
Abstract
The periplasm provides a strongly oxidizing environment; however, periplasmic expression of proteins with disulfide bonds is often inefficient. Here, we used two different tripartite fusion systems to perform in vivo selections for mutants of the model protein bovine pancreatic trypsin inhibitor (BPTI) with the aim of enhancing its expression in Escherichia coli. This trypsin inhibitor contains three disulfides that contribute to its extreme stability and protease resistance. The mutants we isolated for increased expression appear to act by eliminating or destabilizing the Cys14-Cys38 disulfide in BPTI. In doing so, they are expected to reduce or eliminate kinetic traps that exist within the well characterized in vitro folding pathway of BPTI. These results suggest that elimination or destabilization of a disulfide bond whose formation is problematic in vitro can enhance in vivo protein folding. The use of these in vivo selections may prove a valuable way to identify and eliminate disulfides and other rate-limiting steps in the folding of proteins, including those proteins whose in vitro folding pathways are unknown. Antioxid. Redox Signal. 14, 973–984.
Introduction
Frustratingly, despite the extremely high native-state stability of proteins with multiple disulfide bonds (34), heterologous proteins with complex disulfide bond patterns often express very poorly in E. coli (32). One example is the bovine pancreatic trypsin inhibitor (BPTI) (31), which in its mature form contains three nonconsecutive disulfide bonds involving all of its six cysteines. Fully oxidized, native BPTI has a melting temperature of over 100°C (36). However, the protein folds poorly in bacteria, resulting in very low expression yields (31). The in vitro folding pathway of BPTI is probably the most intensively studied of all proteins and is dominated by the formation and isomerization of its disulfide bonds (1, 16, 45). Conversely, much less is known about the in vivo folding of BPTI, especially in heterologous systems (31 –33). It is still not clear whether the folding challenges BPTI faces in vitro are also responsible for its relatively low expression in heterologous systems compared to that of other proteins (31).
In this study, we utilized two independent genetic systems to select for BPTI variants that exhibit improved expression and folding properties in the E. coli periplasm. The nature of the BPTI variants obtained supports the theory that destabilization or elimination of the native disulfide bond Cys14-Cys38 allows for a more productive route of folding toward the native state in vivo. The use of in vivo selections may therefore prove to be a valuable way to identify and circumvent critical rate-limiting steps in the in vivo folding of proteins, including those proteins whose in vitro folding pathways are unknown.
Materials and Methods
Cloning of phages for the protein stability increased by directed evolution system
fd phage derivatives were grown on XL1-Blue E. coli cells (Stratagene, La Jolla, CA). For cloning and sequencing of phage DNA, the replicative form was prepared using a Qiagen Miniprep Kit (Qiagen, La Jolla, CA). To construct tripartite fusions within the g3p protein of fd, a derivative of fd phage called fdP213 was used. This variant carries the g3p protein stabilizing mutations T13I, T101I, Q129H, D209Y, and P213G (29). The gene for BPTI was amplified from pTI103 plasmid (15) using the primers 5′-TAATTAGGGCCCGGCCTGACTTCTGC-3′ and 5′-GCTAATAGGGCCCCACCACAGGTCCTCATGCA-3′. These primers introduced ApaI sites at the 5′ and 3′ ends of the gene for mature portion of BPTI, enabling its insertion into the ApaI site that had been previously engineered into the g3p gene of fd phage by Krebber et al. (25). This allowed the in-frame cloning of BPTI into the g3p gene with no change in the amino acid sequence of BPTI, resulting in the phage PHSG7. It also placed BPTI between the N- and the C-terminal domains of the g3p protein. Inverted inserts were selected against because they created stop codons in g3p, preventing phage infectivity. The resulting fusion protein is referred to as “g3p′-BPTI WT-′g3p.”
Directed evolution of BPTI in the phage system
Mutagenesis of the BPTI gene was performed according to the GeneMorph II EZClone domain mutagenesis kit (Stratagene) with some minor modifications. Briefly, the BPTI sequence was amplified from pHSG7 DNA in an error-prone polymerase chain reaction (PCR) using the Mutazyme II enzyme mix and primers 5′-CTGTCAATGCTCCGTCCGGGGCC-3′ and 5′-GTACCAGAAGCCATGGCCGGCTG-3′, which annealed up- and downstream of the BPTI gene. The PCR was performed as recommended by Stratagene, except that the annealing temperature was 55°C and the elongation time was 1 min. The resulting pool of mutagenized BPTI genes was then used to perform a second, nonmutagenic PCR on pHSG7 DNA, according to the GeneMorph II protocol, to complete the synthesis of the fd phage. After digestion with DpnI (New England Biolabs, Beverly, MA), the remaining PCR product was precipitated. The resuspended DNA pellet was transformed into XL1-Blue cells, and the cultures were grown in LB for 14 h at 37°C. This simple outgrowth served as a growth competition selection that allowed the most infectious phage to outgrow less infectious phages in the culture. This protocol was designed to enable the selection of mutations within the BPTI gene that conferred a growth advantage to the g3p-BPTI tripartite fusion containing fd phage. Subsequently, the cultures were pelleted and the phage containing supernatant was plated using 0.7% top agar LB in different dilutions. Plaques larger than those of the unmutagenized pHSG7 phage (which generated tiny, almost invisible plaques) were picked and used to re-infect 10 ml of an overnight culture of XL1-Blue cells. These cells were again grown for 14 h to grow up the large plaque phage clones. After harvesting, the phage titer [plaque forming units (pfu)/ml] was determined, and the sequence of the BPTI gene determined from the isolated replicative form of the phage.
Directed evolution of BPTI in the β-lactamase system
To construct a tripartite fusion between BPTI WT and β-lactamase, the gene for BPTI was first PCR-amplified from pT1103 using primers 5′-CTGTCAATGCTCCGTCCGGGGCC-3′ and 5′-GTACCAGAAGCCATGGCCGGCTG-3′ and then cloned into the PfoI site within the β-lactamase gene in pBR322*link to obtain pBR322*link-BPTI. The resulting fusion protein is referred to as bla′-BPTI WT-′bla. The BPTI gene was then randomly mutagenized, and clones that showed an enhanced resistance to penicillin V (Pen V) or ampicillin were isolated, and the minimal inhibitory concentration (MIC) of antibiotic necessary to inhibit their growth was determined as previously described (13). Additional specific mutations in BPTI were generated using the QuikChange™ Site-Directed Mutagenesis Kit (Qiagen).
Determination of expression levels
Whole cell extracts were prepared as previously described (13).
For the separation of the soluble and insoluble fractions, mid-log-phase cells were pelleted (13,000 g, 4°C, 5 min) and adjusted to OD600 nm = 7 in lysis buffer (10 mM Tris, 2 mM EDTA, 1 mg/ml lysozyme, and protease inhibitor). One hundred microliters of this mixture was subjected to 10 freeze–thaw cycles and the soluble and the insoluble fractions were separated by centrifugation (13,000 g, 4°C, 10 min). The supernatant after centrifugation was the soluble fraction. The pellet (insoluble fraction) was resuspended in a total volume of 100 μl resuspension buffer (3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, 135 mM NaCl, 2 mM EDTA, and protease inhibitor, pH 7.4).
For the preparation of periplasmic extracts, a volume of mid-log-phase cells corresponding to OD600 nm = 7 was pelleted (13,000 g, 4°C, 10 min) and resuspended in 70 μl osmotic shock buffer (0.2 M Tris pH 8, 0.5 M sucrose, 1 mM EDTA). After incubation on ice for 30 min, the supernatant (periplasmic extract) and the pellet (cytoplasmatic fraction) were separated by centrifugation (13,000 g, 4°C, 10 min).
To obtain samples in which bla′-link-BPTI-′bla would still contain its signal sequence, mid-log phase cells expressing bla′-link-BPTI-′bla under arabinose control were induced with 2% arabinose. After 2 min, the cells were treated with 0.08% sodium azide, incubated for another 18 min, and whole cell extracts were prepared as described above. For the expression of BPTI in the absence of β-lactamase, BPTI was expressed from pET11a (New England Biolabs) under a T7 promoter. Protein production was induced with 5 μM IPTG.
Protein samples were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot analysis. Proteins were detected using a monoclonal rabbit antibody raised against TEM1-β-lactamase (Millipore, Billerica, MA) or reduced BPTI (generous gift from George Georgiou) as a primary and HRP-goat anti-rabbit IgG (Thermo Scientific, Waltham, MA) as a secondary antibody. Various dilutions of the protein samples for constructs containing WT BPTI and the BPTI variants were loaded on the gel. For each dilution, the ratio of band intensities for the mutant BPTI and WT BPTI was determined and averaged.
Results
To select for variants of BPTI that show enhanced expression and folding in the bacterial periplasm, we employed two independent tripartite fusion approaches that link the in vivo folding of BPTI to the activity of a reporter protein. The first approach was a modification of the protein stability increased by directed evolution (PROSIDE) technique (29); the second approach was the split β-lactamase system developed in our lab (13). In the PROSIDE approach, a guest protein is inserted between the N- and C-terminal domains of the g3p capsid protein of the fd phage (Fig. 1A). Both domains must remain covalently linked for g3p to be functional and confer phage infectivity. If the inserted protein is poorly folded or unstable, it becomes proteolytically sensitive. Degradation of the inserted protein will lead to a separation of the N- and C-terminal domains and a loss of infectivity. However, if the inserted guest protein folds well, the two domains of g3p remain covalently linked, and the phage carrying the fusion protein is infectious. This method therefore directly links the in vitro stability of the guest protein with phage infectivity. Improved folding efficiency of the guest protein in the periplasm should result in a higher infectivity of the phage, resulting in larger plaque sizes and increased phage titers.
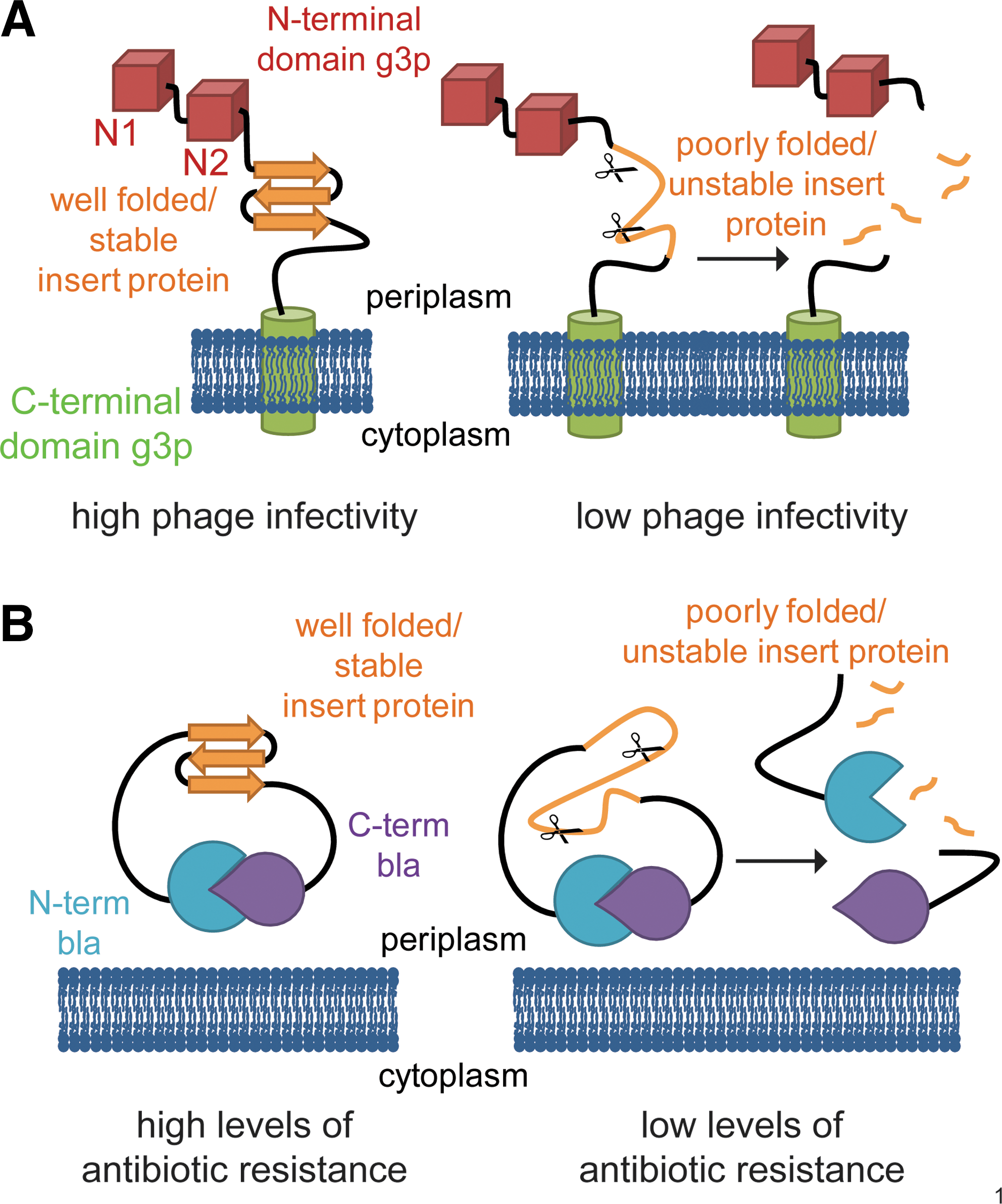
In the β-lactamase system, the guest protein is inserted between the N- and C-terminal halves of TEM1-β-lactamase, a periplasmic enzyme that confers resistance to β-lactam antibiotics (13) (Fig. 1B). We used this technique previously to monitor the in vivo stability of a variety of different proteins and to identify variants with improved thermodynamic and kinetic stability by selecting for increased levels of antibiotic resistance (13). As in the PROSIDE system, poor folding of the guest protein makes it susceptible to proteolytic degradation, leading to a separation of the two halves of the reporter protein. Bacteria expressing such a construct are therefore sensitive to antibiotic stress. A well-folded guest protein, however, ensures that the fusion remains intact, thereby leading to high levels of antibiotic resistance.
Selecting for BPTI variants that lead to increased infectivity in the PROSIDE system
The phage system links the proteolytic stability of a guest protein to phage infectivity and therefore to the rate of phage amplification. Insertion of WT BPTI into the protein g3p of the fd phage (referred to as “g3p′-BPTI WT-′g3p”) resulted in a dramatic decrease in phage titer (from ∼1011 pfu/ml for WT fd phage to ∼106 pfu/ml for g3p′-BPTI WT-′g3p, Table 1). This result is consistent with the poor in vivo folding and expression of BPTI noted previously (33). We reasoned that by mutagenizing the BPTI portion of the fusion and selecting for variants that show improved phage growth, we should be able to identify mutants with improved in vivo expression and folding properties. About 22 independent mutagenesis reactions yielded a total of 42 phage variants that resulted in increased plaque sizes and higher phage titers. Sequence analysis revealed two classes of fd variants that improved phage growth. The first class consisted of 25 independent deletion variants in which all or nearly all of the BPTI encoding sequence was selectively removed from the g3p gene. These deletion variants all showed titers and plaque sizes comparable to the original WT phage (data not shown). The remaining 17 phage variants had acquired single or multiple mutations in the BPTI sequence and showed increased plaque size and titer compared to g3p′-BPTI WT-′g3p (listed in Table 1).
Insertion of WT BPTI into the g3p encoding sequence of the phage decreased the phage titer from ∼1011 pfu/ml (+++++) to 106 pfu/ml (+/−). Titers varied significantly from experiment to experiment; values shown are the average of 3 experiments.++++corresponds roughly to a phage titer of 1010 pfu/ml,+++roughly to 109 pfu/ml. Residues in bold were found to be substituted in multiple independent mutagenesis reactions. In most of these cases, the chemical nature of the amino acid substitution was the same. Underlined are the BPTI exact amino acid substitutions that were previously found to selectively reduce the Cys14-Cys38 disulfide upon adding DTT in vitro (2, 18) and/or are known to destabilize this particular disulfide bond in vitro (17). Dotted underlined variants eliminate the Cys14-Cys38 disulfide bond completely. Variants that have been shown to decrease the accumulation of kinetic traps in the folding pathway of BPTI in vitro are marked with * (50).
BPTI, bovine pancreatic trypsin inhibitor; DTT, dithiothreitol; pfu, plaque forming unit; WT, wild-type.
For the two BPTI variants with single mutations (F33L and G12D), it is clear that the single mutation is sufficient for the enhanced growth phenotype. The vast majority of BPTI variants isolated, however, contained multiple amino acid substitutions, complicating interpretation. Many of the residues substituted were isolated on multiple occasions; 12 of the 33 point mutations identified affected residues that were found to be altered in multiple, independent mutagenesis reactions (shown bold in Table 1). We reasoned that the more often a residue was independently replaced in different BPTI variants, the more likely that alteration of this residue is important for robust growth of the phage expressing the g3p-BPTI variant. Among the isolated BPTI variants, six residues (F4, R17, I18, V34, R39, and R42) were independently found to be mutated twice, four (A16, Y35, A40, and S47) were mutated three times, and two (C14 and C38) were mutated four times. It is striking that of the 15 BPTI variants with multiple amino acid substitutions, the four variants exhibiting the largest increase in phage titer all eradicate the Cys14-Cys38 disulfide by eliminating both of the involved cysteines. Remarkably, seven of the substitutions that do not affect the cysteines are exactly the same as substitutions that have previously been found to destabilize Cys14-Cys38 in vitro (underlined in Table 1) (17).
To confirm that these amino acid substitutions are, on their own, sufficient to increase phage titers compared to phage containing WT BPTI, we attempted to construct the individual mutations in the phage system by site-specific mutagenesis. However, the very strong growth disadvantage conferred upon fd by g3p fusions containing WT BPTI and the resulting low titer greatly decreased the amount of DNA that could be isolated from the WT BPTI construct, making site-specific mutagensis very difficult. When high yields of DNA were obtained, we observed that it was often the result of the spontaneous evolution of phage containing deletions or other mutations within BPTI. The underlying basis of this problem is unfortunately exactly the same as the basis of the genetic selection; namely, the coupling of the growth of the phage to the folding of the test protein.
Selecting for BPTI variants that lead to increased antibiotic resistance in the β-lactamase system
To circumvent the problem of low DNA levels and frequent excisions of the BPTI gene from the phage genome, we decided to establish growth conditions in which the folding of the inserted protein could be selectively uncoupled from the growth of strains containing the selectable marker. This was not possible for the g3p tripartite fusions because g3p is required for phage propagation and consequently phage DNA amplification. We therefore employed a second genetic selection system, the β-lactamase system. Here, the tripartite fusion is expressed from a bacterial plasmid that also encodes for an additional selectable marker, tetracycline resistance. Growing cells containing this plasmid in tetracycline allows for an easy amplification and manipulation of plasmid DNA without any requirement for efficient folding of the tripartite β-lactamase protein. The β-lactamase system further provides a very reproducible readout for the relative level of antibiotic resistance conferred by different tripartite fusions (13).
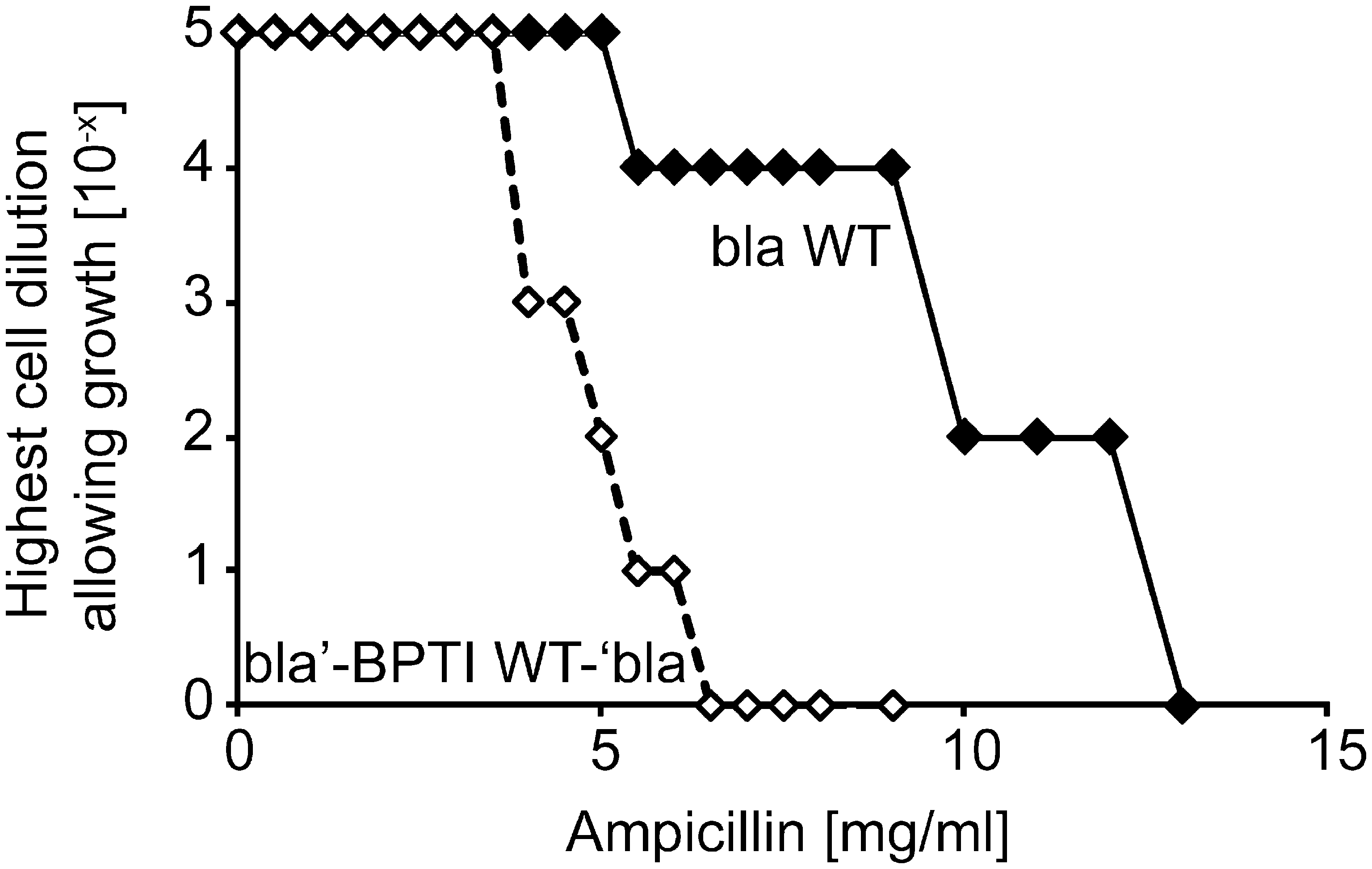
The construction of a plasmid expressing a tripartite fusion between BPTI WT and β-lactamase (referred to as “bla′-BPTI WT-′bla”) and the determination of the resulting level of antibiotic resistance is described in Materials and Methods. Antibiotic resistance is given as the MIC that prevents cell growth. The level of resistance caused by a given BPTI mutant is normalized to the level of resistance conferred by bla′-BPTI WT-′bla. The insertion of BPTI into β-lactamase resulted in a massive drop in antibiotic resistance compared to cells expressing WT β-lactamase, similar to the drop in phage infectivity that accompanied the insertion of BPTI into g3p (Fig. 2). As with the phage selection, we randomly mutagenized the BPTI portion of the tripartite fusion gene; however, in this case, we selected for increased antibiotic resistance to identify BPTI variants that show enhanced protein levels and folding in the periplasm. Using this approach, we isolated 13 point variants and 1 triple variant (P13S C14Y G28R) that showed increased MIC values compared to cells expressing bla′-BPTI WT-′bla (Table 2). We also constructed point variants P13S and G28R in the BPTI WT background by site-specific mutagenesis, and they also showed increased levels of antibiotic resistance. Variants isolated with the β-lactamase system that eliminate or destabilize the Cys14-Cys38 disulfide bond are underlined in Table 2.
The relative level of antibiotic resistance of cells expressing different bla′-BPTI-′bla variants is given as the minimal inhibitory concentration (MIC) that prevents cell growth (see the Materials and Methods section). Residues in bold were substituted more than once in independent mutagenesis reactions. In most of these cases, the chemical nature of the amino acid substitution was the same. Residues underlined were identified in a screen for DTT-sensitive mutants as single point mutations or are known to destabilize the Cys14-Cys38 disulfide in vitro (2, 17, 50). The dotted underlined variants prevent formation of Cys14-Cys38.
P13S and G28R were selected as part of a triple mutant (P13S C14Y G28R) and subsequently constructed as single point mutations via site-directed mutagenesis.
Relative protein levels of [5–55] [14–38], [14–38] [30–51], and [5–55] [30–51] in the β-lactamase system were 0.9 ± 0.1, 1.2 ± 0.2, and 1.6 ± 0.1, respectively.
SD, standard deviation.
For the two point mutations that led to the largest increase in MIC, BPTI G28W and K26M, we also tested the steady-state protein levels in the periplasm. These measurements were made both for BPTI when it was part of the tripartite fusion, as well as the isolated BPTI protein expressed on its own in the absence of β-lactamase. In both these contexts the protein levels of BPTI G28W and K26M in whole cell extracts were significantly increased compared to the corresponding WT constructs (2.5 ± 0.2-fold for BPTI G28W and 2.8 ± 0.3-fold for K26M in the fusion context, compared to the protein level of bla-BPTI WT-bla; 1.5 ± 0.1-fold for BPTI G28W and 1.7 ± 0.2-fold for K26M in the isolated protein in the absence of the fusion, compared to the protein level of WT BPTI itself. This suggests that the β-lactamase system works well for the selection of protein variants with increased expression in E. coli.
Destabilization of Cys14-Cys38 in vitro correlates with increased levels of antibiotic resistance in the β-lactamase system in vivo
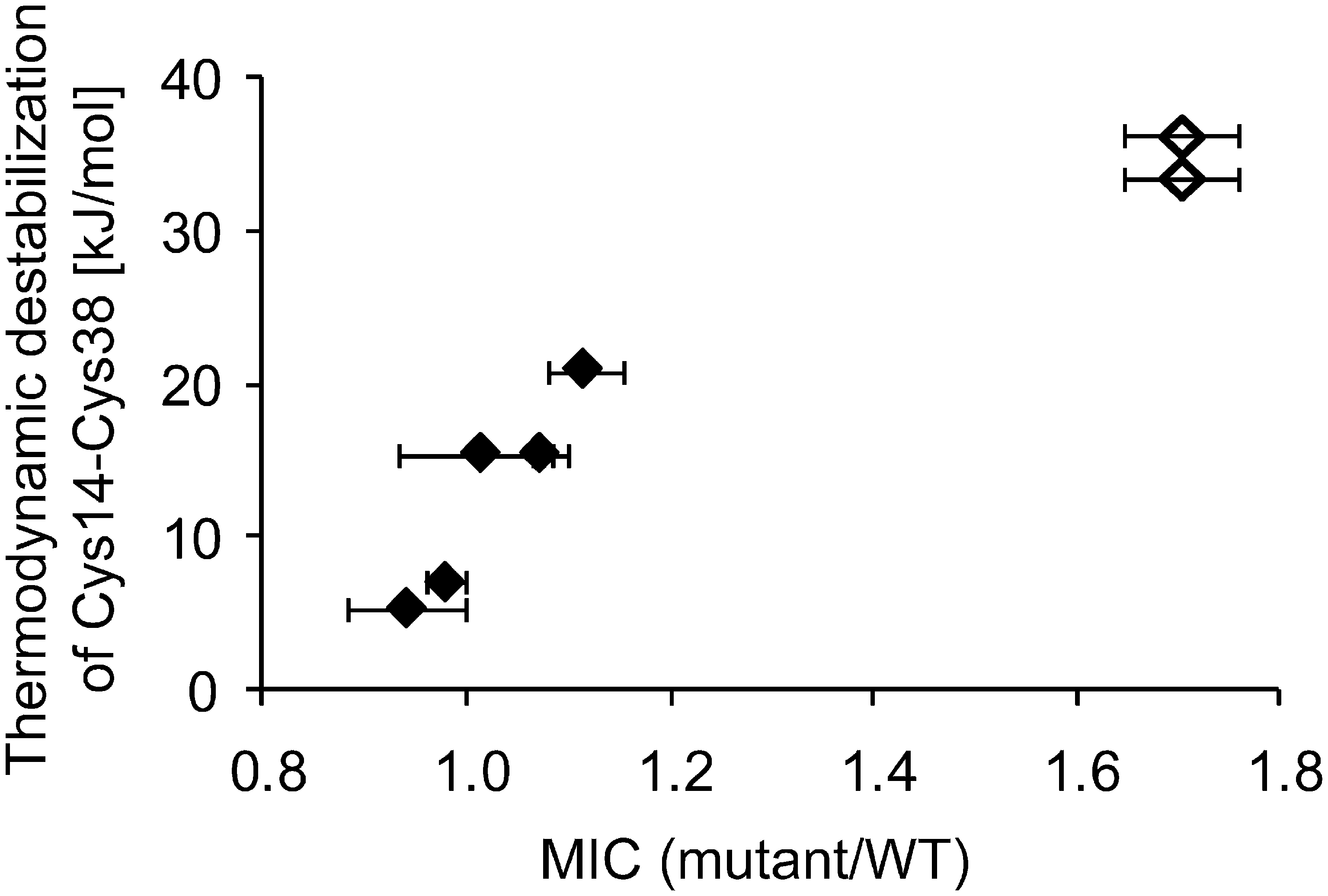
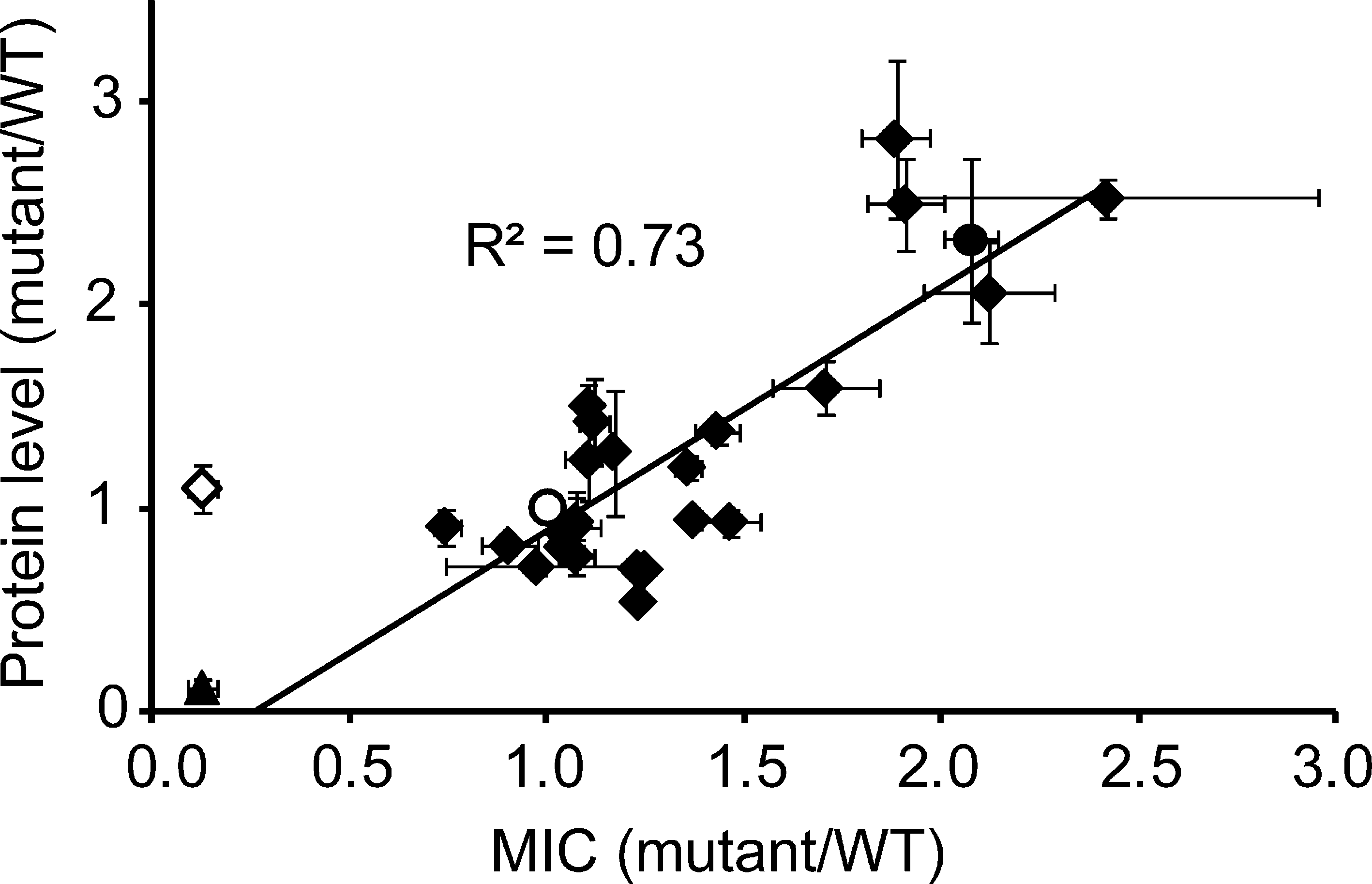
Because disulfide bonds act to stabilize proteins, we were initially surprised that the elimination or destablization of Cys14-Cys38 in BPTI was actually reflected in improved phenotypes in the phage system. We were therefore interested in whether the degree of destabilization of the Cys14-Cys38 disulfide as determined in vitro would correlate with increased protein stability in vivo as reflected by improved phenotypes in the tripartite fusion systems. In contrast to the phage system, which was characterized by a rather high propensity for recombination events that lead to the elimination of the protein inserted into g3p and phage titers that varied significantly from experiment to experiment, the β-lactamase system is genetically very stable and has been shown to provide a very reproducible phenotype for a variety of insert proteins (13). We introduced four of the amino acid substitutions that were obtained in the phage selection as point mutations into the bla′-BPTI WT-′bla background, and one mutant, Y35L, that was derived from the literature (17). All five BPTI variants are known to destabilize this disulfide bond in vitro (17). Remarkably, the extent of thermodynamic destabilization of Cys14-C38 caused by a given point mutation in vitro correlated well with the level of antibiotic resistance in the β-lactamase system (Fig. 3). Consistent with these findings, complete removal of the Cys14-Cys38 disulfide bond leads to an even higher MIC (Fig. 3, open diamonds). Importantly, the elimination of the Cys14-Cys38 disulfide bond increases the protein level of BPTI in E. coli both when it is part of the tripartite fusion as well as when BPTI is expressed on its own (1.6 ± 0.1-fold increase in protein level relative to bla′-BPTI WT-′bla in presence of β-lactamase and 2.3 ± 0.2-fold increase in protein level relative to BPTI WT in absence of β-lactamase, respectively). These results fit nicely with a previous observation that the elimination of this disulfide by serine substitutions leads to a 7-fold increase in the expression level of BPTI in the E. coli periplasm (31).
The stabilities of single disulfide-bonded BPTI species in vitro correlate with the level of antibiotic resistance in the β-lactamase system in vivo
Because the β-lactamase system has been shown to be a good readout for the in vivo stability of a variety of proteins (13), we decided to use it to assess which of the different intermediates that accumulate in BPTI's folding pathway in vitro might be especially problematic for BPTI's folding in vivo. Traditionally, much information about the folding pathway of BPTI has been obtained by studying the properties of analogs of folding intermediates (11) in which particular pairs of cysteines were chemically blocked or replaced with other amino acids, especially serine (6). To create three BPTI variants that can only form a single native disulfide bond, we used site-directed mutagenesis to replace all but one cysteine pair with serines. We list the disulfide mutants we constructed and define our nomenclature in Table 3. Among the single disulfide-bonded species containing native disulfides, BPTI [5–55] has the most native-like conformation in vitro, whereas [14–38] has the least (26, 41). Introduction of [5–55] into the β-lactamase system resulted in the highest MIC, whereas introduction of the least structured and least stable species ([14 –38]) resulted in the lowest MIC, suggesting that the latter intermediate does not form a stable conformation in the bacterial periplasm (Fig. 4A). These results indicate that, for single disulfide-bonded species of BPTI, the extent of native structure in vitro is correlated to the MIC in vivo. Consistently, the levels of antibiotic resistance for BPTI [5–55], [14–38], and [30–51] correlate very well with the contribution of the particular disulfide bond to the stability of the folded conformation, as reflected by the effective concentration of the disulfide bond in the native state (7, 23) (Fig. 4B). Interestingly, there is an inverse correlation between the MIC and the tendency of a disulfide to form in reduced BPTI: the higher the conformational preference for formation of a particular native disulfide in the reduced state, the lower the MIC of the BPTI variant that can only form this particular disulfide (Fig. 4C). An inverse correlation was also found between the MIC and the reactivity of the involved cysteines, with higher reactivites resulting in lower MICs (data not shown) (9). Further, we observed that a single disulfide-bonded variant of BPTI, [5–55], which is substantially less stable than the native protein (43), exhibits a higher MIC than WT BPTI in the β-lactamase system (Table 2), and this is testimony to the severe folding problems WT BPTI faces in the periplasm of E. coli.

CysX-CysY refers to a disulfide bond formed between cysteine residue X and cysteine residue Y. BPTI variants representing folding intermediates found in vitro were obtained by substituting cysteines not involved in disulfide bond formation with serines.
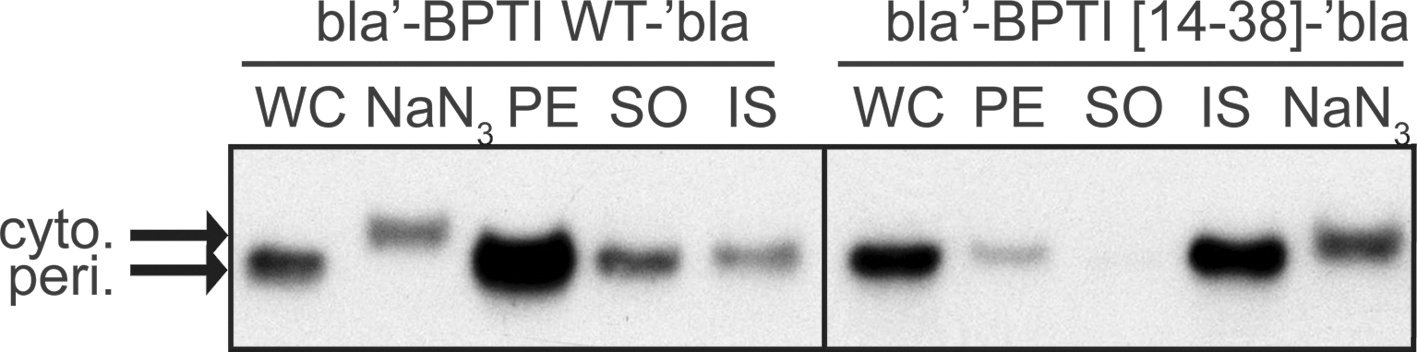
Because the MIC is a measure of how many molecules are successfully exported, we wondered if the low MIC caused by bla′-BPTI [14–38]-′bla was the result of a preferred formation of this disulfide in the cytosol, preventing export. bla′-BPTI [14–38]-′bla showed a protein level in whole cell extracts that was substantially higher than expected from its MIC value (protein level was 1.1 ± 0.1, MIC was only 0.13 ± 0.0 compared to bla′-BPTI WT-′bla). Subsequent analysis of soluble/insoluble and periplasmic/cytosolic fractions of cells expressing bla′-BPTI [14–38]-′bla indicated that the majority of the protein was periplasmic, but insoluble (Fig. 5). This result is in stark contrast to that obtained for the rest of the bla′-BPTI-′bla variants, which showed a very good correlation between level of antibiotic resistance and steady-state levels of the fusion proteins in whole cell extracts (Fig. 6). The latter observation is consistent with previous experiments using the β-lactamase system, which showed that the level of antibiotic resistance is a good measure for the relative level of the fusion protein in the periplasm (13). However, the actual amount of soluble, periplasmic bla′-BPTI [14–38]′-bla reflected the observed level of antibiotic resistance for bla′-BPTI [14–38]-′bla very well (periplasmic level compared to bla′-BPTI WT-′bla was 0.11 ± 0.05).
The β-lactamase system is also suitable to assess the effect of free cysteines on the in vivo stability of a protein
During heterologous protein expression in E. coli, the presence of unpaired cysteines in recombinant, disulfide bond containing proteins can promote the formation of incorrect intra- and intermolecular disulfide bonds, leaving the protein in inactive and aggregation-prone conformations (27). Elimination of free cysteines via site-directed mutagenesis has successfully been used to increase the stability, specific activity, and expression of these proteins and reduce their aggregation propensity (14, 30). However, since the substitution of cysteines has also been shown to decrease protein stability by reducing hydrophobic interactions or generating cavities in the protein core (30), the effect of such mutations is hard to predict and can largely depend on the amino acid the cysteine is replaced with (21).
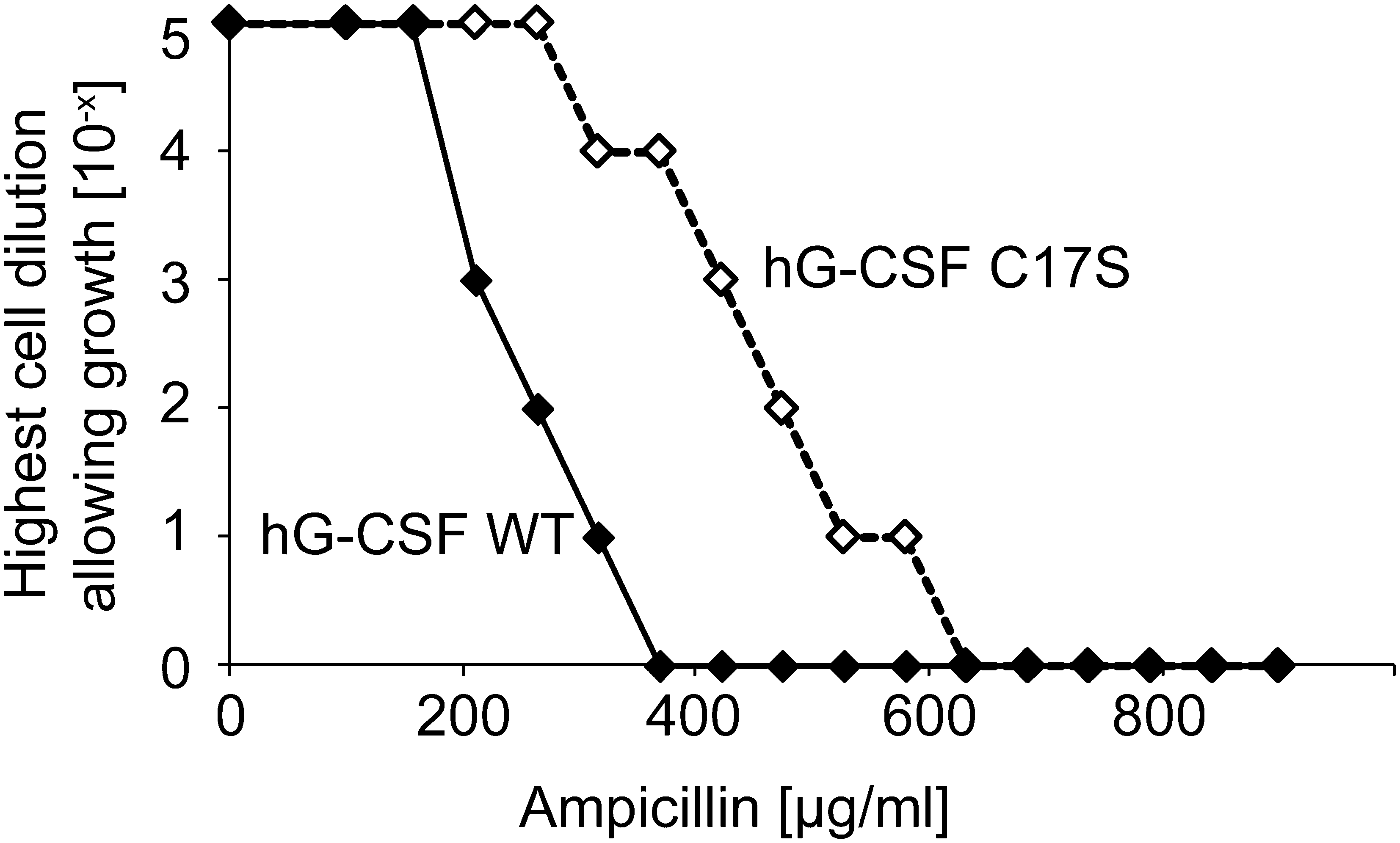
WT human granulocyte-colony stimulating factor (hG-CSF), which forms two natural disulfide bonds and contains one unpaired cysteine (Cys17), has a high propensity to aggregate, even under native-like conditions (35). This aggregation propensity is significantly decreased when the free cysteine is eliminated (35). In agreement with this observation, introduction of hG-CSF C17S into the β-lactamase system leads to an substantial increase in antibiotic resistance over a range of PenV concentrations compared to WT hG-CSF (Fig. 7) (13). This suggests that the β-lactamase technique can be used to identify cysteine mutations that are beneficial for the expression and in vivo stability of the corresponding protein by randomly mutating the proteins cysteines and simply selecting for increased levels of antibiotic resistance.
Discussion
Elimination or destabilization of the Cys14-Cys38 disulfide of BPTI leads to an increased expression in the bacterial periplasm
Despite the increasing number of studies investigating the folding and stability of proteins in the living cell (20), the fundamental question as to what extent in vitro folding experiments reflect the actual folding pathway in vivo has not been sufficiently answered for many proteins. In this study, we chose a multi-disulfide-bonded protein that is particularly poorly expressed in heterologous systems with the aim of selecting for variants that show improved folding and expression in the periplasmic space of E. coli. The test protein BPTI and many of its mutants have already been extensively characterized in vitro, allowing a direct comparison of mutants described in the literature with variants obtained in our in vivo selection.
Both genetic selection systems employed in this study were designed to exclusively identify variants that are successfully exported to the oxidizing environment of the periplasm, ensuring that we isolated BPTI variants showing increased protein levels in the bacterial compartment that would favor the correct formation of the protein's disulfide bonds. Strikingly, BPTI variants that resulted in the strongest phenotypes indicative of improved folding and expression in both selection systems completely eliminated one of BPTI's three native disulfide bonds, Cys14-Cys38. When analyzing our isolated BPTI variants in which no cysteines were mutated, we further noticed a high degree of overlap with BPTI variants sensitive to the reducing agent dithiothreitol (DTT) that were previously isolated in a screen by Coplen (2). This screen identified BPTI variants that are able to fold into an active conformation but, unlike WT BPTI, rapidly lose their protease inhibitory activity upon addition of DTT and are consequently inactivated in the presence of trypsin (2, 18). In total, our selections for increased folding and expression of BPTI in the bacterial periplasm yielded 8 variants (G12D, A16T, Y35N, Y35G, F33L, G28W, P13S, and G36D) that are known to be DTT-sensitive as single amino acid substitutions in vitro and/or were shown reduce the effective concentration of the Cys14-Cys38 disulfide, increase the rate constant for its reduction, and reduce its stability in vitro by up to 21 kJ/mol (BPTI Y35G) (2, 17). Five of the DTT-sensitive mutants (G12D, A16T, Y35N, Y35G, and F33L) have further been shown to selectively make the Cys14-Cys38 disulfide susceptible to reduction by DTT (17). For these variants, it was suggested that the resulting loss of BPTI activity was due to an increased flexibility of the BPTI variants and that the mutations might favor the intramolecular rearrangement of intermediates (2, 17, 18). Altogether, 65% of the BPTI variants obtained in the in vivo phage selection contain amino acid substitutions that either destabilize or eliminate the Cys14-Cys38 disulfide bond in vitro. This is a remarkable overlap, considering that there are 1102 single amino acid substitutions possible for the BPTI sequence. The idea that destabilization or removal of Cys14-Cys38 is key to improving the folding of BPTI in the bacterial periplasm is further supported by our findings that the level of antibiotic resistance in the β-lactamase system measured in vivo directly correlates with the extent of thermodynamic destabilization of the Cys14-Cys38 disulfide caused by a given single point mutation in vitro (Fig. 3) and that the relative protein level of BPTI C14S C38S expressed on its own is significantly enhanced compared to WT BPTI.
Elimination of the Cys14-Cys38 disulfide might prevent its unfavorable, early formation of in vivo
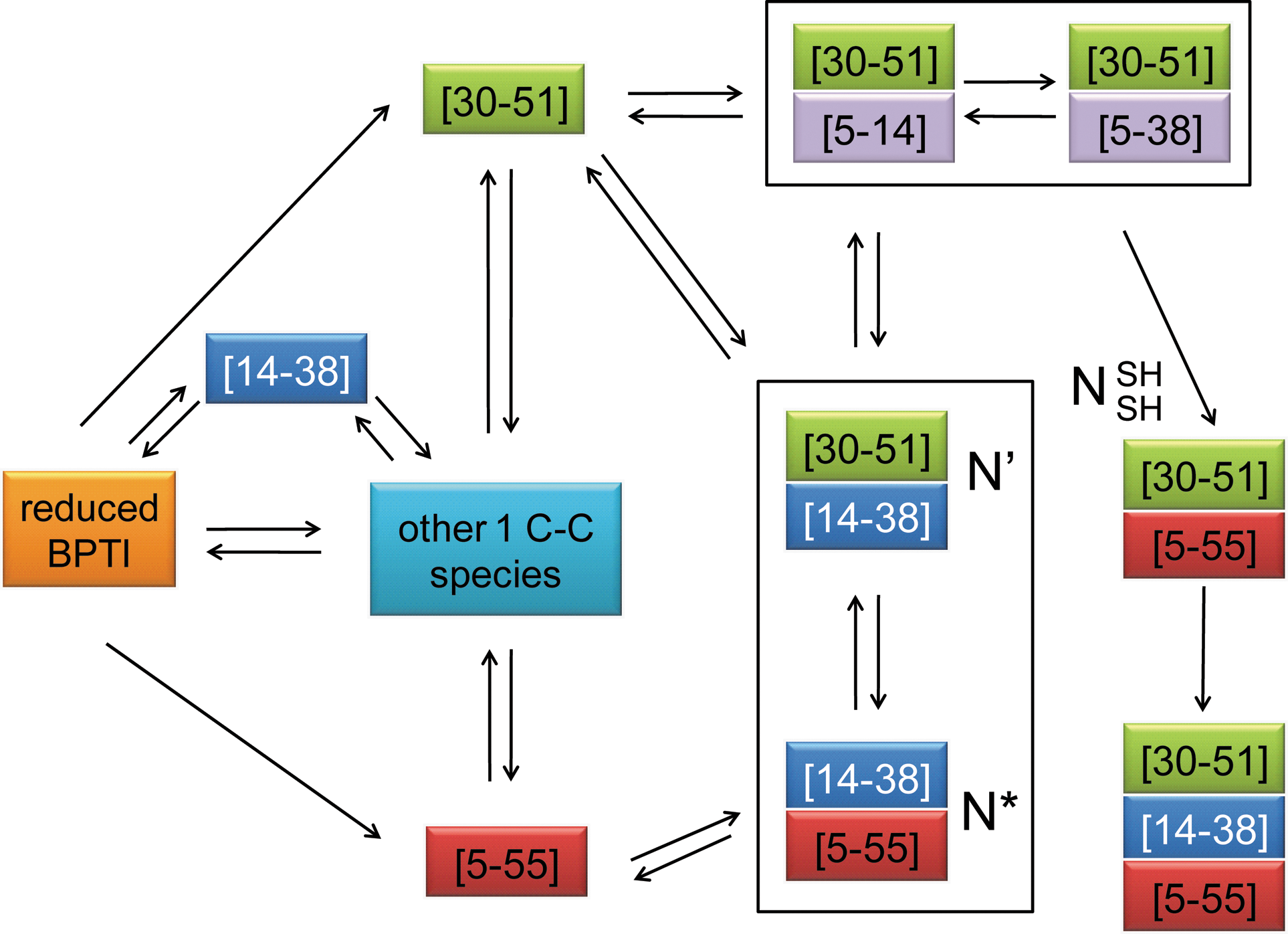
That mutations reducing a protein's in vitro stability lead to an increase in expression in vivo might appear counterintuitive. However, the formation of Cys14-Cys38 is particularly problematic in BPTI's in vitro folding pathway due to its early but nonproductive formation in BPTI folding and its involvement in the rate-limiting step of the overall BPTI folding reaction (Fig. 8) (6, 46, 49). Due to the high reactivity of Cys14 and Cys38 and the significant conformational preference for introducing the Cys14-Cys38 disulfide during BPTI refolding in vitro at neutral pH, the [14–38] intermediate is formed much more rapidly from reduced BPTI than any other species containing a single disulfide bond, native or non-native (1, 8, 9). Despite this, [14–38] is not a very productive folding intermediate. The vast majority of [14–38] molecules are either reduced or isomerized before they accumulate in the form of the more stable single disulfide-bonded species [5–55] and [30–51] in vitro (41, 45, 46). However, the reduction or isomerization of the Cys14-Cys38 disulfide that would allow for this efficient folding pathway might be more difficult in the very oxidizing environment of the periplasm (38) than in in vitro refolding reactions, which tend to be done under less oxidizing conditions. Importantly, in the absence of other stabilizing effects such as an additional disulfide bond, the sole formation of Cys14-Cys38 in reduced BPTI does not substantially stabilize the native conformation in vitro (12). In E. coli, BPTI [14–38] does not appear to adopt a soluble, stable conformation, as reflected by its low MIC and its accumulation in the insoluble fraction in the β-lactamase system. Therefore, the early and rapid formation of Cys14-Cys38 in the periplasm might favor the accumulation of BPTI in an aggregation prone state, reducing the number of molecules that can proceed folding toward [5–55] and [30–51], which are thermodynamically more stable in vitro (1). Consistently, BPTI variants that mimic single disulfide-bonded intermediates [5–55] and [30–51] also show an increased protein level compared to [14–38] in vivo as reflected by their increased MICs in the β-lactamase system. In fact, the stabilities of BPTI's native disulfide bonds as determined in vitro correlate well with the observed antibiotic resistance of the corresponding tripartite fusions in vivo, suggesting that the β-lactamase system is well suited to assess the stabilities of different BPTI variants in the living cell.
Elimination of the Cys14-Cys38 disulfide might further prevent the accumulation of protease-sensitive intermediates in later stages of BPTI folding
Preventing or decreasing the rapid formation of Cys14-Cys38 in the early folding of BPTI in vivo might be sufficient to explain the superior phenotypes of our selected BPTI variants that eliminated or destabilized this disulfide. However, formation and subsequent reduction of Cys14-Cys38 is also critical in later folding stages in vitro, which might also help explain the enhanced antibiotic resistance of tripartite fusions in which both Cys14 and Cys38 are replaced. At neutral pH, a large fraction of BPTI molecules accumulates in the form of the kinetic traps [5–55; 14–38] (N*) or [14–38; 30–51] (N′) (7, 46). For folding to proceed, N* and N′ must reduce Cys14-Cys38 and unfold substantially to isomerize into the native-like
All the double disulfide-bonded intermediates N*, N′, and
Because antibiotic resistance is determined by the steady-state levels of soluble and periplasmic protein, it is possible that the low MIC values of N* and N′ are caused in part by a problematic early formation of Cys14-Cys38, inducing protein aggregation. However, the very good correlation between antibiotic resistance and protein level in whole cell extracts for N*, N′, and
In addition to its impact on the early folding events of BPTI, the elimination or destabilization of Cys14-Cys38 might therefore also be associated with a simplification of BPTI's folding pathway in vivo in later folding stages, in which the accumulation of the proteolytically sensitive intermediates N* and N′ is prevented or decreased. Variants that destabilize Cys14-Cys38 might favor the formation of double disulfide-bonded species with non-native disulfides, which are more likely to undergo intramolecular rearrangements and are part of the most productive folding route of BPTI in vitro, as it has been already shown for several of the BPTI variants isolated in our selection (F33L, Y23L, F4L, and Y35L) (50). This might lead to a preferred formation of the stable and very native-like species
Interestingly, in variants of BPTI in which the formation of the Cys14-Cys38 disulfide bond was prevented, the rate of formation of native-like BPTI decreased from 9 s−1 to 1.6 × 10−3 s−1 at 25°C and pH 8.7 in vitro (15). However, this effect was diminished when experiments were performed at 37°C and at a less basic pH (8.3); under these conditions, only a 3-fold decrease in the overall folding rate for native BPTI was observed (28). Our finding that BPTI C14S C38S shows increased protein levels compared to BPTI WT when expressed in the bacterial periplasm in absence of β-lactamase, an observation also made by (31), suggests that this slightly higher rate of formation of the native or native-like protein for BPTI WT compared to [5–55; 30–51] in vitro cannot, if even relevant in vivo, compensate for the potentially negative impact of the presence of Cys14-Cys38 in the early and later folding stages of BPTI in vivo.
The observation that the elimination of a stabilizing disulfide bond enhances the expression of BPTI might provoke the question why it was not eliminated by nature in the course of evolution. However, BPTI variants in which the Cys14-Cys38 disulfide is eliminated by substituting one or both of the involved cysteines are known to be cleaved by trypsin up to 10,000-fold more rapidly (47) and show decreased secretion efficiencies in yeast by up to 30% (24), suggesting that Cys14-Cys38 is important for the in vivo function and protease resistance of the protein.
Dependence of BPTI folding on the presence of folding factors
Our data suggest that the elimination or reduction of the kinetic traps N* and N′ in vivo might increase the level of BPTI in the bacterial periplasm. In BPTI's natural environment, the endoplasmatic reticulum, these species are likely to be isomerized by the eukaryotic disulfide bond isomerase (protein disulfide isomerase [PDI]), which has been shown to promote isomerization of these kinetic traps and enhance the formation of native BPTI by 27-fold in vitro (48). In fact, co-expression of rat PDI has been successfully used to increase the expression of BPTI in E. coli (32). Compared to its eukaryotic counterpart PDI, the isomerase activity of the bacterial enzyme responsible for disulfide bond isomerization in the periplasm, DsbC, is relatively low (37). Consistent with that, we did not find the level of antibiotic resistance conferred by various BPTI tripartite fusions to be strongly dependent on the presence of native levels of DsbC (data not shown). BPTI folding in E. coli, however, severely depends on the activity of the disulfide bond oxidase DsbA (33). The importance of DsbA for BPTI folding is supported by our finding that dsbA − strains expressing different bla-BPTI fusions showed decreased levels of antibiotic resistance compared to a dsbA + strain (data not shown).
Conclusion
Presumably due to the differences in the redox-environment of the endoplasmatic reticulum and the bacterial periplasm, the heterologous expression of disulfide-rich, heterologous proteins in E. coli is often very challenging (32). Our results for BPTI and hG-CSF indicate that our tripartite fusion systems are well suited to enhance expression and folding of difficult to express heterologous proteins in the bacterial periplasm by random mutation of the target gene and a simple selection for improved phenotypes in our systems. The feasibility of our approach is supported by the good correlation between the relative level of antibiotic resistance and the relative level of the tripartite fusion in the β-lactamase system (Fig. 6) and the enhanced protein levels of selected BPTI variants compared to BPTI WT (determined for double mutant C14S C38S and single mutants K26M and G28W) in absence of β-lactamase we observe. Further, the majority of variants selected for increased expression of BPTI eliminates or destabilizes the Cys14-Cys38 disulfide bond, suggesting that circumvention of Cys14-Cys38-related folding problems in early or later folding stages may improve the expression of BPTI in vivo and compensate for the apparent loss in stability in vitro. For proteins with unpaired cysteines like hG-CSF, our systems can be used to easily identify if and how free cysteines can be substituted to increase expression and in vivo stability of the protein. Our results therefore emphasize the power of genetic selection to identify and circumvent critical steps and residues problematic for in the in vivo folding pathway of proteins without the need to perform time-consuming site-directed mutagenesis experiments. Although we used an extremely well-studied model protein here, this technique should be applicable to a variety of target proteins with potentially unknown folding pathways. In cases where an unmutagenized target protein is preferred, our selection systems also provide the convenient possibility of fine-tuning the redox-environment of the periplasm for the enhanced expression of a target protein by mutating co-expressed folding factors such as oxidoreductases or proteins involved in redox-maintenance.
Footnotes
Acknowledgments
We thank George Georgiou for the generous gift of BPTI antibody. We are further grateful to F.X. Schmid and B. Eckert for providing us with fd phage.
Author Disclosure Statement
No competing financial interests exist.