
Abstract
Introduction
According to the American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias, usual interstitial pneumonia (UIP), including myofibroblasts and fibroblastic foci, is the histopathologial finding of IPF (3). This disease is characterized by fibroproliferation, associated with an excessive extracellular matrix deposition leading to pulmonary architecture distorsion and respiratory failure. Unfortunately, no treatment has shown any benefit in terms of survival (38).
Myofibroblastic differentiation plays a key role in the pathophysiologic process of IPF. Fibroblasts, differentiated to myofibroblasts, acquire a pro-fibrosing phenotype, defined by the expression of alpha-smooth muscle actin (α-SMA), the production of extracellular matrix (collagen I and III), and increased cell proliferation, contraction, and migration abilities (35).
Innovation
We showed a decreased expression of nuclear factor erythroid 2-related factor 2 (Nrf2) and antioxidant “Phase II Enzymes” in human pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) and the key role of Nrf2 modulation in the fibroblast phenotype. Nrf2 inhibition induced myofibroblastic differentiation and increased oxidative stress in vitro. Conversely, nuclear Nrf2 induction restored the oxidant/antioxidant equilibrium and reversed myofibroblastic differentiation. The pro-fibrosis effect of transforming growth factor β was related to decreased nuclear Nrf2 expression and counteracted by the antifibrosis effect of Sulforaphane, an Nrf2 activator. Nrf2 could play a central role in fibroblast differentiation in IPF, leading to new therapeutic strategies.
Oxidative stress, resulting from an imbalance of oxidants and antioxidants, has also been implicated in IPF pathophysiologic features (1, 17). To counteract oxidative stress, lung cells enlist a battery of classical antioxidant systems, such as Superoxide dismutase, Catalase, Glutathion Peroxidase, Glutathion S Transferase, Thioredoxin, Sulfiredoxin, Glutamate Cysteine Ligase (25, 26), and especially Phase II Enzymes (EPII) such as NADPH quinone oxidoreductase 1 (NQO-1), epoxyde hydrolase (EPHX), and Heme oxygenase-1 (HO-1), whose central protective roles against oxidant and inflammatory injuries have been previously identified in lung disease (12, 26).
The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is a key orchestrator of the induction of several antioxidants, especially EPII. Nrf2 belongs to the “cap ’n’ collar” basic leucin zipper family. Under basal conditions, Nrf2 is sequestered in the cytoplasm by its inhibitor Kelch-like erythroid cell-derived protein CNC homology-associated protein 1 (Keap1), then ubiquitinylated, and, finally, degraded by the proteasome. In the presence of oxidative stress, Keap1 releases Nrf2, which can migrate in the nucleus, bind to the antioxidant responsive element sequence, and induce phase II gene transcription (20). Previous studies reported an increase in the expression of oxidant enzymes (NADPH oxidases) in IPF fibroblasts and showed the role of oxidants in myofibroblastic differentiation (1, 17). The important role of Nrf2 in lung fibrosis has previously been published in animal models (8, 22, 33, 41, 43). Cho et al. showed that bleomycin induced earlier and more severe pulmonary fibrosis in Nrf2−/− mice than in wild-type mice, which suggests the involvement of Nrf2 (8). In humans, a potential protective role for Nrf2 against chronic fibrosing lung disease has been reported (30, 31). However, the role of EPII and Nrf2 in lung fibrogenesis in patients with IPF and the potential therapeutic effect of Nrf2 inducers have not been studied.
Sulforaphane (SFN), an isothiocyanate found in cruciferous vegetables from the Brassicaceae family, is a known Nrf2 activator. SFN interacts in a concentration-dependent manner (7) with thiol groups of Keap1, thus leading to its conformational modification, dissociation of the Nrf2–Keap1 cytoplasmic complex, and, finally, Nrf2 translocation into the nucleus (11). McWalter et al. reported that Nrf2 is essential for the induction of EPII expression by SFN (32).
We hypothesized that (i) the imbalance in oxidants and antioxidants, as well as altered EPII and Nrf2 expression, are involved in myofibroblast differentiation observed in IPF and (ii) Nrf2 activation may modulate the fibroblast phenotype in IPF. Thus, we compared Nrf2 expression in human pulmonary fibroblasts from control and IPF patients and investigated the role of activation of Nrf2 in myofibroblast differentiation and fibrosis.
Results
Basal pulmonary fibroblast phenotype, expression of oxidative stress markers, and Nrf2
We characterized the basal pulmonary fibroblast phenotype after a 24-h culture of cells without stimulation. Phase-contrast microscopy revealed control fibroblasts as thin and spindle shaped with prolonged cytoplasmic projections; in contrast, IPF fibroblasts appeared star shaped, with abundant cytoplasm and short projections (Fig. 1A). These morphological features matched the higher expression of α-SMA and collagen I protein in IPF fibroblasts than in controls (both p<0.05; Fig. 1B). Thus, IPF fibroblasts were characterized by a myofibroblastic phenotype.
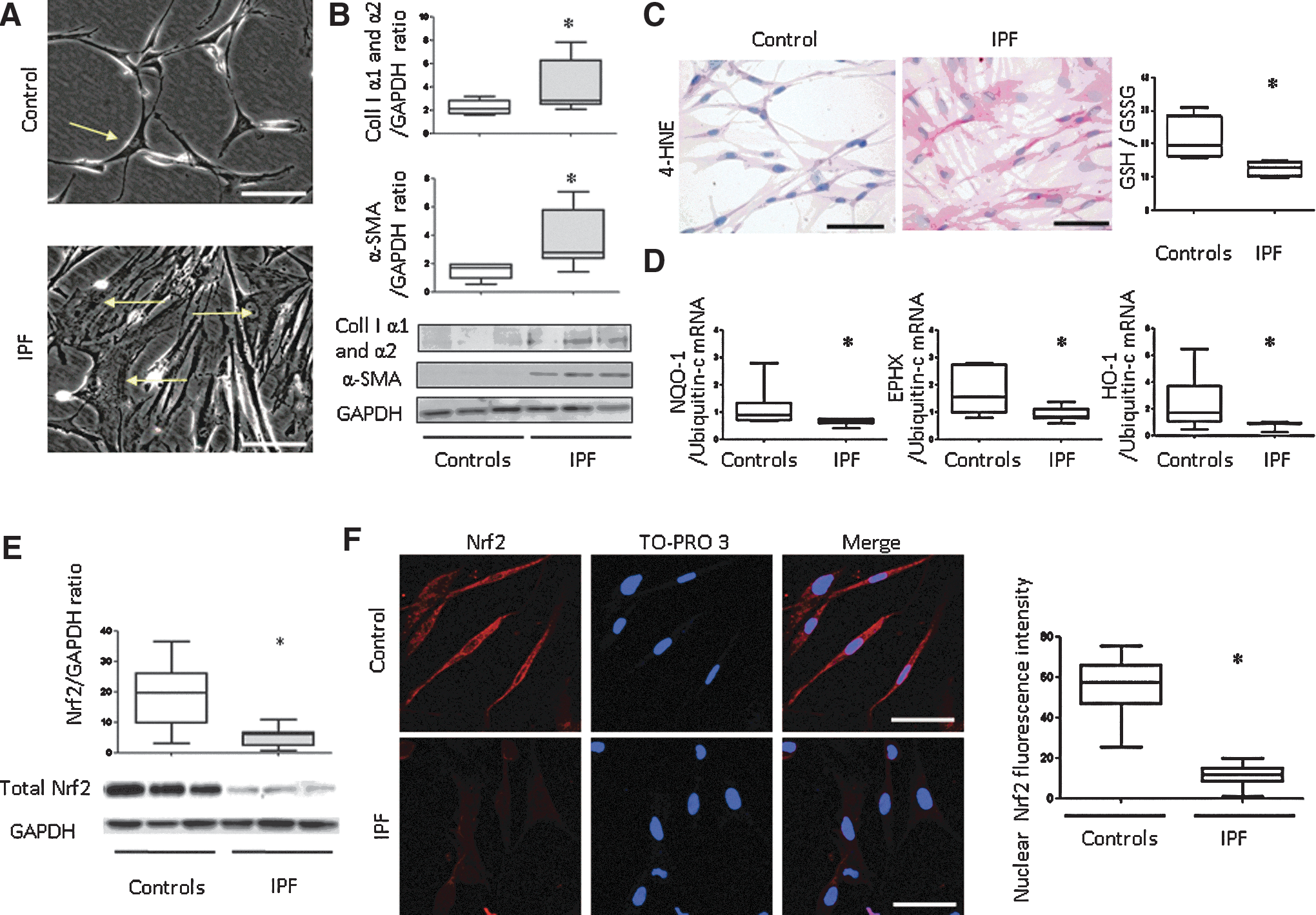
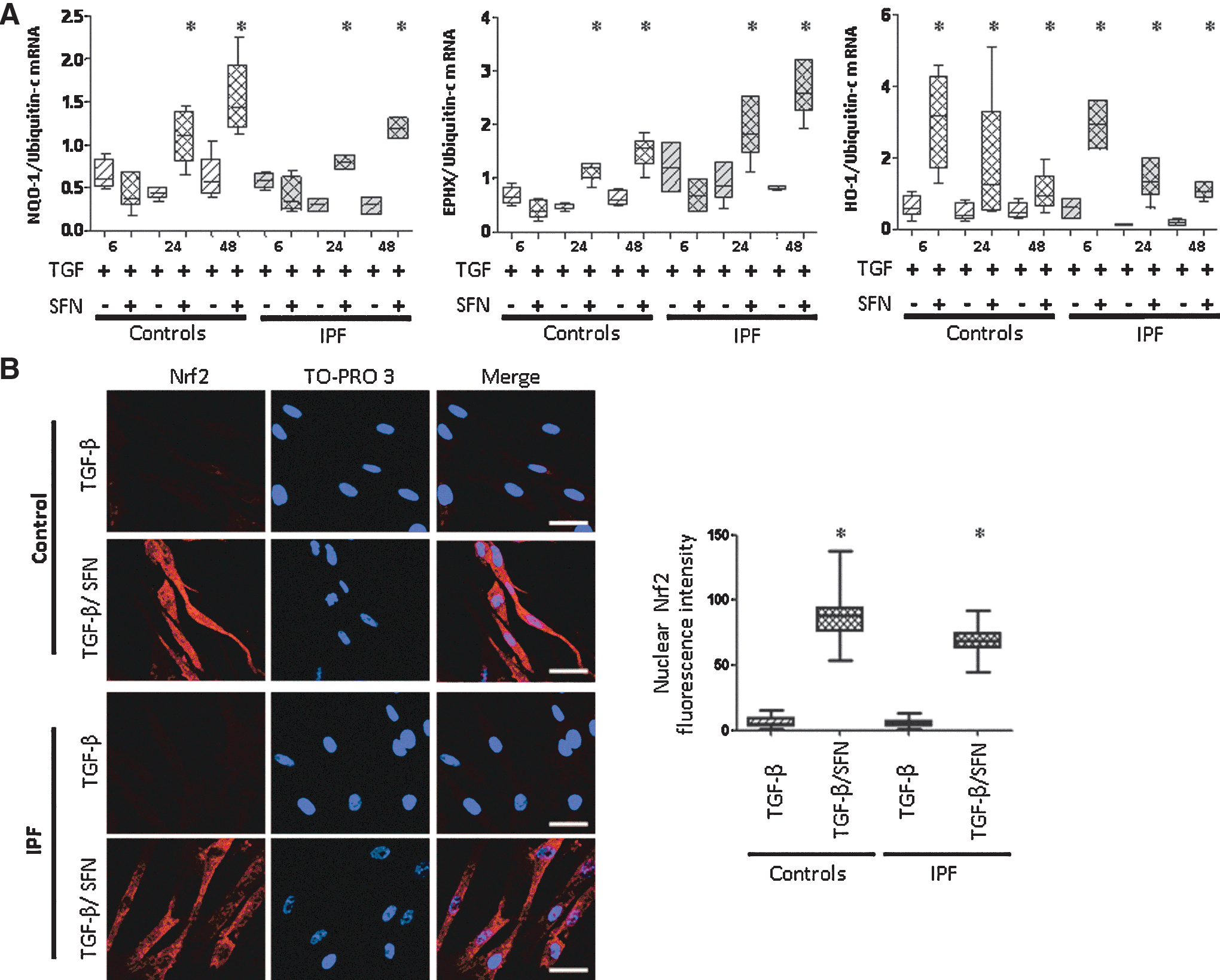
The staining of 4-hydroxynonenal (4-HNE), an oxidative stress lipid peroxidation marker, was higher in IPF fibroblasts than in controls (Table 1), and the quantitative glutathione (GSH)/oxidized glutathione (GSSG) ratio was decreased (p=0.03; Fig. 1C). mRNA expression of NQO-1, EPHX, and HO-1 (all p<0.05; Fig. 1D) and Nrf2 protein were lower in IPF fibroblasts than in controls (p=0.014; Fig. 1E). Moreover, immunostaining for the nuclear fraction of Nrf2 was lower in IPF fibroblasts than in controls (p<0.0001; Fig. 1F). Thus, ex vivo, decreased nuclear Nrf2 expression was associated with decreased EPII expression, GSH/GSSG ratio, and increased 4-HNE staining in IPF fibroblasts as compared with controls.
p<0.05 IPF versus controls.
0, absent staining; +, moderate staining; ++, intense staining; +++, very intense staining; results are presented as median [min-max] percentage, 4-HNE, 4-hydroxynonenal; IPF, idiopathic pulmonary fibrosis.
Effect of siRNA knockdown of Nrf2 expression on oxidative stress and fibroblast phenotype
In control fibroblasts, Nrf2 inhibition by Nrf2 siRNA increased 4-HNE staining (Fig. 2A and Table 2); decreased NQO-1, EPHX, and HO-1 mRNA expression (all p=0.03; Fig. 2B); induced a myofibroblastic dedifferentiation phenotype (Fig. 2E); and increased α-SMA mRNA expression (Fig. 2F; p=0.03). Conversely, in IPF fibroblasts, Nrf2 activation by Keap1 siRNA knockdown decreased 4-HNE staining (Fig. 2C and Table 3); increased NQO-1, EPHX, and HO-1 mRNA expression (all p=0.03; Fig. 2D); and induced a control-like myofibroblastic dedifferentiation phenotype (Fig. 2G). This phenotype was confirmed by decreased α-SMA and Collagen I α1 mRNA expression (p=0.04; Fig. 2H).
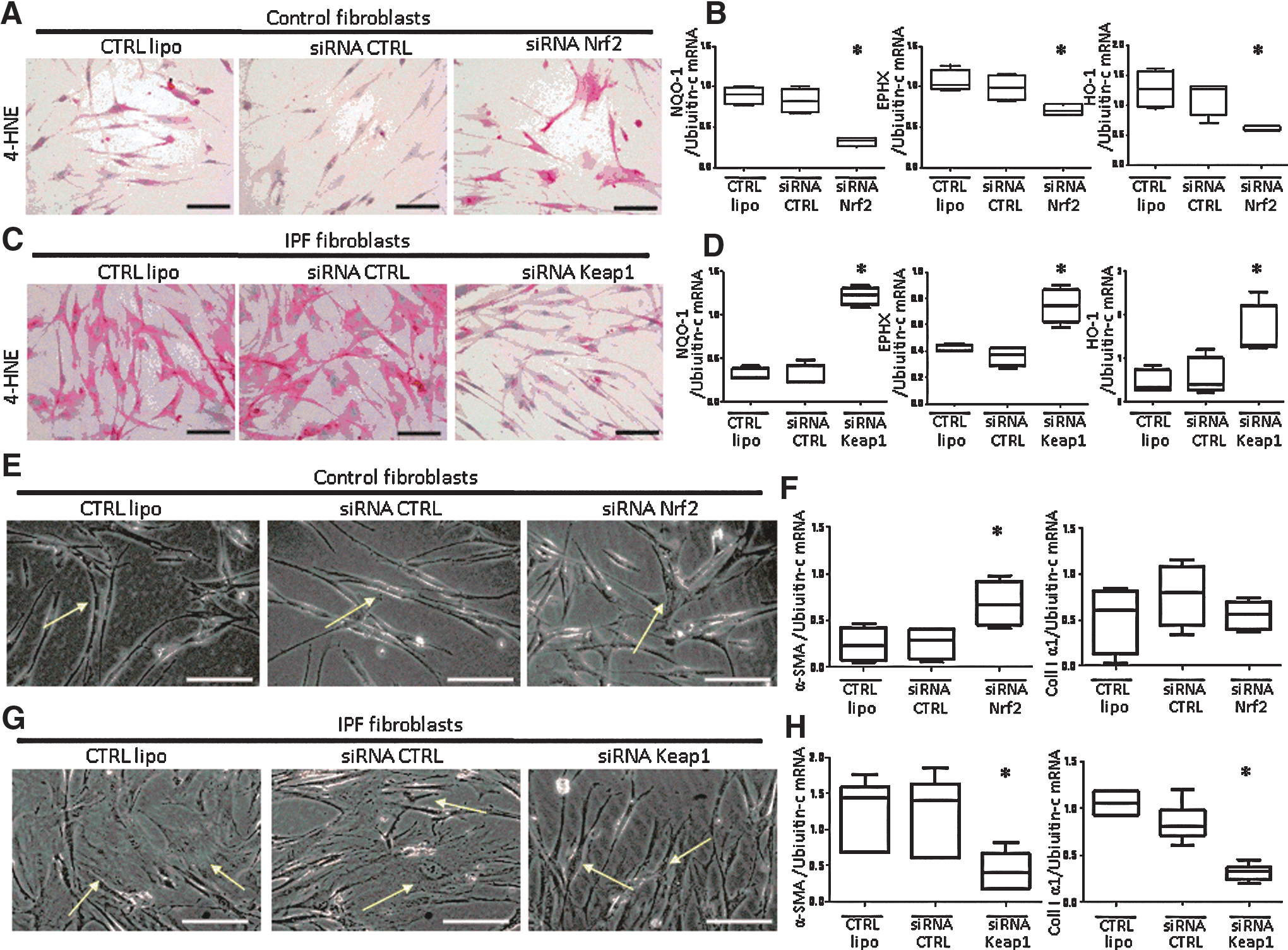
p<0.05 Nrf2 siRNA versus lipofectamine control or control siRNA (siCTRL).
p<0.05 Keap1 siRNA versus lipofectamine control or control siRNA (siCTRL).
Effect of SFN on oxidative stress, Nrf2 expression, and fibroblast phenotype
To assess the effect of pharmacological activation of Nrf2, fibroblasts were exposed for 24 h to SFN, a known Nrf2 inducer. SFN decreased 4-HNE staining (Fig. 3A and Table 4) and increased the mRNA expression of EPII (Fig. 3B) and nuclear Nrf2 expression in both control and IPF fibroblasts (all p<0.001; Fig. 3C).
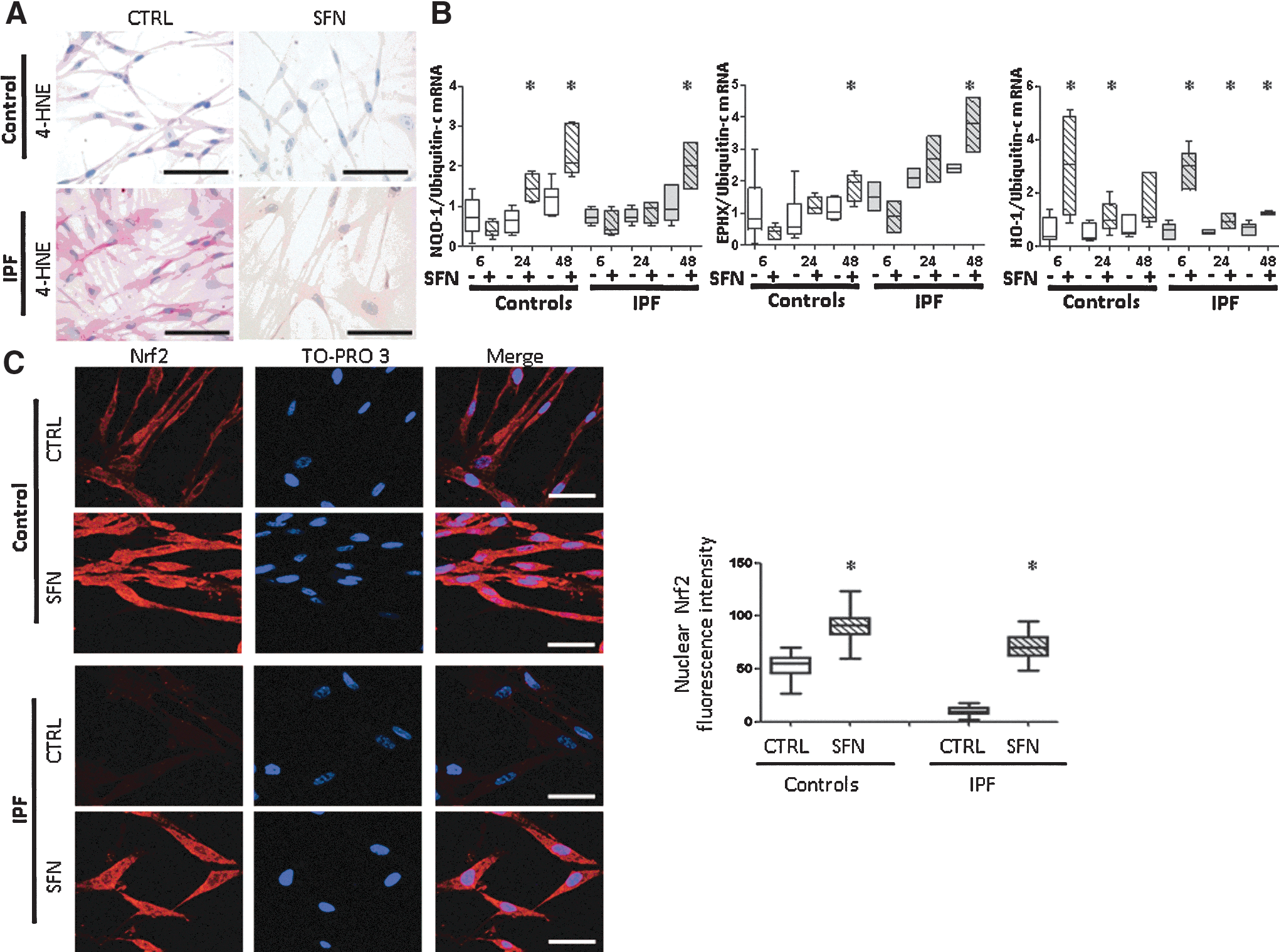
p<0.05 versus CTRL.
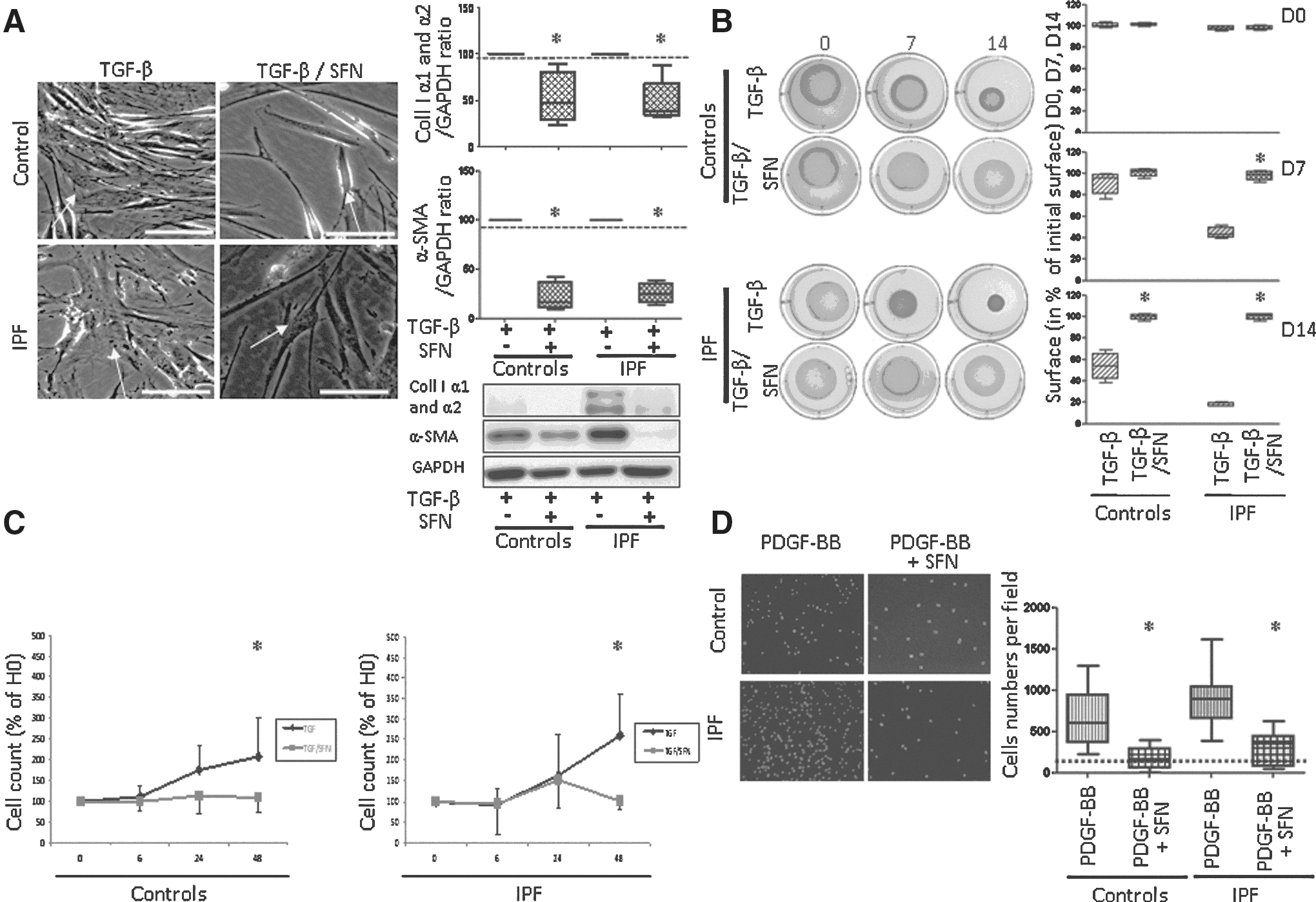
Stimulation for 24 h with SFN reversed myofibroblastic differentiation as confirmed by decreased protein level of α-SMA in control and IPF fibroblasts (both p<0.05) and collagen I (both p<0.01; Fig. 4A). For IPF fibroblasts, SFN decreased contractility from day 7 (p=0.03) to 14 (p=0.03); the same effect was observed but delayed at day 14 in control fibroblasts (p=0.03; Fig. 4B). SFN decreased fibroblast proliferation at 48 h in both control and IPF fibroblasts (both p<0.05; Fig. 4C) and inhibited control and IPF fibroblast migration as compared with basal conditions (both p<0.05; Fig. 4D). Therefore, SFN had antifibrosis effects.
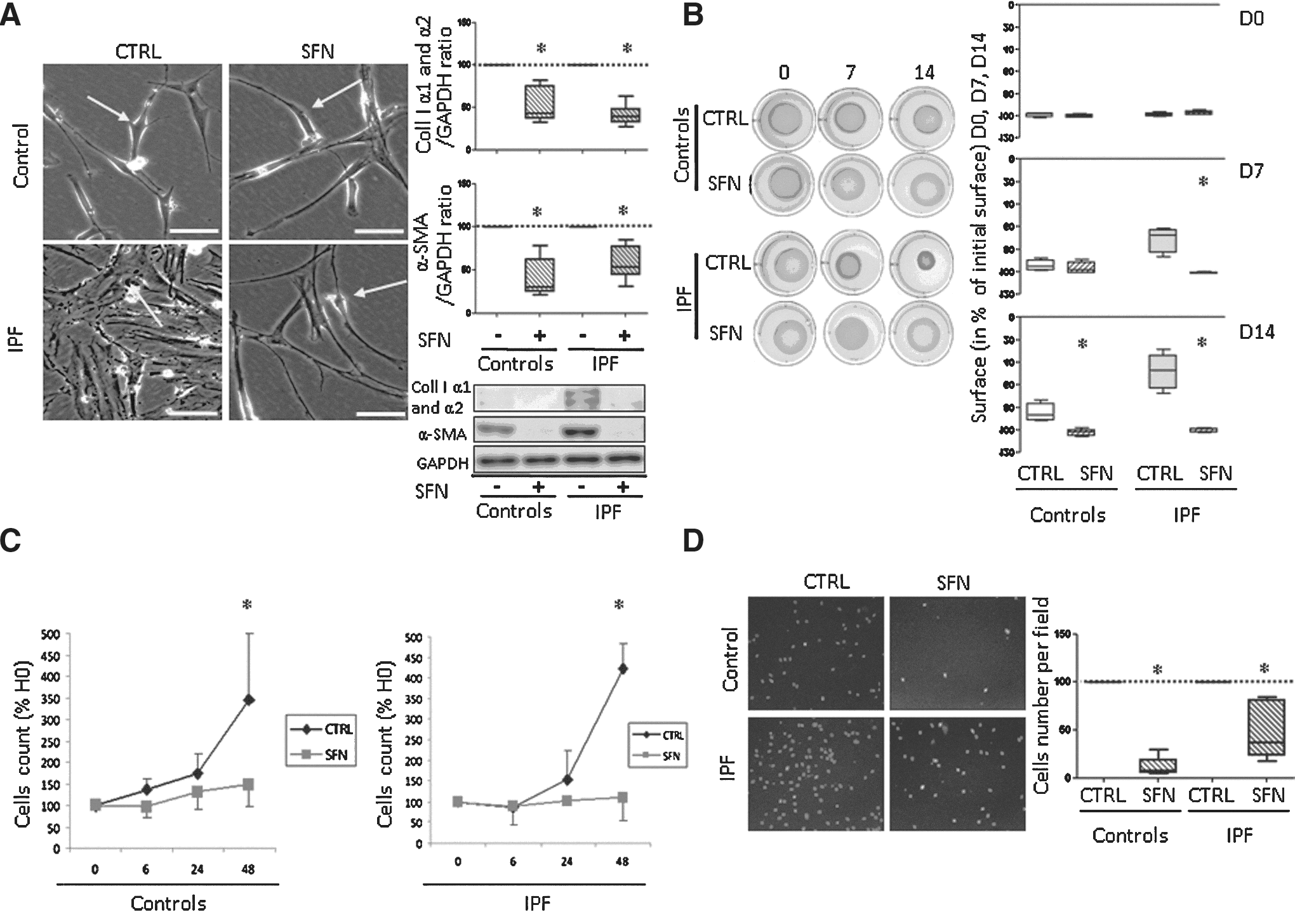
Effect of SFN on EPII and nuclear Nrf2 expression in fibroblasts stimulated with transforming growth factor β
In fibroblasts pretreated with transforming growth factor β (TGF-β) for 24 h, a pro-fibrosing condition, SFN treatment increased the mRNA expression of NQO-1, EPHX, and HO-1 (Fig. 5A) and concomitantly increased the nuclear Nrf2 fraction in both control and IPF fibroblasts (all p<0.0001; Fig. 5B).
Effect of SFN on fibroblast phenotype in fibroblasts stimulated with TGF-β or platelet-derived growth factor BB
We assessed the potential therapeutic effect of SFN in fibroblasts stimulated with TGF-β or platelet-derived growth factor BB (PDGF-BB) for 24 h to differentiate into myofibroblasts. SFN treatment strongly modified fibroblast morphology reversing myofibroblastic differentiation as confirmed by a decreased of α-SMA (controls: p=0.03; IPF: p=0.005) and collagen I (controls: p=0.03, IPF: p=0.02) (Fig. 6A). SFN also decreased the contractility of IPF myofibroblasts at days 7 to 14 (all p=0.03). The same effect was observed in control fibroblasts but was delayed at day 14 (p=0.03; Fig. 6B). Moreover, SFN decreased control and IPF fibroblast proliferation at 48 h (both p<0.05; Fig. 6C), and 24-h SFN stimulation inhibited control and IPF fibroblast migration induced by PDGF-BB (both p<0.05; Fig. 6D).
Antifibrosis effects of SFN were via Nrf2
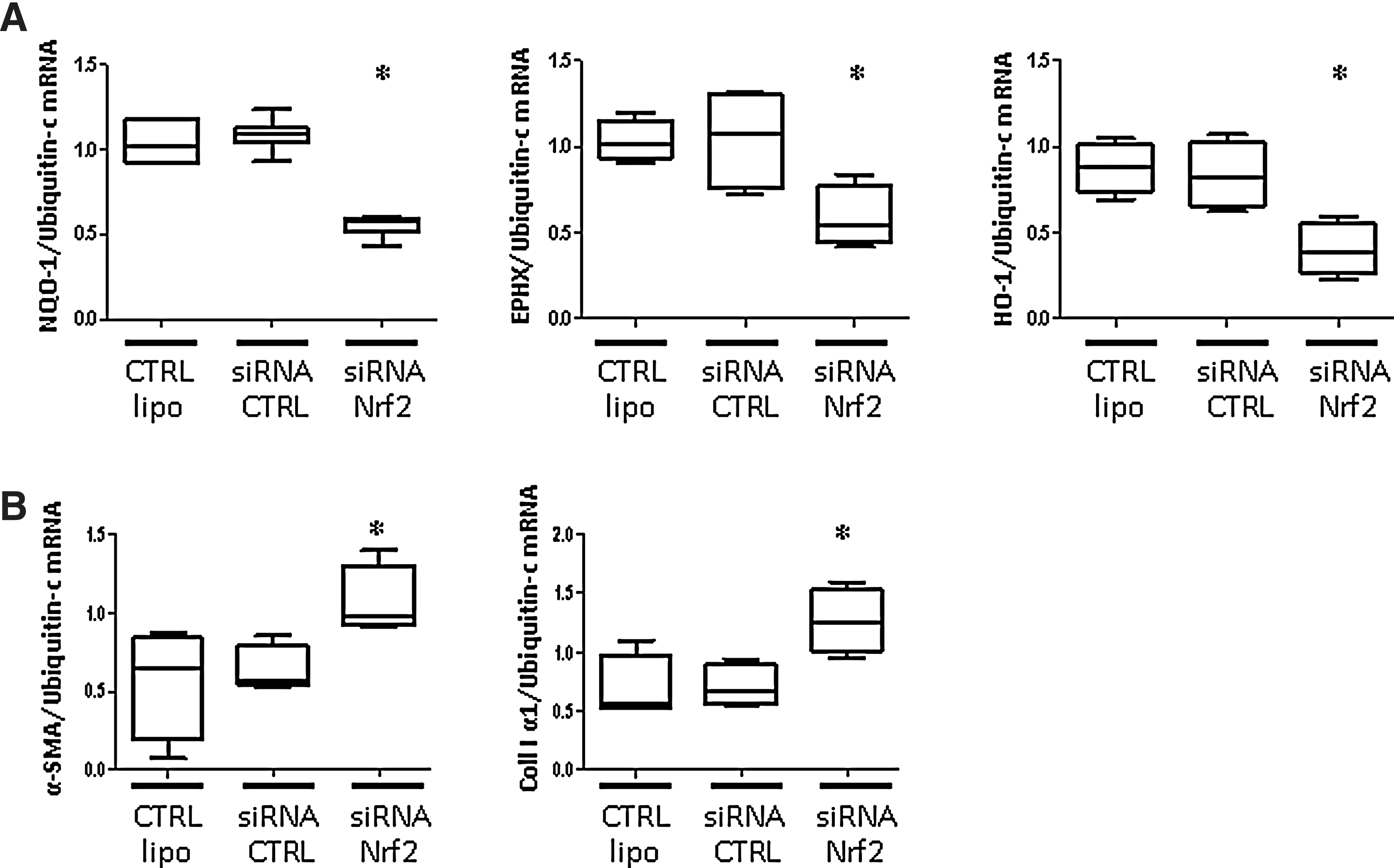
To confirm that SFN effects were via Nrf2, we transfected control fibroblasts with Nrf2 siRNA. With Nrf2 knockdown, SFN stimulation neither induced mRNA expression of NQO-1, EPHX, and HO-1 (all p<0.05; Fig. 7A) nor decreased the protein levels of α-SMA and collagen I (both p<0.05; Fig. 7B). Therefore, the antifibrosis and antioxidant effects of SFN acted via the Nrf2 pathway.
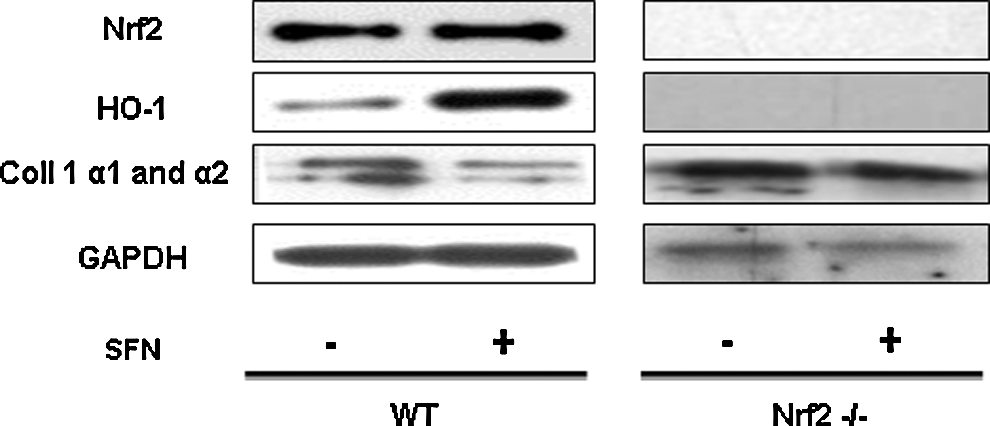
These results were further confirmed in Nrf2 knockout mice. Indeed, as expected, 10 μg/ml SFN inhibited collagen I protein expression in wild-type mice lung fibroblasts and induced concomitant HO-1 expression. These effects of SFN were not observed in Nrf2−/− mice (Fig. 8).
SFN did not prevent lung fibrosis in the murine bleomycin model
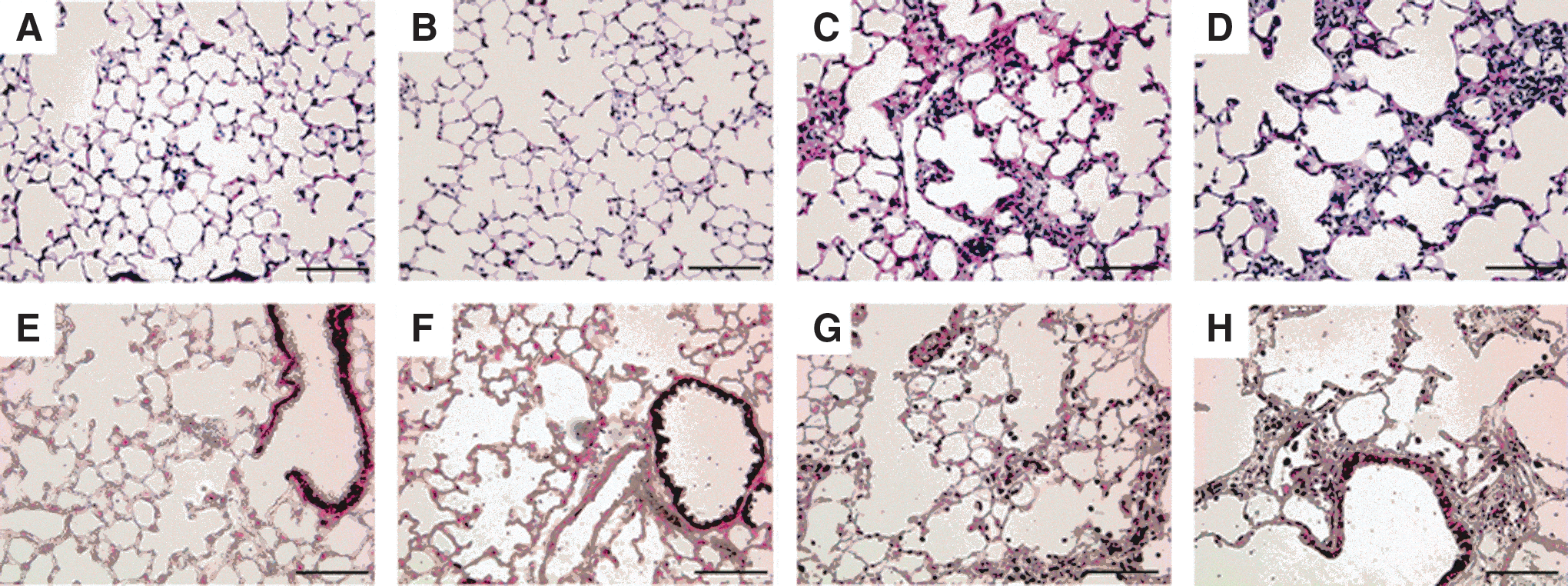
To determine whether SFN treatment could protect mice from bleomycin-induced fibrosis, 8-week-old mice received an intratracheal instillation of either vehicule (NaCl) or bleomycin on Day (D) 0 and were treated by either phosphate-buffered saline (PBS) or SFN for 2 weeks. Lung histology from SFN mice was similar to control mice (Fig. 9A, B), indicating the lack of SFN toxicity in noninjured mouse lungs. Thickened alveoli and fibroblastic foci were observed in bleomycin-treated mice (Fig. 9C, D). SFN treatment did not prevent lung fibrosis after pulmonary bleomycin injury (Fig. 9D). Nrf2 immunostaining was detected in the bronchiolar epithelium (Fig. 9E) and was slightly increased in SFN-treated mice compared with control mice (Fig. 9F). Cytoplasmic Nrf2 staining was increased in the bronchiolar epithelium, alveolar type II cells, and alveolar macrophages in bleomycin-treated mice compared with control mice (Fig. 9G). Nrf2 immunostaining was slightly increased in bleomycin/SFN mice compared with bleomycin-treated mice (Fig. 9H).
Discussion
We showed a decreased expression of nuclear Nrf2 and EPII in human pulmonary fibroblasts from patients with IPF and the key role of altered Nrf2 level in the fibroblast phenotype. Nrf2 inhibition with siRNA-induced myofibroblastic differentiation was associated with increased oxidative stress. Conversely, Nrf2 activation with Keap1 knockdown restored the oxidant/antioxidant balance and reversed the myofibroblastic differentiation. SFN, a known activator of Nrf2, reversed the myofibroblastic differentiation of IPF fibroblasts in vitro: decreased α-SMA and collagen I expression and decreased cell proliferation, migration and contraction abilities, both under basal and profibrosis conditions. These alterations were associated with restored Nrf2 expression and oxidant/antioxidant balance in fibroblasts. Nrf2 may play a central role in fibroblast differentiation in IPF, with the activation by SFN providing antifibrosis effects.
We highlighted increased oxidative stress in human IPF fibroblasts, which confirms previous results showing increased expression of NADPH oxidase in human pulmonary fibroblasts from IPF patients (1, 17). Our study explored the endogenous antioxidant side of this balance. Indeed, we observed decreased EPII expression in IPF fibroblasts, concomitant with increased 4-HNE staining, a lipid peroxidation product. Our results agree with the loss of HO-1 in the fibroblastic foci of the human IPF/UIP lung reported by Lakari et al. (27). This oxidant/antioxidant imbalance in IPF pulmonary fibroblasts was associated with decreased nuclear Nrf2 expression. Our results agree with the recent evidence of decreased total Nrf2 expression in fibroblastic foci in IPF lung biopsies (31).
As recently described (1, 17), a profibrosis condition such as with TGF-β stimulation induces myofibroblastic differentiation of fibroblasts and increases oxidative stress. In our study, TGF-β stimulation decreased the antioxidant defences of fibroblasts. Our results strongly suggest an association of myofibroblastic differentiation and oxidant/antioxidant imbalance, both involving TGF-β. The role of Nrf2 pathway in the differentiation of human pulmonary fibroblasts into myofibroblasts has never been studied. In control fibroblasts, the specific inhibition of Nrf2 by siRNA induced myofibroblastic differentiation associated with increased oxidative stress (increase of oxidant damage with increased 4-HNE staining and defective antioxidant defences). Moreover, in IPF fibroblasts, Nrf2 specific activation by Keap1 siRNA transfection reversed the myofibroblastic differentiation associated with restored antioxidant defences and decreased oxidant damage. These results suggest a potential antifibrosis role of Nrf2 in IPF in humans. In a murine model of bleomycin-induced pulmonary fibrosis, Cho et al. showed increased sensitivity to bleomycin and more severe inflammatory lesions in Nrf2-knockout mice (8). In agreement with our results, Nrf2 was previously found to down-regulate collagen I mRNA level in mice (28). Beyond its clear role in inflammatory respiratory diseases such as chronic obstructive pulmonary disease in humans (6), Nrf2 has been found involved in cellular differentiation, in cancer cell lines in vitro (37). Our study highlights a new role of Nrf2 in myofibroblastic differentiation of fibroblasts during IPF.
The activation of Nrf2 by SFN could decrease oxidative stress damage in IPF and control fibroblasts as previously reported (9, 14, 45). SFN induced EPII expression in cutaneous human fibroblasts (10), Beas2B cells (29), and tubular epithelial rat cells (40). In our study, SFN decreased oxidative stress, increased EPII expression and nuclear Nrf2 not only in control but also in IPF fibroblasts. SFN could also restore EPII expression and nuclear Nrf2 in IPF and control human pulmonary fibroblasts even after a prestimulation with TGF-β. Interestingly, the inhibition of P38 subunits by SFN (21) could block the TGF-mediated Nrf2 degradation reported in alveolar epithelial cells (34). This suggests that SFN can induce nuclear Nrf2 and decrease oxidative stress even in IPF patients and, thus, could lead to new therapeutic strategies.
The pharmacological activation of Nrf2 by SFN was assessed in the murine bleomycin fibrosis model. Unfortunately, SFN did not exhibit antifibrotic effect in vivo. Nrf2 was not expressed in lung fibroblasts in this model in the basal condition. Moreover, both SFN and bleomycin failed to modulate Nrf2 in lung fibroblasts. In the bleomycin model, the major inflammatory response and the oxidant nature of bleomycin contrast with the histological features of human IPF (4, 23). The bleomycin model of lung fibrosis is rather an acute lung injury with major oxidative and inflammatory responses. This probably explains the increased susceptibility of the Nrf2−/− mice to bleomycin (8). In our study, bleomycin increased Nrf2 expression in lung epithelial cells and macrophages. The Nrf2 activator SFN probably represents a small additive effect, compared with bleomycin alone. Finally, this model associated with major oxidative stress did not fit well to address the role of an Nrf2 inducer in lung fibrosis, especially in myofibroblastic dedifferentiation in the absence of Nrf2 expression in mice compared with human lung fibroblasts.
The antifibrosis effect of SFN as an Nrf2 activator on human pulmonary fibroblasts of IPF patients has never been studied. In our study, myofibroblast stimulation by SFN led to their dedifferentiation into fibroblasts concomitant with antioxidant defence restoration, even in pro-fibrosis conditions. The effect of SFN on cell differentiation was reported in a rat model of renal fibrosis (40). SFN inhibited the epithelial–mesenchymal transition of renal tubular epithelial cells, with decreased α-SMA expression (40). Antifibrosis properties have been reported with in vitro stimulation with N-acetylcystein, another Nrf2 activator (42). However, unlike SFN, N-acetylcystein has not been shown to inhibit fibroblast migration. SFN acted specifically via Nrf2 in fibroblasts. Indeed, SFN stimulation in fibroblasts after Nrf2 knockdown did not induce myofibroblastic dedifferentiation. In addition, we demonstrated that the effects of SFN on HO-1 and collagen I expression were mediated by Nrf2 in lung fibroblasts from wild-type and Nrf2 knockout mice.
We report an antiproliferating effect of SFN in human pulmonary fibroblasts, which could be associated with Nrf2 signals. Previous studies showed that SFN reduced the proportion of Barett esophageal adenocarcinoma cells in the S phase (36). SFN blocked the cell cycle in the G0 phase. Moreover, N-acetylcystein inhibited mouse fibroblast proliferation in vitro, not by inducing cell death but by cell cycle arrest in the mid-G1 phase. N-acetylcystein prevented activation of the mitogen-activated protein kinase extracellular-signal-regulated kinase1/2 and inhibited the expression of cyclin D1 (39).
In conclusion, we demonstrated decreased Nrf2 expression concomitant with oxidant/antioxidant imbalance and myofibroblastic differentiation in human pulmonary fibroblasts from IPF patients in vitro. Conversely, the induction of nuclear Nrf2 restored the oxidant/antioxidant equilibrium and reversed the myofibroblastic differentiation. The pro-fibrosis effect of TGF-β was related to decreased nuclear Nrf2 expression and counteracted by the antifibrosis effect of an Nrf2 activator, SFN. Our study identified Nrf2 as a novel potential therapeutic target in IPF and confirmed the in vitro antifibrosis effects of SFN.
Materials and Methods
Patients
This study was approved by the local ethics committee, and stored biopsies were reported to our institutional board (Delegation a la Recherche Clinique, Assistance Publique - Hopitaux de Paris, Carre Historique - Hopital Saint-Louis, 1 avenue Claude Vellefaux, 75475 Paris Cedex 10, France/Comite de Protection des Personnes, Ile de France 1, 1 Place du Parvis Notre-Dame, 75181 Paris cedex 04, France, reference 0811760).
We obtained biopsies from eight patients with IPF according to the American Thoracic Society definition (2), and eight control patients without radiographic, spirometric or histological features of fibrosis.
Control and IPF patients groups did not differ in age.
Cell culture
Processing of human lung samples and fibroblast isolation
Human lung tissue fragments were either immediately frozen in liquid nitrogen and stored at −80°C or used for extraction of fibroblasts. Human lung fibroblasts were cultured from lung explants until passage 4–6 as previously described (1). In some experiments, cells were incubated with recombinant human (rh) TGF-β1, PDGF-BB (R&D Systems) (1), or SFN (10 μM; Sigma Aldrich) (13).
Mouse lung fibroblasts
Mouse lung fibroblasts were obtained as previously described (44). Male C57BL/6 mice (8–12 week old) and Nrf2−/− mice were provided by the RIKEN BRC according a MTA to Dr S. Kerdine-Römer (19).
Cell stimulation
Cells were stimulated with TGF-β or SFN for 24 h. For therapeutic experiments, fibroblasts were prestimulated with TGF- β for 24 h, and then SFN for 6, 24, or 48 h.
Cell migration assay
Cell motility was examined as previously described (1) with use of a modified Boyden chamber (Transwell Costar) in response to 24-h stimulation with PDGF-BB (10 ng/ml), with or without SFN (10 μM) or media alone (with 1% fetal calf serum (FCS)). The chemotactic index was determined as the ratio of cells moving in response to PDGF-BB with or without SFN relative to cells moving in response to medium alone (control). Nuclear cells were stained by 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, 5 mg/ml; Invitrogen Corp.), and cells migrating through the Transwell membrane were automatically counted.
3-D collagen gel culture
Fibroblast-seeded collagen lattices were prepared by mixing collagen I extracted from rat tail (BD Biosciences), with 10×Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) and 10 N NaOH before adding 1.5 ml cell suspension to give a final concentration of 150 000 fibroblasts/ml lattice mixture. Fibroblasts in collagen gels were stimulated with TGF-β or SFN for 0, 7, and 14 days (18). At each time, the lattices were photographed, and rates of gel contraction were calculated by determining the remaining surface area by computer-based analysis and expressed as a percentage of initial or control area.
Cell proliferation
Fibroblasts were seeded in six-well plates (100, 000 cells/well) and stimulated in a complete medium with 10% FCS with TGF-β or SFN for 24 h or TGF-β for 24 h and then SFN for 48 h. Fibroblasts were trypsined, centrifuged, and counted by an automatic cell counter (VI-cell XR; Beckman).
siRNA transfection
Fibroblasts were transfected for 48 h with 50 nM Nrf2 siRNA (NM_006164) or 100 nM Keap1 siRNA (L-012453-00, both Dharmacon SMARTpool siRNA reagents), or negative control (scramble) siRNA (D-0018010-10-20, Dharmacon plus nontargeting pool) by the use of Lipofectamine 2000 according to the manufacturer's instructions (5, 16).
Cellular toxicity
Cellular toxicity and viability were assessed by trypan blue exclusion and lactate dehydrogenase release (1).
Phase-contrast microscopy
Phase-contrast microscopy was used to evaluate the morphological features of fibroblasts. Myofibroblastic phenotype was characterized by star-shaped cells, with abundant cytoplasm and short projections. Fibroblastic phenotype was characterized by thin and spindle-shaped cells with prolonged cytoplasmic projections.
GSH measurement
GSH/GSSG ratio was measured according to the manufacturer's instructions (glutathione assay Euromedex; Arbor Assay).
Western blot analysis
Proteins were extracted, and western blot analysis was performed as previously described (16) with antibodies for Nrf2 (Abcam), α-SMA (Sigma), and collagen I (SouthernBiothec), with expression normalized to that of Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Abcam).
Quantitative analysis of mRNA expression
The mRNA expression of HO-1, NQO-1, EPHX, α-SMA, and collagen I α1 was quantified by a reverse transcriptase–real-time polymerase chain reaction (7500 Software v2.0.1, Applied Biosystem) as previously described with expression normalized to that of ubiquitin-c (15).
Confocal laser scanning microscopy
Simple immunofluorescence labelling was performed to assess the subcellular localisation of Nrf2 (Santa Cruz Biotechnology) in fibroblasts as previously described (16). Nuclear Nrf2 immunofluorescence was quantified by co-localisation with TO-PRO3, a nucleic acid marker (Molecular Probes).
4-HNE immunocytochemistry
Fibroblasts were grown in 8-well Lab-teks (25, 000 cells/well in DMEM with 10% fetal calf serum; Lab-tek Chamber Slide System, 177445 Permanox Slide, NUNC) and stimulated or not with SFN for 24 h. 4-HNE fibroblast staining (Calbiochem) was used as an oxidative stress marker (16). Positive cells were revealed by use of the Vectastain ABC-alkaline phosphatase kit (Vector Laboratories) and the Liquid Permanent Red substrate-chromogen system (Dako). Positive cells were evaluated by two independent observers examining 10 different high-power fields at×400 magnification. Results were expressed as the number of positive cells per field (16). The intensity of protein staining was graded from 0 (absent) to +++ (very intense staining). Complete agreement in scoring was obtained between the two independent observers.
Bleomycin lung fibrosis
All experiments were performed with 8-week-old male C57BL/6J mice (Janvier) according to Institut National de la Santé et de la Recherche Médicale guidelines that complied with national and international regulations. On Day (D) 0, mice received an intratracheal instillation of 0.06 unit of bleomycin hydrochloride (Bleomycine Bellon; Aventis) or vehicle (0.9% sterile saline), in a volume of 50 μl. The animals were killed on Day 14 after instillation, and their lungs were removed for further analysis. Saline-treated mice were used as controls. PBS (20 mM Tris·HCl, pH 7.6, 137 mM NaCl) or SFN (25 mg/kg/day body weight) were administered by a subcutaneous injection to saline-treated or bleomycin-treated mice from D0 to D14 with a 2-day break (D5-D6 and D12-13) twice. This resulted in 4 groups of 12 animals each: Control, Bleomycin, SFN, Bleomycin/SFN.
Tissue preparation and immunostaining
Tissue harvesting was preceded by the administration of anesthetic and then exsanguination by severing the inferior vena cava. Mouse lungs were inflation fixed by gravity with 4% paraformaldehyde (PFA in PBS) at 25 cm hydrostatic pressure for 1 min. The lungs were immersed overnight in 4% PFA at 4°C, washed in PBS followed by dehydration in a series of ethanol solutions before paraffin embedding. Paraffin sections were deparaffinized in xylene, followed by rehydration in ethanol washes. Peroxidase treatment in methanol with 0.5% hydrogen peroxide was followed by heat-assisted antigen retrieval in 0.01 M sodium citrate buffer (pH 6.0). Blocking was performed for 2 h at room temperature using 2% horse serum followed by overnight incubation with primary antibody anti-Nrf2 (1:100; Abcam) at 4°C. Rinsed sections were incubated with biotinylated secondary antibodies directed to primary antibody host IgGs (7.5 μg/ml; Vector Laboratories), visualized with the Vectastain Elite ABC kit (Vector Laboratories) using nickel-diaminobenzidine as a substrate, enhanced with Tris-cobalt, and counterstained with 0.1% nuclear-fast red.
Statistical analysis
Data were analysed by use of Statview software (Abacus Concepts), and displayed as box-and-whiskers plot with median, interquartile range, and minimum and maximum values. Between-group differences were first assessed by a nonparametric analysis of variance (Kruskal–Wallis test) and then the Mann–Whitney U-test or the Wilcoxon paired test. p<0.05 were considered statistically significant.
Footnotes
Acknowledgments
Marcel Bonay was supported by the Chancellerie des Universités de Paris (Legs Poix) and the Fonds de Dotation “Recherche en Santé Respiratoire.” Elise Artaud-Macari was supported by the “Centre d'Assistance Respiratoire à Domicile d'Ile-de-France” (CARDIF).
Author Disclosure Statement
No competing financial interests exist.