
Abstract
Introduction
Here we sought to investigate energy homeostasis in a model of axonal degeneration caused by oxidative stress of a known etiology, the mouse model of X-linked adrenoleukodystrophy (X-ALD: McKusick no. 300100). This is a rare and fatal disease characterized by central inflammatory demyelination in the brain or slowly progressive spastic paraparesis, as a consequence of axonal degeneration in the spinal cord (17, 42). X-ALD is the most frequently inherited leukodystrophy, with a minimum incidence of 1 in 17,000 men. All patients have mutations in the gene encoding the ABCD1 protein (NM_000033), an ATP binding cassette peroxisomal transporter involved in the importing of very long-chain fatty acids (VLCFA, C ≥22:0) and VLCFA-CoA esters into the peroxisome for degradation (66). Defective function of the ABCD1 transporter leads to VLCFA accumulation in most organs and plasma; and elevated levels of VLCFA are used as a biomarker for the biochemical diagnosis of the disease. Classical inactivation of ABCD1 in the mouse results in late onset neurodegeneration with axonopathy in spinal cord, in the absence of inflammatory demyelination in the brain, resembling the most frequent X-ALD phenotype or adrenomyeloneuropathy (49, 50). Oxidative damage has been evidenced in postmortem brain samples from individuals with cerebral ALD (24) and in mouse spinal cords before disease onset (20). Further, we recently reported compelling evidence that a combination of antioxidants halts clinical progression and reverses axonal damage in X-ALD mouse model, thereby providing formal conceptual proof that oxidative injury is a major etiopathogenic factor in this disease (38). The source of this oxidative damage is most likely an excess of saturated and unsaturated VLCFA, which are known to generate free radicals and cause oxidative damage to proteins in cell culture (20, 22).
In this study, we demonstrate by a redox proteomics approach that ABCD1 ablation induces the oxidation of enzymes of glycolysis and TCA cycle in spinal cords. This oxidation inactivates the affected enzymes, and bioenergetic failure is manifested by decreased levels of cellular NADH and ATP, together with decreased levels of GSH. All these changes occur months before disease onset. Additionally, we provide evidence that the combination of antioxidants N-acetylcysteine and α-lipoic acid prevents protein oxidation and metabolic failure in spinal cords.
Results
ABCD1 loss induces the specific oxidation of glycolysis and tricarboxylic acid cycle enzymes in spinal cord
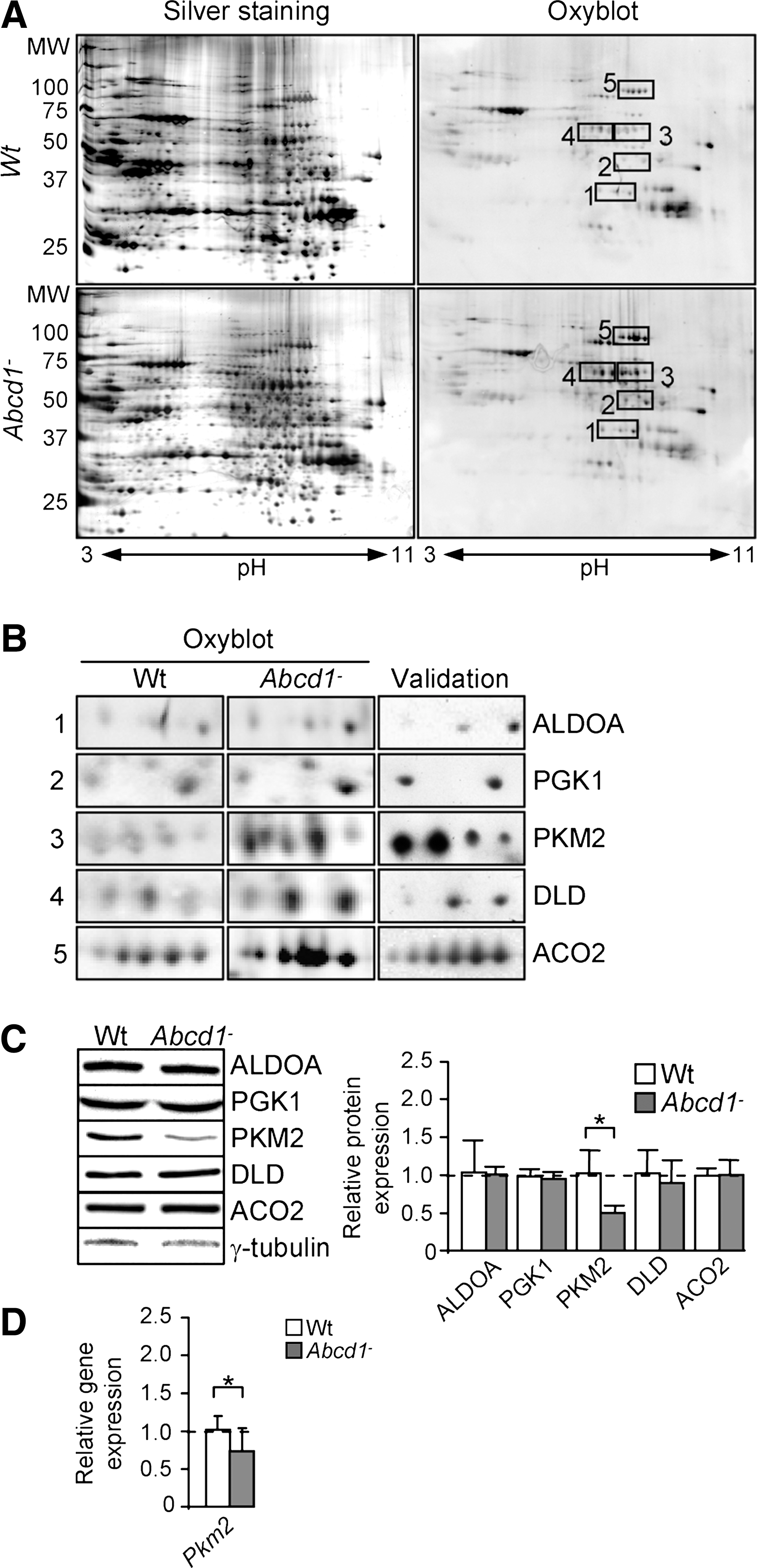
We pointed out earlier that oxidative damage is a main etiopathogenic factor in X-ALD mouse model (20, 38). This damage is characterized by an increase in the markers of lipoxidation to proteins (MDA-lysine), combined with markers of glycoxidation and lipoxidation, carboxyethyl-lysine (CEL) and carboxymethyl-lysine (CML), together with markers of direct carbonylation of proteins (glutamic semialdehyde [GSA] and aminoadipic semialdehyde [AASA]), in spinal cords and in peripheral mononuclear cells or fibroblasts derived from patients with X-ALD (20, 22). Thus, we set out to identify oxidation targets in spinal cords with a redox proteomics approach (54).
Using this methodology (Fig. 1A and Supplementary Table S1; Supplementary Data are available online at
C26:0 excess induces pyruvate kinase oxidative inactivation in human fibroblasts
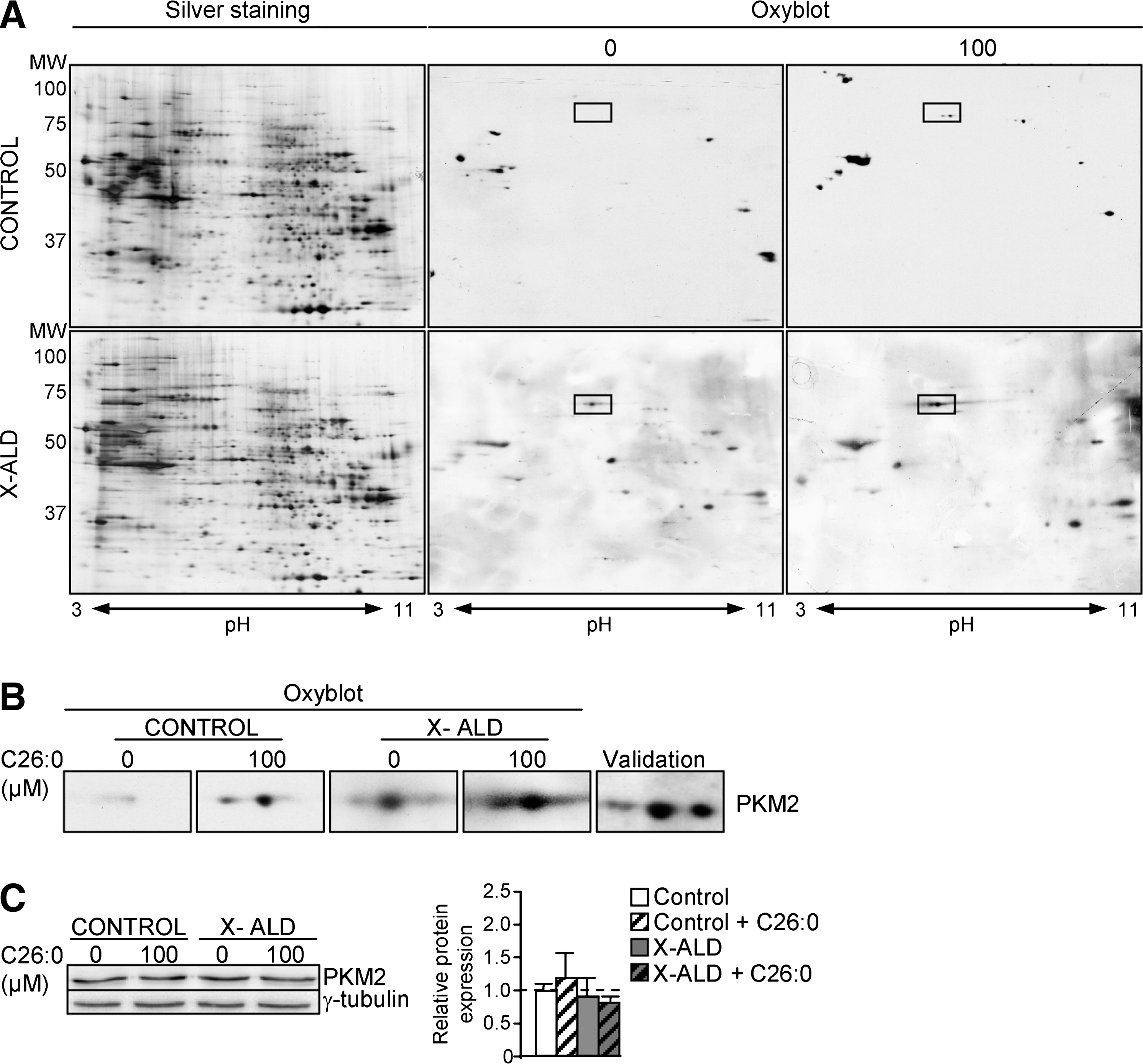
Earlier, we reported that the levels of lipoxidative (MDAL), glycoxidative/lipoxidative (CEL, CML), and protein oxidative (GSA; AASA) markers were about doubled in the fibroblasts derived from patients with X-ALD (20). We also demonstrated that excess C26:0 generates ROS in human fibroblasts (20) and oxidative lesions to proteins in X-ALD fibroblasts. To identify which proteins are oxidation targets in human X-ALD fibroblasts, we performed redox proteomics. We found that PKM2 is more oxidized in human X-ALD than in control fibroblasts (Fig. 2A). To investigate whether C26:0 excess is involved in the PKM2 oxidation, we also performed redox proteomics experiments with cultured human nondiseased and X-ALD fibroblasts that were treated with a pathophysiologically relevant dose of C26:0 (100 μM) for 7 days, as previously described (20). We found that C26:0 excess induces PKM2 oxidation and inactivation in both nondiseased and X-ALD fibroblasts (Fig. 2A). As control, we incubated X-ALD and nondiseased fibroblasts with 100 μM oleic acid (C18:1) and could not detect oxidation increases in 2D gels after DNP exposure (data not shown). No signs of toxicity or reduced proliferation were seen in the cultures. To confirm that the excised spot corresponded to PKM2, we also performed a 2D gel, then a western blot with an antibody against PKM2 (Fig. 2B and Supplementary Table S1). Further, we quantified PKM2 gene expression and found that it was neither affected by genotype nor by C26:0 excess in human fibroblasts (Fig. 2C).
ABCD1 loss provokes specific glycolytic and TCA cycle metabolic signature in spinal cord
Several reports indicate that oxidation of enzymes involved in energy metabolism results in a decrease in their activity (8, 60, 65). To investigate whether enzyme activity is affected in our model, we quantified substrates and/or products of the five oxidized enzymes with a directed metabolomics approach.
We found that the level of fructose 1,6-bisphophate, the substrate of ALDO A, was not modified in Abcd1− spinal cord (Fig. 3A and Table 1). However, its products—dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (GA3P)—were present at significantly lower levels in Abcd1− mice, which suggests that oxidation affected ALDO A activity in vivo (Fig. 3A and Table 1). Lower levels of DHAP and GA3P could also be explained by its nonenzymatic conversion to methylglyoxate (MGO) (48), which can be degraded through glyxolases and/or react with lysine residues in proteins. This is consistent with the reported increase in carboxyethilisyne (CEL) (20). 2-Phosphoglycerate and 3-Phosphoglycerate cannot be distinguished by MS, but an m/z (mass-to-charge ratio) ion compatible with their masses is lowered in spinal cord from Abcd1− mice, which suggests that activities of PGK1 (which produces 3-Phosphoglycerate) and/or Phosphoglycerate mutase (which converts 3-Phosphoglycerate into 2-Phosphoglycerate) were modified (Fig. 3A). Since PGK1 was found to be oxidized in our model, 3-Phosphoglycerate level was likely decreased due to lower PGK1 activity (Fig. 3A).
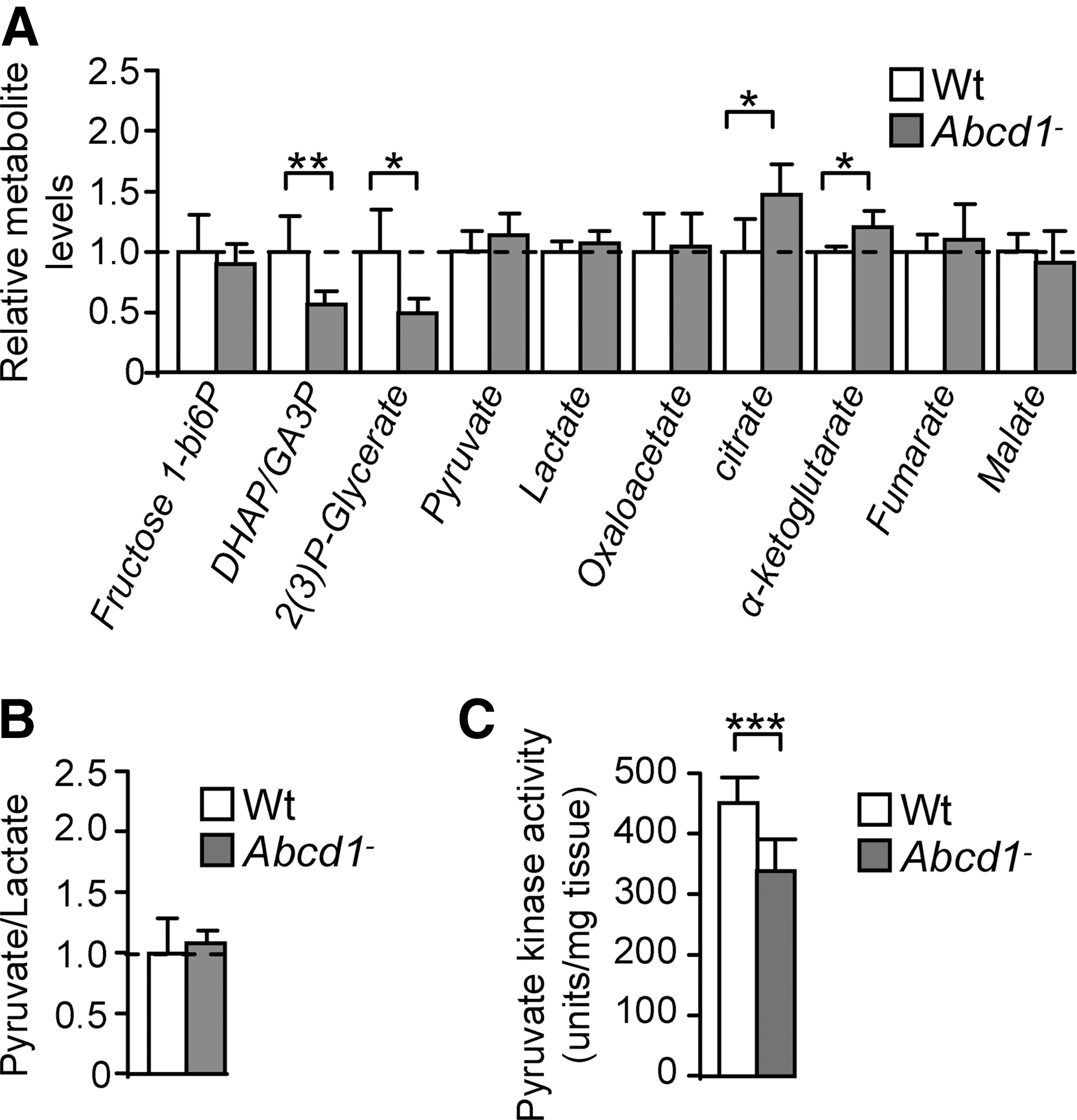
Fructose 1–6 bisphosphate, dihydroxyacetone phosphate and glyceraldehyde 3 phosphate (GA3P), 2 and 3-phosphoglycerate, pyruvate, lactate, oxalacetate, citrate, α-ketoglutarate, fumarate, and malate levels were quantified in Wt and Abcd1− mice.
Oxidized.
n.s. not significant.
Dihydrolipoamide dehydrogenase (DLD), a subunit of α-ketoglutarate dehydrogenase complex (KGDHC) and of four other important mitochondrial enzymes, was found to be oxidized, and the concentration of its substrate α-ketoglutarate was increased in Abcd1− spinal cord (Fig. 3A). Thus, we hypothesized that KGDHC activity was most likely to be decreased in Abcd1-null mice, even if the steady-state level of α-ketoglutarate is also determined by several factors such as glutamate availability and the rate-limiting enzyme in KGDHC is not DLD but α-ketoglutarate dehydrogenase E1k.
Since DLD is also a subcomponent of pyruvate dehydrogenase complex (PDHC), it was expected that pyruvate catabolism would also be altered. However, we found that pyruvate level was not modified in Abcd1-null spinal cord. Although lactate level and the pyruvate/lactate ratio are not modified in Abcd1− spinal cord, we found that PKM2 was oxidized and its expression decreased in Abcd1− spinal cord (Fig. 3A, B). These results indicate that steady-state level of pyruvate is maintained through a balanced decrease in its synthesis and degradation, without lactate accumulation. This could be due, for instance, to concerted decreased glycolytic production of pyruvate and its decreased uptake or oxidation in mitochondria.
We also found that citrate concentration was increased in spinal cord from Abcd1-null mice, which suggests a defect in citrate catabolism (Fig. 3A), likely due to the oxidative damage to ACO2 in Abcd1− mice (Fig. 3A).
Moreover, we also observed that (i) oxaloacetate (OAA), fumarate, and malate levels are not affected and (ii) the enzymes involved in the production or degradation of these metabolites are not oxidized.
Altogether, these results demonstrate for the first time pronounced bioenergetic dysfunction in spinal cord from Abcd1-null mice, at presymptomatic stages (Fig. 3A).
ABCD1 loss decreases pyruvate kinase activity in spinal cord
The results just shown suggest that the pyruvate steady-state level is not modified by ABCD1 loss, because its synthesis and catabolism may be reduced. In addition, we demonstrated that pyruvate kinase (PKM2) was highly oxidized and its expression reduced in spinal cord from Abcd1− mice (Fig. 3C). To investigate whether pyruvate synthesis was affected, we assessed the activity of pyruvate kinase and found that it was decreased in spinal cord from 12 month-old Abcd1− mice (Fig. 3C). No significant dysregulation of pyruvate kinase activity was found in 12 month-old mouse brain cortex or liver and in spinal cords at an earlier stage (3 months) (Supplementary Fig. S1).
ABCD1 loss disturbs NADH, NADPH, GSH, and ATP levels in spinal cord
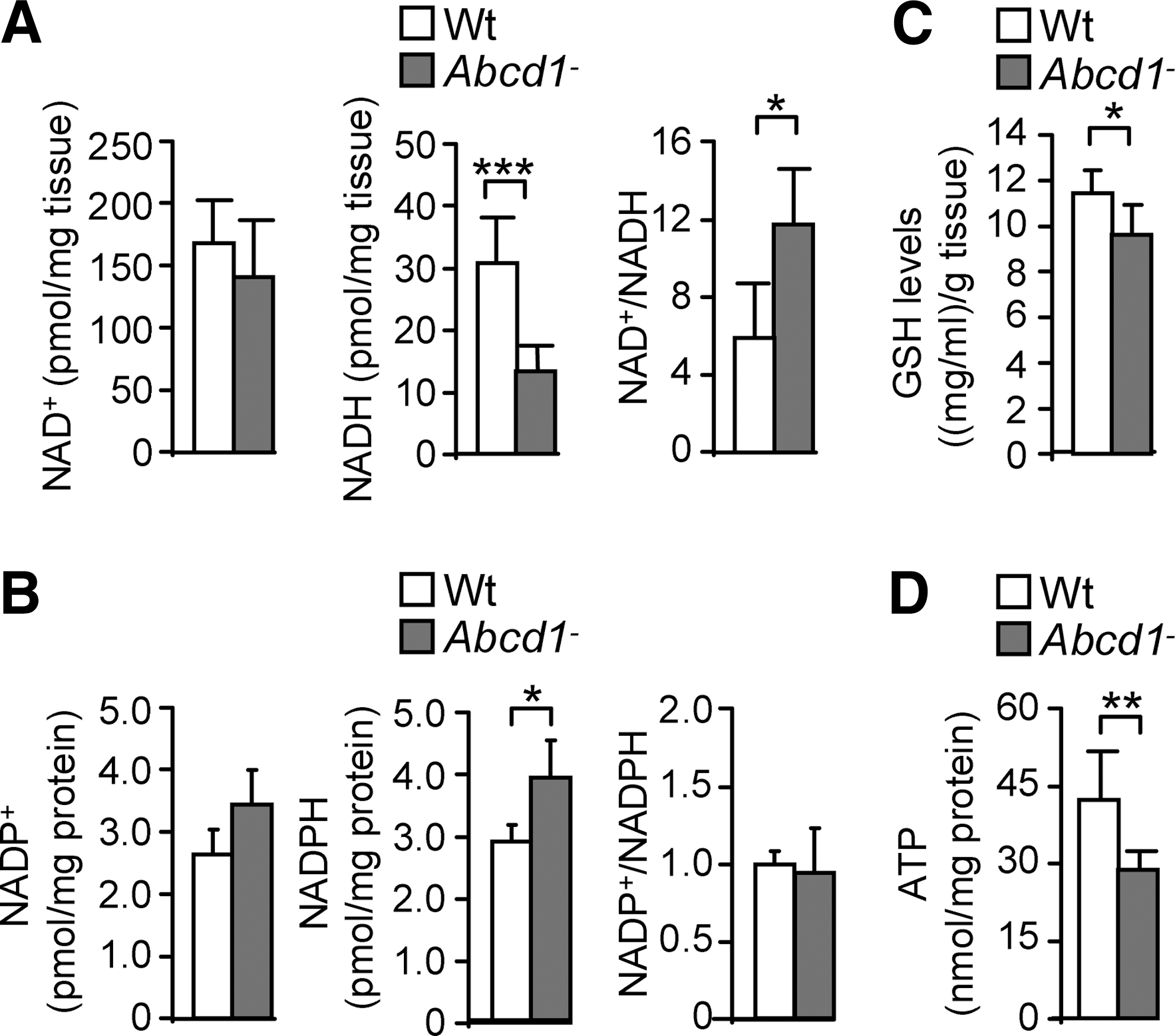
To study possible global consequences of protein oxidation on energy homeostasis, we quantified NAD+, NADH, NADP+, NADPH, and ATP contents (60, 65) in Abcd1− spinal cords at 12 months. NADH but not NAD+ levels were reduced on ABCD1 loss compared with Wt samples (Fig. 4A). Consequently, the NAD+/NADH ratio was increased (Fig. 4A), thus reflecting an abnormal redox status Moreover, we observed that NADPH was elevated in Abcd1− spinal cords at 12 months (Fig. 4B). The levels of reduced glutathione (GSH) are intimately related to NADPH, because the GSH/GSSG ratio is determined by the NADPH consuming enzyme Glutathione Reductase (GR) (31, 69). We have, therefore, quantified GSH levels in whole spinal cord extracts, to find out that GSH levels were reduced in Abcd1− mice, a situation consistent with increased oxidative stress (Fig. 4C). In addition, we found that ATP is also reduced in these samples (Fig. 4D), which directly indicates bioenergetic failure. However, NAD+, NADH, and ATP levels were not affected in brain cortex or liver at 12 month of age or in spinal cord at 3 months of age in Abcd1-null mice, indicating organ specificity and progressive nature of the metabolic impairment (Supplementary Fig. S2). Altogether, these results point out the specific importance of ABCD1 in the energy metabolism of the spinal cord of aged, but still presymptomatic, Abcd1− mice.
To investigate whether the decrease of ATP could be due to mitochondrial respiratory chain impairment, we measured the activity of respiratory chain complexes in spinal cords extracts from 12 month-old Abcd1− mice. We quantified the activities of complex I plus complex III, complex II plus complex III and complex IV. Activities did not vary between Abcd1-null and wild type littermates (Supplementary Fig. S3).
A combination of antioxidants prevents oxidative stress and metabolic failure
We have previously shown that a cocktail of antioxidants including N-acetylcysteine (NAC), α-lipoic acid (LA), and vitamin E reversed oxidative damage to proteins and DNA, immunohistological signs of axonal degeneration and associated locomotor disability in an X-ALD mouse model (38).
To investigate whether these antioxidants were effective in counteracting oxidative stress to the specific proteins identified, we treated 8 month-old Abcd1−
mice with a combination of NAC and LA for 4 months. We have previously reported that excess of VLCFA decreases reduced glutathione, and X-ALD cells are more sensitive to glutathione depletion (20). N-acetylcysteine was chosen, because it can regenerate reduced glutathione (GSH) and scavenge several ROS species including OH
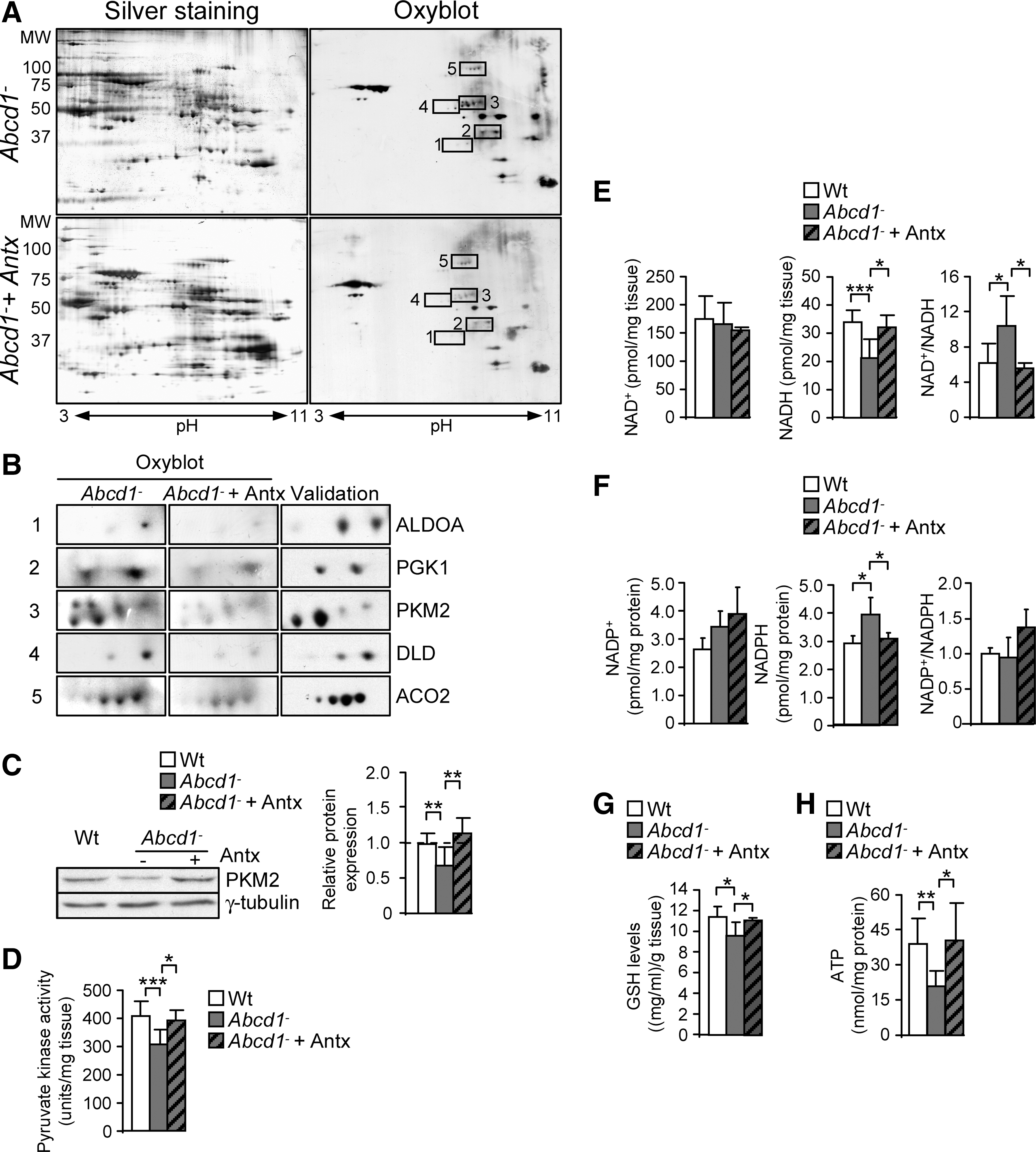
We also observed that the Abcd1-associated loss of pyruvate kinase activity and expression was prevented by NAC and LA, thereby suggesting that in vivo, the level of oxidation of this protein may directly correlate with its expression and enzymatic activity (Fig. 5C, D).
The reduction of NADH, GSH, and ATP levels and the increase of NADPH were all prevented by antioxidants (Fig. 5E–H), indicating that the treatment was effective in preventing bioenergetic failure.
Discussion
We have previously suggested that oxidative damage could be a significant factor contributing to X-ALD pathogenesis (20, 38, 58). This study corroborates our hypothesis and points to how energetic failure manifested by diminished levels of NADH and ATP is most likely due to inhibition of glycolysis and TCA cycle resulting from the early oxidative damage to proteins,. Further, our findings strongly suggest that energetic failure plays a major role in the physiopathogenesis of X-ALD, because (i) is progressive and appears presymptomatically at 12 months of age, in a mouse that exhibits first locomotor disabilities at 20–22 months of age; (ii) in vivo antioxidant treatment prevented motor disability and axonal degeneration (38), and also oxidative damage of important glycolytic and TCA cycle proteins normalizing NADH, NADPH, GSH, and ATP levels (Fig. 6).
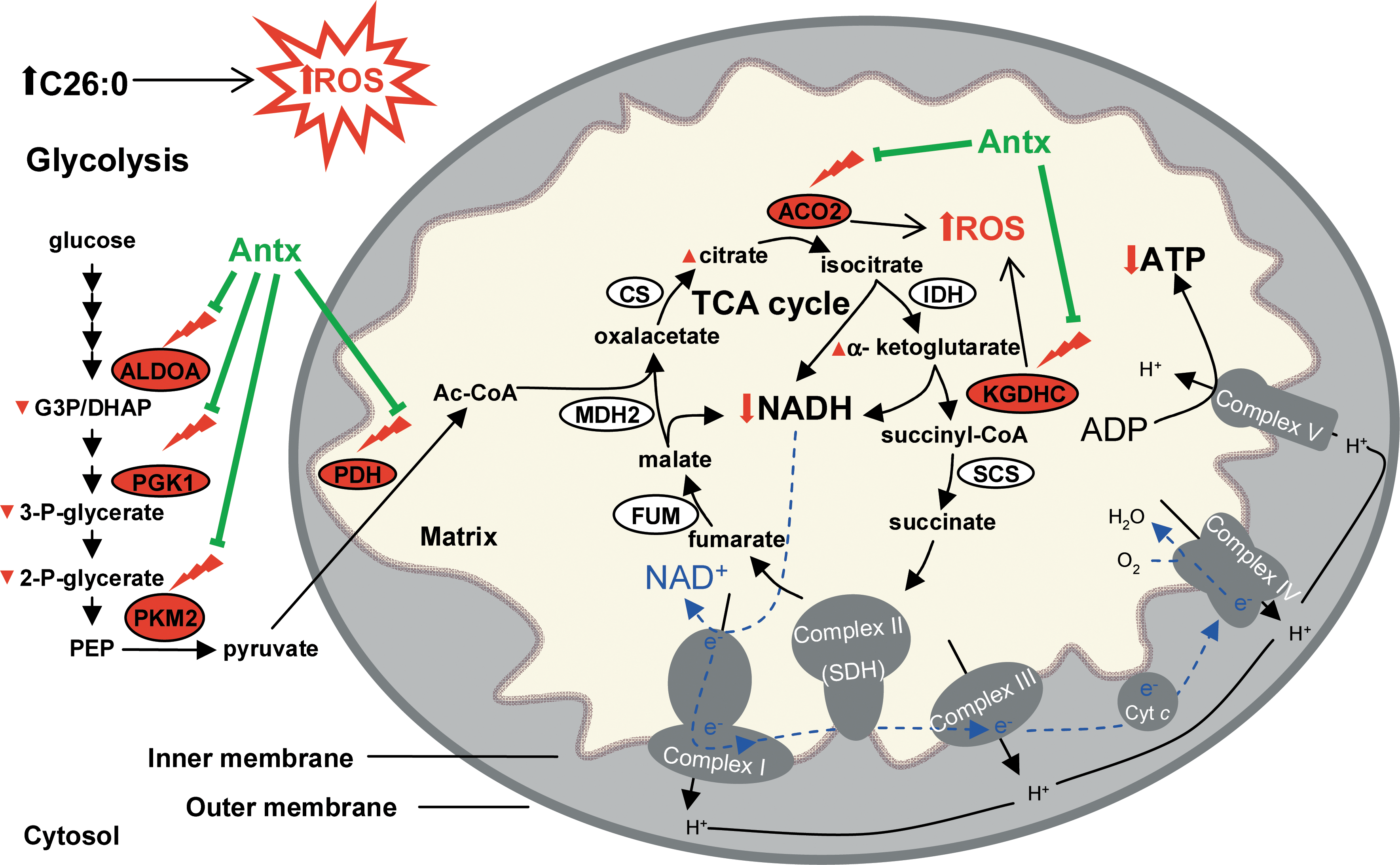
It has been suggested that oxidative modification of key mitochondrial TCA enzymes such as pyruvate and α-ketoglutarate dehydrogenases and aconitase may be an important pathophysiological factor in neurodegenerative diseases, by causing mitochondria dysfunction and bioenergetic failure (8, 32, 43, 60, 65). Indeed, aconitase and KGDHC are known targets and sources of ROS and their enzymatic activity is impaired by ROS-induced oxidation (8, 32, 43, 60, 65). Further, increased mitochondrial ROS generation is thought to induce and stimulate pathogenic feedback cycle by inactivating sensitive TCA enzymes and impairing NADH and NADPH generation, which, in turn, results in further impairment of mitochondrial ROS defense capacity, disturbed Ca2+, and ion homeostasis, eventually causing a decrease in ATP production and overall bioenergetic failure (43, 64, 65).
Of note, we have demonstrated that activities of respiratory chain are not modified; NADH and ATP levels are lowered and NADPH is elevated in X-ALD, in vivo in mouse spinal cord extracts. This tissue contains a mixture of gray and white matter, where neurons represent ∼10% of the total amount of cells, whereas glia is about 90%, with astrocytes being the most abundant cell type. Energy is mainly produced by mitochondria in neurons, whereas in astrocytes most of ATP is generated by glycolysis (30). It was reported that, in the gray matter of the brain, astrocytes export lactate (derived from glucose or glycogen) to neurons to power their mitochondria. In the white matter, lactate can support axonal function under conditions of energy deprivation (53). We, therefore, cannot exclude the possibility that the results reflect only a sum of effects in whole tissue and do not reflect a specific disturbance of a given metabolite or pathway in a particular cell type. For instance, OXPHOS activities could be impaired in, that is, neurons. Nevertheless, these results suggest that metabolic failure is most likely due to impairment in glycolysis and/or TCA cycle rather than due to damaged mitochondrial respiratory chain. In astrocytes, ATP reduction is probably due to a defective glycolysis. In neurons, the decrease in ATP levels could be due to a reduction in NADH generation in mitochondria caused by the damage to KGDHC and aconitase and/or some other unidentified catabolic enzymes. In addition, the elevation of NADPH could have been caused by an increased production via Pentose Phosphate Pathway (30) and/or by an inhibition of Glutathione Reductase (GR) (70). Indeed, it has been reported that the low glycolytic rate in neurons results in increased flux through the pentose-phosphate pathway, thus providing NADPH necessary to regenerate antioxidant glutathione (30).
The GSH reduction observed, and its recovery on antioxidant treatment, is in line with a major role of GSH in oxidative stress scenarios, and consistent with previous results: (i) we formerly showed that X-ALD cells were more sensitive to GSH depletion and more prone to undergo cell death due to the oxidative stress-induced damage than the passage-matched control fibroblasts (20); (ii)Glutathione peroxidase (GPX1) protein expression was increased in Abcd1− spinal cord (20, 38) and normalized by antioxidant treatment (38). Therefore, GSH reduction is most likely to be caused by an increased consumption by GPX1 due to an ongoing oxidative stress process in Abcd1 spinal cords. Oxidative stress has been classically considered a common event in the neurodegenerative cascade in a variety of conditions (37, 40). Ample evidence demonstrates that energy metabolism also plays a major role in cell death (16, 23). Glycolytic and TCA cycle proteins such as ALDO A, PGK1, PKM2, DLD, and ACO2 can be considered as classical targets of oxidative stress, because the oxidation of these proteins is commonly detected in several neurodegenerative disorders (9, 40, 47, 59), although no “oxidation prone consensus” has yet been identified. ALDO A is oxidized in Parkinson mouse model (59), in amnestic mild cognitive impairment (MCI), in early onset Alzheimer disease (EOAD), and in Alzheimer (AD) human brain (9, 40). Oxidized ALDO A has also been detected in progressive supranuclear palsy (PSP) and infantile Parkinson disease (iPD) (40). PGK1 is oxidized in AD and PD mouse model (59), in MCI brain (9), and in PSP (40). Moreover, PKM2 is oxidized in MCI (9) and in Alzheimer brains (9, 40), and a correlation has been observed between levels of oxidation and activity of PKM2 in MCI brain (10), and in a rat hepatoma cellular model (27). In agreement with this observation, we demonstrate here that PKM2 is oxidized and its activity decreased in Abcd1-null mice. Nevertheless, as PKM2 expression is also decreased in our samples, we cannot determine whether this reduction in activity is due to oxidative stress or PKM2 protein levels. Unfortunately, no information on PKM2 expression is available in patients with MCI or patients with Alzheimer (9, 10, 40), but our result suggest that its expression levels would be worth checking. The mechanisms by which reduction in expression of PKM2 occur deserve further investigation. The decrease in ATP levels that we are reporting in this study is likely due to a mitochondria dysfunction. Indeed, ATP is mainly produced by mitochondria in nonproliferating, postmitotic cells; whereas it is preferentially generated by glycolysis in proliferating, for example, cancer cells (19). Evidence of ultrastructural anomalies of mitochondria in spinal neurons of Abcd1− mice have been reported (18); this is consistent with previous findings of mitochondria alterations in liver of peroxisomal deficient models (5). According to the findings in a mouse model of AD, glycolysis induction could be a mechanism to compensate for mitochondria dysfunction as a metabolic reprogramming (68). However, this reprogramming cannot be efficient in Abcd1− spinal cord, because ALDO A, PFK1, and PKM2 are oxidized and their activity might be altered. It was also reported that aconitase is oxidized in both PD and AD mouse models (59), in AD (9), and in Huntington's disease (40). Further, the oxidation of DLD had been shown in two PD mouse models (59). Although some specific oxidized proteins have been identified in neurodegenerative disorders, a large number of oxidized proteins are more commonly found (9, 40, 59). This could be due to limitations of proteomics experiments. Indeed, only proteins that are very well expressed can be identified by 2D gel proteomics (56). Indeed, the Western-blot anti-DNP detect as little as 1 pmol carbonyl in a protein sample and require a minimum of as little as 50 ng protein oxidized to the extent of 0.5 mol carbonyl/mol protein. Moreover, intrinsic limitations to 2D gels techniques include, for instance, that proteins migrating outside a pH scale from 3 to 11 and having a molecular weight outside the range of 25–100 kD cannot be detected. Similarly, membrane-located or highly hydrophobic proteins cannot be resolved by this technique. Low-abundance proteins cannot be identified due to lowered sensitivity of MALDI-TOF sequencing. Moreover, many of these proteins are generally considered as house-keeping agents, having an essential function in maintenance of cell viability. In addition, in the case of neurons, for instance, it was shown that oxidative injury affects a large number of substrates including enzymes of the glycolysis and TCA cycle, thus resulting in a weakened energy metabolism (23). As a result, reduced ATP production in affected neurons reduces their capacity to respond to physiological energy demands such as synaptic input or axonal transport, which might lead to axonal degeneration in our particular disease scenario, or to progressive neuronal dysfunction in the most frequent neurodegenerative diseases (16).
Ample evidence indicates that oxidative damage to proteins related to energy metabolism is accompanied by the corresponding detriment of their function and impaired bioenergetics as observed in Alzheimer's and Parkinson's diseases (9, 16, 40, 59). Most commonly, this failure is manifested by a decrease in cellular ATP, as in AD (52), PD (25), and ALS (7). Less evidence is available about NADH levels. A significant decrease in both reduced and oxidized forms of NAD and an increase in NAD+/NADH ratio was reported in the brains of Ataxia-telangiectasia mouse model (61). It was also shown that oxidative products generated by dopamine were able to reduce NADH levels in isolated mitochondria (6). Both NADH and NADPH levels were decreased in neurons cultured from aged (24 month-old) rats (46). Moreover, it was shown that H2O2 decreased NADH levels in nerve terminals (64) and increased NAD+/NADH ratio in neonatal heart muscle cells (33), but their ratios have not been systematically measured in most prevalent neurodegenerative diseases.
The widely used antioxidants LA and NAC have been shown to increase the level of GSH and affect the regulation of various redox signalling pathway in cells (15, 44). Combined antioxidant therapy aims at reproducing the multistep, combined response, which is observed in vivo leading to recovery after an oxidative challenge (35). Some studies have shown that combinations of antioxidants can be beneficial for pathologies associated with increased oxidative stress (55) and that such a strategy might be advantageous over higher doses of single antioxidants for treating mitochondriopathies (62, 63), reproducing what it is already present in nature; that is, a combination of antioxidant systems rather than a single one. In particular, it has been recently demonstrated that several combinations of antioxidants {[LA, NAC and vitamin E (4)], [LA and NAC (41)], or [LA and acetyl-L-carnitine (1, 57)]} are able to prevent oxidative damage and improve mitochondrial ultrastructural decay or dysfunction in Alzheimer disease mouse models (57), and even in some clinical studies (13, 51). Further, since LA is an essential cofactor of PDHC and KGDHC, its supplementation could protect and increase the enzymatic activity of KGDHC (2), and, therefore, help bring about an increase in NADH and ATP production. Indeed, we demonstrate in this study that the improvement in disability and axonal degeneration in X-ALD mice by LA and NAC as shown elsewhere (38) correlates with a decrease in oxidation damage of key proteins involved in metabolic homeostasis, and with preserved NADH and ATP levels. Thus, our results provide new insights into the molecular mechanisms of action of antioxidants in X-ALD.
Moreover, we have identified new markers of pathology in the Abcd1− mice such as PKM2 expression level and activity, NADH, and ATP levels. These markers may become very useful to monitor the efficiency of treatments in preclinical trials in X-ALD mice. Monitoring of the biological effects of the drugs in patients would be further facilitated by the recent identification by MS/MS of quantitative biomarkers of oxidative damage to proteins in the peripheral blood mononuclear cells from patients with X-ALD (22). Therapeutic implications derived from this work could be extrapolated to other diseases in which energy metabolic failure due to oxidative stress is a main or early contributing pathogenic factor.
Materials and Methods
Antibodies
The following antibodies were used for western blots: anti-rabbit DNP (D9659, [Sigma]), dilution 1/500; anti-mouse γ-tubulin, dilution: 1/5000 (T6557, clone GTU-88 [Sigma]); anti-rabbit pyruvate kynase, dilution 1/500 (ab-38237 [Abcam]); anti-rabbit aldolase, dilution 1/1000 (NB600-915 [Novus Biologicals]); anti-rabbit-phosphoglycerate kinase 1, dilution 1/250 (AB38007 [Abcam]); anti-rabbit aconitase 2, dilution 1/1000 (ACO2-AP1936c [Abgent]); and anti-rabbit lipoamide dehydrogenase, dilution 1/1000 (L2498-05 [US Biological]). Goat anti-rabbit IgG linked to horseradish peroxidase, dilution: 1/15000 (P0448 [Dako]) and Goat anti-mouse IgG linked to horseradish peroxidase, dilution: 1/15000 (G21040 [Invitrogen]) were used as secondary antibodies.
Mouse breeding
The generation and genotyping of Abcd1− mice has previously been described (39, 49, 50). Mice used for experiments were of a pure C57BL/6J background, all male. Animals were sacrificed, and tissues were recovered and conserved at −80°C. All methods employed in this study were in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publications No. 85–23, revised 1996) and with the ethical committee of IDIBELL and the Generalitat de Catalunya.
Treatment of mice
α-Lipoic acid (LA) (0.5% w/w) was mixed into AIN-76A chow from Dyets (Bethlehem, PA). N-acetylcysteine (1%) was dissolved in water (pH 3.5) (38).
Eight-month-old animals were randomly assigned to one of the following dietary groups for 4 months. Group I (Wt): Wt mice (n=8) received only normal AIN-76A chow, Group II (Abcd1− ) Abcd1− mice (n=8) received only normal AIN-76A chow, and Group III (Abcd1− +Antx [Antioxidant]) Abcd1− mice (n=6) were treated with chow containing LA and with NAC in drinking water (38).
Cell culture and treatments
Control from healthy donors (n=5) and X-ALD human fibroblasts (n=5) were obtained after informed consent at the Bellvitge University Hospital. Cells were treated in medium containing FCS (10%) at 37°C in humidified 95% air/5% CO2. After the growing period, the medium was changed to serum-free medium supplemented with 100 μM BSA (free fatty acid)-bound fatty acid for 7 days at a 2:1 C26:0 fatty acid/BSA ratio. Untreated cells received an amount of BSA equal to that grown with the BSA-fatty acid complex. No significant changes in morphology were observed during incubation in serum-free medium (Supplementary Fig. S4). Unless otherwise stated, experiments were carried out with cells at 95% of confluence. Lines were used on passages 12–18.
Monodimensional electrophoresis and western blotting
Tissues were removed from euthanized mice and flash-frozen on liquid nitrogen. Frozen tissues and human fibroblasts samples were homogenized in RIPA buffer using a motor-driven grinder (Sigma-Aldrich) and then sonicated for 2 min at 4°C in an Ultrasonic processor UP50H (Hielscher-Ultrasound Technology). Ten to 100 μg were loaded on to each lane of 10% polyacrylamide gels for 60 min at 120 mV. Resolved proteins were transferred onto nitrocellulose membrane. Proteins were detected with ECL western blotting analysis system followed by exposure to CL-XPosure Film (Thermo Scientific). Autoradiographs were scanned and quantified using GS800 Densitometer (Bio-Rad).
Two-dimensional electrophoresis and western blotting
Spinal cord and human fibroblast samples were homogenized in a lysis buffer (180 mM KCl, 5 mM MOPS, 2 mM EDTA, 1 μM butylated hydroxytoluene, and protease inhibitor cocktail [Roche Diagnostics GmbH]) using a motor-driven grinder, sonicated for 2 min at 4°C in a Ultrasonic processor UP50H, and then centrifuged for 5 min at 1000 g. Afterward, the supernatant was collected and a new centrifugation (5 min, 1000 g) was carried out. After quantification, 1 mg of protein was precipitated with 20% TCA, and the pellet was resuspended in 200 μl of a denaturizing buffer (9 M urea, 4% CHAPS). Proteins were newly quantified, and 100–200 μg of proteins was dissolved in IEF (Isoelectric focusing) buffer (9 M urea, 4% CHAPS, 1% bromophenol blue, 50 mM DTT, and 0.5% ampholites (pH 3-11NL (GE Healthcare Bio-Sciences AB) up to 340 μl. This solution was applied overnight to 3–11 NL 18 cm IPG strips (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Isoelectric focusing migration was performed as follows: 250 V for 5h, followed by a linear gradient to 60,000 V, and then 250 V for 1 h in a Bio-Rad system. Strips were derivatized in a solution of 0.2% DNPH in HCl 2 N for 10 min and then equilibrated in 2 M TrisHCl-30% glycerol buffer for 15 min. For the last two steps of re-equilibration, strips were first incubated in a buffer containing 6 M urea, 2% SDS, 20% glycerol, 0.13 M DTT, and 0.375 M TrisHCl pH 8.8 for 10 min and then in the same buffer containing 2.5% iodoacetamide for 10 min. The equilibrated strips were loaded in a 10% SDS-PAGE gel (20×20 cm) and run at 250 V for 4 h at RT. For oxyblot, proteins were transferred on to nitrocellulose membranes and then detected with ECL western blotting analysis system followed by exposure to CL-XPosure Film (Thermo Scientific). This method is based on the formation of a hydrazone (DNP) resulting from the reaction of protein-bound carbonyl and 2,4-dinitrophenylhydrazine. An antibody against DNP is used to detect carbonylated proteins (54). Thereby, we performed two-bidimensional (2D) gels in parallel. The first gel was silver stained to detect whole proteins, and the second one was transferred on to nitrocellulose membrane to detect oxidized protein. However, before the second electrophoresis migration and the transfer on to nitrocellulose membrane, samples were derivatized on to the strips with 2,4-dinitrophenylhydrazine (DNPH) after the first isoelectric focusing migration (Fig. 1A). Then, differentially oxidized cut spots were digested with trypsin (DigestPro MS), and peptides were analyzed by MS. Peptide Mass Fingerprinting database was used to identify proteins from a spectrum generated by MS (Supplementary Table S1). For silver staining, 2D gels were fixed for 30 min in a solution containing 30% ethanol and 70% glacial acetic acid. The fixing solution was replaced with sensitizing solution consisting of 30% ethanol, 0.2% w/v sodium thiosulphate, and 6.8% w/v sodium acetate. After 30 min, the sensitizing solution was removed, and the gels were washed thrice with distilled water for 5 min. The gels were stained with a silver solution containing 2.5 g/l of silver nitrate for 20 min and then washed twice with distilled water for 1 min. The gels were put in a solution of sodium carbonate 2.5% w/v. To arrest the developing process, solution was removed and the reaction was stopped with a solution containing EDTA-Na2 1.26% w/v.
Fold difference was statistically compared among the five independent experiments. Proteins for further investigation were selected on the basis of their higher oxidation values when comparing Wt versus Abcd1− (Supplementary Fig. S5).
Protein identification
Proteins were identified in the Proteomic Unit of Institut de Recerca Vall d'Hebron (Barcelona) (Supplementary Table S1). Detailed methodology is described in the Supplementary Methods.
Metabolomics
Metabolites were extracted from homogenate tissues with methanol as previously described (67). Briefly, 60 μl of cold methanol was added to 20 μl of homogenate (containing 1.85 μg protein), vortexed for 1 min, and incubated at −20°C for 1 h to precipitate proteins. Samples were centrifuged for 3 min at 12,000 g, and the supernatant was collected. The supernatant was dried in a SpeedVac and resuspended in 50 μl of water. The sample was filtered in an eppendorf UltraFree 5 kDa filter. Four microliters of extracted sample was applied to a reverse-phase column (C18 Luna 3n pfp(2) 100A 150*2 mm, Phenomenex). The flow rate was 200 μl/min with solvent A composed of water containing 0.1% formic acid for positive ionization or 0.1% acetic acid for negative ionization, and solvent B composed of 95% acetonitrile and 5% water containing corresponding counterions. The gradient consisted of a gradient of solvent B from 5% to 100% in 20 min, held at 100% solvent B for 5 min, and re-equilibrated at 5% solvent B for 6 min. Data were collected in positive electrospray mode in a QTOF (Agilent) operated in full-scan mode at 100–3000 m/z. The capillary voltage was 3500 V with a scan rate of 1 scan/s. N2 was used as a gas nebulizer (flow was 5 l/min and temperature was 350°C). We used the MassHunter Data Analysis Software (Agilent) to collect the results and the MassHunter Qualitative Analysis (Agilent) to perform the integration and metabolite quantitation. The identity of metabolites was confirmed by identity of mass, isotopic distribution, and coelution with authentic standards. The m/z values used for quantification were m/z 394.9781 [2M+Na]+ for 2 (or 3) phosphoglycerate, m/z 176.0546 [2M+NH4 +-H2O]+ for pyruvate, m/z 229.0133 [M+CH3COO] − for DHAP/GA3P, m/z 193.0343 [M+H]+ for citrate, m/z 292.0662 [2M+NH4]++[-H2O] for α-ketoglutarate, m/z 338.9888 [M-H − ] − for fructose-1,6-biphosphate, m/z 115.0038 [M-H − ] − for fumarate, m/z 133.0136 for malate, m/z 112.5880 [M-H − ] − for oxalacetate, and m/z 179.0547 [2M-H − ] − for lactate. In all cases, Δ between calculated molecular weight (M.W.) and detected masses was lower than 0.001 Da. The identity of all metabolites was confirmed by identical chromatographic and mass spectrometric properties (molecular weight and isotopic distribution) of the quantified metabolites in comparison with authentic standards.
ATP levels
Mice were sacrificed by cervical dislocation, and spinal cords were immediately frozen in liquid nitrogen and stored at −80°C. ATP was extracted with cold perchloric acid (10%) from 10 mg of spinal cord, neutralized with KOH, and centrifuged (34). Then, ATP concentrations were quantified in triplicate per animal using the ATPlite 1 step (PerkinElmer) according to the manufacturer's protocol. Data were normalized to mg of proteins. All assays were performed in triplicate.
NAD-NADH and NADP-NADPH determinations
NAD+, NADH and NADP+, NADPH were, respectively, quantified by the NAD and NADP cycling assay. Detailed methodology is described in the Supplementary Methods.
Q-TOF based GSH analyses
Spinal cord samples were homogenate with a buffer containing 200 mM methane sulphonic acid with 5 mM DTPAC. Detailed methodology is described in Supplementary Methods.
Respiratory chain activity
We quantified the activities of complex I plus complex III, complex II plus complex III, complex IV, and Citrate synthase in the spinal cord samples from 12 month-old Abcd1− mice. Detailed methodology is described in Supplementary Methods.
Pyruvate kinase activity
Pyruvate kinase activity was determined by a spectrophotometrical method as previously described (26). 15 μg of mitochondria-free supernatant was added to a 0.2 ml of reaction buffer (50 mM TrisHCl pH 7.4, 100 mM KCl, 20 mM MgCl2, 0.3 mM NADH, 4 mM ADP, 1 mM phosphoenolpyruvate (PEP), and 5 units/ml of lactate dehydrogenase [LDH]). NADH was spectrophotometrically recorded after 6 min at 340 nm in a microplate spectrophotometer (PowerWave Microplate Spectrophotometer, BioTek). All assays were performed in triplicate at room temperature. Results were expressed as units (μmol/min) per mg tissue.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using RNeasy Kit (Qiagen), and Q-PCR experiments were performed according to manufacturer's instructions (LightCycler, Roche Diagnostics) as previously described (21). PCR were carried out with 36b4 (also called Rpl0) used as a standard gene. The nucleotide sequences of primers are available (Supplementary Table S2). Data are given as mean±SD.
Statistical analysis
Data are given as mean±SD. Significant differences were determined by one-way ANOVA followed by Tukey HSD post hoc test after verifying normality (*p<0.05, **p<0.01, ***p<0.001) or Student's t test (*p<0.05, **p<0.01, ***p<0.001). Statistical analyses were performed using SPSS 12.0 program.
Footnotes
Acknowledgments
This study was supported by grants from the European Commission [FP7-241622], the European Leukodystrophy Association [ELA2009-036C5; ELA2008-040C4], the Spanish Institute for Health Carlos III [FIS PI080991 and FIS PI051118], and the Autonomous Government of Catalonia [2009SGR85] to A.P. The CIBER de Enfermedades Raras is an initiative of the ISCIII. The study was developed under the COST action BM0604 [to A.P.]. J. L-E. was a fellow of the Department of Education, Universities, and Research of the Basque Regional Government [BFI07.126]. S.F. was a fellow of the European Leukodystrophy Association [ELA 2007-018F4], and J.G. was a fellow of the IDIBELL program of PhD-student fellowships.
Work carried out at the Department of Experimental Medicine was supported in part by R+D grants from the Spanish Ministry of Science and Innovation [AGL2006-12433 and BFU2009-11879/BFI], the Spanish Ministry of Health [RD06/0013/0012 and PI081843], the Autonomous Government of Catalonia [2009SGR735], and COST B35 Action of the European Union.
The authors are indebted to Professor Isabel Fabregat for scientific discussion.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.