disease after RSV bronchiolitis (25). BHA administration significantly decreases leukotriene C4 (LTC4) secretion, with parallel reduction of AHR after a methacholine challenge, suggesting a casual relationship of the two phenomena, although we cannot exclude that inhibition of other important mediators could be responsible for AHR reduction after BHA treatment (40).
In terms of viral replication, BHA administration slightly increases RSV replication in the lung, likely due to the observed reduction in inflammatory cells recruited to the lungs of infected mice (40). A similar finding is observed after inhibition of NO production in a mouse model of RSV infection, in which iNOS inhibition results in decreased lung inflammation, but in increased viral titers (222).
F. SOD and SOD mimetics
The use of recombinant SOD and SOD mimetics has been explored as therapeutics in a variety of disease models either in vitro or in vivo. A number of SOD mimetics based around organomanganese complexes have been developed. They include metalloporphyrin-based compounds, such as AEOL10113 and 10150, cyclic polyamine-based molecules, such as M40403 and 40419, and the Salen compounds, such as EUK8, 134, and 189, the latter ones possessing also significant catalase and peroxidase activity [reviewed in (20)].
Administration of SOD1 and 2 in a mouse model of influenza infection has been shown to be protective against lethal disease, and SOD3 overexpression in a similar model of infection significantly decreases influenza-induced lung injury [reviewed in (236)]. Both SOD1 and 2 have been tested for antiviral activity in a cotton rat model of RSV infection, and they both significantly reduce pulmonary viral titers when administered either parenterally or intranasally (253), although the authors of the study did not investigate their effect on disease or lung inflammation.
Although EUKs have been used in a variety of disease models, there is no reported literature about their use in models of viral infections. In recent in vitro studies, we have shown that treatment of A549 cells with EUK-134 significantly inhibits RSV-induced chemokine secretion (106). In addition, we also found that EUK8 and EUK189 are also effective in reducing RSV-induced cytokine and chemokine production in A549 cells (Casola A, unpublished observation). EUK8 and EUK189 treatments of A549 cells can effectively scavenge RSV-induced intracellular ROS generation, measured by DCFDA oxidation, and reduce lipid peroxidation markers, such as MDA and 4-HNE, in infected cells (Casola A, unpublished observation).
G. Nrf2-inducing agents
A variety of other compounds that stimulate ARE-driven transcription have been identified from natural and dietary sources, metabolites, and synthetic agents. These compounds have been extensively reviewed in a recent review article (111), and we classify them in our brief summary based on this review. Although none of them have been tested in the context of RSV infection, either in vitro or in vivo, they hold great potential for modulating RSV-induced oxidative stress.
1. Triterpenoids
Extensive reviews on the state of the art of triterpenoids have been published, and we mainly refer to these review articles for an incomplete description of these compounds (150, 220). More than 20,000 triterpenoids exist in nature, representing one of the largest groups of plant products. Triterpenoids are synthesized in plants by cyclization of squalene and are included among others squalenes, lanostanes, fusidanes, dammaranes, euphanes, lupanes, oleananes, ursanes, hopanes, and tetranortriterpenoids. Two of these natural compounds, oleanolic acid and ursolic acid, which have weak anti-inflammatory and antitumorogenic activity in vivo, represent the base for the initial screen and subsequent synthetic modifications of over 300 derivatives that have been conducted at the Dartmouth College (104). These agents have been tested for their anti-inflammatory activity, mainly by measuring their ability to block synthesis of iNOS in IFN-γ-stimulated macrophages. Two potent synthetic oleanane triterpenoids, 2-cyano-3,12-dioxooleana-1,9,-dien-28-oic acid (CDDO) and its methyl ester (CDDO-Me), are currently in phase I clinical trials for the treatment of leukemia and solid tumors. Further derivatization of CDDO to yield imidazolides (CDDO-Im), amides (methyl amide, CDDO-MA; ethyl amide, CDDO-EA), or a dinitrile (Di-CDDO) significantly increases biological activity (150). In addition to suppressing inflammation, these CDDO can activate cellular pathways that are associated with cytoprotection. This is the result of the induction of the coordinated response of Nrf2-induced, ARE-dependent cytoprotective proteins that include the enzymes of GSH synthesis and transfer, NQO, TRX, catalase, superoxide dismutase, and HO-1. The carbonyl groups on rings A and C of CDDO are critical functional groups to enable Michael addition with a nucleophilic target, such as Keap1 (a protein with multiple nucleophilic −SH residues), resulting in the release of Nrf2. The anti-inflammatory properties of CDDO also involve binding of CDDO to other protein targets with Michael addition on active cysteine residues. Among these targets, there are known signaling proteins such as IKKs, JAK and PTEN, and proteins associated with actin cytoskeleton [reviewed in (220)].
This class of agents has been tested in numerous experimental animal models for a variety of diseases, mainly those with important inflammatory and oxidative components (220). Relevant to airway diseases, these agents have been used in animal models of cystic fibrosis and emphysema induced by cigarette smoke (CS). In one study, mice carrying the R117H Cftr mutation of cystic fibrosis showed significantly reduced airway inflammatory responses to both LPS and flagellin when treated with CDDO before inflammatory challenge (181). Anti-inflammatory effects observed include reduced airway neutrophilia, reduced concentrations of proinflammatory cytokines and chemokines, and reduced weight loss. Mode of action of CDDO appeared to be due to an Nrf2-mediated increase in expression of a number of proteins with antioxidant properties. Parallel experiments performed with primary human bronchial cells after CDDO exposure and stimulation with TNF-α were consistent with changes observed in the mouse model. Using a mouse model of chronic exposure to CS, another study evaluated whether the strategy of activation of Nrf2 and its downstream network of cytoprotective genes with a small molecule would attenuate smoke-induced oxidative stress and emphysema (228). Nrf2 +/+ and Nrf2 −/− mice were fed a diet containing CDDO-Im, while being exposed to CS for 6 months. CDDO-Im significantly reduced lung oxidative stress, alveolar cell apoptosis, alveolar destruction, and pulmonary hypertension in Nrf2 +/+ mice caused by chronic exposure to CS. This protection from CS-induced emphysema depended on Nrf2, as Nrf2 −/− mice failed to show significant reduction in alveolar cell apoptosis and alveolar destruction after treatment with CDDO-Im. Similar protective effects of CDDO have been reported in studies of hyperoxia-mediated acute lung injury (198).
Although direct studies on the effect of CDDO in RSV infection have not been conducted so far, a randomized single-blind trial of the Chinese herbs Shuang Huang Lian was evaluated for the treatment of acute bronchiolitis (143). Shuang Huang Lian consists mainly of the three Chinese herbs, shuanghua, huangqin, and lianqiao, which are derived from plants and is composed of at least 29 known ingredients. Interestingly, the lianqiao herb contains oleanolic acid, ursolic acid, and triterpenoid saponin, the three major precursors of CDDO. In that study, children with acute bronchiolitis were randomized into three treatment groups: herbs, herbs with antibiotics, and antibiotics alone. The herbs were administered daily by intravenous infusion for 7 days. Main outcomes, assessed blindly, were symptomatic improvement in cough, fever, wheezing, chest signs, and duration of stay in hospital. The mean duration of symptoms from the beginning of treatment was 2.4 days shorter in the two groups treated with herbs compared to the group treated with antibiotics alone, suggesting a beneficial effect of one or more of the compounds contained in these specific Chinese herbs on RSV disease.
2. Sulforaphane and other isothiocyanates
Isothiocyanates are organosulfur compounds found in cruciferous vegetables (broccoli and cabbage), and a diet rich in such vegetables has been shown to reduce the risk of developing certain types of cancer. Sulforaphane is the best-known ARE inducer in this class, and as such, it modifies a number of Keap1 cysteine residues through formation of carbamodithioate (111). Recent reports have shown that supplementation with sulforaphane affects respiratory viral infections, both in vitro and in vivo. In one study, nasal epithelial cells treated with sulforaphane before infection with influenza virus show significant inhibition of viral replication, demonstrated by marked decreased hemagglutinin gene expression, and increase protein levels of Nrf2 and HO-1 (133). In a mouse model of RSV infection, sulforaphane treatment significantly decreases the RSV gene expression level and reduces numbers of BAL neutrophils and eosinophils (46).
3. Polyphenols
Polyphenols, in addition to their ability to scavenge ROS, can also react with thiols in Keap1 and elicit ARE-dependent responses. Among them are the flavonoids quercetin and EGCG, the nonflavonoids curcumin and resveratrol, and the phenolic acid and diterpens rosmarinic and carnosic (carnasol) acids. Curcumin has been tested in clinical trials for certain cancers and in degenerative neurological disease (18). Curcumin has been shown to induce Nrf2- and ARE-dependent genes in human cell lines and in mice fed with a diet containing curcumin (80). EGCG, as well as other catechins contained in green tea, has been shown to modulate Nrf2-dependent gene expression [reviewed in (227)]. Carnasol, a catechol-type antioxidant from Rosmarinus officinalis, increases levels of Nrf2 and the target gene HO-1 in cells, and transgenic mice expressing an ARE-driven luciferase reporter gene, treated by oral gavage with this compound, showed robust ARE activation in different organs (15). Quercetin is capable of activating Nrf2-dependent phase II enzymes (NQO1) in cell lines by both inhibiting ubiquitination of Nrf2 and increasing Nrf2 mRNA [reviewed in (67)]. A 2-week regimen of dietary quercetin has been shown to suppress colorectal carcinogenesis in F344 rats (61). Several studies have suggested that resveratrol activates Nrf2-dependent antioxidant gene expression in a number of cell types, including hepatocytes, epithelial cells, and endothelial cells, by activating Nrf2 (67, 227). In a study using A549 cells, resveratrol-attenuated CS extracts induced ROS and restored GSH level by upregulating GCL via Nrf2 (141).
As mentioned earlier in this review, BHA and BHT are phenolic compounds with known ROS-scavenging properties. One of the major metabolites of BHA is the demethylated product tert-butylhydroquinone (tBHQ), which exhibits its chemopreventive effects in a similar manner to those by BHA, including the modulation of the enzyme systems responsible for metabolic activation or deactivation of chemical carcinogens. Different studies have shown that BHA and tBHQ have the capacity to induce some phase II enzymes via Nrf2. For example, a study has shown that BHA and tBHQ treatments increased HO-1, NQO1, and Nrf2 proteins in both primary-cultured rat and human hepatocytes (134). BHA- and tBHQ-mediated induction of HO-1 and NQO1 proteins occurred through transcriptional activation in primary-cultured rat hepatocytes and involved activation of ERK1/2 and JNK1/2. In preliminary experiments, treatment of airway epithelial cells with tBHQ significantly increases ARE-dependent gene transcription and Nrf2 protein expression (Casola A, unpublished observation) (Fig. 14), suggesting the possibility that the beneficial effect we observed for BHA treatment on RSV-induced lung inflammation and lung diseases could be in part ascribed to the ability of these phenolic compounds to modulate Nrf2-dependent gene expression, in addition to directly scavenging ROS formed in response to the viral infection.
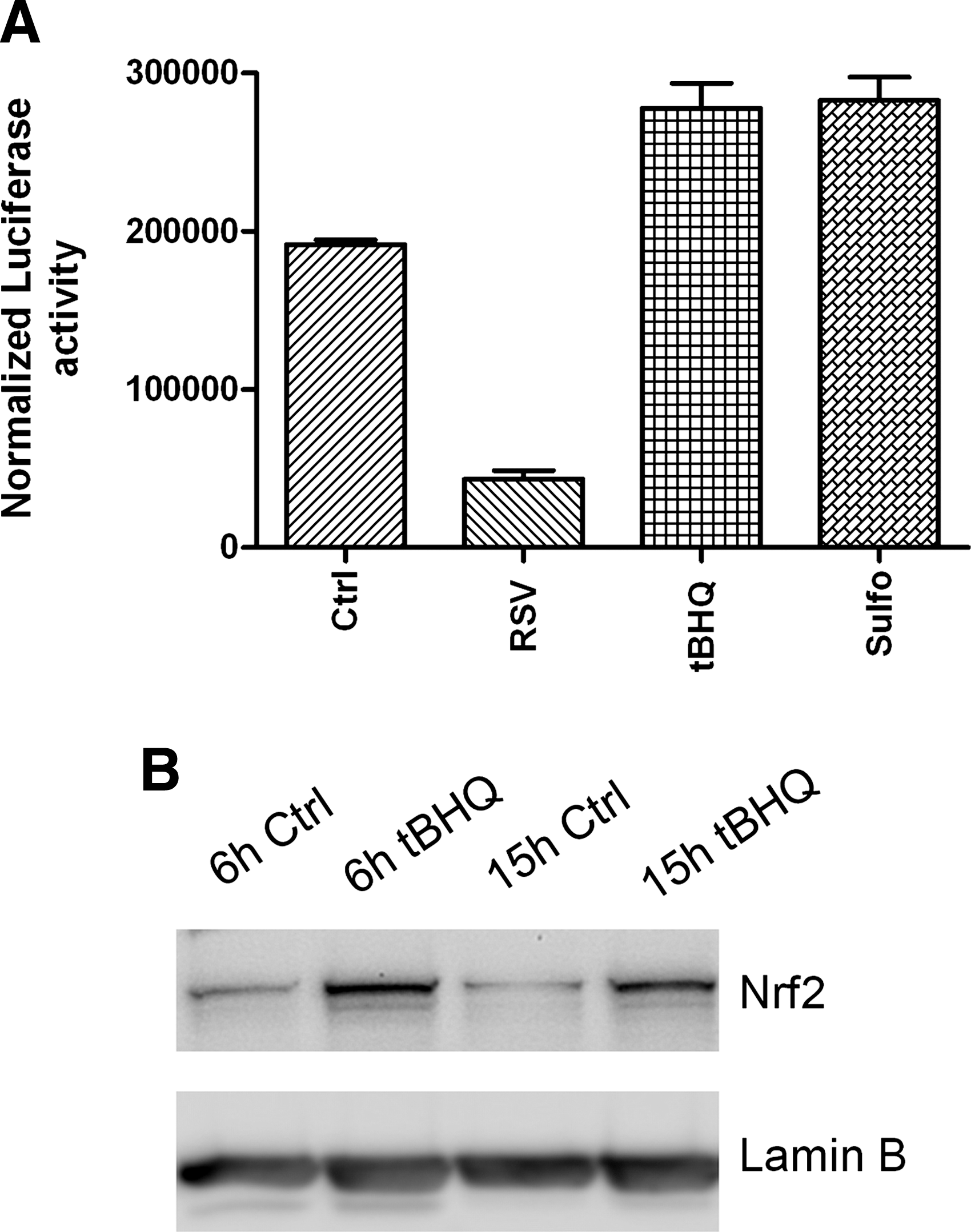
4. Other classes of Nrf2 inducers
15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is a prostaglandin metabolite produced by the action of COX-2, whose activity, including its anti-inflammatory activity, is mediated through its covalent reaction with the cysteine residues in target proteins. 15d-PGJ2 has been shown to increase the levels of HO-1 and GSH in various cell lines. In mouse peritoneal macrophages, 15d-PGJ2 activated Nrf2 by forming adducts with Keap1, resulting in an Nrf2-dependent induction of HO-1 and Prdx I (Prx1) gene expression (115). Administration of the COX-2 inhibitor NS-398 to mice with carrageenan-induced pleurisy caused persistence of neutrophil recruitment and, in macrophages, attenuated the 15d-PGJ2 accumulation and PrxI expression (115). Administration of 15d-PGJ2 into the pleural space of NS-398-treated wild-type mice largely counteracted both the decrease in PrxI and the persistence of neutrophil recruitment. In contrast, these changes did not occur in the Nrf2 −/− mice, suggesting that Nrf2 regulates the inflammation process downstream of 15d-PGJ2 (115).
As an example of ARE inducers identified using high-throughput screening of unbiased small-molecule library described by Hur et al., AI-1 is a quinolinone–scaffold compound that exhibits a single-digit micromolar EC50 for inducing ARE luciferase reporter gene activity and Nrf2 stabilization in cells (112). AI-1 has attractive features such as a potent Nrf2 activation, low cytotoxicity, and a versatile chemistry for derivatization. A biotin-modified AI-1 was used to reveal that AI-1 alkylates C151 of Keap1 both in vitro and in cells, weakening the interaction between Cul3 and Keap1, thereby inhibiting Nrf2 ubiquitination and activating ARE-mediated gene transcription.
VI. Conclusions
RSV-induced respiratory disease is associated with increased ROS/RNS generation and oxidative stress that are likely to play a key role in initiating and amplifying lung injury and inflammation. In animal models, modulation of ROS/RNS production has been shown to be beneficial in ameliorating viral-induced lung inflammation and clinical disease, although intervention is associated with some increase in viral replication. Approaches that combine scavenging ROS/RNS together with the inhibition of viral replication would be likely the most effective in modulating severe lung disease associated with RSV infection. This could be achieved by administration of antioxidant compounds that possess antiviral activity (resveratrol, for example, has been shown to reduce RSV replication in vitro and in vivo), in addition to ROS-scavenging properties, or by combining old/novel antivirals for RSV with compounds able to increase lung antioxidant defenses, such as AOE mimetics or Nrf2 inducers. Antioxidant supplementation would be successful only if available at the site of infection/inflammation; therefore, route of administration, bioavailability, tissue distribution are all important parameters that will need to be taken into consideration when planning future therapeutic interventions. The incomplete understanding of the pathophysiology of RSV bronchiolitis and its genetic components remains a barrier to the development of targeted immunomodulatory, anti-inflammatory, or antioxidant therapeutics. As an example of our incomplete understanding of the disease process and its clinical progression, there are studies showing that a robust inflammatory response driven by certain cytokines and chemokines might be beneficial in the early symptomatic phases of RSV infection, yet detrimental at later times of disease (23). Therefore, potential anti-inflammatory interventions in RSV infections may be either detrimental or insufficient to prevent progression to the lower respiratory tract depending on the stage of infection. Ideally, the decision process for anti-inflammatory or antioxidant treatment for RSV infections will have to be guided by the integration of a number of clinical and laboratory criteria, including early at point-of-care viral diagnostics, assessment of risk factors, and identification of biomarkers of disease progression and genetic susceptibility. The latter may also involve regulation of the antioxidant response, since functional polymorphisms in the Nrf2 gene promoter, leading to reduced gene transcription, have been shown, for example, to associate with severity of certain airway diseases, including COPD and lung injury after trauma (107, 160).
As we explore new treatment and prophylactic regimens for viral bronchiolitis that target the oxidant response, other important aspects will have to be considered, including whether RSV is indeed unique compared to other respiratory viral pathogens in its ability to alter the expression of AOEs, to trigger the generation of ROS and the magnitude and type of oxidative damage in the airways, including the presence of other lipid peroxidation products or protein modifications, which characterize the biological targets of ROS formation. The role of possible co-infections in altering the pro-oxidative/antioxidative balance remains unexplored. Also, the known developmental process of the AOE system that starts during fetal life and that is characterized by a certain degree of immaturity in the neonatal period and early infancy may contribute to the severity of RSV infections that occur during this vulnerable period of life (57). All these factors are likely to contribute to the oxidative-mediated pathogenesis of viral bronchiolitis and perhaps to the relative greater effect of RSV compared to other pathogens. Therefore, in these earlier phases of postnatal life, the well-known antioxidant capacity of human milk in combination with the specific anti-RSV antibodies that are present in human milk may play a critical role in preventing or reducing the risk of severe RSV infections, as suggested by multiple studies of breastfeeding and bronchiolitis (214). Although the complete list of antioxidants present in human milk is not known, it is has been shown to contain a number of AOEs such as those that were found to be affected by RSV infection (including SOD and catalase) (88). Overall, human milk demonstrates an increased total antioxidant capacity compared to formula, and even preterm infants, which are at a higher risk for severe RSV infections, show evidence of lower oxidative damage when fed exclusively mother's milk compared with formula-fed infants (148).
With even broader implications, generation of such an oxidative stress environment in the airways, along with the impaired antioxidant response as result of RSV infection, may induce critical chemical modifications of bystander antigens and their immunogenicity. Such possibility has been suggested by studies of aldehyde-mediated carbonylation of protein allergens resulting in enhanced Th2 responses, perhaps via enhanced immune priming (171). Thus, treatment or prophylactic regimens aimed to reduce the RSV-induced pro-oxidative may have even longer protective effects by potentially reducing the risk of the development of allergic diseases and asthma.
Last but not least, therapeutic strategies aimed to increase the antioxidant airway capacity by increasing Nrf2 activity should probably be used for short periods of time with an appropriate therapeutic dose, avoiding continuous Nrf2 activation. In fact, although Nrf2 exhibits protective effects against several types of chronic airway injury, its overexpression might be harmful. There is increasing concern about the potential detrimental effects of Nrf2 overexpression in cancer and high doses of cancer chemopreventive phenolic antioxidants, such as BHA, which activates Nrf2, and have been reported to promote tumors.
Footnotes
Acknowledgments
Supported by NIH grants AI062885, AI30039, and HV28184, and by Flight Attendant Medical Research Institute (FAMRI) Clinical Innovator Awards to AC (# 072147) and RPG (# 42253). The authors would like to thank Mrs. Cynthia Tribble for her assistance in manuscript submission.