
Abstract
Introduction
Innovation
It has not been investigated if Prx-2 is unregulated at the transcriptional level in response to oxidative stress. In this study, we first showed that Prx-2 is upregulated during muscle differentiation and regeneration. We further demonstrated that Prx-2 is upregulated by the PI3K/NF-κB pathway during muscle differentiation. Interestingly, the overexpression of Prx-2 prevents both myogenesis and ROS production. In contrast, the inhibition of Prx-2 expression induces the compensatory expressions of other antioxidant enzymes, and enhances muscle differentiation and ROS production. In conclusion, our findings provide a critical insight into the transcriptional regulatory mechanisms of Prx-2 via PI3K/NF-κB during muscle differentiation and regeneration.
Peroxiredoxins (Prxs) are a novel group of peroxidases with high antioxidant activity, and some members of this family can affect cell proliferation, immune responses (31), and apoptosis and differentiation (13). In mammals, six family members (Prx1–6) have been identified; these Prxs can be classified as typical 2-Cys, atypical 2-Cys, or 1-Cys Prxs based on the locations of their cysteine residues. Prx-2 belongs to the typical 2-Cys class and is a predominantly cytoplasmic Prx (29, 30); Prx-2 has redox-active cysteine residues that participate in the reduction of H2O2. Therefore, in the catalytic cycle, the conserved peroxidatic cysteine (Cp) residue of Prx-2 is oxidized to cysteine sulfenic acid (Cys-SOH), which subsequently interacts with the resolving cysteine (Cr) of another Prx-2 to form disulfide bonds between the two molecules, resulting in a homodimer that is subsequently reduced by thioredoxin (Trx). In general, Prx-2 is present in its reduced monomeric form together with Prx-2 disulfide catalytic cycle intermediates. In contrast, under oxidative stress, Prx-SOH is further oxidized by peroxide to sulfinic (-SO2H) and sulfonic acids (-SO3H), thereby causing the inactivation of its peroxidase activity. Prx-SO2H and Prx-SO3H, which are hyperoxidized Prx-2, can be slowly converted back to the reduced state by sulfiredoxin (Srx) (26).
H2O2 is an important regulator of signal messenger molecule for various pathways (34). We have previously demonstrated that H2O2 is an essential mediator of muscle differentiation (7). However, it is still not clear whether myogenesis is associated with the function of Prx-2 as an H2O2 scavenger; therefore we investigated the potential roles of Prx-2 in myogenesis.
In addition, the previous study showed that the H2O2 that is generated during muscle differentiation regulates nuclear factor kappa B (NF-κB) activation (17, 27, 28). During redox events, NF-κB is regulated by oxidative stress (12). NF-κB consists of five subunits: p65, RelB, c-Rel, p105/p50, and p100/p52, and these subunits are conserved and exist as hetero- or homodimers (8). The p65/p50 heterodimer is the most abundant NF-κB complex in the cell and is present in the cytoplasm as an inactive heterodimer with its inhibitor protein subunit (IκB). After stimulation, the IκB kinase induces the IκB phosphorylation, which promotes its degradation and causes the NF-κB complex to translocate into the nucleus (22). NF-κB induces the expression of genes involved in key cellular processes including proliferation, survival, and differentiation (25).
PI3K is a major myogenic signaling molecule that also activates NADPH oxidase, which generates reactive oxygen species (ROS) and leads to NF-κB activation. Many antioxidant enzymes are upregulated under conditions of oxidative stress. In contrast to other antioxidant enzymes, it is still unclear whether Prx2 is upregulated at the transcriptional level during oxidative stress. In this study, we hypothesized that Prx-2 is upregulated by the PI3K/NF-κB pathway during muscle differentiation because ROS are more commonly generated during muscle differentiation than proliferation. Initially, we demonstrated that Prx-2 is induced via the PI3K/NF-κB pathway during muscle differentiation and regeneration. We showed that Prx-2 plays an important role during myogenesis both in vitro and in vivo.
Results
Prx-2 expression increases during muscle differentiation
To observe the muscle differentiation phenotype in C2C12 cells, we examined the morphological changes that occurred when the cells were cultured in differentiation medium (DM) for up to 96 hours. We also measured the fusion index to semi-quantify the differentiation levels. As shown in Figure 1A, muscle differentiation progressed in a time-dependent manner. ROS levels were determined using dichlorofluorescein diacetate (DCFH-DA) after the cells were cultured in DM for 24 hours. ROS generation was significantly higher in cells cultured in DM than in proliferation medium (PM) (Fig. 1B, left). The semi-quantification of H2O2 generated during muscle differentiation was also performed using the Amplex UltraRed assay on the basis of a standard H2O2 dose curve. The results indicated that ROS generation was significantly higher in cells cultured in DM than in PM (Fig. 1B, right). The expression levels of muscle differentiation markers, such as myogenin and MHC, were also increased during muscle differentiation (Fig. 1C). To determine whether Prx-2 expression was induced during the myogenic process, we observed the expression levels of Prx-2 under nonreducing or reducing conditions. Compared with cells cultured in PM, the expression levels of the dimeric and monomeric forms of Prx-2 were increased in a time-dependent manner in DM-cultured cells (Fig. 1C). To investigate further the oxidative state of Prx-2, we evaluated Prx-SO3H levels by Western blot analysis and found that the hyperoxidized forms of Prxs were more abundant during differentiation (Fig. 1C). The same results were observed in H9c2 rat cardiac myoblasts (data not shown). Next, we treated proliferating and differentiating cells with 100 mM N-ethylmaleimide (NEM, a thiol-blocking agent) prior to lysis to prevent dimerization during the sample preparation. These samples were analyzed by Western blot analysis under nonreducing conditions. As shown in Figure 1D, higher expression levels of the monomeric and dimeric forms of Prx-2 were observed during muscle differentiation.
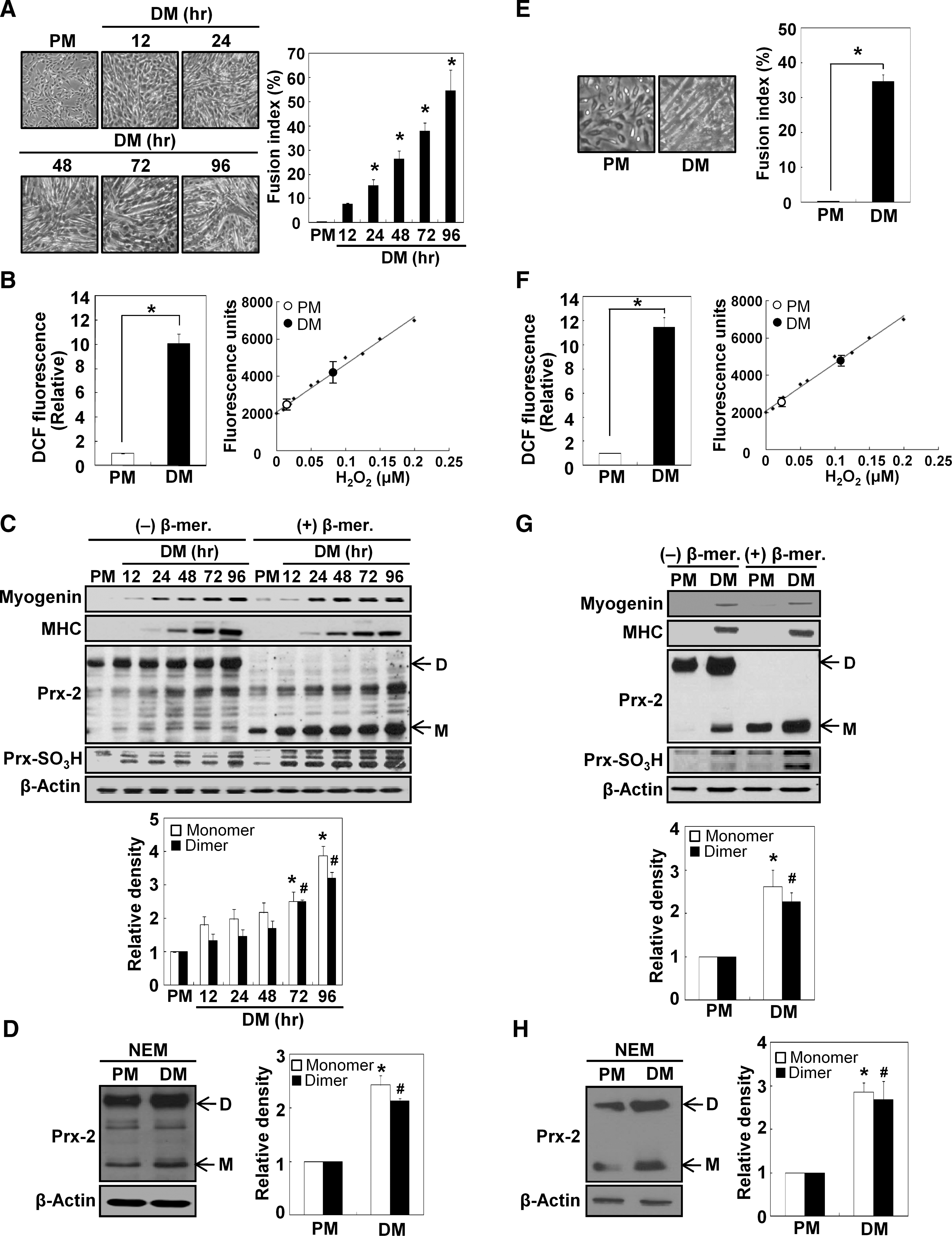
To verify the results observed in C2C12 cells, we isolated satellite cells from the mouse hind limb muscles, and monitored their differentiation by observing the morphological changes and measuring the fusion index. Primary cultured myoblasts were differentiated effectively in response to DM (Fig. 1E). H2O2 levels were significantly higher in cells cultured in DM rather than PM, which was in accordance to our observations in C2C12 cells (Fig. 1F). The expression levels of the dimeric and monomeric forms of Prx-2 also increased in primary cultured myoblasts after differentiation was induced for 48 hours (Figs. 1G and 1H). Therefore, we concluded that Prx-2 expression increases during muscle differentiation.
Prx-2 is upregulated at the transcriptional level during muscle differentiation
We hypothesized that the increase in Prx-2 expression may occur at the transcriptional level. To test this hypothesis, we performed semi-quantitative RT-PCR (Fig. 2A) and real-time quantitative RT-PCR (Fig. 2B). Both experiments revealed that Prx-2 mRNA level increased during muscle differentiation. To rule out the possibility of mRNA stabilization, we first induced the upregulation of Prx-2 mRNA by incubating the cells in DM for 24 h and subsequently treated cells with actinomycin D to inhibit de novo mRNA synthesis in PM or DM for up to 24 h to assess the mRNA decay rates by semi-quantitative RT-PCR (Fig. 2C) and real-time quantitative RT-PCR (Fig. 2D). We observed that the transcription of Prx-2 mRNA was completely inhibited 24 h after actinomycin D treatment. More importantly, the decay rate of Prx-2 mRNA was similar for the cells cultured in both PM and DM, indicating that the induction of Prx-2 expression during muscle differentiation is caused by transcriptional activation.
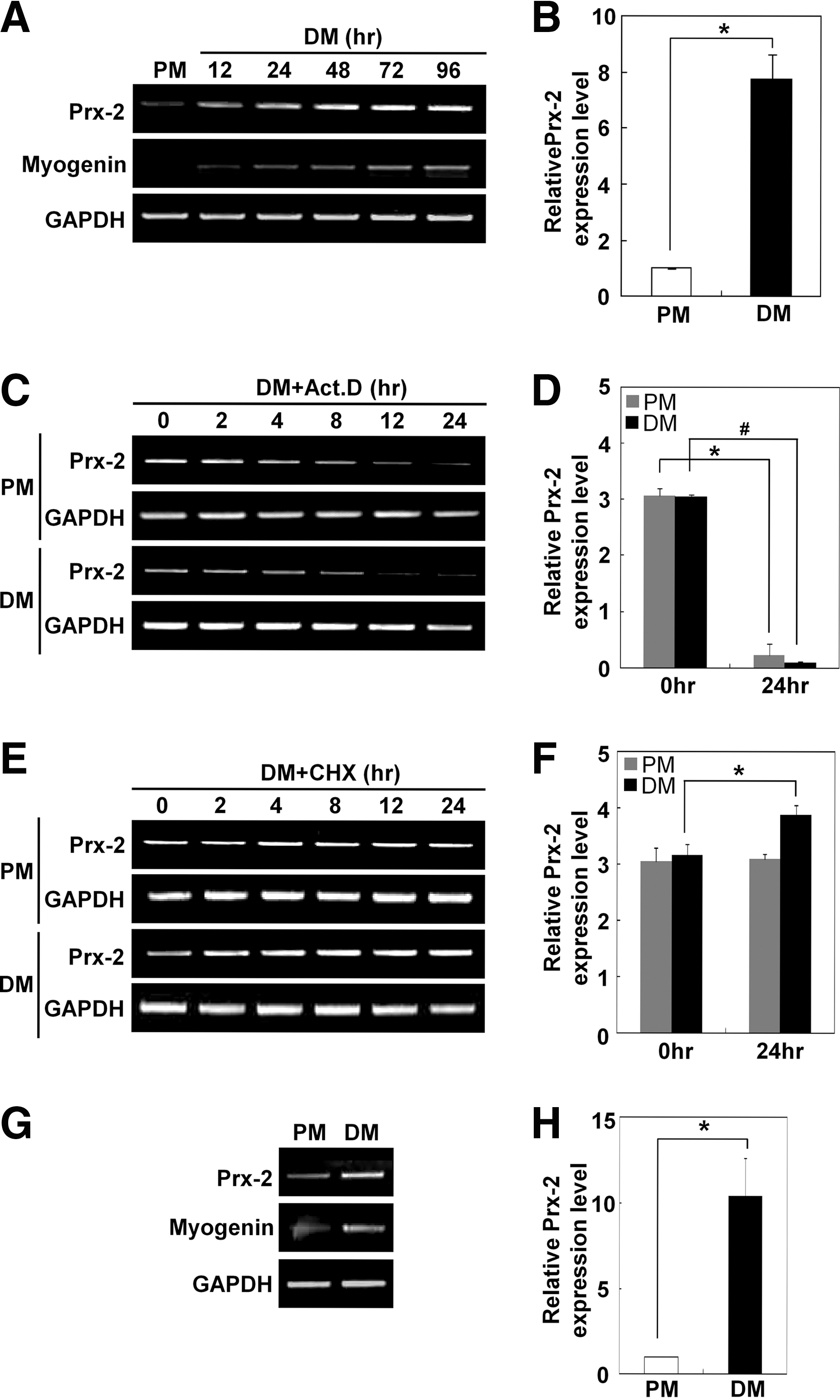
To determine whether the induction of Prx-2 during muscle differentiation is dependent on the synthesis of new proteins, we treated the cells with cycloheximide to inhibit protein synthesis in PM or DM for up to 24 h after induction of Prx-2 under DM culture conditions for 24 h. As shown in Figures 2C and 2D, cycloheximide did not block the increase in Prx-2 mRNA levels. Instead, cycloheximide increased Prx-2 mRNA levels in DM-cultured cells compared to PM-cultured cells as measured by semi-quantitative RT-PCR (Fig. 2E) and real-time quantitative RT-PCR (Fig. 2F). Using semi-quantitative RT-PCR (Fig. 2G) and real-time quantitative RT-PCR (Fig. 2H), we also found that Prx-2 mRNA level was upregulated at the transcriptional level during the differentiation of primary myoblasts. Taken together, these data suggested that Prx-2 is transcriptionally upregulated during muscle differentiation and that this mRNA induction is independent of new protein synthesis.
Upregulation of Prx-2 by H2O2 occurs only in myoblasts
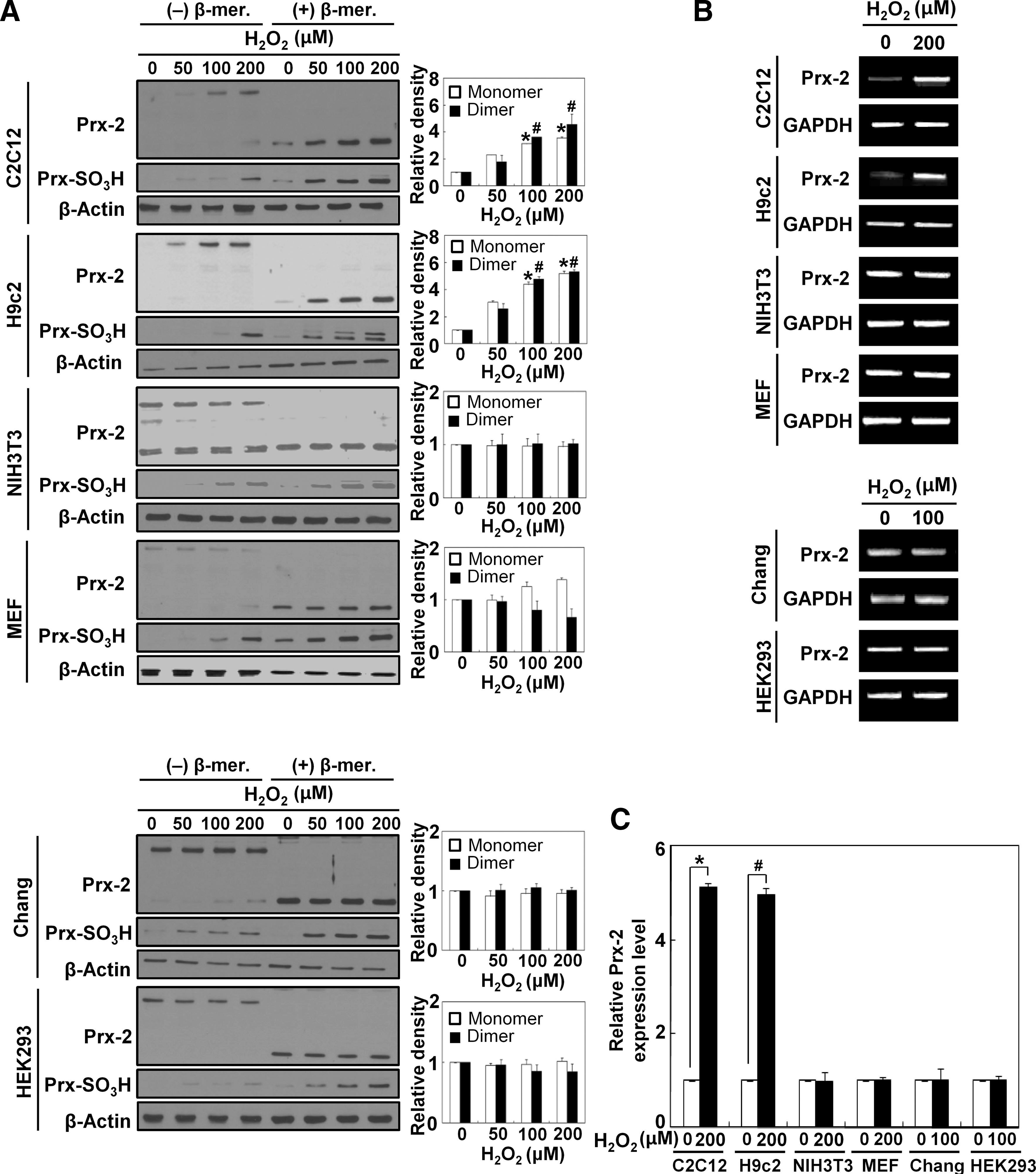
We next investigated whether Prx-2 expression also increases in response to exogenous ROS as observed during muscle differentiation. For these experiments, we treated several cell lines with H2O2 for 6 hours. The appropriate concentrations of H2O2 were determined using MTT assay as shown in Supplementary Fig. S1 (Supplementary Data are available online at
Antioxidants prevent the upregulation of Prx-2 and attenuate myogenesis
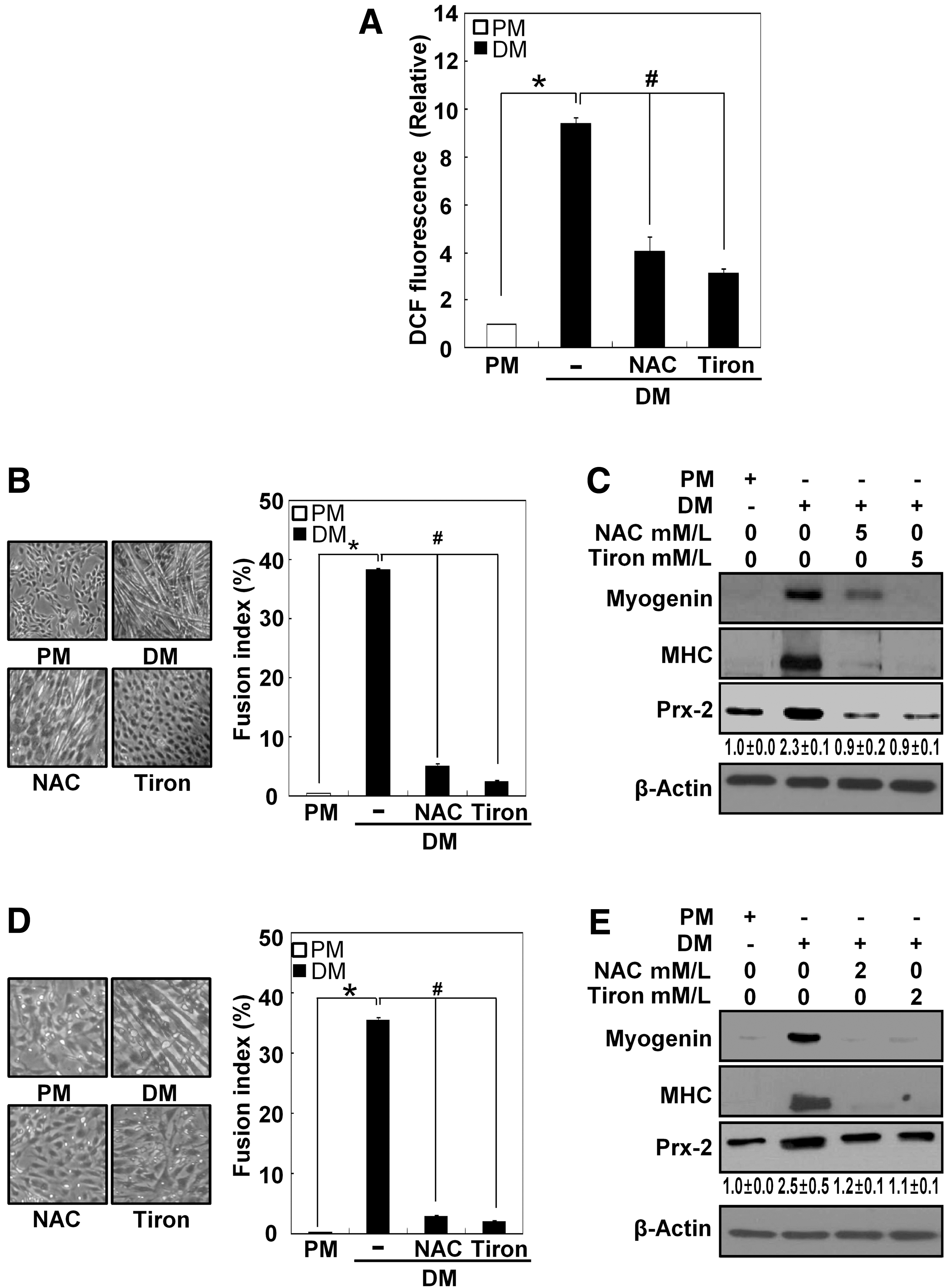
To determine whether the ROS that are generated during muscle differentiation stimulate Prx-2 upregulation, we treated differentiating cells with 5 mM N-acetyl L-cysteine (NAC) and 5 mM 4,5-dihydroxy-1,3-benzenedisulfonic acid (Tiron, a superoxide scavenger) for 24 hours and then measured ROS levels. As shown in Figure 4A, both antioxidants diminished the higher ROS levels in DM-cultured cells. They also both suppressed myotube formation, lowered the expression levels of myogenin, MHC, and Prx-2 (Figs. 4B and 4C), and attenuated both myogenesis and Prx-2 induction in primary myoblasts as well (Figs, 4D and 4E). To further determine the expression levels of the dimeric and the monomeric forms of Prx-2 and Prx-SO3H in response to antioxidants, we analyzed their expression levels under nonreducing and reducing conditions (Supplementary Fig. S2).
Transcriptional induction of Prx-2 is regulated by NF-κB during muscle differentiation
To determine whether NF-κB regulates Prx-2 transcription during muscle differentiation, we analyzed Prx-2 promoter with the Genomatix program. As shown in a schematic representation of potential transcription factor binding sites (Fig. 5A), we found that Prx-2 promoter contains two putative NF-κB binding sites that are located between 3361 and 3352 bp (candidate #1) and between 3254 and 3245 bp (candidate #2) upstream of Prx-2 start codon.
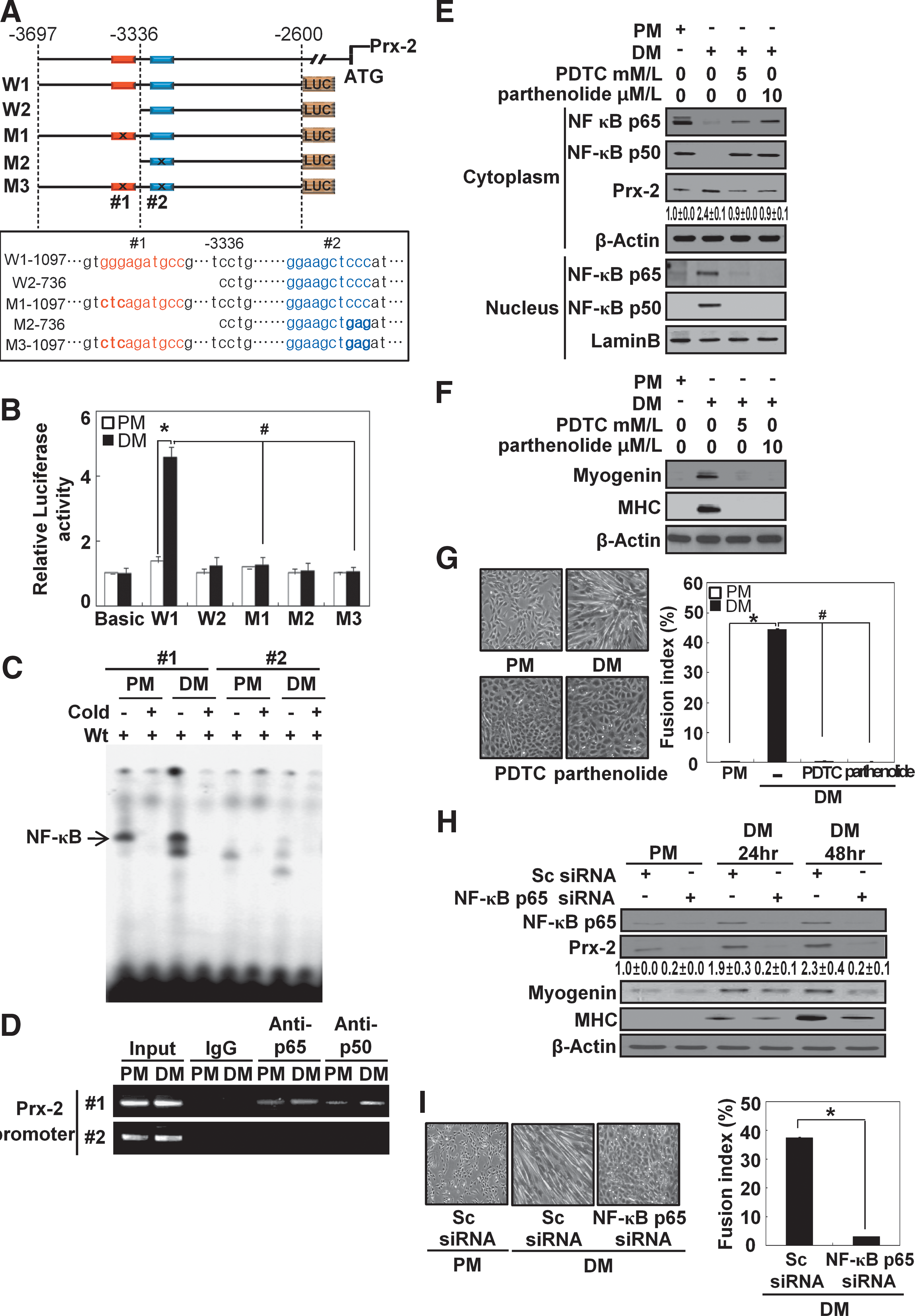
To test whether NF-κB is required for Prx-2 transcription, we first constructed wild-type luciferase plasmids (W1-1097 and W2-736). The W1-1097 plasmid contained both putative NF-κB binding sites (candidates #1 and #2), while the W2-736 plasmid contained only one putative NF-κB binding site (candidate #2). We also made constructs containing either a point mutation each putative NF-κB binding site (M1-1097 and M2-736) or in both binding sites (M3-1097) to identify the functional NF-κB binding site on Prx-2 promoter. These resultant reporter plasmids were transiently introduced into C2C12 cells, and the activity of Prx-2 promoter activity was monitored by measuring luciferase activity during the differentiation process. The luciferase activity increased with the transfection of the −3697 to −2600 sequence of Prx-2 upstream promoter located upstream of the start codon. When the candidate site #1 was mutated (M1-1097), the transcriptional activity of NF-κB was dramatically abolished. However, the mutation in candidate site #2 (M2-736) did not change the level of luciferase activity from wild-type construct in the reporter assay. The double mutation constructs (M3-1097) also displayed basal levels of luciferase activity. We did not observe any luciferase activity in cells cultured under PM or DM conditions when empty pGL3-basic vector was used (Fig. 5B).
Next, we used oligonucleotides containing each putative NF-κB binding sites to perform an electrophoretic mobility shift assay (EMSA). Only the oligonucleotide containing candidate site #1 showed strong binding, while the oligonucleotide containing candidate site #2 did not show any significant binding (Fig. 5C). The binding was abolished by a hundred-fold excess amount of unlabeled competitor oligonucleotides. A Chromatin Immunoprecipitation (ChIP) assay also showed that candidate site #1 is associated with NF-κB (Fig. 5D). Therefore, we concluded that the NF-κB binding site that is located between 3361 and 3352 bp upstream of Prx-2 start codon is indispensable for the induction of Prx-2 mRNA.
To further investigate the effect of the NF-κB on Prx-2 induction, we treated cells with NF-κB inhibitors PDTC (pyrrolidinedithiocarbamate acid) and parthenolide and observed the translocation of NF-κB (Fig. 5E). As expected, NF-κB p65 and NF-κB p50 were both translocated from the cytosol to the nucleus in cells cultured with DM, but NF-κB inhibitors prevented this nuclear translocation. We also observed that NF-κB inhibitors decreased the protein levels of Prx-2, myogenin, and MHC, and inhibited myotube formation (Figs. 5F and 5G). Also, siRNA knockdown of NF-κB p65 produced the same results as with the inhibitors (Figs. 5H and 5I).
Antioxidants prevent NF-κB from binding to Prx-2 promoter during muscle differentiation
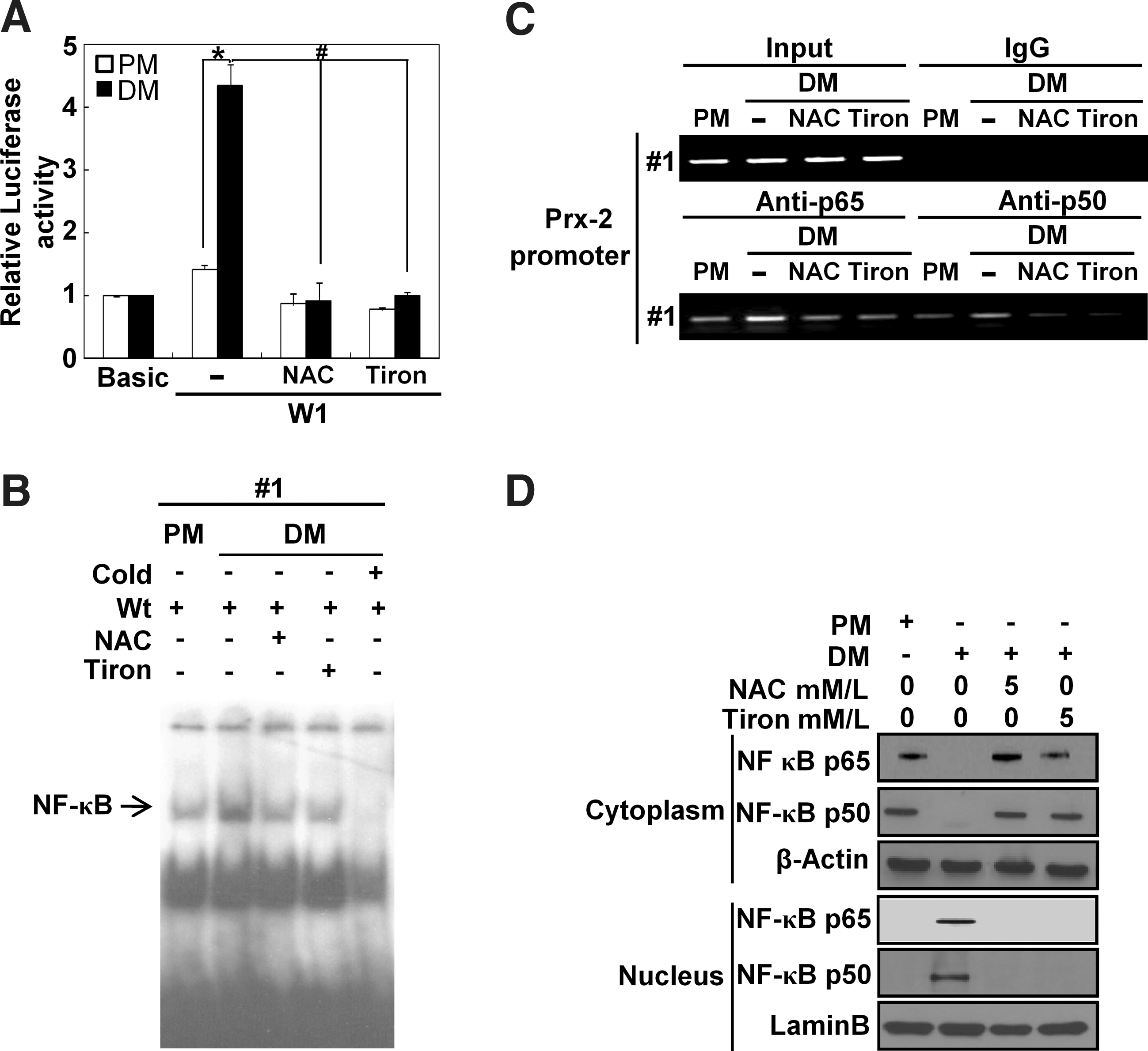
To further investigate whether NF-κB binding to Prx-2 promoter was suppressed by antioxidants during muscle differentiation, we treated the differentiating cells with NAC and Tiron and then observed NF-κB activity. The luciferase activity of Prx-2 promoter was significantly reduced by antioxidants, compared with the untreated transfectants (Fig. 6A). Both antioxidants dramatically decreased NF-κB binding to Prx-2 promoter as determined by both the ChIP assay and the EMSA (Figs. 6B and 6C). In addition, they both prevented nuclear translocation of NF-κB p65 and NF-κB p50 from the cytosol to the nucleus in cells cultured with DM (Fig. 6D).
PI3K is involved in NF-κB activation during muscle differentiation
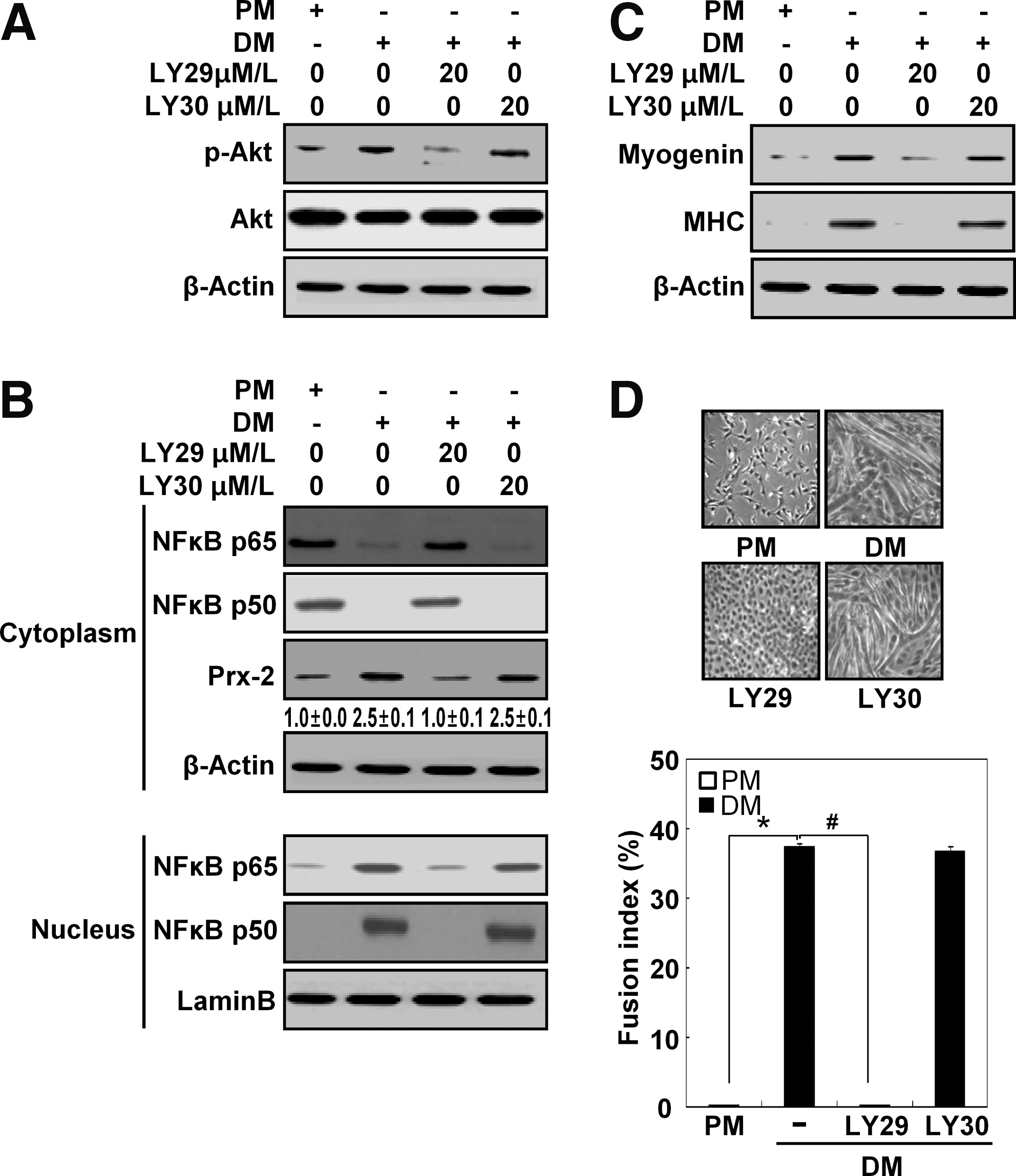
The PI3K signaling pathway is crucial for muscle differentiation (11). To examine whether the PI3K pathway is also involved in muscle differentiation, we treated the differentiating cells with LY294002 (a PI3K inhibitor) or LY303511 (a negative control for LY294002). In an effort to assess the effectiveness and specificity of LY294002, we monitored p-Akt level during muscle differentiation and observed that the level of p-Akt decreased when PI3K was inhibited (Fig. 7A). Previous reports have indicated that NF-κB can be activated downstream of PI3K during muscle differentiation (11, 28). Therefore, we tested whether inhibition of PI3K suppresses NF-κB activation and lower Prx-2 protein levels during muscle differentiation. As shown in Figure 7B, inhibiting PI3K prevented the nuclear translocation of NF-κB p65 and NF-κB p50 in cells cultured with DM. Additionally, Prx-2 expression, which was regulated by NF-κB, was decreased in the presence of LY294002; this inhibitor also blocked muscle differentiation (Figs. 7C and 7D). As expected, LY303511 did not have the effects as LY294002. Therefore, we concluded that the PI3K/NF-κB signaling pathway performs a key function in the induction of Prx-2 expression during muscle differentiation.
Prx-2 overexpression attenuates muscle differentiation
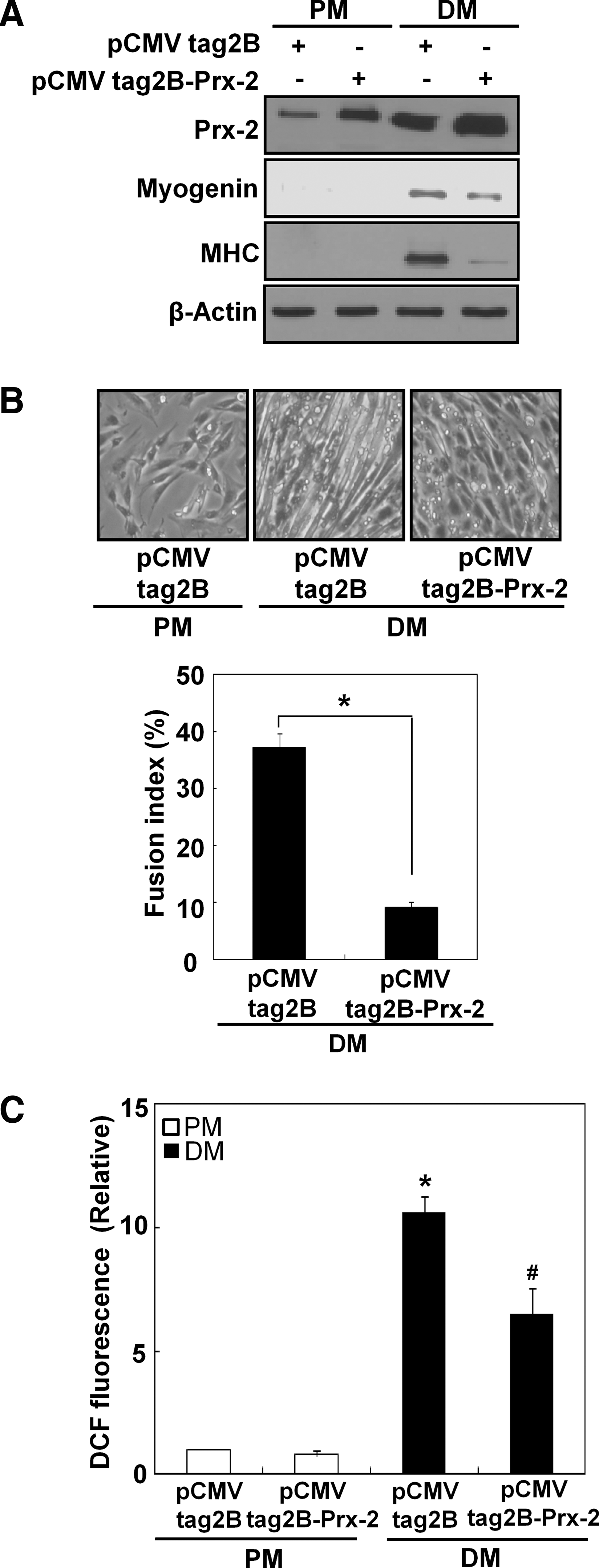
To understand further the role of Prx-2 in myogenesis, we transfected C2C12 cells with either an empty pCMV-Tag2B vector or the pCMV-Tag2B-Prx-2 plasmid and then cultured the cells using DM. As shown in Figure 8A, Prx-2 was highly overexpressed after transient transfection with pCMV-Tag2B-Prx-2, and Prx-2 overexpression attenuated myogenesis (Figs. 8A and 8B). The levels of intracellular ROS were lower in C2C12 cells transfected with pCMV-Tag2B-Prx-2 than in control pCMV-Tag2B-transfected cells (Fig. 8C). Collectively, our data showed that overexpression of Prx-2 attenuates myogenesis.
Prx-2 knockdown upregulates the expression levels of other antioxidant enzymes
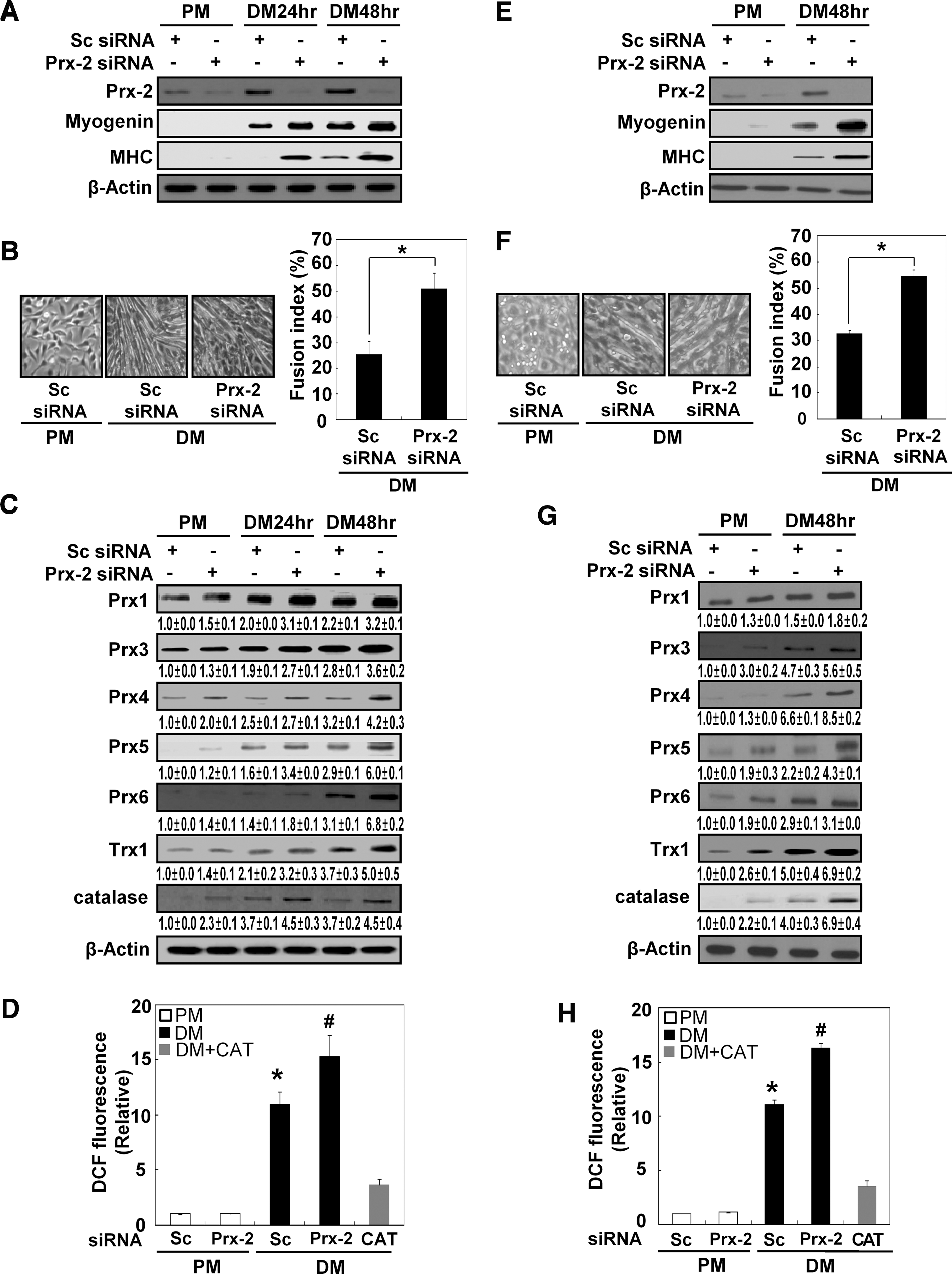
To assess the effects of Prx-2 on muscle differentiation, we used Prx-2-specific siRNA to completely eliminate its expression (Fig. 9A). Inhibiting Prx-2 resulted in a more distinct myotube formation and an increase in the expression levels of both myogenin and MHC (Figs. 9A and 9B). The loss of Prx-2 also increased the expression levels of other antioxidant enzymes, including all other Prx family members, Trx1 and catalase, which are cyotosolic antioxidants (Fig. 9C). Moreover, the levels of intracellular ROS were higher in cells transfected with Prx-2 siRNA than in cells transfected with scrambled siRNA. The levels of intracellular ROS were significantly reduced after treatment with catalase (1000 U/mL) in cells cultured with DM (Fig. 9D). In primary myoblasts, Prx-2 expression was also completely suppressed by treatment with Prx-2-specific siRNA (Fig. 9E). As observed using C2C12 cells, the suppression of Prx-2 expression induced a more distinct myotube formation and an increase in the expression levels of myogenin, MHC and other antioxidant enzymes, including Prx family members, Trx-1, and catalase (Figs. 9F and 9G). In addition, the levels of intracellular ROS were higher in cells transfected with Prx-2 siRNA than in cells transfected with scrambled siRNA. The levels of intracellular ROS were reduced by the addition of catalase to DM-cultured C2C12 cells, as observed in C2C12 cells. In summary, we concluded that Prx-2 plays an important role in controlling the H2O2 that is generated during muscle differentiation.
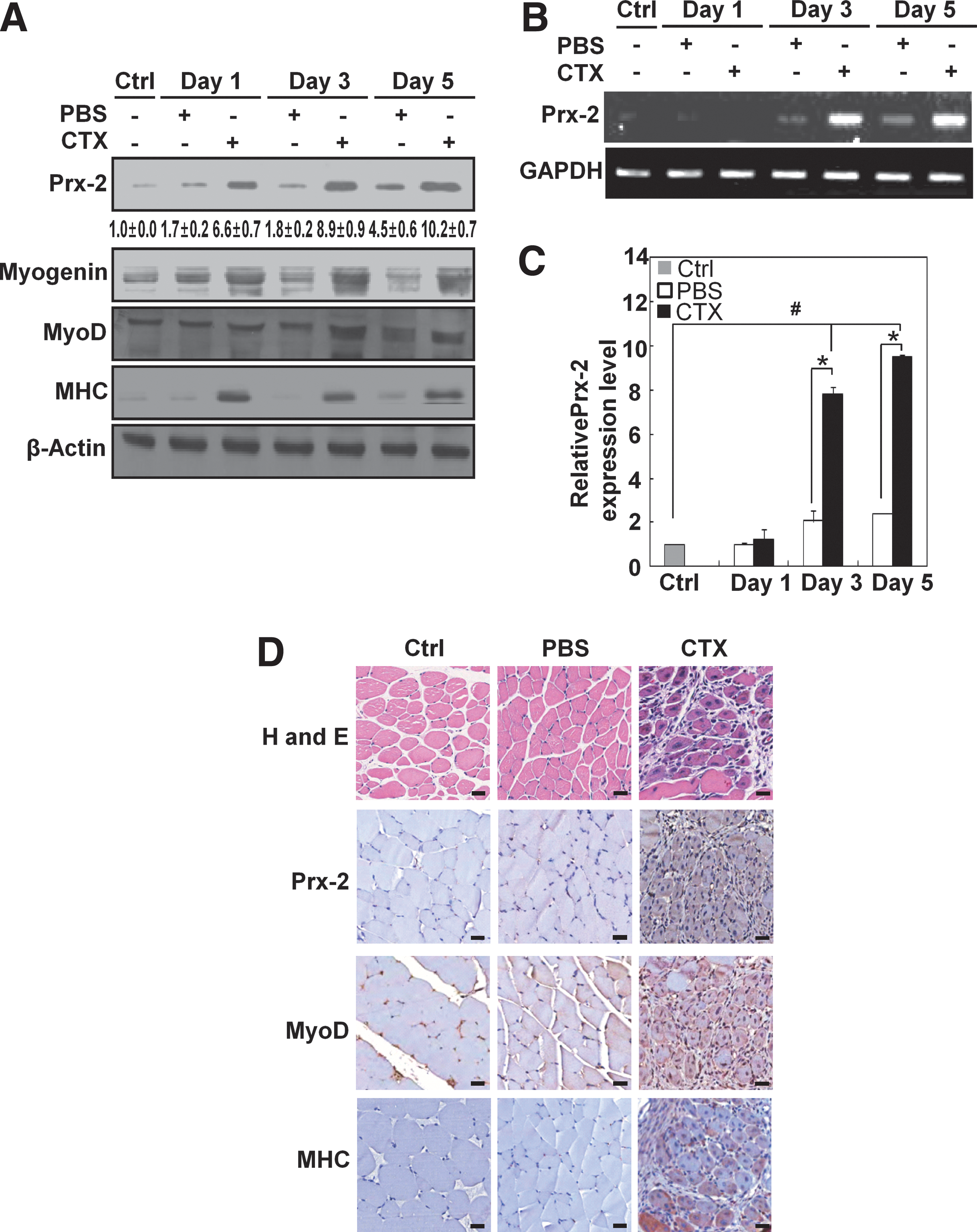
Prx-2 expression is upregulated in skeletal muscle upon cardiotoxin injury
To determine whether Prx-2 participates in skeletal muscle differentiation in vivo, we examined its expression in a model of skeletal muscle regeneration. We triggered skeletal muscle injury by injecting cardiotoxin (CTX) into the tibialis anterior (TA) muscles. The tissues were analyzed at days 1, 3, and 5 (n=3–5). As shown in Figure 10A, the expression levels of Prx-2, myogenin, MyoD, and MHC were higher in CTX-injected TA muscles than in uninjured or PBS-injected TA muscles. Both semi-quantitative RT-PCR (Fig. 10B) and real-time quantitative RT-PCR (Fig. 10C) revealed that Prx-2 mRNA levels were remarkably upregulated after injury. The H and E stained cross-sections of TA muscle showed typical signs of injury and regeneration, 5 days after the CTX injection as indicated by the presence of newly formed myofibers with centralized nuclei. An immunohistochemical analysis showed that Prx-2, MyoD, and MHC were strongly expressed by muscle precursors and regenerating muscle fibers (Fig. 10D). In conclusion, Prx-2 expression is upregulated during injury-induced muscle regeneration in vivo.
Discussion
In this study, we first noted that Prx-2 expression was upregulated by NF-κB during muscle differentiation in C2C12, H9c2, and primary myoblast cells. We also found that Prx-2 was upregulated during the muscle regeneration process in vivo. In addition, the binding of NF-κB to Prx-2 promoter was induced by the H2O2 that is generated during muscle differentiation. Prxs are present in all subcellular compartments and are expressed at various levels. The changes in the expression levels of the diverse family of Prxs have been extensively investigated in recent reports. For example, Prx-1 is the most widely studied and experiences the greatest transcriptional upregulation following treatment with a variety of factors that cause oxidative stress. NF-E2-related factor 2 (Nrf2) regulates of Prx-1 gene expression in response to electrophilic ROS-producing agents and hypoxia (10, 16). In addition, the stress-dependent induction of Prx gene expression has also been observed for other Prx family members. Previous studies have shown that Forkhead box O3 (FOXO3A) is required for the upregulation of Prx-3 under serum deprivation conditions in cardiac fibroblasts (2), and the Nrf2/Nrf1 pathway plays a role in the expression of Prx-3 and Prx-5 in response to quercetin (23). Also, Nrf2 is a key regulator of transcription of the Prx-6 gene under conditions of oxidative stress (4). However, the transcriptional regulatory mechanisms of Prx-2 have rarely been studied. A few reports have shown that Prx-2 is upregulated at the protein level during keratinocyte differentiation (36), and in response to cytokines or H2O2 in pancreatic β-cells (1). Therefore, we investigated Prx-2 promoter to identify a binding site for its transcription factor. We found that typical consensus NF-κB binding sequences exist in the human, rat, and mouse Prx-2 promoters. One of these, which was located between 3361 and 3352 bp upstream of the mouse Prx-2 start codon, bound directly to NF-κB during muscle differentiation.
We also demonstrated that Prx-2 expression only increased in response to exogenous H2O2 in myoblasts and not in NIH3T3, MEF, Chang, and HEK293 cells. These data are supported by previous reports that demonstrated that only the hyperoxidized form of Prx-2 increases in response to H2O2 in the MEF and HEK293 cell lines (3, 15), while the total amount of Prx-2 remains the same. It is currently unclear why Prx-2 is not upregulated in the NIH3T3, MEF, HEK293, and Chang cell lines. Furthermore, we showed that NF-κB was activated either by endogenous H2O2 generated during muscle differentiation or by exogenously added H2O2 and was involved in the upregulation of Prx-2 expression at the transcriptional level; this upregulation was also specific to myoblasts. Because the previous studies have shown that ROS-activated NF-κB promotes muscle differentiation and that its suppression blocks muscle differentiation and the transcriptional upregulation of MnSOD and iNOS (28,32), it is likely that NF-κB plays diverse roles in muscle differentiation.
Myoblasts proliferate under high serum conditions, and confluent cells spontaneously differentiate after several days in low serum due to the autocrine expression of IGF acting through the IGF receptor (6). Moreover, IGF-1 and −2 stimulate muscle differentiation by activating several signaling pathways. These IGFs stimulate the myogenic process via PI3K. Previous reports have demonstrated that PI3K leads to the activation of NADPH oxidase, which, in turn, produces ROS during muscle differentiation (17, 28). In addition, NF-κB is a target for NADPH oxidase or mitochondria-derived ROS in skeletal myoblasts (5, 18, 28). In this study, we showed that the inhibition of PI3K blocked NF-κB activation, suppressed the expression of Prx-2, and impaired muscle differentiation. Therefore, we concluded that Prx-2 upregulation was induced by NF-κB, which is downstream of the PI3K pathway during muscle differentiation. The scheme presented in Supplementary Fig. S3 depicts how Prx-2 is upregulated in the PI3K/NF-κB-dependent myogenic signaling pathway.
The overexpression of Prx-2 prevented both myogenesis and ROS production. In contrast, Prx-2 knockdown improved muscle differentiation and upregulated the expression of other antioxidant enzymes, including all other Prx family members, Trx1, and catalase. Furthermore, intracellular ROS levels were higher in cells transfected with Prx-2-siRNA than in cells transfected with scrambled siRNA, suggesting that other antioxidant enzymes may compensate for the loss of Prx-2. These findings are supported by previous reports indicating that the expression of Prx-3 was increased in Prx-5-knockdown mice under conditions of hypoxia, suggesting that the upregulation of Prx-3 compensates for the knockdown of Prx5 (35). Also, these previous reports have shown that excessive H2O2 not only causes cell death but also impairs myogenesis, while physiological levels of H2O2 are a major signaling mediator for muscle differentiation (7, 19). Therefore, the enhanced levels of H2O2 following treatment with Prx-2-siRNA may accelerate myogenesis instead of impairing it. Our data showed that although Prx-2 is dispensable due to the compensatory upregulation of other antioxidant enzymes, it is an important antioxidant enzyme that is used to control excessive ROS during muscle differentiation.
In summary, we have provided novel evidence that Prx-2 is upregulated at the transcriptional level via the NF-κB/PI3K pathway during muscle differentiation. Also, Prx-2 knockdown induced the compensatory expressions of other antioxidant enzymes, including all other Prx family members, Trx1, and catalase, but it also increased intracellular ROS levels. Taken together, these results suggest that Prx-2 is an important molecule for the control of ROS homeostasis in myoblasts. It remains unclear why Prx-2 is upregulated only in myoblasts and not in other cell lines. It is possible that one or more myogenic transcription factors may cooperate with NF-κB to transcriptionally upregulate Prx-2 during muscle differentiation.
Materials and Methods
Materials
Dulbeco's modified Eagle's Medium (DMEM), Dulbecco's modified Eagle's medium/F-12 (DMEM/F-12), fetal bovine serum (FBS), and calf serum were purchased from GIBCO-BRL (Grand Island, NY). 2′7′-dichlorofluorescein diacetate (DCF-DA), actinomycin D, cycloheximide, pyrrolidine dithiocarbamate acid (PDTC), parthenolide, H2O2 solution, catalase, β-mercaptoethanol, and collagenase type I were acquired from Sigma (St. Louis, MO). Basic fibroblast growth factor was purchased from R & D systems (Minneapolis, MN). LY294002 and LY303511 were obtained from Tocris Cookson, Ltd (Bristol, UK). [γ-32P] ATP was purchased from Amersham Biosciences (Little Chalfont, UK). N-ethylmaleimide (NEM), NF-κB probe for electrophoretic mobility shift assays, and antibodies specific to MHC, myogenin, Prx-1, NF-κB p50, Akt, and lamin B were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies specific to Prx 2-6, Prx-SO3H and Trx1 were obtained from Ab Frontier (Seoul, Korea). The antibody specific to catalase was acquired from Calbiochem (Darmstadt, Germany). The antibody specific to NF-κB p65 was provided by Stressgen Biotechnologies (San Diego, CA). The antibody specific to β-actin was purchased from Abcam (Cambridge, MA). The antibody specific to phospho-Akt (S473) was acquired from Cell Signaling Technology (Beverly, MA).
Satellite cell isolation
C57BL/6 female mice were purchased from the Central Lab Animal Inc. (Seoul, Korea) and allowed to acclimate for a week before being used in the study. Animal experimentations were conducted according to the protocol approved by the Institutional Animal Care and Use Committee of Kyung Hee University. Hind limb muscles were surgically removed from 6–8-week-old C57BL/6 female mice, washed three times in Dulbecco's PBS, chopped in 0.025% of collagenase type I with fine scissors, incubated at 37°C for 90 min with stirring, and centrifuged at 350 g for 5 min. The pellet was resuspended in 10 mL of complete medium (Ham's F10 supplemented with 20% FBS and 1% penicillin-streptomycin), and the suspension was passed through a 100 μm filter to remove muscle debris. Cells were centrifuged at 350 g for 5 min, resuspended in complete medium, and seeded into 100-mm gelatin-coated dishes. Basic fibroblast growth factor was added twice a day at a concentration of 4 ng/mL. After 48 h, cells were transferred to noncoated plates and incubated for 45–60 min to allow the fibroblasts to attach to plates. The floating cells were resuspended in complete medium (Ham's F10 supplemented with 2% FBS, 5% Ultroser-G. and 1% penicillin-streptomycin) containing 4 ng/mL of basic fibroblast growth factor, and then seeded into gelatin-coated dishes. When cells were confluent, they were placed in alpha minimum essential medium supplemented with 2% horse serum and 1% penicillin-streptomycin for differentiation.
Cell culture
C2C12 mouse myoblasts were grown in DMEM supplemented with 10% FBS (proliferation medium, PM). Cells were induced to differentiate by placing them in DMEM containing 1% (v/v) FBS (differentiation medium, DM), and full differentiation was achieved 6 days after induction of differentiation. Differentiation was evaluated on the basis of the morphological appearance of cells, fusion index, and the expression levels of the differentiation markers such as myogenin and MHC. NIH3T3, mouse embryonic fibroblasts (MEF), Chang human liver, and human embryonic kidney (HEK) 293 cell lines were maintained in PM. H9c2 rat cardiac myoblasts were grown in DMEM/F-12 containing 10% (v/v) donor calf serum. Unless specified, the culture medium was replaced with fresh medium every 2 days.
Measurement of fusion index
Fusion index was determined as the ratio of the nuclei number in cells containing two or more nuclei to the total nuclei number in hematoxylin-stained C2C12 and satellite myotubes. These cells were washed in PBS, fixed with 10% formaldehyde in PBS for 10 min, and permeabilized with 0.5% Triton–X-100 in PBS for 5 min. Next, they were stained with 10% hematoxylin solution for 5 min and then washed in distilled water. The number of nuclei was measured at least from the six different fields observed under microscope. Fusion (%)=(number of nuclei in myotubes)/(total number of nuclei in myoblasts and myotubes) X 100.
Cardiotoxin-induced muscle injury and regeneration
A 10 μM solution of cardiotoxin from Naja mossambica mossambica was injected into left tibialis anterior (TA) muscles of 6–8- week-old C57BL/6 female mice at 2 μL/g body weight using an insulin syringe. The equal volume of PBS was injected into right TA muscles. The uninjected TA muscles served as the control. After 1, 3, and 5 days, animals were euthanized, and the TA muscle was removed for analysis.
Histology and immunohistochemistry
Paraffin sections (5 μm) were prepared for hematoxylin and eosin (H and E) staining or immunohistochemical analysis. Paraffin sections were deparaffinized with xylenes and hydrated with a series of ethanol. Endogenous peroxidases were quenched with a short treatment of 1% hydrogen peroxide. To enhance the signal, we used antigen retrieval in citrate buffer and signal amplification with biotinylated tyramide. The deparaffinized and rehydrated specimens were incubated overnight at 4°C with antibodies against Prx-2 (1:200; Ab Frontier, Seoul, Korea), MyoD (1:50; Santa Cruz Biotechnology), and MHC (1:50; Developmental Study Hybridoma Bank, IA). The immunostained section was visualized with an EnVision Detection Kit (Dako, CA). Routine H and E-stained sections were examined to ensure the structural integrity of the tissues.
Measurement of reactive oxygen species
Intracellular ROS levels were measured with DCF-DA using FACS analysis as previously described (7). Cells were loaded with 20 μM DCF-DA, and fluorescence was measured with a flow cytometer (FACSCalibur, Becton-Dickinson, Franklin Lakes, NJ). The mean DCF fluorescence intensity was measured with excitation at 488 nm and emission at 525 nm.
Intracellular ROS was also measured by Amplex UltraRed assay as previously described (24). Briefly, cells were cultured in PM or DM, and washed twice with Krebs-Ringer phosphate buffer and incubated with 5 μM Amplex UltraRed reagent (Molecular Probes, Eugene, OR) and 0.1 unit/mL horseradish peroxidase type II (Sigma, St. Louis, MO) in Krebs-Ringer phosphate (Sigma) for 40 min. To identify the hydrogen peroxide-specific signal, control samples were co-incubated with 500 U/mL catalase. Triplicate readings were taken in a 96-well plate using 100 μL samples of medium, and fluorescence was detected via a plate reader at excitation and emission of 530 and 580 nm, respectively. H2O2 levels were calculated in terms of catalase-inhibitable signal and were normalized to cellular protein as measured by the Bio-Rad DC assay. H2O2 concentration was estimated using a standard curve.
Reverse transcription-PCR (RT-PCR)
For the analysis of Prx-2 mRNA, total RNA was prepared using the TRIzol reagent (Invitrogen, San Diego, CA), and total RNA (2 μg) was amplified by a two-step protocol using AMV reverse-transcriptase (Promega) and Taq polymerase. The PCR products were amplified using the following specific primers:
Prx-2 forward, 5′-GACTACAGAGGGAAGTACGTGG-3′;
Prx-2 reverse, 5′-TCAGTTGTGTTTGGAGAAGTATTCC-3′;
myogenin forward, 5′-ATGGAGCTGTATGAAACATCC CCC-3′;
myogenin reverse, 5′-TTCCCTTGCTGGGCTGGGTGT TAG-3′;
GAPDH forward, 5′-CAACTTTGGCATTGTGGAAGGG-3′; and GAPDH reverse 5′-ACACATTGGGGGTAGGAACA-3′.
The amplified products were visualized on a 1% agarose gel, and the amplified GAPDH fragment was used as an internal control for RT-PCR.
Real-time quantitative RT-PCR
The mRNA levels for Prx-2 and GAPDH were measured using SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) and the ABI PRISM® 7300 real-time PCR system (Applied Biosystems) according to the manufacturer's instructions, after primer sequences were designed with the Primer 3 (Rozen and Skaletsky 2000;
Prx-2 mouse forward, 5′-CACGGAGATCATCGCTTTT AGC-3′;
Prx-2 mouse reverse, 5′-TCAACACGCCGTAATTGTGT GAC-3′;
GAPDH mouse forward, 5′-CAACTTTGGCATTGTGGAA GGG-3′;
GAPDH mouse reverse 5′-ACACATTGGGGGTAGGA ACA-3′;
Prx-2 rat forward, 5′-CACCTGGCCTGGATCAATAC-3′;
Prx-2 rat reverse, 5′-TTGACTGTGATCTGGCGAAG-3′;
GAPDH rat forward, 5′-CATGTTTGTGATGGGTGTGA-3′;
GAPDH rat reverse, 5′-TCCACAGTCTTCTGAGTGGC-3′;
Prx-2 human forward, 5′-CGTCTCGGTGGACTCTCAGT-3′;
Prx-2 human reverse, 5′-ATAAAGAGGCCCCTGTAGGC-3′;
GAPDH human forward, 5′-TGACCACAGTCCATGCCAT-3′;
and GAPDH human reverse, 5′-TTCTAGACGGCAGGTC AGGT-3′.
Calculations were based on the ‘‘Delta–Delta method’’ using the following equation: R (ratio)=2-[ΔCT sample – ΔCT control] (21). The data were expressed as fold changes of the treatment groups relative to the control. GAPDH was amplified as a control for real-time quantitative RT-PCR.
Construction of plasmids for Prx-2 promoter analysis
The Prx-2 promoter sequence was analyzed using the MatInspector of Genomatrix (
pGL3-Prx-2/W1-1097 forward, 5′-AGG TAC CGT CTC CTA CTA CCT CCA TTC-3′;
pGL3-Prx-2/W1-1097 reverse, 5′-TTG AGC TCG CAA TGC TAG TCA GAA CTG-3′;
pGL3-Prx-2/W2-736 forward, 5′-ATG GTA CCC CTG TTT AGG CTC TGC A-3′;
pGL3-Prx-2/W2-736 reverse, 5′-TTG AGC TCG CAA TGC TAG TCA GAA CTG-3′;
pGL3-Prx-2/M1-1097 forward, 5′-TAC CAC TGT CTC AGA TGC CGG-3′;
pGL3-Prx-2/M1-1097 reverse, 5′-CCG GCA TCT GAG ACA GTG GTA-3′;
pGL3-Prx-2/M2-736 forward, 5′-TTA GGA AGC TGA GAT CTC CCG GA-3′;
and pGL3-Prx-2/M2-736 reverse, 5′-TCC GGG AGA TCT CAG CTT CCT AA-3′.
Luciferase assay
Cells were transfected with 0.5 μg of the pGL3-basic derived plasmids along with the internal control plasmid, pSV-β-gal (Promega). Cells were incubated in PM or DM, and harvested in lysis buffer. Luciferase and β-gal activities were measured by a microplate reader (Bio-Rad, Richmond, CA), using 20 μL of each cell lysate. The luciferase activity was normalized on the basis of β-galactosidase (β-gal) activity.
Preparation of nuclear extracts
Nuclear extracts were obtained as previously described with minor modifications (28). Confluent cells were detached by trypsinization and centrifuged at 90 g at 4°C for 5 min. Cells were resuspended in PBS, centrifuged at 20,000 g at 4°C for 5 min, and once again resuspended in 200 μL of buffer (10 mM Hepes, pH 7.9, 10 mM KCl, 1 mM DTT, 0.5 mM PMSF, 0.1 mM EDTA). After incubation on ice for 10 min, cells were lysed by addition of 12.5 μL of 10% NP-40. Harvesting of the nuclei was accomplished by centrifugation at 20,000 g at 4°C for 2 min. The supernatant (cytosolic extract) was transferred to a new tube, and the nuclear pellets were resuspended in 50 μL of extraction buffer (20 mM Hepes, pH 7.9, 0.4 M NaCl, 1 mM DTT, 1 mM PMSF, 1 mM EDTA, 1% NP-40) and incubated on ice for 10 min. Nuclear debris was removed by centrifugation at 20,000 g at 4°C for 10 min, and the supernatant was used as nuclear protein extract.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed as previously described (27) using oligonucleotides. The following oligonucleotides for the NF-κB consensus-binding sequences were used for EMSA: candidate 1 (5′-ACTGTGGGAGATGCCGGGCC-3′) and candidate 2 (5′-TTTAGGAAGCTCCCATCTCC-3′). Sense and antisense strands of oligonucleotides were annealed into double-stranded oligonucleotides and labeled with [γ-32P] ATP. For a competition study, a 100-fold molar excess of unlabeled oligonucleotides was added to the reaction mixture prior to the addition of radiolabeled probe.
Chromatin immunoprecipitation (ChIP)
Conventional ChIP was conducted as previously described (14), except that cross-linked C2C12 chromatin was subjected to immunoprecipitation with antibodies against NF-κB p65 and NF-κB p50. The Prx-2 promoters containing the NF-κB binding sites were amplified by following primers:
candidate 1 forward, 5′-GCTGAGGCCTTCTGACTCACA TTT-3′;
candidate 1 reverse, 5′-TAA ACAGGAAGCAGCTGG GGC-3′,
candidate 2 forward, 5′-AGCTGCTTCCTGTTTAGGCT CTG-3′;
and candidate 2 reverse, 5′-CGGGAGATGGGAGCTTC CTAAAA-3′.
Prx-2 overexpression
The wild-type Prx-2 gene was inserted into the pCMV-tag2B vector with XhoI and EcoRI (Stratagene, La Jolla, CA). C2C12 cells were transiently transfected with pCMV-tag2B-Prx-2 or an empty pCMV-tag2B vector using TurboFect™ in vitro Transfection Reagent (Fermentas, Burlington, Ontario, Canada) according to the manufacturer's instructions.
RNA interference
Small interfering RNA (siRNA) against Prx-2 and NF-κB p65, and scrambled siRNA (control) were purchased from Dharmacon (Chicago, IL). Transient transfection of C2C12 cells with siRNAs targeting Prx-2 (50 nmol/L) and NF-κB p65 (100 nmol/L), or scrambled siRNA (50 nmol/L or 100 nmol/L) was carried out with the Lipofectamine 2,000 transfection reagent (Invitrogen, San Diego, CA). Transient transfection of primary myoblasts with siRNAs targeting Prx-2 (25 nmol/L) or scrambled siRNA (25 nmol/L) was carried out with the Lipofectamine RNAiMAX reagent (Invitrogen). When cells reached 30%–50% confluence, they were transfected with each siRNA, and further incubated for 24–48 h. When cells reached 80% confluence, they were induced to differentiate in DM.
Western blot analysis
Cells were lysed with RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Na-deoxycholate, 1% NP-40), supplemented with protease inhibitors (200 mM PMSF, 200 mM Na2VO4, 200 mM NaF). The TA muscles were lysed with protein extraction solution (iNtRON Biotechnology, Chinju, Korea). The lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. After blocking, the membranes were incubated with the indicated primary antibodies, followed by incubation with secondary antibody. Samples were detected with enhanced chemiluminescence reagents (Santa Cruz Biotechnology). Quantification of Western blots was performed by densitometry using Image J software. All Western blot analyses were performed three times to ensure that the results were repeatable, and the representative blots were shown. β-actin was used as a loading control.
Statistical analysis
Results were expressed as the means±standard error (SE). Error bars represent the means±SE of at least three independent experiments. The difference between two mean values was analyzed by Student's t- test, and considered statistically significant when p<0.05.
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No. 2011-0030721).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.