
Abstract
Transcriptional coactivators and corepressors often have multiple targets and can have opposing actions on transcription and downstream physiological events. The coactivator peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α is under-expressed in Huntington's disease and is a regulator of antioxidant defenses and mitochondrial biogenesis. We show that in primary cortical neurons, expression of PGC-1α strongly promotes resistance to excitotoxic and oxidative stress in a cell autonomous manner, whereas knockdown increases sensitivity. In contrast, the transcriptional corepressor silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) specifically antagonizes PGC-1α-mediated antioxidant effects. The antagonistic balance between PGC-1α and SMRT is upset in favor of PGC-1α by synaptic activity. Synaptic activity triggers nuclear export of SMRT reliant on multiple regions of the protein. Concommitantly, synaptic activity post-translationally enhances the transactivating potential of PGC-1α in a p38-dependent manner, as well as upregulating cyclic-AMP response element binding protein-dependent PGC-1α transcription. Activity-dependent targeting of PGC-1α results in enhanced gene expression mediated by the thyroid hormone receptor, a prototypical transcription factor coactivated by PGC-1α and repressed by SMRT. As a consequence of these events, SMRT is unable to antagonize PGC-1α-mediated resistance to oxidative stress in synaptically active neurons. Thus, PGC-1α and SMRT are antagonistic regulators of neuronal vulnerability to oxidative stress. Further, this coactivator–corepressor antagonism is regulated by the activity status of the cell, with implications for neuronal viability.
Introduction
Transcription factors do not exist in isolation: most signal-dependent transcription factors require association with broad-specificity coactivators to promote transcription through the recruitment of chromatin remodeling complexes. The transcriptional coactivator peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC-1α) controls important physiological and metabolic processes in a diverse array of tissues. These include glucose metabolism, energy homeostasis, adaptive thermogenesis, and mitochondrial biogenesis (11, 37). In neurons, PGC-1α regulates mitochondrial density, antioxidant defenses, and vulnerability to excitotoxic insults (45, 47). Moreover, it is under-expressed in Huntington's disease (HD) as a direct result of mutant Huntingtin (mtHtt)-mediated repression, potentially contributing to HD pathogenesis (6, 27, 49). PGC-1α mediates its effects through the coactivation of a number of transcription factors, such as nuclear respiratory factors 1 and 2, the myocyte enhancer factor 2 (MEF2) family, and nuclear receptors, including PPARγ, estrogen receptors (ER), thyroid hormone receptor (TR), and the retinoic acid receptor (RAR) (37, 38). This coactivation involves the recruitment of chromatin-modifying complexes to the promoter, which may contain histone acetyl transferases, methyltransferases, and nucleosome destabilizing enzymes (23).
As well as associating with coactivators when in an active state, many signal-regulated transcription factors when in their basal, uninduced state are not merely inactive but mediate active repression
Here we have studied how PGC-1α and SMRT control cortical neuronal vulnerability to oxidative and excitotoxic insults, and the influence of neuronal activity on these transcriptional coregulators. We find that SMRT can block the ability of PGC-1α to protect neurons against oxidative insults, but cannot antagonize the antiexcitotoxic effect of PGC-1α expression. Further, we find that neuronal activity, by activating PGC-1α and repressing SMRT function, upsets this antagonistic balance in favor of the neuroprotective PGC-1α.
Materials and Methods
Neuronal cultures and stimulations
Cortical neurons from E21 Sprague Dawley rats were cultured as described (2) except that the growth medium was comprised of Neurobasal A medium + B27 (Invitrogen), 1% rat serum, and 1 m
Western blotting and antibodies
Total cell lysates were boiled at 100°C for 5 min in 1.5 × sample buffer (1.5
RNA isolation, reverse transcriptase-polymerase chain reaction, and quantitative polymerase chain reaction
RNA was isolated using the Qiagen RNeasy isolation reagents (including the optional DNAse treatment) following passage of the cells through a QiaShredder column. For quantitative polymerase chain reaction (qPCR), cDNA was synthesized from 1 to 3 μg RNA using the Stratascript qPCR cDNA Synthesis kit (Stratagene, Amsterdam, Netherlands) according to the manufacturer's instructions. Briefly, the required amount of RNA (up to 3 μg) was diluted in RNase-free water (up to 7 μL final volume) and mixed on ice with 1 × cDNA Synthesis master mix (10 μL), random primers: oligo-dT primers 3:1 (total 2 μL–200 ng) and either 1 μL RT/RNase block enzyme mixture (for RT reactions) or 1 μL water (for No RT control reactions). Reaction mixtures were mixed and spun down and incubated for 2 min at 25°C, 40 min at 42°C, and 5 min at 95°C. cDNA was stored at −20°C. Dilutions of this cDNA were subsequently used for real-time PCR (cDNA equivalent to 6 ng of initial RNA per 15 μL qPCR reaction except GAPDH; cDNA equivalent to 2 ng initial RNA per 15 μL reaction for GAPDH). qPCR was performed in an Mx3000P QPCR System (Stratagene) using Brilliant SYBR Green QPCR Master Mix (Stratagene) according to the manufacturer's instructions. Briefly, the required amount of template was mixed on ice with 1 × Brilliant SYBR Green Master Mix, the required concentration of forward and reverse primers, 30 nM ROX passive reference dye, and water to the required reaction volume. Technical replicates as well as no template and no RT negative controls were included and at least three biological replicates were studied in each case. The sequence of the primers used are as follows: Rat PGC-1α–F: 5′-gaatgcagcgctcttagc-3′, -R: 5′-gct ttt gct gtt gac aaa tg-3′ rat GAPDH–F: 5′-aga agg ctg ggg ctc acc-3′ 200 nM, -R: 5′-agt tgg tgg tgc agg atg c-3′ 100 nM; 18s rRNA–F: 5′-gtg gag cga ttt gtc tgg tt-3′, -R: 5′-caa gct tat gac ccg cac tt-3′. The cycling programme was 10 min at 95°C; 40 cycles of 30 s at 95°C, 40 s at 60°C with detection of fluorescence and 30 s at 72°C; 1 cycle (for dissociation curve) of 1 min at 95°C and 30 s at 55°C with a ramp up to 30 s at 95°C (ramp rate: 0.2°C/s) with continuous detection of fluorescence on the 55°C–95°C ramp. The data were analyzed using the MxPro QPCR analysis software (Stratagene). Expression of the gene of interest was normalized to GAPDH, a commonly used control, which is invariant against a second control [18s rRNA (32)].
Plasmids
TR-Luc and GFP-SMRTα full length was a gift from Martin Privalsky [UC Davis (17)]. For construction of GFP-SMRT1233–2472, GFP-SMRT1523–2472, GFP-SMRT1–590, GFP-SMRT1–1854, and GFP-SMRT1–1018, GFP-SMRTα was digested with
Immunofluorescence
Immunofluorescence was performed as described (26). Pictures of GFP-SMRT construct-expressing neurons were taken on a CCD camera driven by Openlab software, and the subcellular distribution scored. Distribution was scored as nuclear if localization was exclusively nuclear, whereas significant observable GFP immunofluorescence in the cytoplasm counted as a cell that exhibited SMRT export. Anti-GFP antibody (Invitrogen) was used (1:600) and observed using biotinylated secondary antibody/cy3-conjugated streptavidin. Nuclei were counter-stained with DAPI. For each treatment, ∼150–200 cells were analyzed within 3–5 independent experiments.
Transfections and gene expression analysis
Neurons were transfected using Lipofectamine 2000 (Promega) and stimulated 24–48 h after transfection. For gene expression analysis cells were then incubated overnight or for 8 h and luciferase activity read out. Luciferase assays were performed using the Dual Glo assay kit (Promega) with Firefly luciferase-based reporter gene activity normalized to the Renilla control (pTK-RL plasmid) in all cases. Mean ± SEM of 3–6 experiments is shown. Where used, siRNA (PGC-1α or Dharmacon's control nontargeting siRNA #2 siRNA) was at 100 nM. siRNA sequences used were as follows: (i) 5′-caa uga aug cag cgg ucu u-3′; (ii) 5′-gaa caa gac uau uga gcg a-3′.
Following the fate of transfected cells
This was performed essentially as described (43). Briefly, pictures of GFP-expressing neurons were taken using a Leica AF6000 LX imaging system, with a DFC350 FX digital camera. Neurons were then treated with 100 μ
Results
PGC-1α is strongly protective against oxidative and excitotoxic insults
To assess the extent of neuroprotection afforded by PGC-1α in primary cortical neurons, we transfected neurons with a vector encoding PGC-1α driven by a constitutively active promoter, plus an eGFP vector to identify transfected neurons. The fate of these neurons was monitored both before and after the application of an oxidative insult (100 μ
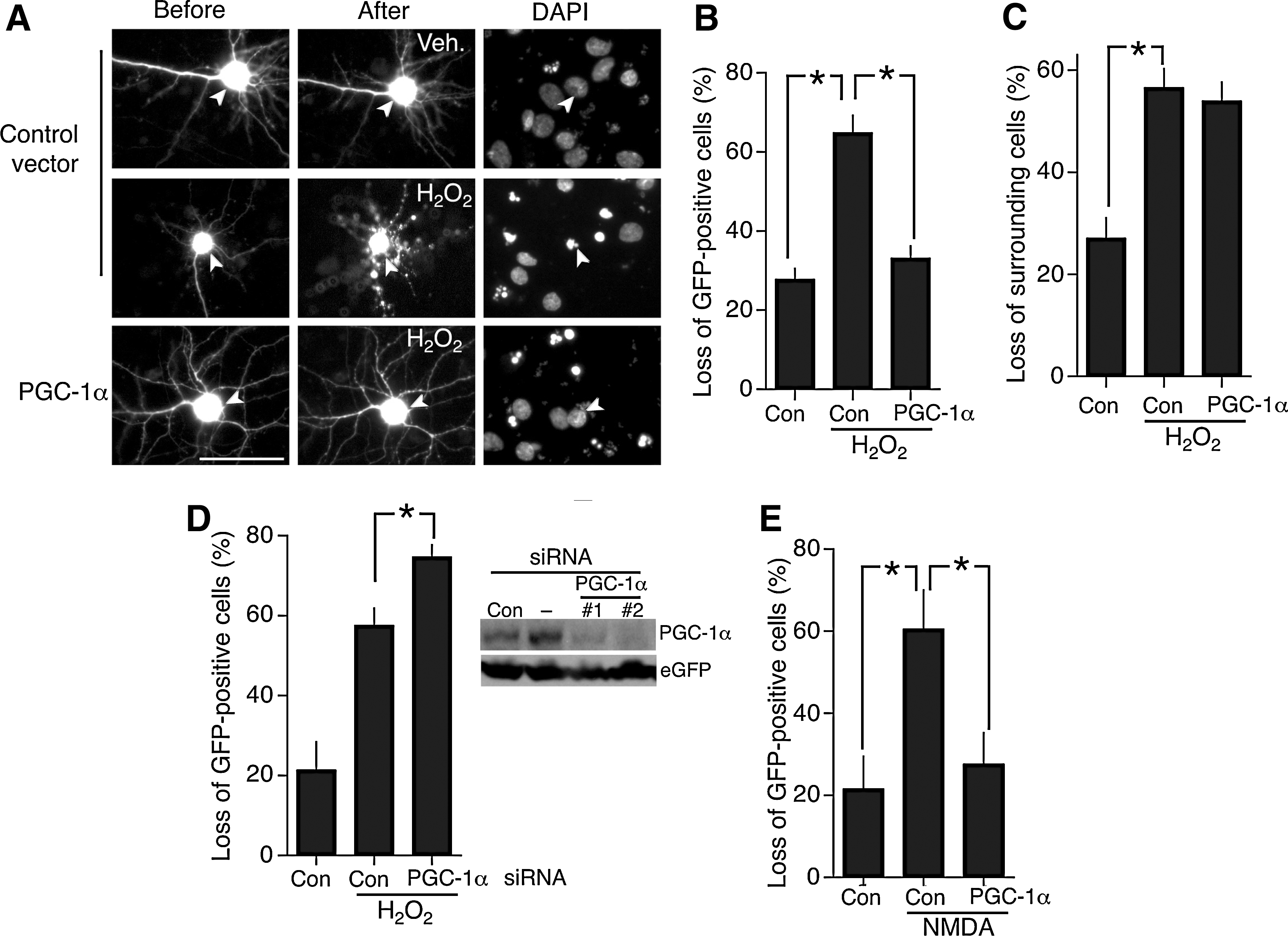
PGC-1α has also been reported to confer protection against excitotoxic insults such as kainate-induced seizure, oxygen-glucose deprivation, and mtHtt-induced death (6, 25, 31, 45). While excitotoxic insults undoubtedly have an oxidative component, other routes to neuronal death are also triggered that are not associated with oxidative stress (13). We therefore wanted to know whether PGC-1α protected against an excitotoxic insult in our system as effectively as it did for oxidative insults. We exposed control- and PGC-1α-expressing cortical neurons to an excitotoxic dose of NMDA and once more saw near complete resistance to this insult (Fig. 1E).
SMRT specifically represses the antioxidant boosting effects of PGC-1α
As a coactivator, PGC-1α promotes the transactivating potential of many transcription factors, including nuclear receptors and MEF2s. These and other transcription factors associate with PGC-1α-containing coactivator complexes when active but also are capable of mediating active transcriptional repression when associated with corepressor complexes in their inactive state. Taking the example of nuclear hormone receptors, their coactivator- and corepressor-associated forms exist in an equilibrium that is pushed in favor of the active state by presence of cognate ligand but can also be influenced by the level of corepressors or coactivators available (41).
SMRT is a transcriptional corepressor that exerts its effects by binding target transcription factors and recruiting other repressor molecules such as HDACs. It shares several targets with PGC-1α, including nuclear receptors and MEF2s, raising the possibility that SMRT could exert opposite effects to PGC-1α with respect to vulnerability of neurons to oxidative and excitotoxic insults. We therefore investigated the effect of SMRT expression on vulnerability to these trauma. We found that expression of SMRT alone did not affect basal viability (data not shown), nor did it increase neuronal vulnerability to H2O2-induced oxidative death (Fig. 2A). However, SMRT completely blocked the protective antioxidant effects of PGC-1α expression (Fig. 2A). We next looked at the effect of SMRT expression on the vulnerability of neurons to NMDA receptor (NMDAR)-dependent excitotoxic death. In this case, SMRT expression alone did not exacerbate cell death but neither did it antagonize the protective effect of PGC-1α expression (Fig. 2B). Thus, SMRT antagonizes the antioxidant effects of PGC-1α but not the antiexcitotoxic effects. This indicates that these two PGC-1α-driven neuroprotective effects are mechanistically different, and that SMRT specifically antagonizes those PGC-1α-mediated events that ultimately boost resistance to oxidative insults.
Synaptic NMDAR activity boosts PGC-1α transcription in vitro and in vitro
Synaptic activity, acting in part by triggering synaptic NMDAR-dependent Ca2+ transients, is able to control the activity, expression, and subcellular localization of several broad specificity transcriptional coactivators and corepressors. We next investigated the influence of synaptic activity on PGC-1α and SMRT function and its implications for the antagonistic effect of SMRT on PGC-1α-dependent antioxidative effects.
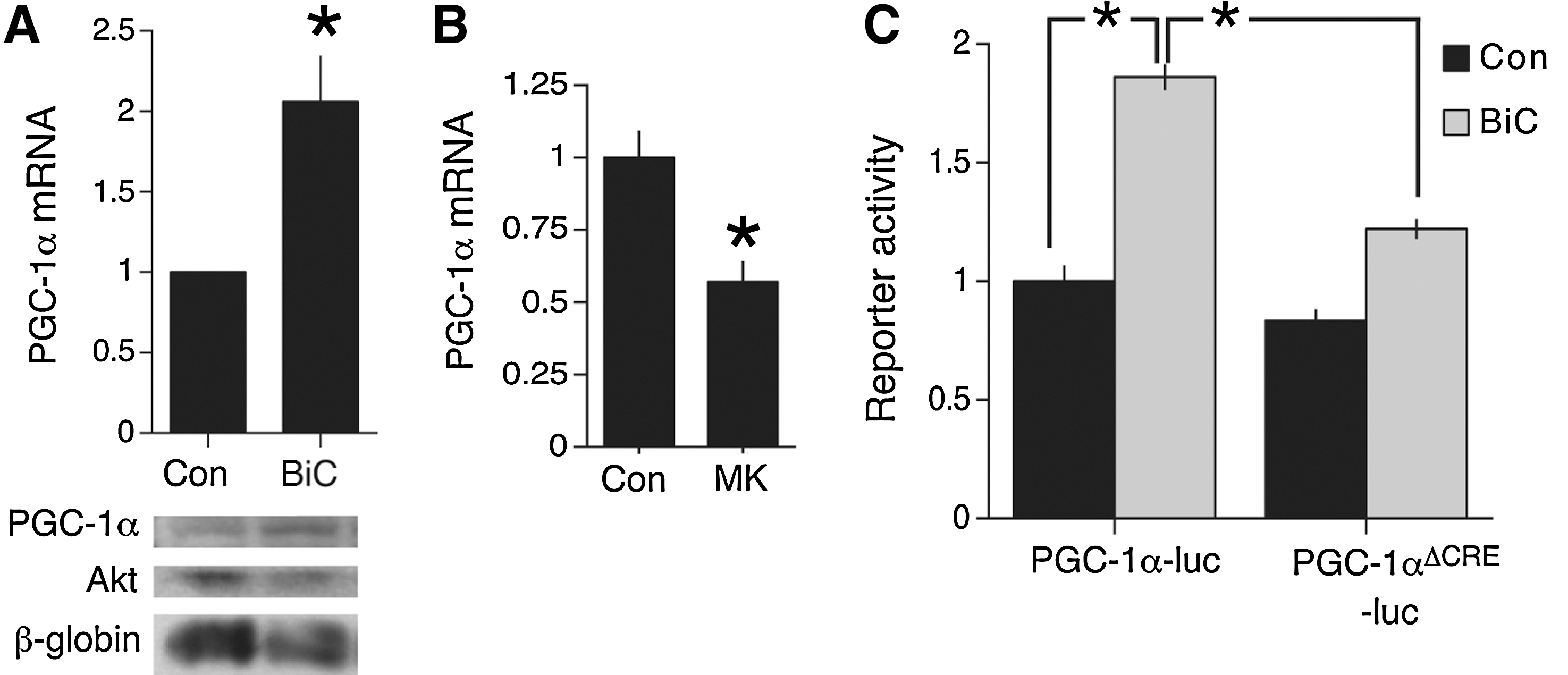
Membrane depolarization, triggering Ca2+ entry, is known to upregulate the transcription of PGC-1α (29), indicating that synaptic activity is likely to do the same. We used an established method of network disinhibition to enhance synaptic activity, by applying the gamma-aminobutyric acid A receptor antagonist BiC. This treatment results in synchronized bursts of action potentials that are associated with intracellular Ca2+ transients dependent on influx through NMDARs, as well as release from internal stores and some influx through voltage-gated channels (14). The K+ channel antagonist 4-aminopyridine (4-AP) was coapplied with BiC to enhance burst frequency (14) (hereafter BiC/4-AP). BiC/4-AP treatment resulted in the upregulation of PGC-1α mRNA and protein expression (Fig. 3A).
Synaptic activity boosts PGC-1α activity post-translationally via p38.
As well as being subject to transcriptional regulation, PGC-1α has also been reported to be subject to post-translational control. Cytokine stimulation of muscle cells has been shown to induce PGC-1α's intrinsic activity
Since synaptic activity can trigger p38 activity (44) we considered the possibility that PGC-1α could be subject to activity-dependent post-translational control. To study the post-translational regulation of PGC-1α directly we tested the ability of PGC-1α, tethered to the promoter by means of fusion to a GAL4 DNA binding domain, to confer activity-dependent inducibility on a luciferase-based reporter containing multiple GAL4 binding sites. Expression of GAL4-PGC-1α resulted in an 11-fold activation of the reporter compared to expression of the GAL4 domain alone (Fig. 4A), consistent with a significant degree of basal activity. However, triggering synaptic activity with BiC/4-AP resulted in a further fivefold enhancement of PGC-1α-dependent gene expression, indicating that PGC-1α's intrinsic activity is enhanced by neuronal activity (Fig. 4B). To investigate this further we looked at the role of p38 in this activation process. We found that pretreatment with the p38 inhibitor SB202580 strongly inhibited the potentiating effect of synaptic activity on GAL-PGC-1α-mediated reporter activity (Fig. 4B).
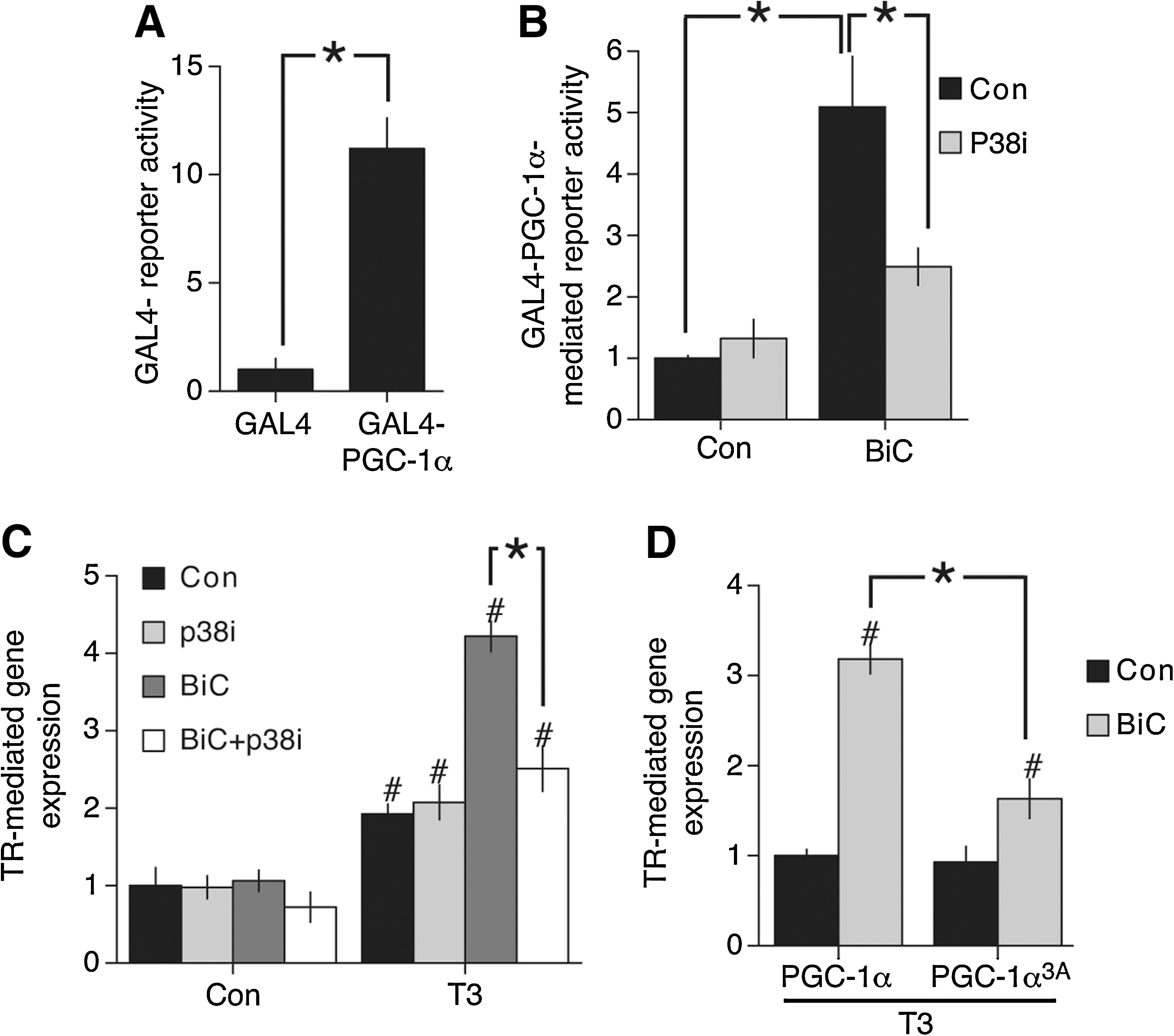
PGC-1α promotes the activation of nuclear receptors, including the TR (37, 38). TR-mediated gene expression is suppressed by SMRT, and so represents an example of one of several transcription factors that could integrate the opposing actions of PGC-1α and SMRT. We recently found that synaptic activity can super-induce T3 hormone activation of the TR. This was associated with the activity-dependent nuclear export of SMRT and the dissociation of SMRT from TR promoter elements (28). Our observation that PGC-1α can also be regulated by synaptic activity raised the possibility that activity-dependent activation of PGC-1α
We wanted to know if PGC-1α is a target of the p38 pathway in boosting TR-mediated gene expression. p38-dependent activation of PGC-1α is mediated by phosphorylation on three different sites (36). Mutation of these residues to alanine abolishes p38-inducibility of PGC-1α-mediated processes without reducing basal PGC-1α activity. We investigated the effect of this mutant (PGC-1α3A) on the activity-mediated, p38-dependent boosting of TR-mediated gene expression. Compared to expressing wild-type PGC-1α, expression of PGC-1α-3A had the effect of specifically inhibiting the potentiating effect of activity on TR-mediated gene expression (Fig. 4D). Basal T3-induced, TR-mediated gene expression (in the absence of synaptic stimulation) was not affected (Fig. 4D). We also confirmed that PGC-1α3A has similar basal activity to wild-type PGC-1α with regard to its ability to protect against H2O2-induced death (data not shown). The effect of PGC-1α3A in specifically interfering with activity-mediated, p38-dependent boosting of TR-mediated gene expression suggests that phosphorylation of these sites may contribute to the p38-mediated effects observed.
SMRT's RD3 domain confers nuclear localization in neurons
We previously reported that synaptic activity promotes the nuclear export of SMRT and dissociation of SMRT from target transcription factors including the TR (28). Activity-dependent redistribution of SMRT to the cytoplasm is observed in the presence of the protein synthesis inhibitor cycloheximide (data not shown), confirming that the pre-existing SMRT is exported (rather than,
We first investigated the region(s) of SMRT that confer nuclear localization under normal conditions in neurons. A C-terminal portion of SMRT (SMRT1223–2472) was nuclear-localized, but this was lost in the SMRT1523–2472 portion, implicating the 1223–1523 region (Fig. 5B, C). Consistent with this, an internal deletion of region 1018–1523 (SMRTΔ(1018–1523)), which encompasses SMRT's RD3 domain, caused a reduction (but not complete loss) in nuclear localization (Fig. 5C). In contrast, deletion of a region spanning SMRT's RD4 domain (SMRTΔ(1523–1854)) did not affect nuclear localization. Further, a portion of SMRT containing the RD3 region (SMRT1025–1523) is nuclear when expressed (Fig. 5C), as has been reported in Hela cells (50). Thus, the RD3 domain is an important determinant of SMRT nuclear localization in neurons.
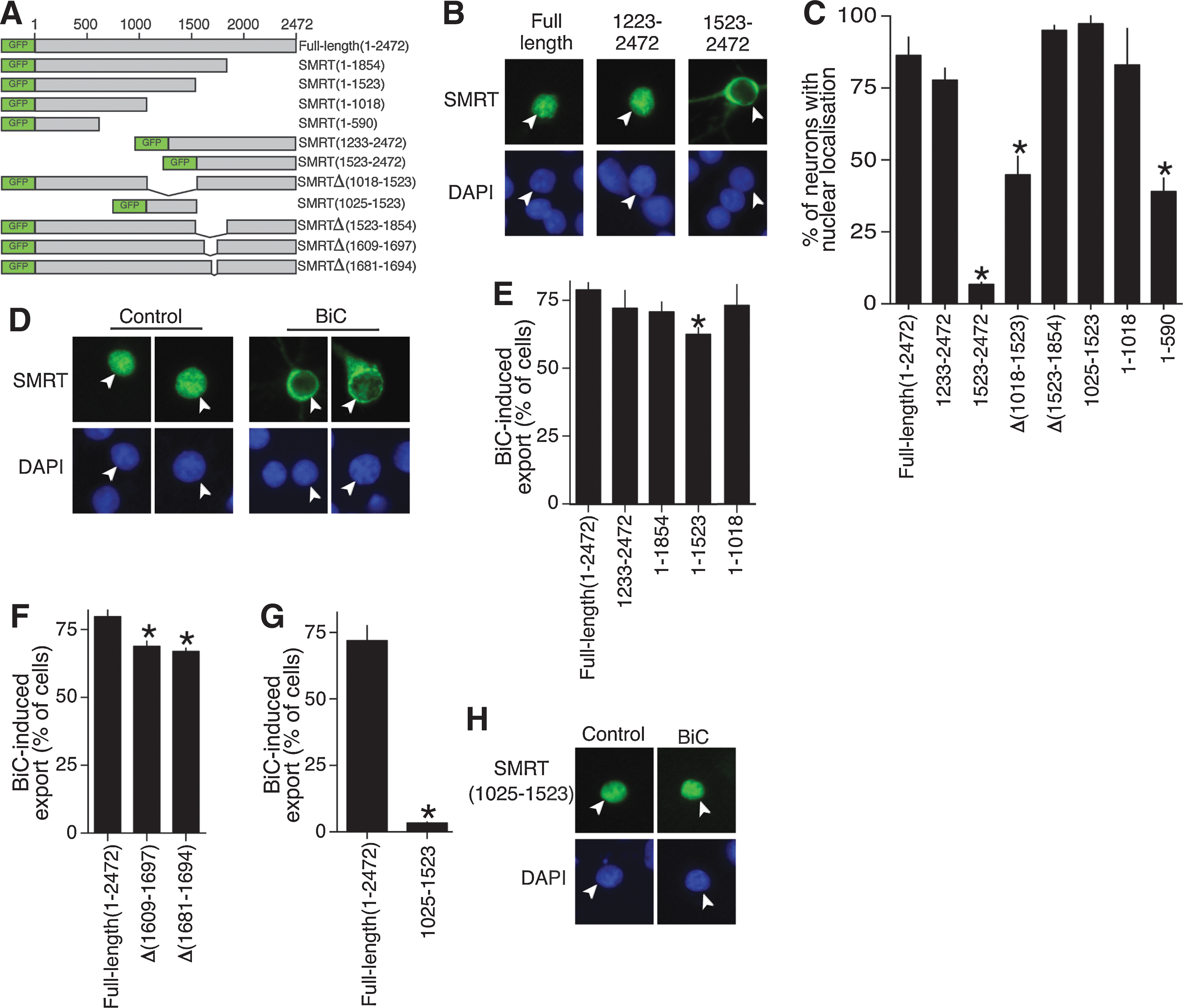
However, the fact that SMRTΔ(1018–1523) retained some nuclear localization (Fig. 5C) is suggestive of further sequences conferring nuclear localization in the N-terminal 1018 amino acids. Indeed, the N-terminal portion of SMRT (SMRT1–1018) was also strongly nuclear when expressed, although a smaller region of SMRT (SMRT1–590) was not, appearing to be distributed evenly throughout the nucleoplasm (Fig. 5C). Collectively these data indicate that the RD3 region is important for conferring SMRT nuclear localization, but that the N-terminal region can also localize to the nucleus independent of this domain.
SMRT N- and C-terminal sequences can mediate activity-dependent nuclear export
Study of the BiC/4-AP-induced export (Fig. 5D, E) revealed that SMRT1–1854 undergoes export as effectively as full-length SMRT but that SMRT1–1523 is exported slightly (but significantly) less well, implicating the region between 1523 and 1854 in contributing to nuclear export (Fig. 5E). Deletion of a smaller piece of this region (SMRTΔ1609–1697) also showed a similar significant reduction in activity-dependent export (Fig. 5F). Further examination of this region revealed a potential nuclear export site (NES) (20) with sequence LRGLSPRESSLA, which was deleted (SMRTΔ1681–1694), once again showing a small but significant reduction in activity-dependent export (Fig. 5F).
Surprisingly, however, further C-terminal deletion beyond position 1523 (creating SMRT1–1018) restored activity-dependent export to normal levels (Fig. 5E), suggesting that the 1018–1523 region of SMRT (containing the RD3 domain) acts as a partial break on nuclear export. This portion of SMRT (SMRT1025–1523) does not undergo export in response to synaptic activity, remaining in the nucleus (Fig. 5G, H). Since both N-terminal SMRT1–1018 and C-terminal SMRT1233–2507 undergo normal activity-dependent export (Fig. 5E), we conclude that no single domain is likely to be critical for this process, although the putative NES region may play a partially nonredundant role.
Synaptic activity renders SMRT unable to suppress PGC-1α-mediated antioxidative effects
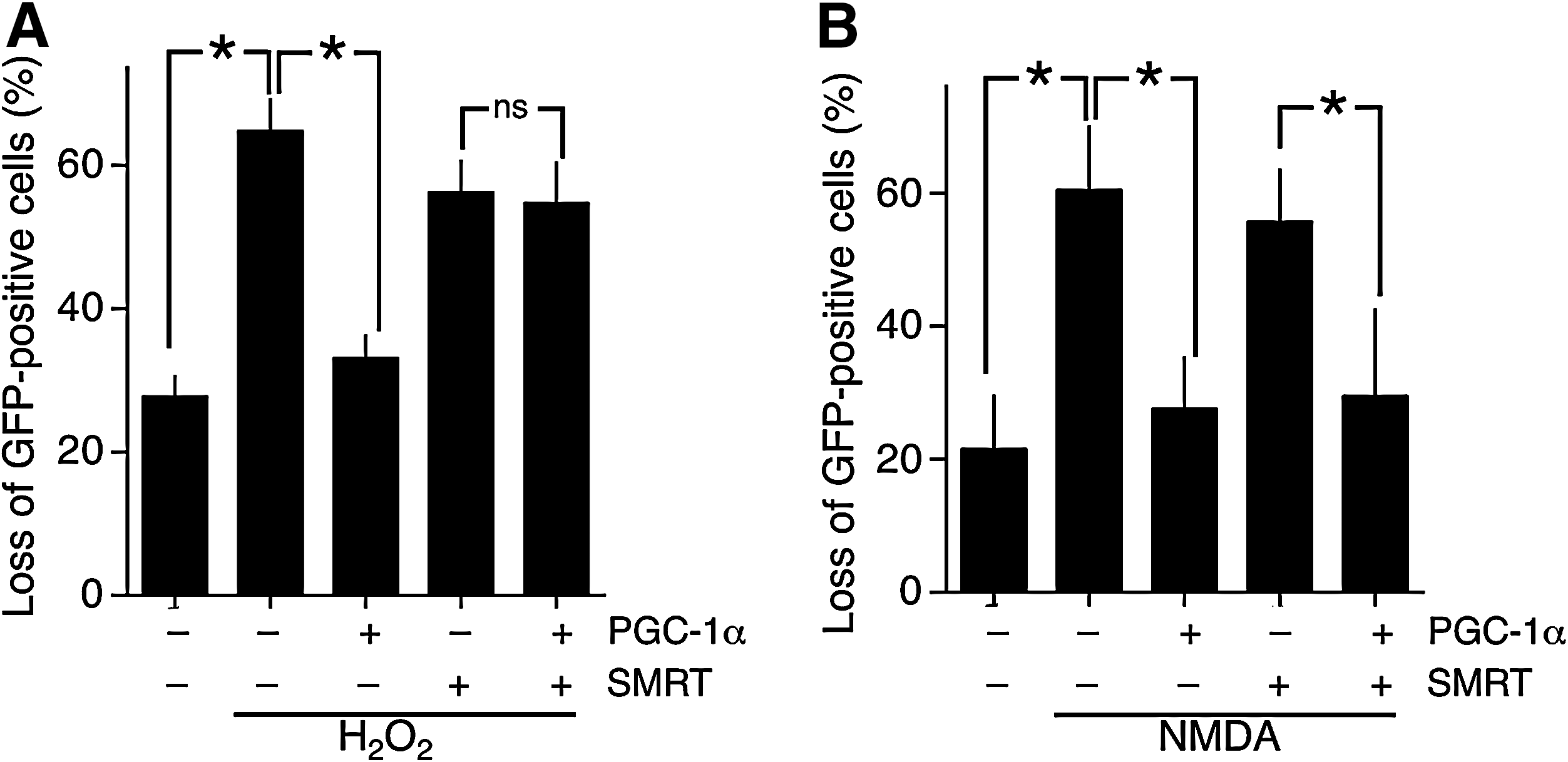
We observed that coexpression of SMRT resulted in the antioxidant effects of PGC-1α expression being inhibited (Fig. 2A). However, our findings that synaptic activity both induces the activity of PGC-1α as well as triggering nuclear export of SMRT (Figs. 3
–5) suggests that the capacity of SMRT to antagonize PGC-1α effects will be reduced. We found that in BiC/4-AP-stimulated neurons, death induced by 100 μ
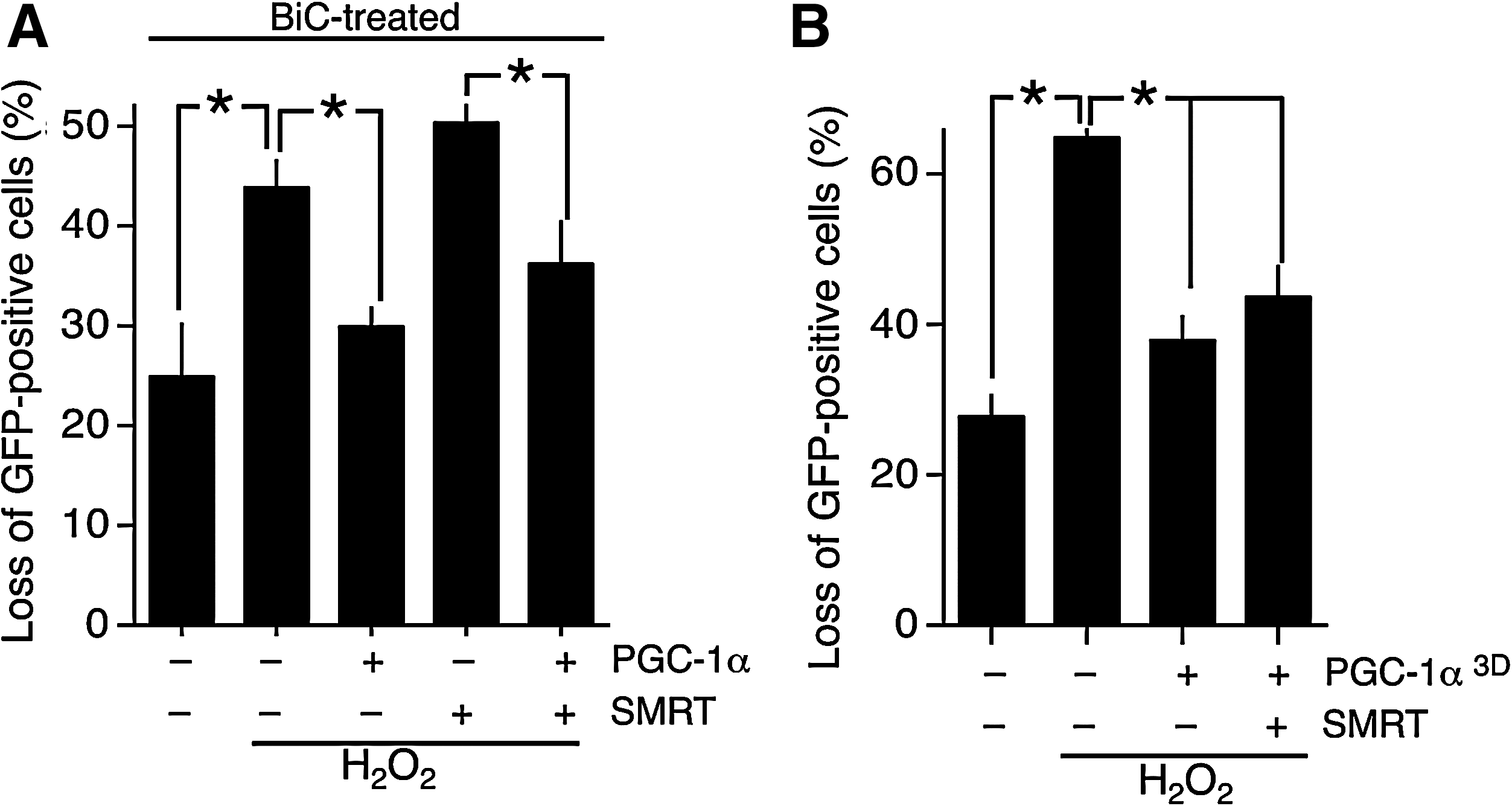
Importantly, PGC-1α-mediated protection against oxidative insults was still observed in SMRT-expressing neurons (Fig. 6A). BiC/4-AP-induced synaptic activity rendered the antagonizing effect of SMRT ineffective. This is consistent with the effect of synaptic activity in enhancing PGC-1α activity and suppressing SMRT activity. As to which of these is most important, it is possible that both contribute. Unfortunately, because the activity-dependent export of SMRT is mediated by multiple domains it was not possible to create a nonexportable mutant of SMRT that could be tested for its ability to antagonize PGC-1α-mediated effects in synaptically active neurons. As such, the importance of SMRT export can be predicted but is difficult to test experimentally.
To test whether PGC-1α phosphorylation could counteract the suppressive effects of SMRT, we expressed a mutant form of PGC-1α (PGC-1α3D) in which the three p38 phosphorylation sites have been mutated from serine/threonine to aspartate to mimic the p38-phosphorylated form of PGC-1α (8). The effect of this mutation is to render PGC-1α resistant to p160MBP-mediated repression and thus enhance its basal activity (8). We found that expression of PGC-1α3D in neurons offered protection even when coexpressed with SMRT (Fig. 6B). The fact that SMRT could not completely suppress the effects of PGC-1α3D indicates that phosphorylation on these residues may be a contributing factor in regulating the antagonistic balance between SMRT and PGC-1α with regard to controlling antioxidant defenses.
Discussion
We have shown here that the transcriptional coactivator PGC-1α is positively regulated by neuronal activity
PGC-1α directs neuroprotective gene expression and its under-expression may contribute to neurodegenerative diseases
As a transcriptional coactivator, PGC-1α controls the activity of many transcription factors and acts as a master regulator of several important physiological processes in neurons. PGC-1α-deficient mice exhibit lowered expression of certain antioxidant genes in the brain, including Sod1, Sod2, and catalase (45). Further, they show increased loss of dopaminergic neurons in response to treatment with MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, which is metabolized to the mitochondrial toxin MPP+ in dopaminergic neurons). PGC-1α-deficient mice also show heightened levels of hippocampal neuronal loss in a kainate seizure model of excitotoxic cell death (45) and suffer from spontaneous striatal lesions (22). Conversely, overexpression of PGC-1α in neuroblastoma cells and also striatal progenitor cells protects against oxidative insults (45). PGC-1α also promotes protection in models of ischemia (5, 25). Overexpression of PGC-1α boosts mitochondrial density in cortical neurons, leading to increased ATP levels (47). Thus, as a regulator of mitochondrial function and antioxidant defenses, PGC-1α is neuroprotective against a variety of insults.
It is possible that different (potentially overlapping) subsets of PGC-1α regulated genes are responsible for protection against different types of insult. For example, protection against a purely oxidative insult may rely heavily on the upregulation of antioxidant genes, whereas PGC-1α-mediated resistance to excitotoxic insults may rely more on enhanced mitochondrial function and energy production. Indeed, the fact that SMRT can antagonize the antioxidative effects of PGC-1α expression but not the antiexcitotoxic effects indicates that different genes may be responsible for these two types of neuroprotection.
Not only is PGC-1α neuroprotective against oxidative and excitotoxic insults, but there is evidence that perturbation in PGC-1α expression is a contributor to neurodegenerative disease, particularly HD. mtHtt interferes with CREB-dependent PGC-1α expression, leading to mitochondrial dysfunction and metabolic defects (6, 49). PGC-1α expression is lower in the striata of human HD patients, but not in the cerebellum or hippocampus, consistent with the brain region-specific effects of HD. As a further exacerbating factor, the striatum appears to particularly vulnerable to loss of PGC-1α expression above other brain regions (22). Overexpression of PGC-1α protects both cortical and striatal neurons from mtHtt- and 3-NPA-induced cell death, whereas lentiviral transduction of PGC-1α reduces striatal loss in a mouse model of the disease (6, 31, 47, 49). As well as being implicated in HD, a recent study hints at involvement in Alzheimer's disease (39) where PGC-1α is reported to promote nonamyloidogenic Amyloid Precursor Protein processing. Further, PGC-1α expression is lowered in human Alzheimer's disease patients as a function of dementia (39).
Dynamic control of PGC-1α by neuronal activity
Expression of PGC-1α in the brain is controlled by CREB (6, 31, 45) and boosted by synaptic NMDAR activity [this study and ref. (31)]. In contrast, activation of extrasynaptic NMDAR activity, which inhibits CREB activity (16) can suppress PGC-1α expression in mtHtt-expressing neurons (31). The activity-dependent nature of the PGC-1α gene suggests that PGC-1α levels will mirror the activity history of the neuron and could represent a way by which the elevated metabolic and antioxidative demands placed on a neuron by high levels of electrical activity could be met. Moreover, our study indicates that PGC-1α activity can be regulated by post-transcriptional mechanisms. Studies in myoblasts revealed that PGC-1α contains a negative regulatory domain that recruits p160MBP, resulting in lowered transactivation function (8). This association is disrupted by p38-mediated phosphorylation (8) representing a potential point of rapid post-translational control. We have shown here that p38 activity also boosts PGC-1α activity in neurons and that a mutant nonphosphorylatable form of PGC-1α inhibits p38-dependent enhancement of gene expression mediated by the TR, a nuclear receptor coactivated PGC-1α. Moreover, the antioxidative effects of a phospho-mimetic mutant of PGC-1α is resistant to suppression by SMRT, consistent with its higher transactivating potential (although an alternative explanation is that phosphorylation renders PGC-1α refractory to SMRT repression). As discussed below, the physiological effects of PGC-1α are also subject to indirect activity-dependent regulation
However, that p38-mediated phosphorylation of overexpressed PGC-1α is not essential for protection by insults in the absence of SMRT coexpression. Overexpression of a mutant nonphosphorylatable form of PGC-1α confers resistance to both oxidative and excitotoxic insults (data not shown). Thus, any requirement for PGC-1α phosphorylation may be in cases where SMRT expression is significant, or PGC-1α levels are low.
Antagonism of PGC-1α's antioxidative effects by SMRT is inhibited by synaptic activity
It remains unclear exactly which transcription factors are responsible for PGC-1α-mediated protection against oxidative insults, and it is likely that several are involved in coordinating a general elevation in ROS-metabolizing capacity. The fact that SMRT is able to antagonize the antioxidative effects of PGC-1α suggests that key transcription factor mediators include those that are targets for both these coregulators. Such factors include (but are not restricted to) PPARγ, ER, TR, RAR, and MEF2s (19, 35, 46, 50). All the aforementioned transcription factors have been reported to promote neuroprotective gene expression and/or resistance to oxidative insults (3, 9, 30, 34, 40).
One can envisage a scenario whereby the balance of PGC-1α coactivator
The finding that neuronal activity renders SMRT unable to antagonize the antioxidative effects of PGC-1α could conceivably be due to either activity-dependent SMRT export or activity-dependent PGC-1α activation. The fact that the phospho-mimetic PGC-1α3D is refractory to SMRT-repression suggests that phosphorylation is sufficient in the context of overexpression at least. Although this may be sufficient in cases of strong activity or high PGC-1α expression, SMRT export may also contribute in cases of modest levels of activity or low PGC-1α expression. Neuronal activity, acting partly
Analysis of the regions of SMRT responsible for nuclear localization and activity-dependent nuclear export in this study have revealed that multiple domains are likely to be responsible in both cases. Nuclear localization of SMRT is disrupted in a mutant lacking the RD3 domain, which is the region of SMRT that interacts with Class IIa HDACs, which include HDAC4 and 5 (19). It is not currently clear whether this region contains a bone fide nuclear localization signal (NLS) or whether SMRT is localized to the nucleus through association with nuclear factors such as Class IIa HDACs. Of note, class IIa HDACs also undergo activity-dependent export that occurs in advance of SMRT export (28) and so while SMRT is clearly not being dragged out of the nucleus by class IIa HDACs, their export may leave SMRT more sensitive to export (28). Analysis of the effect of nonexportable mutants of HDAC4 and HDAC5 on SMRT export (underway in the laboratory) will elucidate the interdependency of SMRT and Class IIa HDAC export by synaptic activity. The direct investigation of activity-dependent redistribution of multiple deletion and truncation mutants of SMRT in this study indicates that more than one domain must be responsible, since nonoverlapping N- and C-terminal regions of SMRT exhibit activity-dependent export (Fig. 5). Once again, we cannot currently distinguish between autonomous export and “piggy back export”
Concluding Remarks
It is clear that the influence of activity-dependent Ca2+ signaling extends beyond the direct regulation of Ca2+-responsive transcription factors. By influencing the function of broad specificity coactivators and corepressors, activity-dependent signals have the capacity to influence broad programs of gene expression, including genes controlled by transcription factors that are not classically Ca2+-responsive. In demonstrating that neuronal activity can regulate the ability of PGC-1α to protect neurons against oxidative insults, we have increased our understanding of how endogenous signals control the product of this important disease-associated gene.
Footnotes
Acknowledgments
The authors thank Martin Privalsky, Bruce Spiegelman, Marc Montminy, Pere Puigserver, and Akiyoshi Fukamizu for plasmids. The authors thank Chrysanthy Ikonomidou for samples taken from MK-801-treated mice. This work was funded by the Biotechnology and Biological Sciences Research Council, the Wellcome Trust, The Royal Society (to G.E.H.) the Medical Research Council, the EMBO Young Investigator Programme (to G.E.H.), Canadian Institute of Health Research (to K.F.S.B.), and The European Network of Neuroscience Institutes.
Author Disclosure Statement
No competing financial interests exist.