
Abstract
Endothelial injury and dysfunction are critical events in the pathogenesis of cardiovascular disease. During these processes, an impaired balance of nitric oxide bioavailability and oxidative stress is mechanistically involved. Circulating angiogenic cells (including early and late outgrowth endothelial progenitor cells (EPC)) contribute to formation of new blood vessels, neovascularization, and homeostasis of the vasculature, and are highly sensitive for misbalance between NO and oxidative stress. We here review the role of the endothelial nitric oxide synthase and oxidative stress producing enzyme systems in EPC during cardiovascular disease. We also focus on the underlying molecular mechanisms and potential emerging drug- and gene-based therapeutic strategies to improve EPC function in cardiovascular diseased patients. Antioxid. Redox Signal. 15, 933–948.
Circulating Angiogenic Cell Characterization and Clinical Relevance
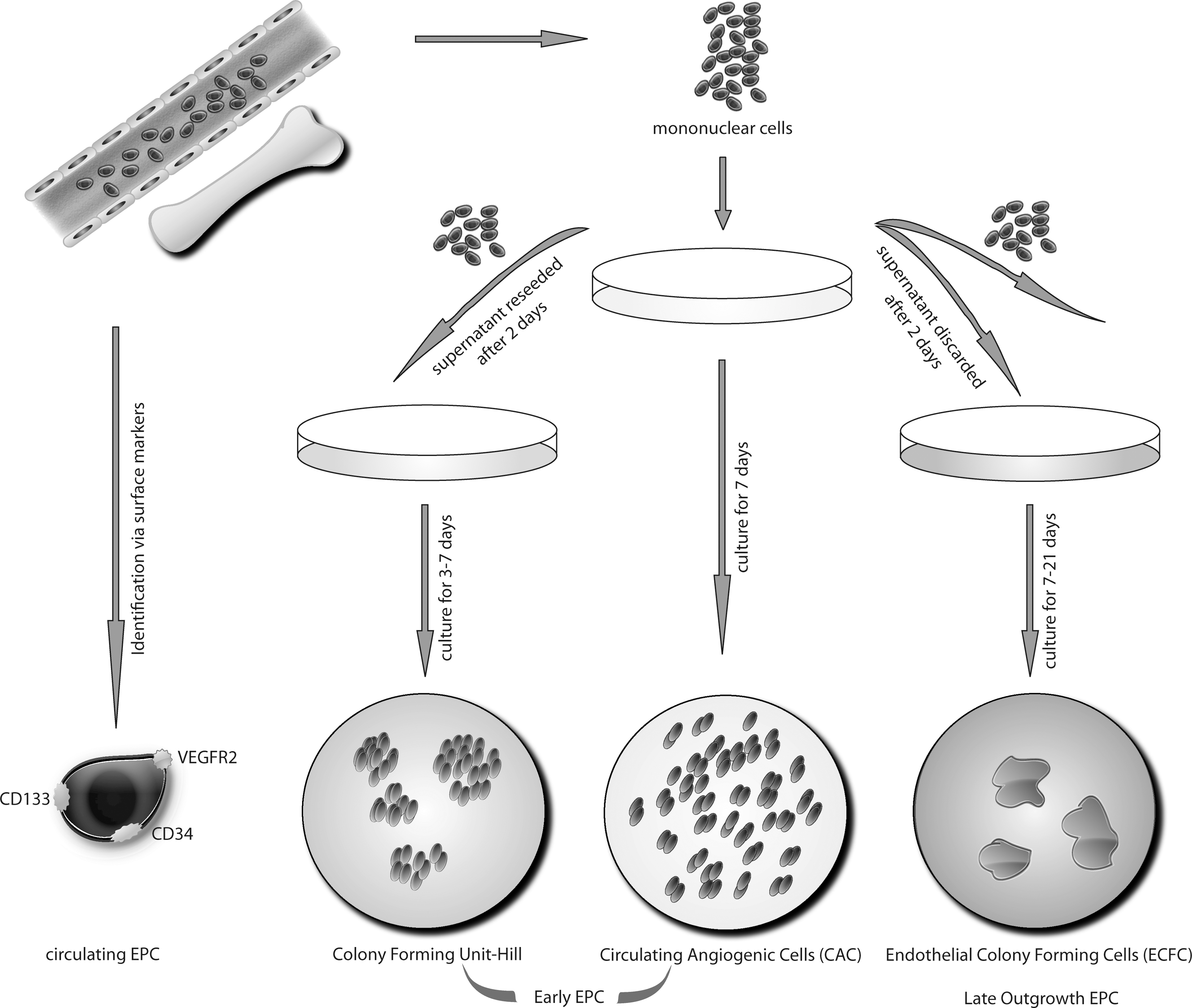
Circulating Angiogenic Cells
First described by Asahara et al., there are blood-derived cells that express markers of the endothelial lineage such as CD31, VE-cadherin, KDR (VEGFR-2), and the von Willebrand factor (vWF) (6). These cells also show putative endothelial behaviour such as migration towards pro-angiogenic factors such as stromal cell-derived factor-1 (SDF-1), vascular endothelial growth factor (VEGF), and such cells take up acetylated low density lipoproteins (LDL) as do mature endothelial cells (5). These early EPC are also referred to as circulating angiogenic cells (CAC) (40), monocytic EPC, early outgrowth EPC or angiogenic progenitor cells (APC). Such cells are usually obtained by an adhesion-related isolation method by plating peripheral blood (or bone marrow) mononuclear cells to fibronectin-coated dishes for several days (see Fig. 1). It should be noted that the endothelial characteristics of such “early EPC” may also be acquired due to uptake of platelet microparticles (69).
Endothelial Colony Forming Units
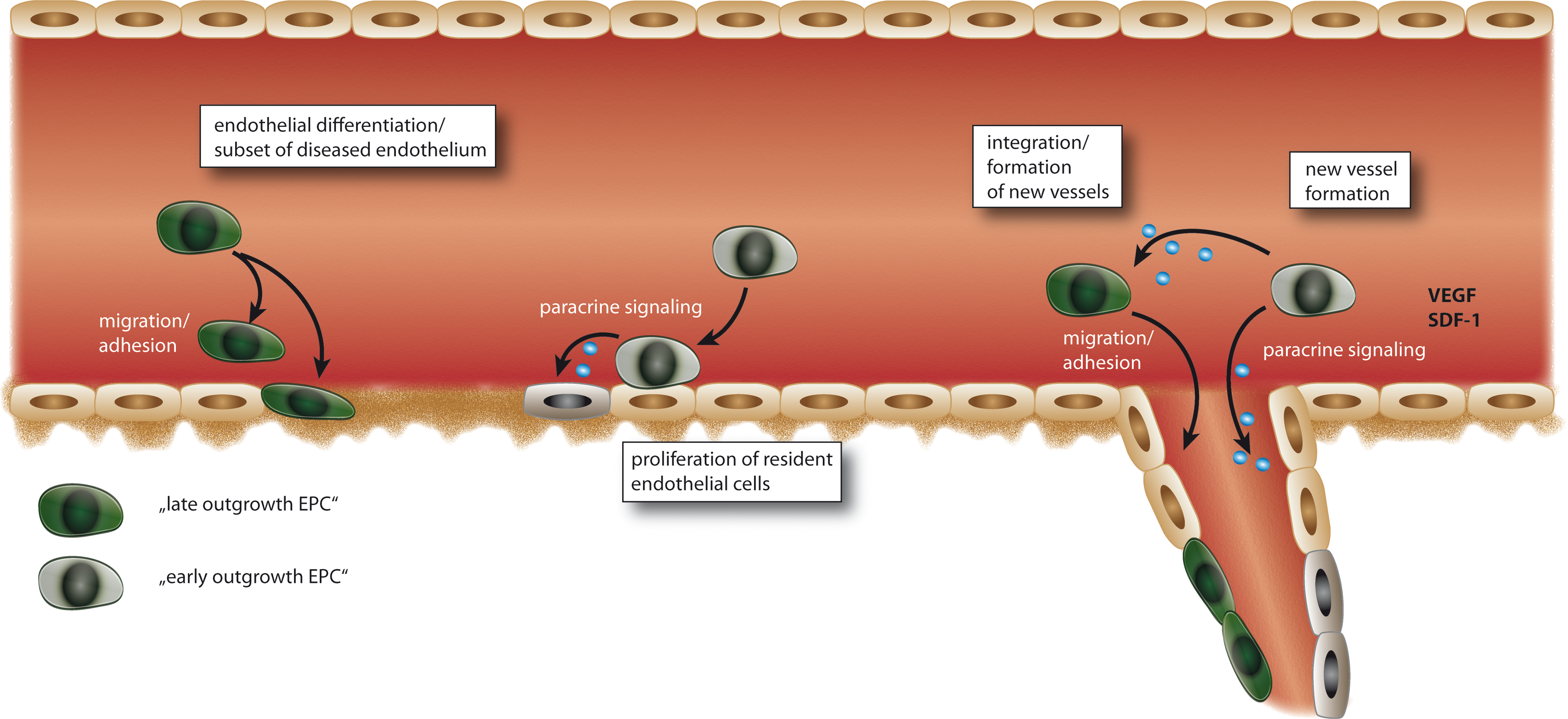
In contrast to the protocols gaining the above mentioned circulating angiogenic cells, Hill et al. used a method where after 48 h nonadherent cells are seeded onto fibronectin-coated dishes in a special differentiation medium (39). These cells form colony forming units (CFU) over time that stain positive for several endothelial markers such as eNOS, VEGFR and others, but also express markers of hematopoietic lineage such as CD45 (Fig. 1). These cells are different from circulating angiogenic cells. However, there are also common features; there is experimental evidence that both circulating angiogenic cells and CFU forming cells are important for vasculature homeostasis by either direct or indirect mechanisms. Hill et al. and Werner et al. could detect a significant correlation between CFU numbers and risk for cardiovascular events and coronary artery disease (39, 124). However, the exact mechanism of the contribution of early EPCs to upkeep vascular integrity remains uncertain. Early EPCs secrete various pro-angiogenic factors, such as VEGF, stromal SDF-1, and NO (105, 117), thus contributing to angiogenesis and vascular repair by paracrine mechanisms (Fig. 2). There is also a continuous shift in expression of stem and endothelial surface markers during differentiation and/or transdifferentiation of EPC. Some characteristics used for phenotyping EPC such as uptake of acetylated LDL or binding to specific lectins are also observed in non-endothelial cells such as monocytes or macrophages (71). Whether such hematopoetic cells also contribute to the formation of new vessels remains to be determined. There are also circulating EPC subtypes that can be directly identified in the blood by specific markers such as CD34, CD133, and VEGFR-2 (107) (Fig. 1).
Late Outgrowth EPC
In contrast to circulating angiogenic cells or endothelial CFU, late outgrowth EPC are highly proliferative, and feature the ability to directly form tubes and intact vascular vessels (Figs. 1 and 2). At later stages, these cells do not express markers of the hematopoietic lineage such as CD45 or CD14. They represent a fraction of cells expressing markers of endothelial cells such as VEGF, VEGFR-1, VEGFR-2, and eNOS (Table 1). Some of these markers are identically expressed in early EPCs; however, early EPC tend to loose these characteristics during culture (45). Late outgrowth EPC represent cells closer to mature endothelial cells; whether such cells can be produced only under artificial in vitro conditions or whether they also have importance for in vivo endothelial homeostasis is unknown. Interestingly, transplantation of more differentiated or mature endothelial cells into ischemic regions result in lower proangiogenic effects than transplantation of early EPC.
Importance of eNOS-Derived NO and Oxidative Stress for EPC Function
eNOS-derived NO is a major regulator of both EPC mobilization and function. Here we summarize preclinical as well as clinical findings and discuss the importance of eNOS and oxidative stress for EPC function in various cardiovascular diseases.
Functional Significance of eNOS for EPC
A common feature of the characterization of various EPC subtypes is eNOS expression. Indeed, most groups have identified eNOS mRNA or protein expression as well as eNOS activity in certain types of EPC (Table 1). CD133-CD14+ cells isolated from human umbilical cord blood showed a strong increase in expression of endothelial markers, including eNOS, whereas CD14 expression decreased during culture (52). Late outgrowth EPC that display a cobblestone-like morphology comparable to that of mature endothelium seem to express eNOS at even higher levels (33). The underlying regulatory events remain unclear, but transcriptional regulation of eNOS in various EPC types and stages of differentiation might be different (see also Section Regulation of eNOS in EPC). Vascular progenitor cells derived from mouse or primate embryonic stem cells also show an increase in eNOS expression over time both at the gene and protein level (84, 132). Although eNOS expression seems to be a major feature of differentiating EPC, most of the results were observed with EPC cultured in vitro under conditions that favor endothelial differentiation. The potential of progenitors in vivo to express eNOS under certain conditions, for example, during homing of EPC to sites of ischemia and subsequent initiation of neovascularization, is less clear and so far little information is available. EPC from eNOS-KO mice have a tremendous reduction in functional capacity in vivo (2), whereas treatment of progenitors with the eNOS enhancer AVE9488 improves neovascularization after transplantation to ischemic tissues (28, 74). This indicates eNOS is also expressed in EPC in vivo with important functional consequences. eNOS-mediated NO improved migratory capacity of EPC by direct effects on vasodilator-stimulated phosphoprotein (VASP) and cytoskeletal changes (82; Fig. 3).
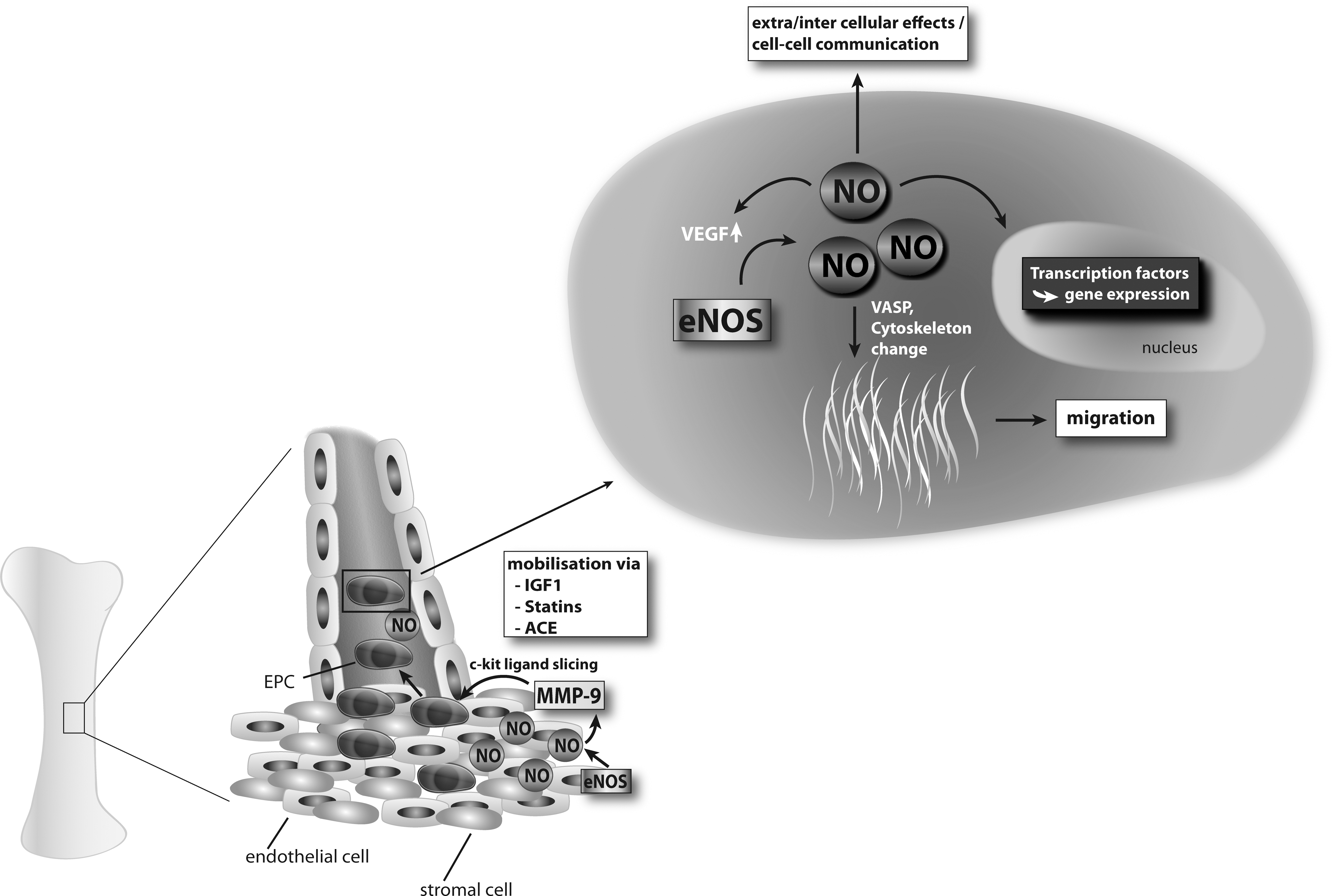
Regulation of eNOS in EPC
There is strong evidence that EPC-expressed eNOS is regulated under various physiological and pathophysiological conditions. Compounds or molecules that increase eNOS expression improve EPC function, whereas eNOS inhibitory substances have deleterious effects (99, 111). It was shown that growth hormone (GH) and insulin-like growth factor 1 (IGF-1) mediated increase of eNOS expression and activity in endothelial cells and EPC led to an increase in migratory potential in vitro (24, 108, 112). In contrast, functional knockdown of eNOS in EPC blocked cellular migration (107). Treatment of EPC with insulin-like growth factor binding protein-3 (IGFBP3) was able to enhance NO bioavailability and subsequent EPC function, leading to enhanced vascular repair (51). These effects on eNOS were phosphatidylinositol 3-kinase/Akt pathway dependent (51). During insulin resistance, the downregulation of the PI3 kinase/Akt pathway in skeletal muscle and endothelium leads to impaired EPC mobilization and differentiation. In Akt-1 knockout mice, EPC mobilization in response to ischemia and VEGF is impaired (1). The positive effects of statins, IGF-1, and erythropoietin on eNOS expression can be abolished by inhibition of the Akt pathway (21, 99, 107). In contrast, hyperoxia leads to decreased growth, reduced VEGFR-2 and eNOS expression, and impaired NO production of CFU-EPCs of preterm infants.
Treatment of EPC with the endogenous NOS inhibitor asymmetric dimethylarginine (ADMA) impaired CFU formation, incorporation of EPC into functional networks, as well as differentiation of EPC (109). Tube formation of cultured late-outgrowth EPC is likewise partly dependent on eNOS function (33). OxLDL treatment reduces eNOS expression both in mature endothelium (102) and EPC, which resulted in decreased EPC survival and impaired adhesive properties (61). Likewise, LDL lipid apheresis increased peripheral EPC function in patients with refractory hyperlipidemia (66). Physical activity upregulates eNOS expression in EPC isolated from the spleen and bone marrow of mice (57). Following experimental brain ischemia, number of EPC and neovascularization of ischemic tissue was enhanced in animals subjected to physical activity (30). The protective effect of running on neoangiogenesis and general outcome was completely abolished when animals were treated with a NOS inhibitor or in animals lacking eNOS expression (30). In contrast, overexpression of eNOS in EPC improves cellular function. Transplantation of eNOS transduced early EPC restored pulmonary hemodynamics and increased microvascular perfusion in a rat monocrotaline model of pulmonary arterial hypertension significantly more as when compared to transplantation of non-eNOS transduced EPC (137). Transplantation of eNOS engineered EPC in a rabbit balloon angioplasty model resulted in significantly reduced neointima formation when compared with transplanted control GFP-transduced EPC (54). Thus, transplantation of autologous EPC overexpressing eNOS to injured vessels enhances the vasculoprotective properties of the reconstituted endothelium. Direct infusion of wild-type progenitor cells, but not of eNOS-KO cells, was able to reverse the defective neovascularization in eNOS-KO mice, indicating that eNOS is required at the site of vessel formation (2). Besides overexpression of eNOS, carbon monoxide (CO) enhanced EPC proliferation. This effect is mediated via phosphorylation of Akt, eNOS phosphorylation, and an increase in NO generation by endothelial cells, leading to an increase in Tie2-positive EPCs (121). In addition, overexpression of SDF-1alpha led to upregulation of eNOS activity and in turn enhanced vasculogenesis, whereas NOS inhibition prevented SDF-1α-mediated effects (38).
Importance of Oxidative Stress to EPC Function
Reactive oxygen species (ROS) play a pivotal role in the homeostasis of the vasculature. In general, low levels of ROS may activate preventive pathways in EPC and even can function in a pro-angiogenic manner, whereas higher ROS levels significantly impair EPC function. Accordingly, in many diseases, which result in increased ROS levels such as diabetes type I or type II, EPC numbers and function are impaired (Fig. 4). Incubation of EPC with hydrogen peroxide significantly reduces the number of EPC by induction of apoptosis (118). Gluthation deficiency in mice leads to diminished EPC mobilization in response to ischemia or VEGF and impaired function of EPC (29). In patients with diabetes, an induction in NADPH oxidase activity and subsequent increase in ROS levels leads to impaired EPC number and function (88, 104). This is also observed in insulin resistance settings (22). Heme oxygenase-1 (HO-1), a heme degradation enzyme with multiple vasoprotective functions, is induced by oxidative stress (9). After vascular injury, HO-1 enhances mobilization of circulating EPC via increased levels of VEGF and SDF-1 (59). In line, Wu et al. identified HO-1 as a potent stimulus for increasing circulating EPC numbers; however the underlining mechanisms remain unclear (129). An increase in advanced glycylation endproducts (AGEs) or oxidized LDLs can impair endothelial and EPC function by increasing ROS levels (15, 101, 102). However, this effect could be blocked by siRNA against the receptor for advanced glycation endproducts (RAGE) or blockade of the oxLDL receptor LOX-1 (94, 101). RAGE is also regulated by C-reactive protein (CRP), which is considered as a proatherosclerotic protein, resulting in impaired EPC function (15). Accordingly, AGE treatment leads to a downregulation of eNOS and Bcl-2 expression, as well as an elevation in cyclooxygenase-2, Bax, NF-kappaB, and caspase-3 in a MAPK (ERK/P38/JNK)-dependant manner (83). Exposure to high glucose levels results in an upregulation of p66ShcA, a gene that regulates the apoptotic responses to oxidative stress (20). Accordingly, p66ShcA knockout mice display decreased ROS production and increased resistance to ROS-induced cell death. Whereas high glucose leads to increased p66ShcA protein expression, a decrease leads to reduction of bone marrow-derived EPCs (20). A reduction in ROS levels improves EPC function. For example, Nox2-containing NADPH oxidase deficiency improves neovascularization via enhanced activation of the VEGF/NO angiogenic pathway with subsequent improved function of EPC (34). ”Uncoupling” of eNOS itself can also increase ROS levels (Figs. 4 and 5). This occurs usually when associated with decreased tetrahydrobiopterin (BH4) levels, for example in diabetes (80, 105; Fig. 5). Targeting BH4 therefore might be a valuable therapeutic strategy for the future to prevent eNOS uncoupling in the vasculature and maybe also EPC dysfunction.
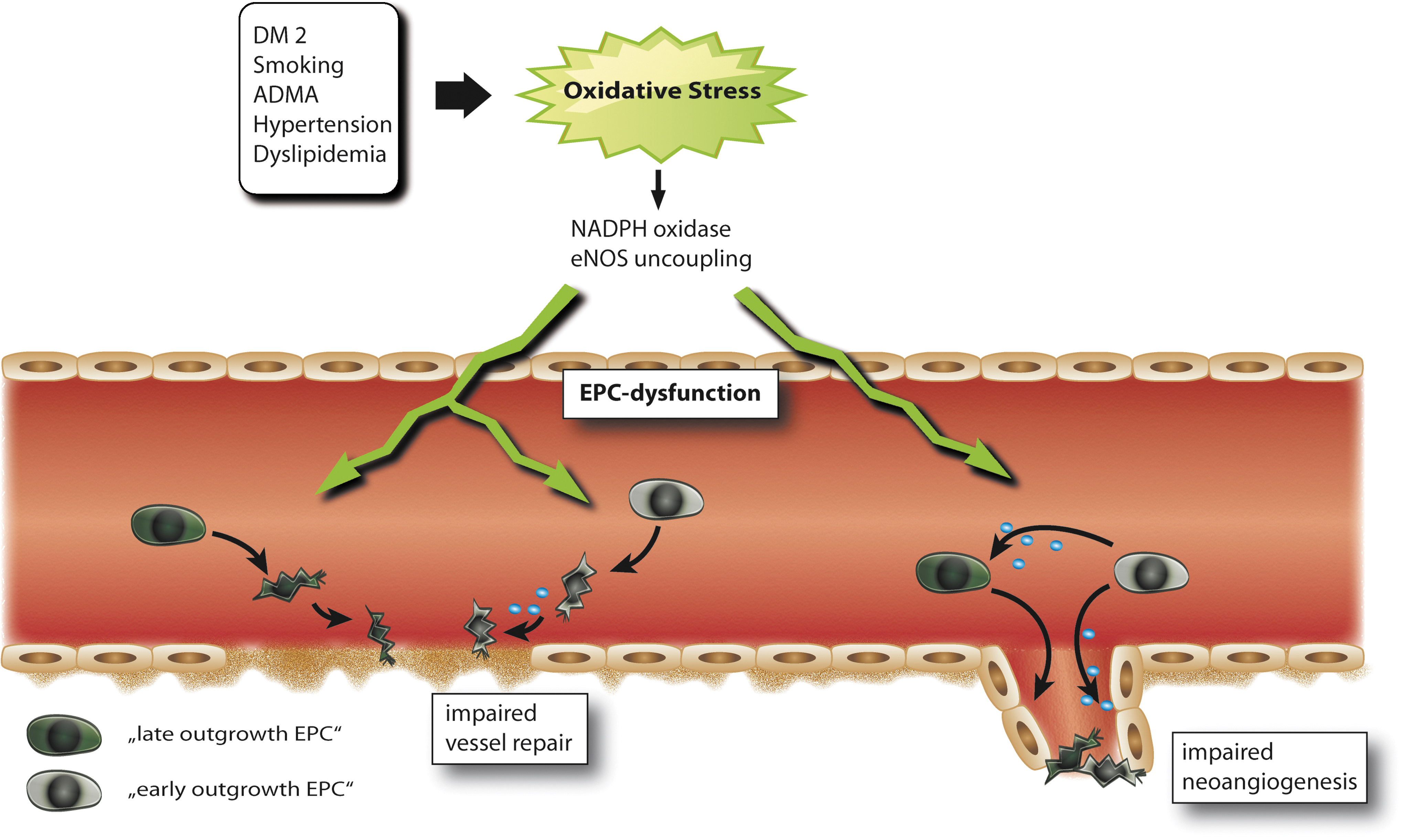
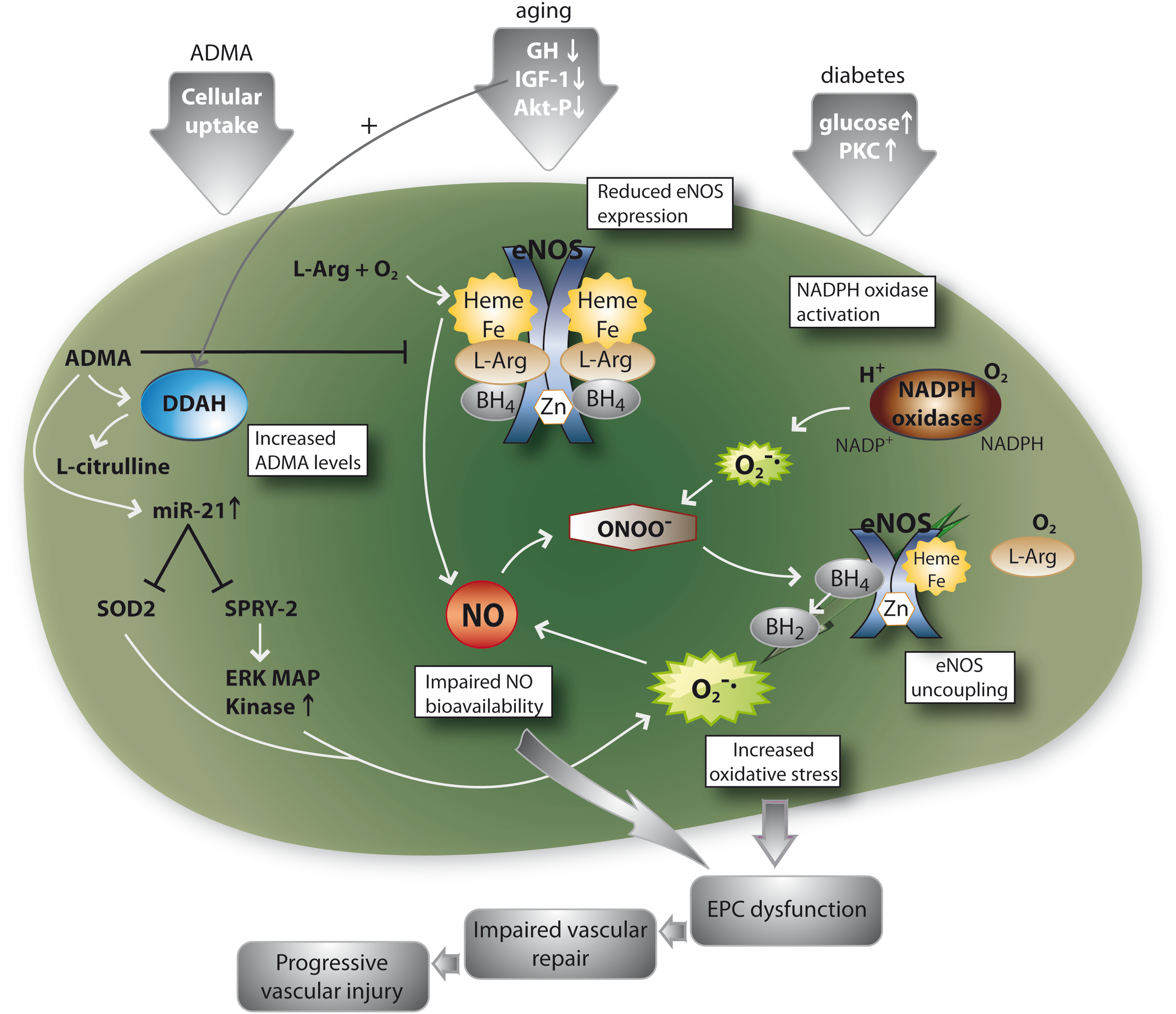
Importance of NO and ROS in EPC from Patients with Cardiovascular Disease
There is ample evidence that both NO and oxidative stress, and particularly the balance between both, regulate number and function of EPC by direct and indirect mechanisms. We here highlight the role of eNOS-mediated NO formation and oxidative stress in some exemplifying diseases.
Diabetes
Diabetic patients often suffer from severe and progressive cardiovascular complications where limited number and function of circulating EPC play an underlying role (60, 95). In the diabetic vascular system, eNOS uncoupling may occur with the consequence that eNOS switches its NO production to release of ROS which in turn can react with NO to the vascular toxicant peroxynitrite (26, 27). It was recently shown that EPC from diabetic patients or in vitro after glucose challenge also undergo eNOS uncoupling, leading to increased intracellular reactive oxygen and nitrogen species, which in turn hamper EPC function (105; Fig. 5). Potentially of therapeutic interest, an underlying molecular cause is a markedly reduced concentration of tetrahydrobiopterin (BH4), a molecule that stabilizes the eNOS dimer and prevents eNOS uncoupling (27; Fig. 5). Treatment of diabetic EPC with BH4 led to a coupling of former uncoupled eNOS, normalized intracellular ROS levels, and improved EPC function (105). Accordingly, EPCs from diabetic patients generate more ROS, at least in part due to eNOS uncoupling and increased NADPH oxidase activity, leading to a reduction in NO bioavailability (88, 105). Treatment of glucose-stressed EPCs with superoxide dismutase in vitro attenuated superoxide anion (O2 -) generation, restored NO production, and partially normalized their ability to form colonies (35). Treatment of diabetic mice with the HO-1 inducer and thus reducer of oxidative stress cobalt protoporphyrin could in part restore EPC function (72).
Patients with diabetes lack the usual increase of circulating EPCs occurring after myocardial infarction in nondiabetic patients (166). High-density lipoprotein (HDL) from patients with metabolic syndrome does not have a similar protective effect on EPCs as HDL from healthy volunteers (87). Indeed, HDL from healthy subjects stimulated endothelial NO production, reduced endothelial oxidant stress, and improved endothelium-dependent vasodilatation and early EPC-mediated endothelial repair. In contrast, these beneficial endothelial effects of HDL were not observed in HDL from diabetic patients, suggesting markedly impaired endothelial-protective properties of HDL in diabetes, maybe due to oxidative modifications of the HDL molecule. Interestingly, niacin therapy improved the capacity of HDL to stimulate endothelial NO, reduced superoxide production, and promoted EPC-mediated endothelial repair (87). Transplantation of bone marrow-derived progenitor cells from healthy into diabetic mice enhanced insulin sensitivity, improved acetylcholine-dependant endothelial relaxation, and partially restored endothelial function (14). Oxidized LDL compounds lead to eNOS downregulation in mature endothelium to induce endothelial dysfunction. In a parallel manner it was demonstrated that oxLDL treatment also decreases both eNOS expression and function in EPC by induction of oxidative stress (61).
Aging, senescence, and exercise
During aging, there is an increasing misbalance between reduced NO production and increased oxidative stress. Indeed, eNOS expression in the vascular system is reduced with age that regularly results in the development of endothelial dysfunction (93, 133). Therapeutically interesting, treatment with eNOS inducers such as AVE9488 (eNOS transcription enhancer; see above), statins, ACE inhibitors, or IGF-1 may revert EPC dysfunction (28, 74, 106, 120; Figs. 5 and 6). During aging, expression and activity of glutathione peroxidase-1 (GPX1), a main effector enzyme to protect the organism from oxidative damage, decreases in EPCs, leading to impaired oxidative stress resistance (36).
Detraining in healthy volunteers with a history of moderate-to-high intensity exercise training lead to a markedly decrease in EPC (CD34+/VEGFR2+) numbers which was correlated with a decrease in hyperemic forearm blood flow (128). In contrast, even a short training treadmill exercise lead to increased eNOS mRNA expression in endothelial CFU and cardiovascular tissues and reduced NADPH oxidase expression, resulting in increased NO bioavailability (48, 122).
Hypertension
Prehypertension and established hypertension have been described to be associated with significantly reduced numbers of circulating EPCs (31). In addition, hypertensive rats with high levels of aldosterone showed impaired function and reduced numbers of EPC via downregulation of VEGF and the VEGF receptor (55). Moreover, aldosterone impairs EPC function in mice and patients with primary hyperaldosteronism by a protein kinase A- and mineralocorticoid receptor-dependent mechanism (96). Patients with high blood pressure show an increased number of circulating senescent EPCs concomitantly with a decreased plasma level of the calcitonin gene-related peptide (CGRP) and a decreased CGRP mRNA expression in EPCs (138). Increasing the concentration of CGRP by rutaecarpine (a CGRP stimulator) in plasma and its expression in EPC reversed EPC senescence induced by oxidative stress, inducing angiotensin II transfusion (138). The importance of CGRP is further underlined by the fact that transplantation with CGRP-overexpressing EPCs could attenuate pulmonary vascular remodeling and lower blood pressure in pulmonary hypertensive rats (135).
Inflammatory diseases
Inflammatory diseases are often paralleled by enhanced oxidative stress, including heart failure, rheumatoid arthritis, or preeclampsia. Cytokines such as Interleukin-1β, SDF-1, and GCSF have been shown to be involved in EPC recruitment, homing or mobilization from bone marrow (3,92). Microvascular endothelial cells challenged with inflammatory stimuli expressed the membrane-bound form of KitL and recruited EPCs via a c-Kit-mediated activation of the Akt signaling pathway to inflamed endothelium (18). However, chronic inflammatory processes or excessive inflammatory stimuli have deleterious effects, resulting in decreased EPCs in the circulation (4, 119, 126). Low levels of circulating CD133/KDR+ EPC also predicted occurrence of coronary atherosclerosis in rheumatoid arthritis (134).
Underlying Mechanisms of EPC dysfunction
The examples given above highlight the importance of a crucial balance between NO and ROS levels in the regulation of EPC function. NO influences, at least in part via Akt phosphorylation, a cascade of genes resulting in improved EPC function (2). NO-derived mobilization processes of EPC from bone marrow via inducing MMP-9, include cleavage of the sKit ligand to mobilize cKit+ EPCs into the circulation (10, 37; Figs. 3 and 5). In addition to NO, carbon monoxide (CO) also has important effects on endothelial function and EPC. Whereas it has stimulatory effects on endothelial cells, it has diametrically opposite effects on vascular smooth muscle cell proliferation (121). This results in possible beneficial effects of CO during balloon angioplasty-induced restenosis that regularly is associated with endothelial dysfunction and increased proliferation of smooth muscle cells. CO enhanced endothelial cell proliferation via activation of RhoA and subsequent phosphorylation of Akt, eNOS phosphorylation, and an increase in NO production. Moreover, CO yielded a 4-fold increase in the number of mobilized green fluorescent protein-Tie2-positive EPC versus controls, with a corresponding accelerated deposition of differentiated green fluorescent protein-Tie2-positive endothelial cells at the site of injury. In contrast, CO was ineffective in augmenting endothelial cell repair and the ensuing development of intimal hyperplasia in eNOS(-/-) mice (121).
Understanding mechanisms in diseases that drive EPC senescence and apoptosis may result in new treatment approaches of impaired angiogenesis. For instance, the apoptosis signal-regulating kinase-1 (ASK-1), which belongs to the MAPKKK family, whose members respond to various external stimuli and initiate the MAPK cascade, regulates senescence in EPC. Interestingly, ASK-1 expression is elevated by glucose treatment and in diabetic patients, leading to EPC dysfunction (50). ASK-1 also influences plasminogen activator-1 (PAI-1), leading to enhanced atherosclerosis and vascular dysfunction in diabetes. Diabetic ASK-1 knockout mice do not show increased levels of PAI-1 resulting in attenuated vascular dysfunction (65). Treatment with antioxidative N-acetylcysteine (NAC) suppressed TNF-α-induced ASK1 activation and apoptosis, suggesting that the generation of ROS is necessary for the TNF-α-induced ASK1 apoptosis (115). This points to ASK-1 as an interesting target to improve EPC function. As mentioned above, glutathione peroxidase type 1 (GPX-1) is a pivotal protein in protection of endothelial cells from oxidative stress. GPX-1 reduces both H2O2 and lipid peroxides to their corresponding alcohols using glutathione as cofactor (116). In response to ischemia, GPX-1 knockout mice do not show an increase in circulating EPC (26). In concordance, impaired angiogenesis in glutathione peroxidase-1-deficient mice is associated with EPC dysfunction (29).
The endogenous NOS inhibitor ADMA decreased both eNOS activity and cellular function of EPC (98 –100,109; Fig. 5). ADMA levels correlate with circulating EPC function and number, explaining in part the increased risk for cardiovascular events in patients with high ADMA levels. Goette et al. showed that during G-CSF treatment the increase in circulating EPC is accompanied by an increase in ADMA levels via upregulation of myeloperoxidase in leucocytes. However, the increase in ADMA and therefore decrease in NO bioavailability seems to diminish the potential beneficial effects of increased number of circulating EPC (32).
Drug-Mediated Treatment of EPC Dysfunction (Current and Future Aspects)
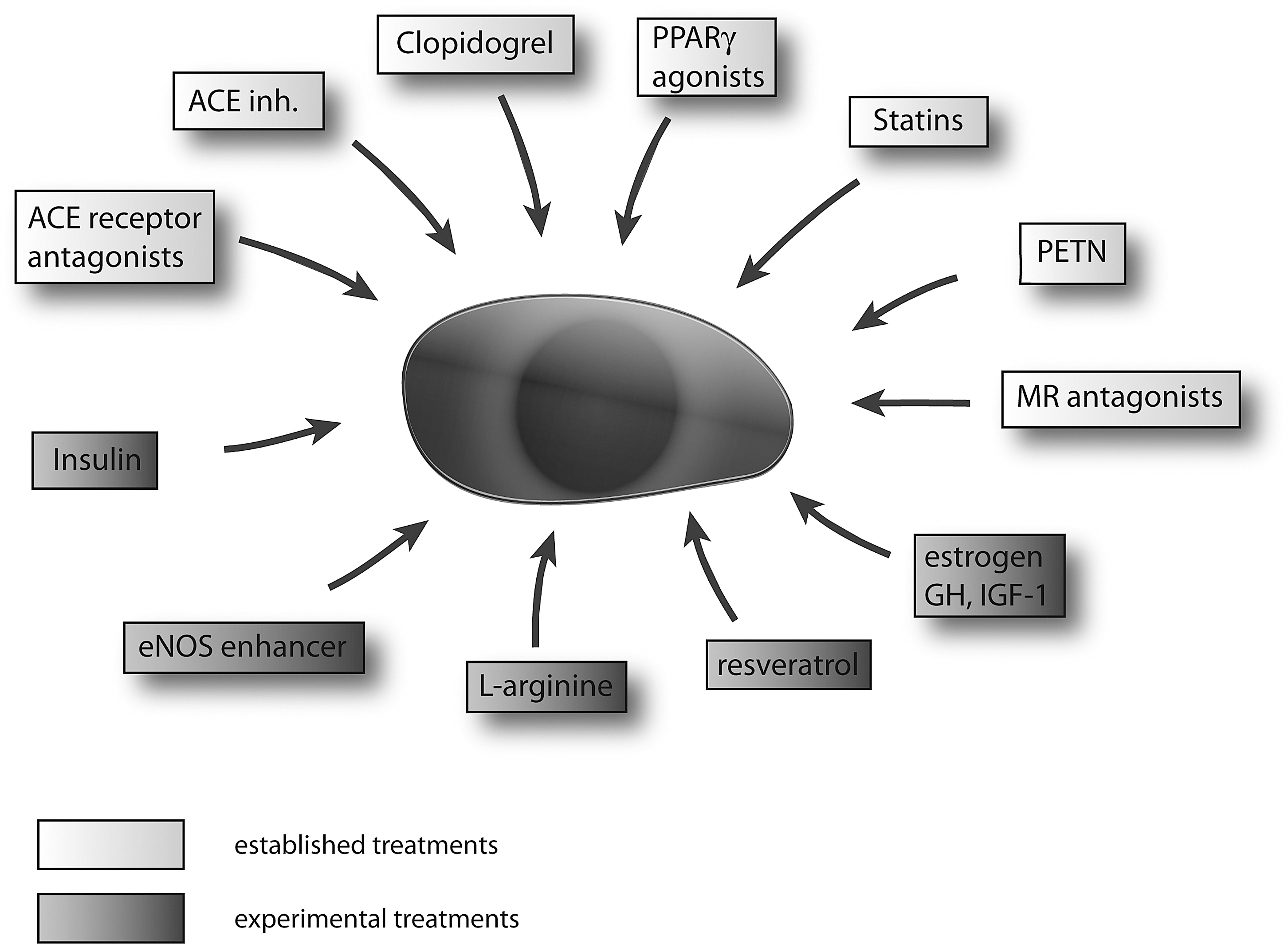
Impaired number and function of EPC occur in various forms of cardiovascular diseases and its correction by drugs, local or systemic EPC transplantation improves specific diseases in animal models or patients (90, 107, 120, 123; Fig. 6). In humans, low numbers of EPC are predictive for future cardiovascular events (77, 124) and as discussed above, impaired EPC function is tightly associated to cardiovascular disease. It is likely that as in animal models improvement of EPC function may also have cardiovascular protective effects in cardiovascular diseased patients.
Renin/angiotensin/aldosterone-mediating drugs
The renin–angiotensin pathway represents a major regulator of not only blood pressure but has also a role in the development of atherosclerosis, oxidative stress, inflammation, and endothelial dysfunction (78). It is therefore interesting that treatment of patients with stable angina with the angiotensin converting enzyme (ACE) inhibitor ramipril leads to increased EPC numbers and might play a role in the beneficial effects of ACE inhibitors on cardiovascular outcome, besides lowering blood pressure (63). Likewise, ACE inhibition increases EPC mobilization by increasing MMP-9 activity in bone marrow after myocardial infarction (104). In patients with type II diabetes, the angiotensin II type I receptor blocker olmesartan lead to an increase in EPC numbers independent of the blood pressure lowering action (7; Fig. 3). Treatment of EPCs with angiotensin II induced senescence and impaired differentiation via upregulation of gp91phox, the heme binding subunit of the superoxide-generating NADPH oxidase, whereas treatment with an AT-blocker rescued the function and differentiation of EPCs (42). Whether newly developed renin inhibitors such as aliskiren impact on EPC biology is currently under investigation. Aldosterone treatment decreased NADPH oxidase p22phox, p47phox, and gp91phox in EPC, whereas the aldosterone antagonist eplerenone attenuated aldosterone-mediated EPC dysfunction (53, 96; Fig. 6).
Statins
HMG-CoA reductase inhibitors (statins) were amongst the first drugs for which effects on EPC numbers and function were described (120, 125). Statin treatment significantly increased circulating EPCs. This effect was independent of its LDL reducing effects (56). Another study showed direct effects of statin treatment on NO formation directly by enhancing eNOS expression in the bone marrow, fostering EPC mobilization (104). Statins modulate EPC function at least in part via activation of the PI3-kinase/protein kinase Akt pathway, which leads to multiple effects on cell signaling, cell cycle, and VEGF and eNOS protein expression, resulting generally in improved EPC function and numbers (2, 56). As statins reduce LDL oxidation, it may also protect EPCs from toxic oxidized LDL that leads to increased cell senescence and EPC dysfunction (43). Recent findings also demonstrated that LDL reduction by lipid apheresis in patients with refractory hyperlipidaemia increases EPC function (66).
Acetylsalicylic acid (aspirin) impaired EPC function in vitro via downregulation of iNOS or possibly decreased Akt phosphorylation in EPCs (17). In contrast, clopidogrel enhances levels of endothelial NO and improved EPC function by increased EPC Akt phosphorylation in peripheral blood of patients with type 2 diabetes mellitus (62; Fig. 6).
Growth hormone, insulin-like growth factor-1, and estrogens
Administration of growth hormone increases NO bioavailability and enhanced circulating EPC numbers in healthy volunteers (103, 107). This increase in NO availability is mediated partly by an increase in IGF-I and subsequent Akt phosphorylation. Besides IGF-1, GH directly may also increase eNOS expression in the vascular system (19, 108, 112). The effect may be more pronounced in patients with growth hormone deficiency or in the elderly where GH treatment can successfully improve EPC number and function (107). Finally, estrogen supplementation also increases bone marrow-derived EPC production and diminishes neointima formation (46, 91) which may at least in part explain the cardiovascular protective properties of this hormone.
Resveratrol
Resveratrol is a polyphenol compound enriched in grapes and grape products and mediates its protective effects on vascular biology via enhancing NO bioavailability and reduction of oxidative stress (70). Red wine enriched with resveratrol leads to increased numbers of circulating EPC, improved NO bioavailability, and reduced levels of the eNOS inhibitor ADMA (41; Fig. 6). Resveratrol also attenuates tumor necrosis factor-alpha-induced EPC senescence and improves EPC function, including tube formation in vitro (8, 41). The increase of telomerase activity in EPCs might also contribute significantly to the reduced senescence of EPC by resveratrol treatment (131). Resveratrol is a known stimulator of sirtuin-1 (Sirt1), which in turn promotes angiogenesis (68). Indeed, SIRT1 is a critical modulator of EPCs dysfunction (8) and knockdown of Sirt1 by siRNA results in diminished EPC angiogenesis and increased senescence (136). Thus, activation of SIRT1 by resveratrol may contribute to the vasoprotective and angiogenic effects of this compound.
L-Arginine
L-Arginine consumption improved markers of NO bioavailability, as well as EPC numbers and potentiated the positive effect of moderate exercise training on EPC function and numbers in animal models (23, 47). However, in patients with peripheral arterial disease, long-term L-arginine supplementation did not increase nitric oxide synthesis or improved vascular reactivity (127). Thus, a contribution of L-arginine supplementation for vascular protection needs further investigations.
Antidiabetics and organic nitrates
Thiazolidinediones (TDZ) show pleiotropic effects that also included effects on EPCs. For instance, the peroxisome proliferator-activated receptors y (PPARgamma) agonist rosiglitazone increased EPC function via upregulating the Akt-eNOS signal pathways (58). Pioglitazone increased EPC adhesion to arteries from diabetic patients via an upregulation of CXCR4, the receptor for SDF-1. These effects are partly explainable due to pioglitazone-mediated decrease in oxidative stress (86). In addition, angiotensin II-induced oxidative stress and subsequent EPC dysfunction could be attenuated by treatment with TDZ, resulting in lowered peroxynitrite levels and increased telomerase activity (44). Insulin treatment increases reendothelialization and the number of circulating progenitor cells and inhibits cell migration and neointimal growth after arterial injury (12).
Since NO serves as an important factor for mobilization of EPC, use of organic nitrates, which are powerful NO donators, should enhance the number and function of circulating EPC. However, different organic nitrates have partly opposing effects on the endothelium and EPCs. For instance, many nitrates are known to induce endothelial dysfunction due to induction of oxidative stress (64, 79). An interesting exception seems to be the organic nitrate pentaerythritol tetranitrate (PETN) that does not induce endothelial dysfunction nor increase oxidative stress in animals or humans (97, 106).
Gene-Mediated Treatment of EPC Dysfunction
There have been attempts to increase eNOS expression in EPC by transient or permanent gene therapeutic approaches. Strategies to genetically overexpress eNOS in EPC have already been discussed in the section “Functional Significance of eNOS for EPC”. An eNOS enhancer compound AVE9488 was used to increase eNOS gene expression in EPC and indeed, transplantation of AVE9488-pretreated EPCs to ischemic tissues enhanced hindlimb reperfusion by improved angiogenesis (74). This treatment also enhanced circulating EPC levels in rats with heart failure after myocardial infarction (28). Transplantation of EPC where the glycogen synthase kinase–beta was genetically inhibited significantly improved efficacy of cell-based therapeutic vasculogenesis (16; Fig. 7). Transfection of the hepatocyte growth factor (HGF) gene enhances EPC function and improves EPC transplant efficiency for balloon-induced arterial injury in hypercholesterolemic rats (85). Interestingly, HGF also attenuates angiotensin II-induced EPC senescence (73).
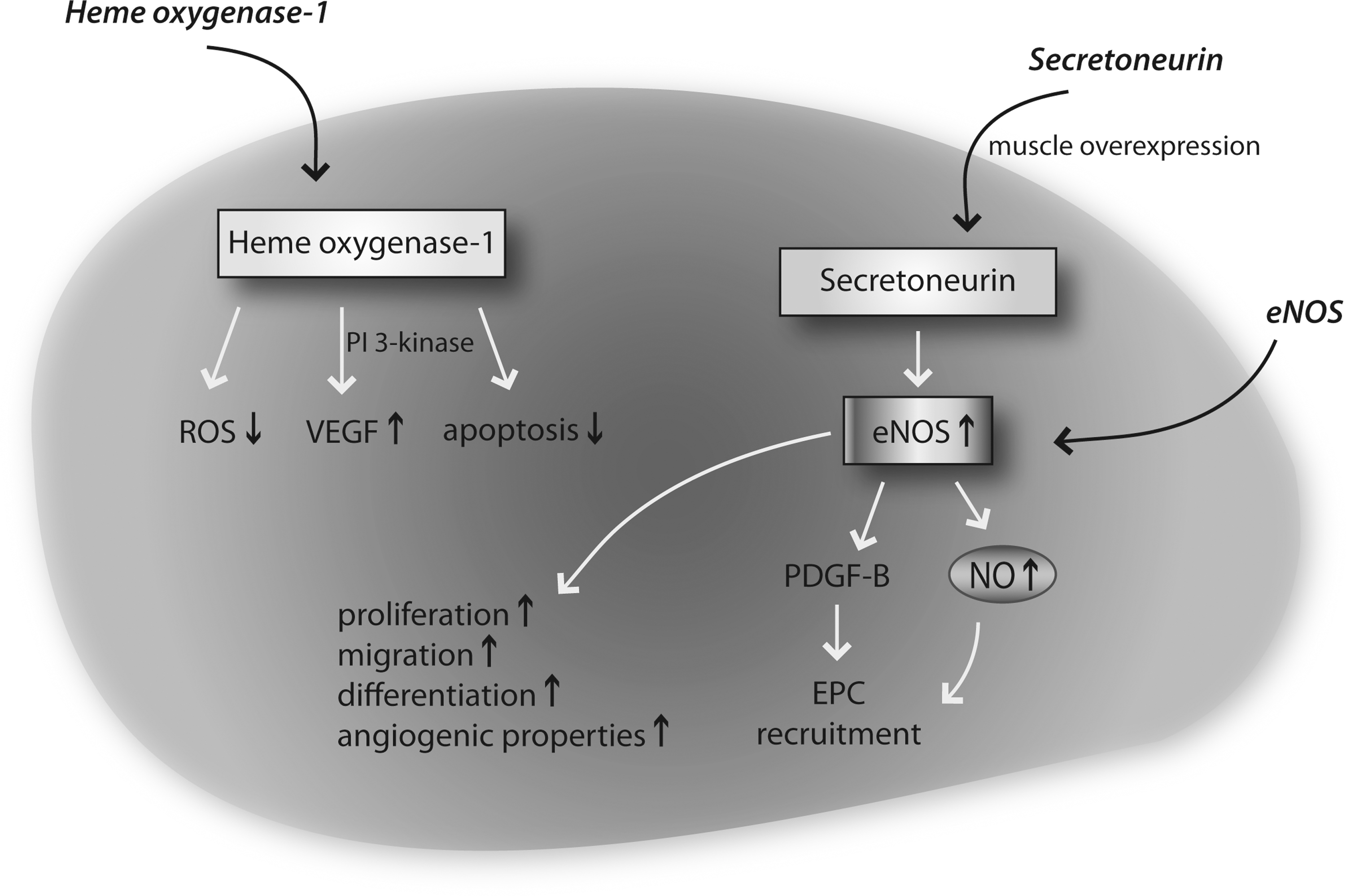
Gene therapeutic approaches to improve EPC function. Increase of heme oxygenase expression leads to reduced ROS levels, enhanced VEGF levels, and reduced apoptosis. HIF-1 and glycogen synthase kinase beta overexpression lead to increased VEGF levels. Secretoneurin overexpression leads to eNOS induction and subsequent improved NO bioavailability, resulting in better EPC recruitment, enhanced EPC function, proliferation, and differentiation. HIF-1; hypoxia-inducible-factor-1; PDGF, platelet derived growth factor; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor.
Another strategy to improve EPC function is modulation of the hypoxia-inducible-factor 1 (HIF-1) pathway. Transplantation of HIF-1alpha-overexpressing EPC into ischemic tissue of a hindlimb ischemia model resulted in significantly enhanced VEGF expression and improved neovascularization (49). In contrast, blockade of HIF-1alpha resulted in reduced expression of endothelial cell markers CD31, VEGF receptor 2 (Flk-1), and eNOS, as well as NO formation in EPC followed by impaired vascularization. Recruitment of EPC with subsequent induction of postnatal vasculogenesis was enhanced by secretoneurin gene therapy, which induced therapeutic angiogenesis, arteriogenesis, and vasculogenesis in the hindlimb ischemia model by a NO-dependent mechanism (75; Fig. 7). Future therapeutic aspects may also include microRNAs (miRNA), which consist of 22 nucleotides short, non-encoding regulatory RNA molecules. MiRNAs regulate about 50% of the genome either by direct inhibition and subsequent degradation of messenger RNAs or translational inhibition of protein expression. MiRNAs play a crucial role in the onset and progression of cardiovascular diseases (11, 13, 113, 114). Of importance, miRNAs emerged as promising candidates for novel and powerful gene therapeutic strategies (110). Recently, a first functional relevance of miRNAs for EPC biology was described; EPC dysfunction induced by the NO synthase inhibitor asymmetric dimethylarginine (ADMA) resulted in upregulation of miR-21 and subsequent reduced NO bioavailability (Fig. 7). This effect could be attenuated by miR-21 specific antagonism and improved EPC dysfunction in EPC from patients with coronary artery disease (25).
Conclusion and Outlook
A disturbed balance between NO formation and increased oxidative stress is observed in cardiovascular disease and aging processes. This also affects the number and function of endothelial-regenerating cells such as EPCs. Indeed, many common drugs in cardiovascular medicine improve the number and function of EPCs such as statins, ACE inhibitors, or certain organic nitrates, and thus may explain at least in part some of their favorable effects in the disease process. Understanding of the underlying mechanisms and normalization of the NO/ROS misbalance by new strategies will probably result in better therapeutic manipulation of impaired EPC function and thus improvement of cardiovascular disease. Whether such strategies may also have preventive cardiovascular effects remains to be determined.
Footnotes
Acknowledgment
We thank Yvonne Görzig for her help in preparing the figures of the manuscript. This work was supported by the IFB-Tx (BMBF, 01EO0802 to TT) and a grant of the Deutsche Forschungsgemeinschaft (TH903/7-2 to TT).