
Abstract
Increased production of reactive oxygen species (ROS) as a result of decreased activities of mitochondrial electron transport chain (ETC) complexes plays a role in the development of many inflammatory diseases, including atherosclerosis. Our previous studies established that paraoxonase 2 (PON2) possesses antiatherogenic properties and is associated with lower ROS levels. The aim of the present study was to determine the mechanism by which PON2 modulates ROS production. In this report, we demonstrate that PON2-def mice on the hyperlipidemic apolipoprotein E−/− background (PON2-def/apolipoprotein E−/−) develop exacerbated atherosclerotic lesions with enhanced mitochondrial oxidative stress. We show that PON2 protein is localized to the inner mitochondrial membrane, where it is found associated with respiratory complex III. Employing surface-plasmon-resonance, we demonstrate that PON2 binds with high affinity to coenzyme Q10, an important component of the ETC. Enhanced mitochondrial oxidative stress in PON2-def mice was accompanied by significantly reduced ETC complex I + III activities, oxygen consumption, and adenosine triphosphate levels in PON2-def mice. In contrast, overexpression of PON2 effectively protected mitochondria from antimycin- or oligomycin-mediated mitochondrial dysfunction. Our results illustrate that the antiatherogenic effects of PON2 are, in part, mediated by the role of PON2 in mitochondrial function. Antioxid. Redox Signal. 14, 341–351.
Introduction
PON2 is one member of the PON gene family that consists of three proteins (PON1, PON2, and PON3). PON2 is an intracellular membrane-associated protein that is widely expressed in vascular cells and many tissues. Results from various laboratories using both in vitro and in vivo models suggest that PON2 protects against oxidative stress (22, 26).
The primary physiological function of the mitochondrion is to generate adenosine triphosphate (ATP) through oxidative phosphorylation via the ETC. The ETC is one of the major sources of cellular ROS production. Respiratory chain dysfunction leads to increased production of superoxide, which in turn leads to aging and inflammatory diseases (14, 15).
In this report, we demonstrate that PON2-deficient mice on the hyperlipidemic apolipoprotein E (apoE)−/− background (PON2-def/apoE−/−) develop exacerbated atherosclerotic lesions with elevated lipoprotein oxidation and increased mitochondrial oxidative stress. We show that PON2 protein is localized to the inner mitochondrial membrane, where it is found associated with respiratory complex III. We further show that PON2 directly binds to coenzyme Q10, an essential constituent of the ETC, in a calcium-dependent manner. Our results suggest that PON2 deficiency leads to dysfunction of mitochondria together with an increase in mitochondrial oxidative stress, which may in part be responsible for the increased oxidative modification of LDL and the pro-atherogenic effects of PON2 deficiency.
Materials and Methods
Mice and diet
Female PON2-def mice and littermate controls were placed on an atherogenic diet consisting of 15.8% fat, 1.25% cholesterol, and 0.5% cholic acid (TD90221; Harlan Teklad), and sacrificed after 15 weeks, and mitochondria were isolated from livers as described (20). In addition, PON2-def mice were crossed with apoE−/− mice on the C57BL/6J background to generate PON2-def/apoE−/− and PON2+/+/apoE−/− littermate controls. Mice were either maintained on a standard chow diet for 16 weeks or started on a Western diet containing 0.15% cholesterol and providing 42% calories as fat (TD 88137; Harlan Teklad) at 6 weeks of age and maintained on this diet for an additional 16 weeks. At the end of the study, mice were fasted overnight, and serum and organs were collected. All animal protocols were approved by the Animal Research Committee at University of California at Los Angeles.
Atherosclerosis studies
Aortic root lesion analysis, En face lesion analysis CD68 quantification, and macrophage-mediated LDL oxidation and lipid peroxide in the lipoprotein were analyzed as described earlier (21, 29).
Mitochondrial nicotinamide adenine dinucleotide reduced form–cytochrome c reductase activity assay
For nicotinamide adenine dinucleotide reduced form (NADH)-cytochrome c reductase (complexes I and III) activity, 100 μg liver mitochondrial protein was added from experimental groups (after freeze and thaw) with final a concentration of 20 μM NADH as substrates, 50 μM cytochrome c 3+, and 250 μM of potassium cyanide, and the enzymatic activity was determined at 550 nm (ɛ = 18.5 mM−1/cm) and expressed as nanomole cytochrome c reduced per minute per milligram protein.
Assessment of mitochondrial oxidative stress in mice
Mitochondrial malondialdehyde and reduced glutathione levels were measured by commercial methods (Cayman Chemical Company) according to manufacturer's protocol using 100 μg of liver mitochondrial proteins. Liver mitochondrial superoxide assay was measured as described (17).
Quantification of ATP level
Wet liver tissues were extracted with 3.5% TCA containing 2 mM ethylene glycol tetraacetic acid, and centrifuged at 800 g for 10 min. The supernatants were adjusted to pH 7.4 using Tris-acetate-ethylenediaminetetraacetic acid buffer and ATP was quantified from 100 μl of sample from each experiential group as described by the manufacturer's protocol (Bioassay System). The results were normalized by tissue weight.
Assessment of mitochondrial dysfunction in peritoneal macrophage
Cellular metabolic rates were measured using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). One day before analysis, macrophages were harvested from PON2-def/apo E−/− mice (maintained on standard chow diet) and cells were counted using trypan blue; 4 × 105 viable macrophages per well were seeded in V7 plates (Seahorse Bioscience). Immediately before measurement, macrophages were washed with unbuffered complete Dulbecco's modified Eagle's medium as described (32). Mixing, waiting, and measurement times were 0.5, 2, and 3 min, respectively (an extra 0.5 min was added after each injection). For mitochondrial superoxide detection, peritoneal macrophages from experimental groups maintained on a standard chow diet were incubated with Mitosox (Molecular Probes–Invitrogen) at a final concentration of 5 μM for 30 min at 37°C, followed by washing three times using Dulbecco's modified Eagle's medium and analyzing the intensity of mitosox by flow cytometry as described by Mukhopadhyay et al. (18); 2 × 106 viable cells were taken and ATP level was quantified as described above under the section Quantification of ATP Level.
Superoxide detection in whole aorta
The aorta of apoE−/− and PON2-def/apoE−/− mice were homogenized in 25 mM Tris buffer containing 250 mM sucrose (pH 7.4), and centrifuged at 800 g at 4°C for 10 min. The supernatant (15 μg of total protein) containing the cytosol and other organelles including mitochondria was used to measure superoxide levels as described (17), except that mitosox was used instead of dihydroethidum.
Isolation and purity assessment of submitochondrial particles and localization of PON2
Mitochondria were isolated from the HeLa cell line, and the livers and hearts obtained from C57BL/6J mice as described (7, 33). The submitochondrial particles were isolated as described (25). Mitochondrial respiratory complexes, including complexes I, II, III, and IV, were immunoprecipitated as instructed by the manufacturer (Mitosciences). The immunocapture antibodies to complexes I, II, and III are specific for both mouse and human complexes, whereas the complex IV immunocapture antibody is specific only for human complex IV. All procedures were performed at 4°C. Mitochondria and submitochondrial particles were resolved by 4–15% SDS-PAGE, transferred onto nitrocellulose membranes, and blocked in Tris-buffered saline containing 3% milk protein for 1 h. PON2 antibody was used at 1:500 (The R&D Systems antibody was used for detection of human PON2, and Genscript USA, Inc., antibody was used for mouse PON2), Histone 1 antibody was used at 1:250 (Santa Cruz Biotechnology), Calnexin antibody was used at 1:1000 (Assay Designs, Ann Arbor, MI), antivoltage-dependent anion channel 1 was used at 1:500 (Cell Signaling Technology), and human cytochrome oxidase antibody was used at 1:500, whereas mouse cytochrome oxidase antibody was used at 1:250 dilution (Cell Signaling Technology). Primary antibodies were diluted in Tris-buffered saline containing 3% milk protein at 4°C overnight. The membranes were probed with their respective secondary antibodies (1:5000) for 1 h and proteins illuminated using an enhanced chemiluminescence Western blotting kit (GE Healthcare).
Coenzyme Q10 binding studies
Binding studies were performed by surface plasmon resonance on a BIAcore 3000 system (BIAcore AB). Human recombinant PON2, expressed and purified as described previously (5), was immobilized on a BIAcore CM5 sensor chip activated per the manufacturer's protocol with N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminoisopropyl) carbodiimide. After achieving adequate immobilization, the sensor surface was deactivated with ethanolamine. Analyte solutions were prepared in a standard BIAcore buffer (HBS-N), containing 10 mM HEPES, pH 7.4, and 150 mM NaCl in the presence or absence of 1 mM CaCl2. Lipid stock solutions (phosphatidyl ethanolamine and coenzyme Q10) were prepared in ethanol, so that analyte-containing, HBS-N binding buffer contained up to 1.0% ethanol. Lipid binding was measured by observing the change in the surface plasmon resonance signal as 150 μl of lipid (various concentrations) in HBS-N buffer flowing over the chip for 3 min at 50 μl/min. Equilibrium affinity constant (KD) values were calculated from assays performed with different concentrations that gave binding responses of 30 to >500 resonance units. The calculations were done with BIAcore's BIAevaluation software, version 4.1, assuming a molar ratio of 1:1 lipid:protein binding. All experiments were done at the Surface Plasmon Resonance Core at the University of California, Los Angeles.
PON2 overexpression studies
A HeLa cell line stably overexpressing the human PON2 gene under the control of a tetracycline inducible promoter (23), and control HeLa cells containing an empty vector were treated first with doxycycline for 48 h, followed by 20 μM antimycin treatment for 1 h to induce the mitochondrial superoxide or 50 nM oligomycine treatment for 2 h to inhibit the ATP synthesis. Mitochondrial superoxide was quantified using 10 μg of mitochondrial protein by fluorimeter as described (17). After the oligomycin treatment, 2 × 106 viable cells were taken and ATP level was quantified as explained above under the section Quantification of ATP Level.
Statistical analysis
Statistical significance was determined using Student's t-test. A p-value < 0.05 was considered statistically significant.
Results
Increased atherosclerosis in PON2-def/apoE−/− mice
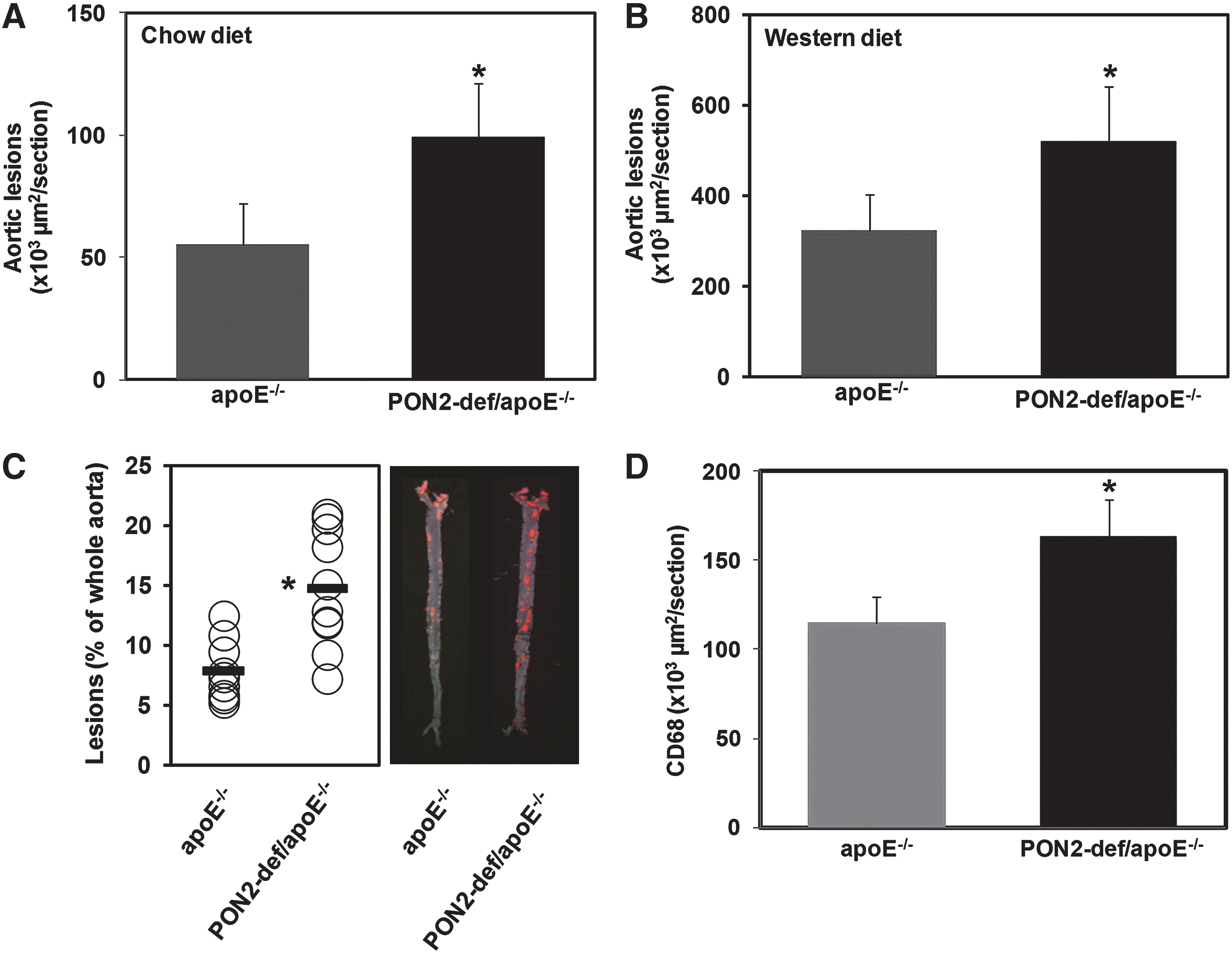
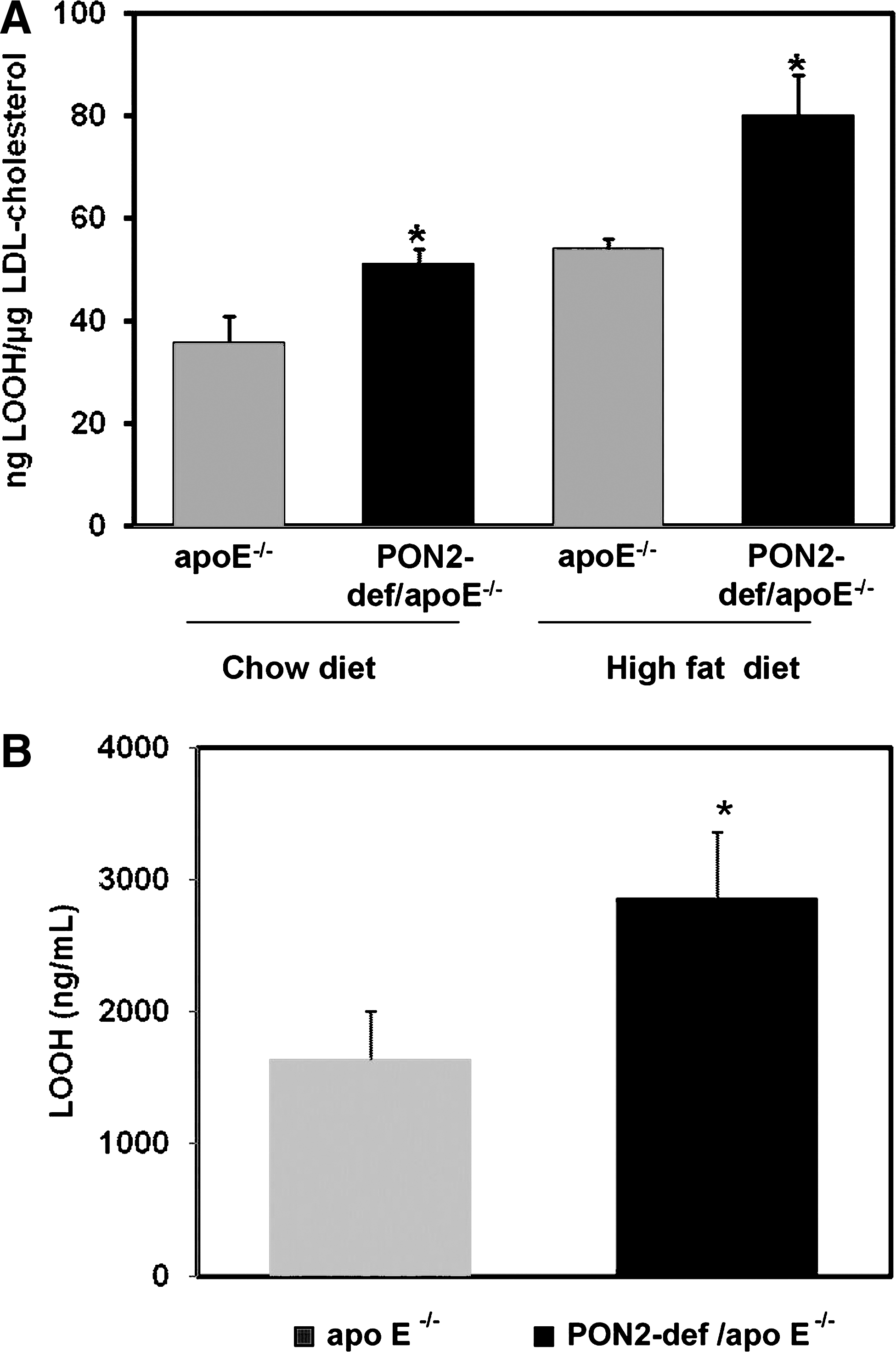
We previously reported that PON2-def mice on the C57BL/6J background are highly susceptible to diet-induced atherosclerosis when administered a high-fat, cholate-containing atherogenic diet (21). To further characterize the physiological function and explore the mechanism of action of PON2 in the context of atherosclerosis, PON2-def mice were backcrossed onto the hyperlipidemic apoE−/− background. Eight-week-old female PON2-def/apoE−/− mice were placed either on the chow diet or on a high-fat Western diet for 16 weeks. PON2-def/apoE−/− mice developed significantly larger atherosclerotic lesions in the proximal aorta (Fig. 1A, B) than in the control groups on both diets. Moreover, on the Western diet PON2-def/apoE−/− mice developed significantly more en face lesions along the whole aorta (Fig. 1C) than apoE−/− mice. There were also higher levels of macrophage immunoreactivity in the aortic sections of PON2-def mice (Fig. 1D) than in their wild-type controls. Accordingly, LDL isolated from PON2-def/apoE−/− mice was associated with increased lipid hydroperoxide content, relative to control mice (Fig. 2A). Figure 2B shows that PON2 deficiency on the apoE−/− background oxidized control LDL significantly above that already elicited by apoE deficiency alone.
PON2 deficiency impairs respiratory complex activity and mitochondrial oxidative stress in liver, peritoneal macrophages, and aorta
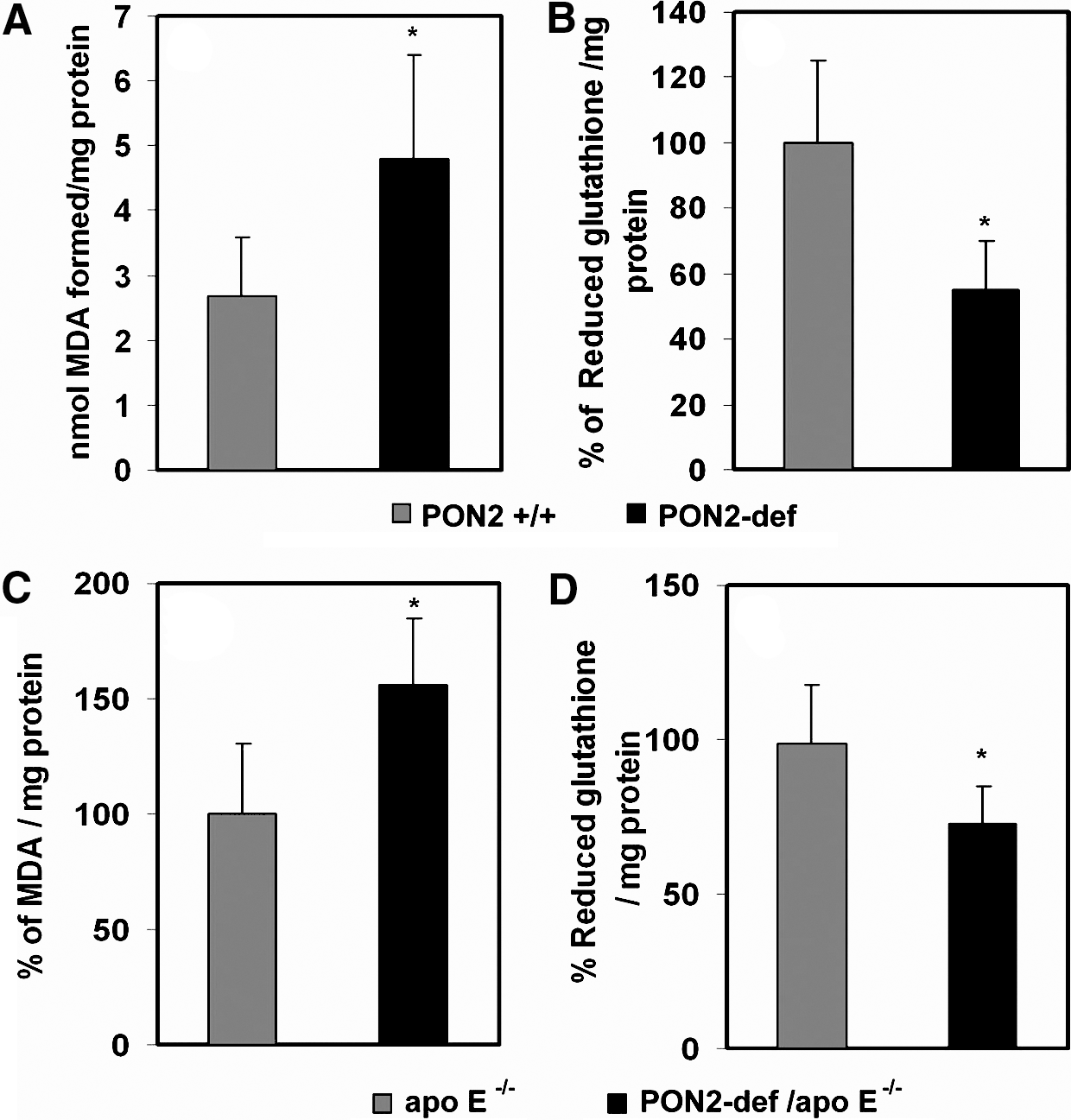
We have demonstrated that PON2 protects against atherogenesis in vivo by modulating lipoprotein oxidation through the reduction of intracellular oxidative stress (21). The mechanism by which it modulates oxidative stress is unknown. As the principal source of cellular free radicals and oxidative stress, ROS generated by mitochondria play a fundamental role for many of the signaling pathways contributing to cardiovascular pathologies (14). We hypothesized that PON2 deficiency may be influencing mitochondrial oxidative status. Therefore, we examined the role of PON2 on mitochondrial function using multiple approaches. First, we evaluated the activities of mitochondrial ETC complexes from the livers of PON2-def and control C57BL/6J mice administered an atherogenic diet, and results revealed that complex I + III activity was more than 50% lower in PON2-def mice than in controls on a corresponding diet (Fig. 3A). Moreover, the mitochondrial superoxide levels were significantly increased in PON2-def mice fed an atherogenic diet, and the ATP levels were reciprocally decreased when compared to control mice (Fig. 3B, C). Figure 4A shows a significant increase in malondialdehyde content in liver mitochondria obtained from PON2-def mice fed on an atherogenic diet. The elevated oxidative stress was further associated with a concomitant decrease in reduced glutathione levels (Fig. 4B). Similarly, mitochondria isolated from PON2-def/apoE−/− mice had significantly higher malondialdehyde, with an associated decrease in reduced glutathione levels relative to mitochondria isolated from control apoE−/− mice (Fig. 4C, D).
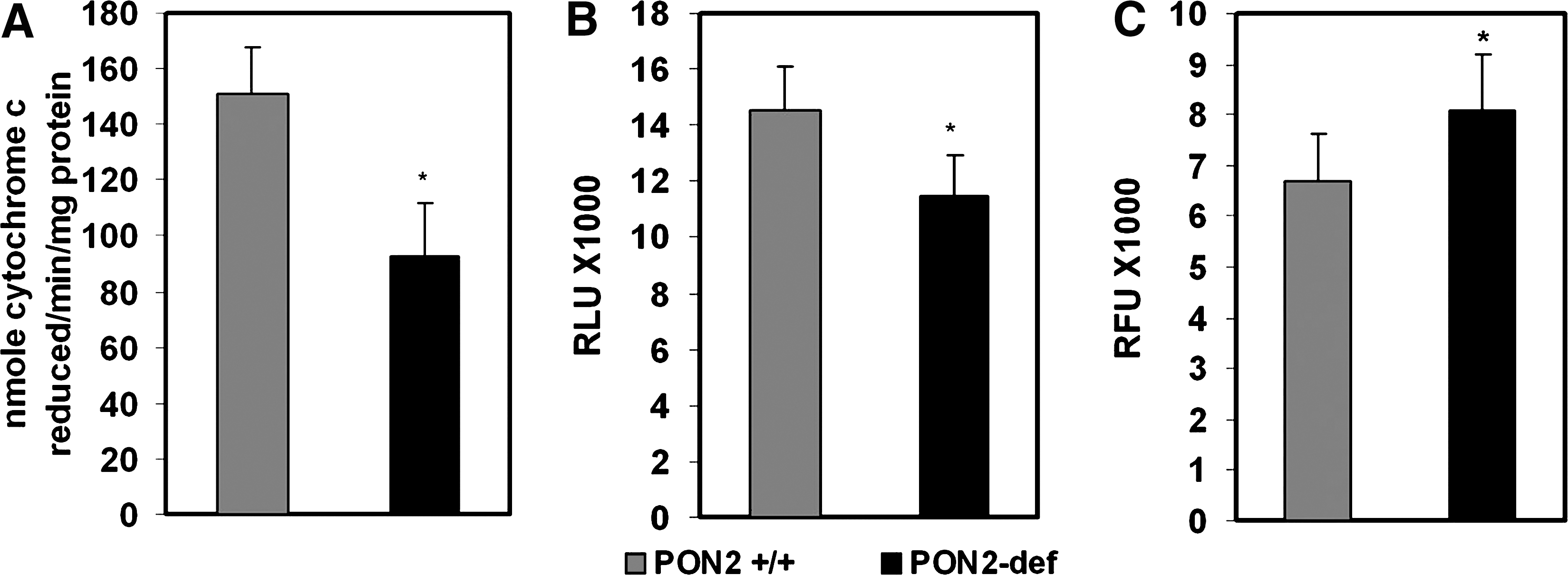
We next evaluated the level of oxygen consumption in peritoneal macrophage isolated from control and PON2-def/apo E−/− mice. As shown in Figure 5A and B, PON2-def mice showed a significantly lower level of basal mitochondrial oxygen consumption than the control peritoneal macrophage. The response to 1 μM oligomycin was also lower, suggesting a defect in ATP synthesis in PON2-def mice (Fig. 5C). Even after the addition of 0.5 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone, which raises the respiratory activity to maximal levels, the oxygen consumption rate was found to be substantially lower in PON2-def mice (Fig. 5D). Mitochondrial superoxide levels in peritoneal macrophages from PON2-def mice were significantly higher (p < 0.05) and ATP levels were significantly lower compared with control mice (Fig. 5E–H).
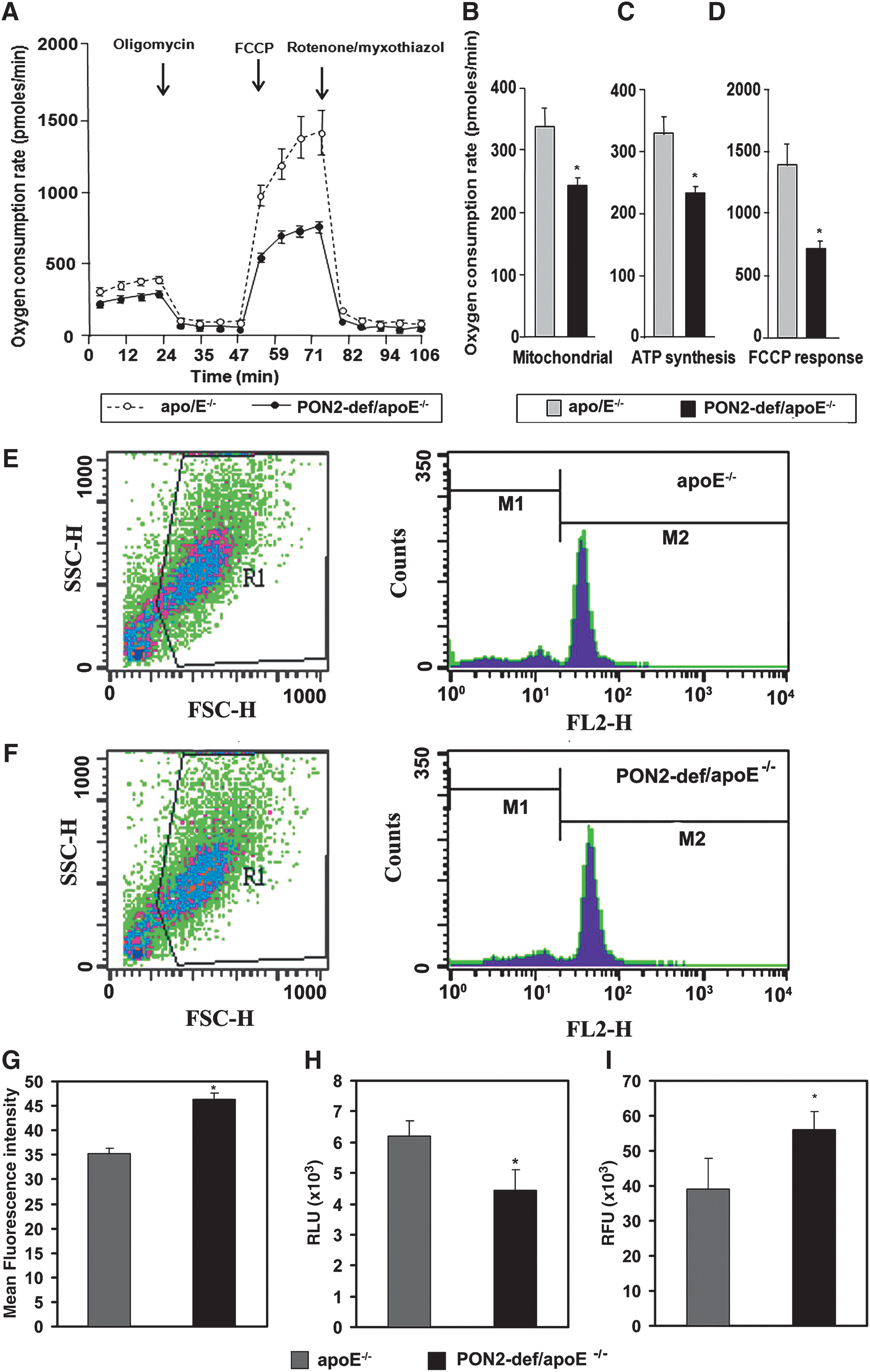
Further, superoxide levels were quantified in the supernatants of whole aorta lysates using Mitosox. The results revealed significantly higher superoxide levels in the aorta of PON2-def/apoE−/− mice relative to controls (Fig. 5I).
PON2 localized in the mitochondria
To determine whether the changes in mitochondrial oxidative stress are due to a direct or indirect effect of PON2 on mitochondria function, we isolated and analyzed mitochondria from HeLa cells for the presence of PON2. PON2 protein was found highly expressed in percoll-purified mitochondria from HeLa cells (Fig. 6A). This preparation showed negligible cross-contamination of calnexin, an endoplasmic reticulum marker, and was not immunoreactive for histone H1, a nuclear marker, confirming the purity of the mitochondrial preparations. To further determine the precise submitochondrial localization of PON2, we prepared inner and outer mitochondrial membrane preparations from the livers of C57BL/6J mice. Subsequent western blot analyses revealed that PON2 is predominantly localized to the inner mitochondrial membrane (IMM) (Fig. 6B, C). Western blot analyses for COX IV, an IMM associated protein, and VDAC, an outer mitochondrial membrane-associated protein, showed negligible cross-contamination of the two preparations in these experiments. Similar results were observed with mitochondria isolated from mouse heart tissue (data not shown).
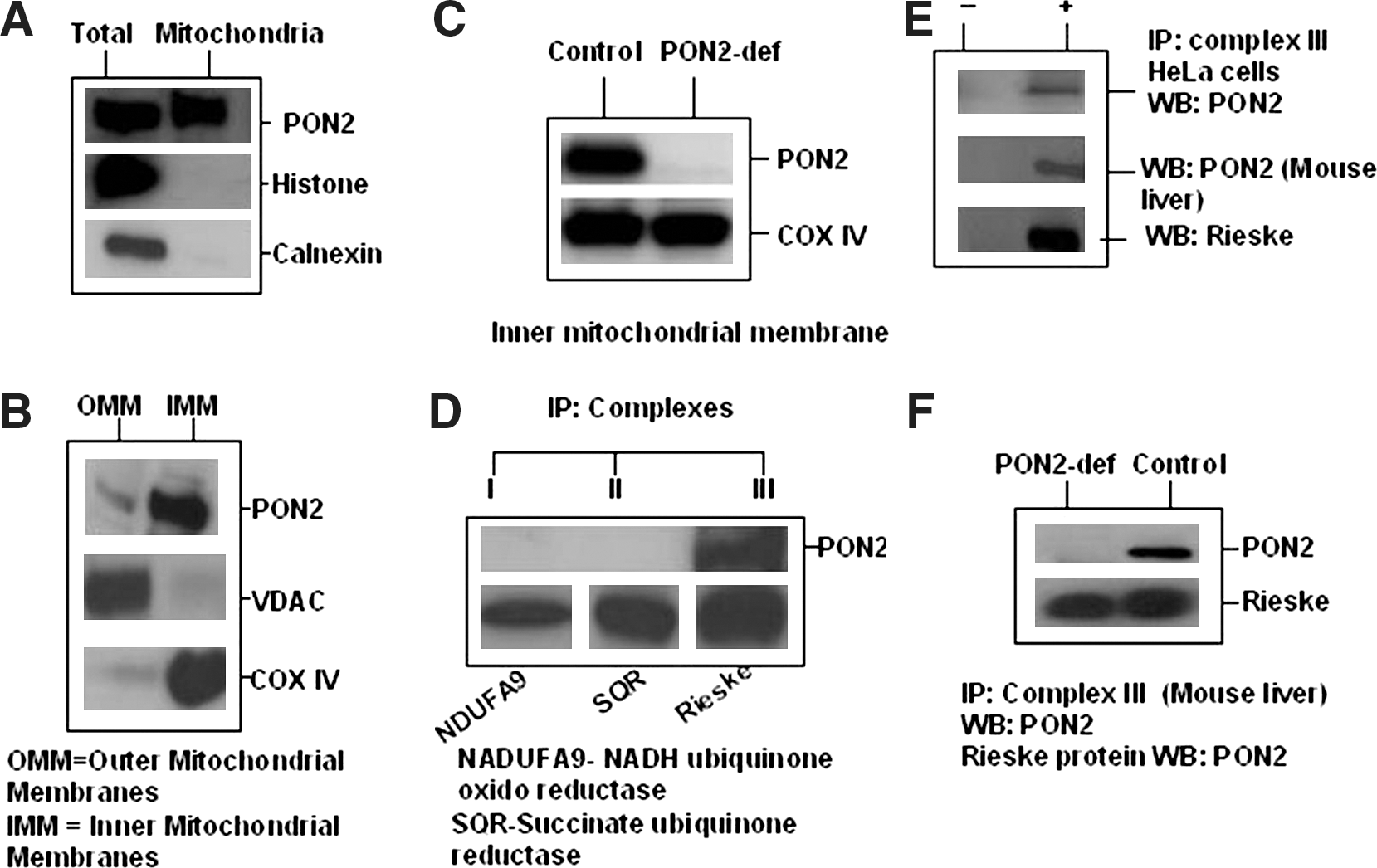
We have identified that PON2 interact with Rieske iron sulphar protein by Yeast Two Hybrid system (data not shown). This Rieske iron-sulfur protein present in the respiratory complex III (ubiquinol cytochrome c oxidoreductase) on the inner mitochondrial membrane. To determine if PON2 is associated with respiratory complexes of the mitochondria, we immunoprecipitated individual mitochondrial respiratory complexes using immunocapture antibodies from crude mitochondrial preparations of HeLa cells and C57BL/6J liver extracts. Western blot analysis with an anti-PON2 antibody of immunoprecipitated complex III preparations further support that PON2 is potentially associated with ETC complex III and otherwise undetectable in complexes I (NADH-ubiquinone oxidoreductase), II (Succinate dehydrogenase) (Fig. 6D–F), and IV (cytochrome oxidase; data not shown).
PON2 directly binds coenzyme Q10
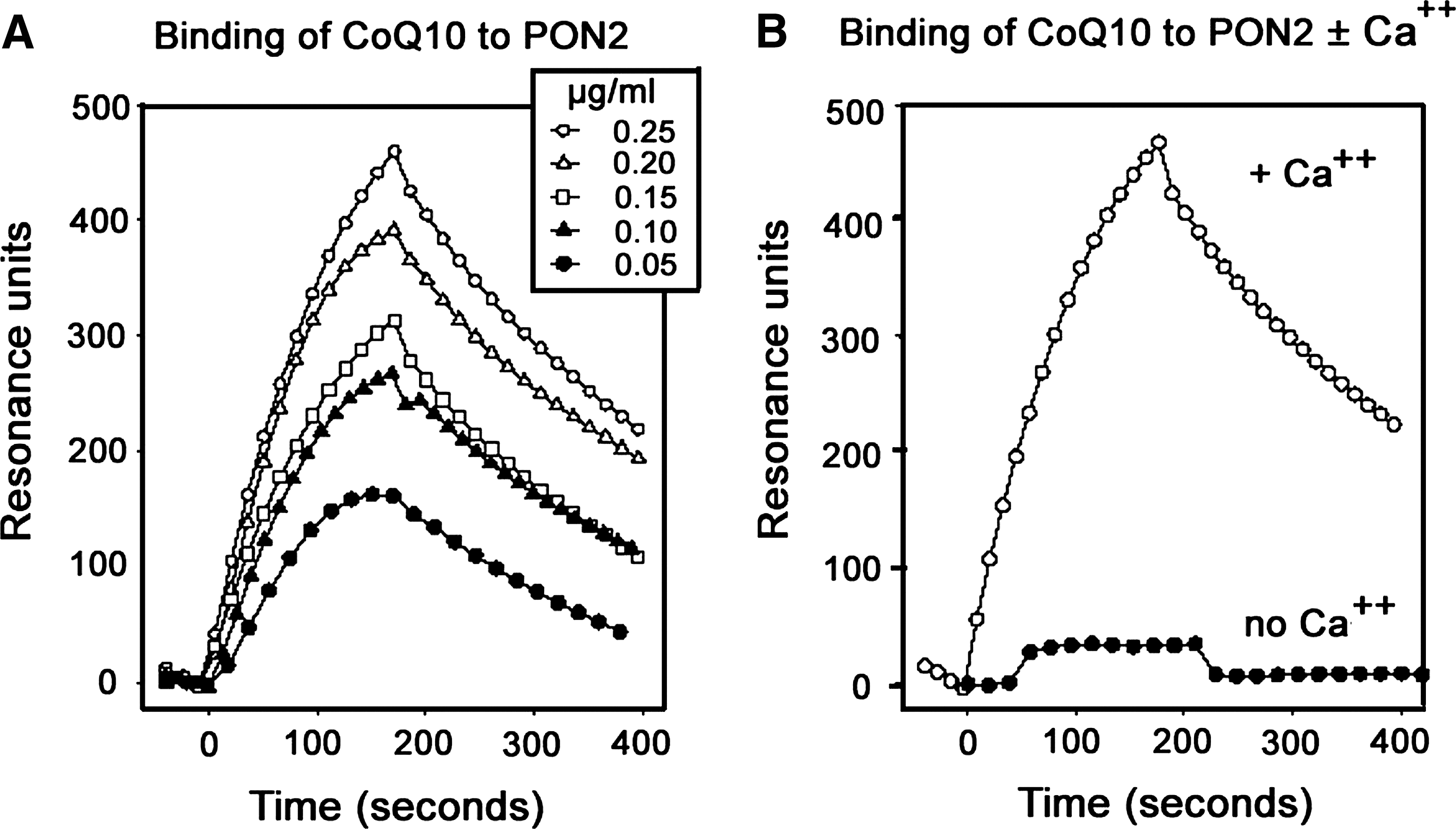
It has been shown that an unstable ubisemiquinone (coenzyme Q10 [CoQ10−] is a free radical resulting from the removal of one hydrogen atom with its electron during the process of dehydrogenation of a hydroquinone to quinone or the addition of a single H atom to a quionone) is one of the ETC intermediates that are involved in both ETC and the formation of ROS (13, 30). Recent biochemical studies reveal that a lactonase activity is shared by all PONs, and x-ray crystallography studies further suggest that several features of PON1 are reminiscent of lipases, and of secreted PLA2 in particular (8), and PON proteins exhibit lactonase activity toward longer fatty acid-derived lactones (e.g., eicosatetraenoic acid lactone). Since PON2 is localized to the IMM and associates with complex III similar to CoQ10 and since CoQ10 possesses a 50 carbon-long decaprenyl tail that folds into a compact structure (4), we investigated whether PON2 binds with Q10. To this end, we performed surface-plasmon-resonance analysis of CoQ10 binding to human recombinant PON2 (rPON2). We report that CoQ10 has an affinity toward human rPON2 with a KD of 4.4 × 10−8 M in the presence of 1 mM calcium chloride (Fig. 7A, B). Specific binding was not observed with phosphatidyl ethanolamine common to the mitochondria (data not shown).
PON2 overexpression protects against mitochondrial dysfunction
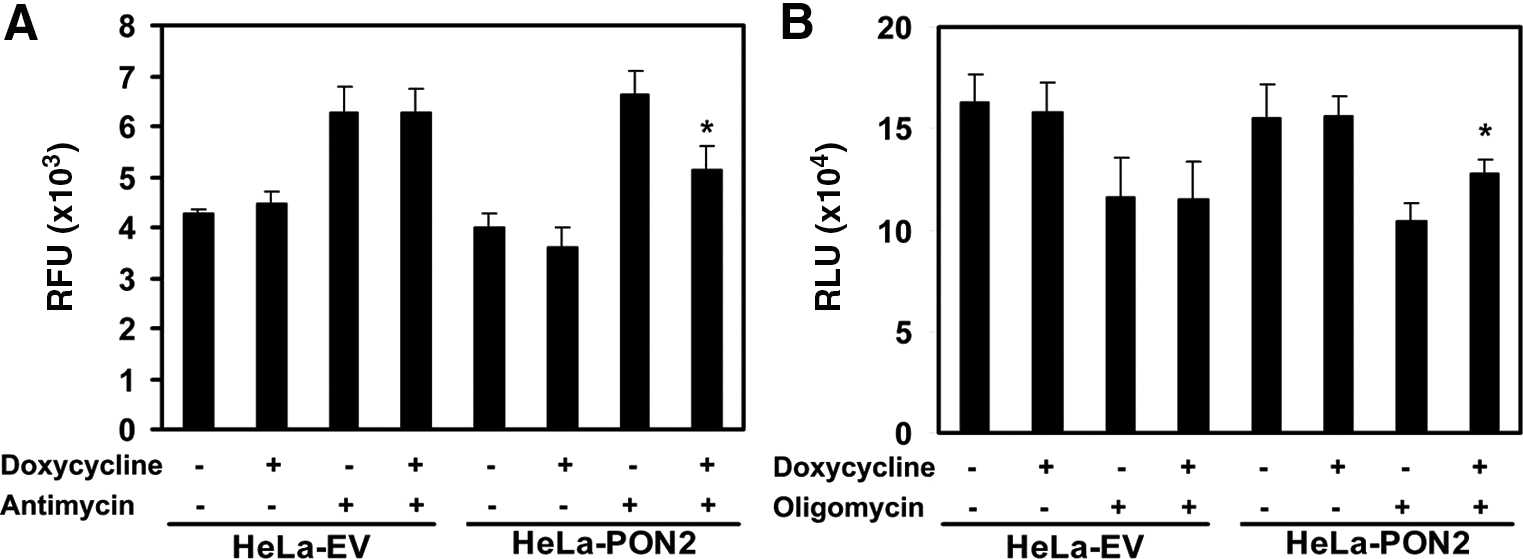
To further validate the role of PON2 in mitochondrial function, we questioned whether overexpression of PON2 could protect against oxidative stress-induced mitochondrial dysfunction. HeLa cells overexpressing human PON2 under the control of a tetracycline-inducible promoter as described previously (23) were treated with antimycin, a compound known to release ubisemiquinone from the ETC, thereby generating mitochondrial superoxide (24), or with oligomycin, a compound known to inhibit ATP synthesis (11). As expected, induction with doxycycline induced PON2 expression in the mitochondria of HeLa-PON2 cells, but not in the empty vector (data not shown). As shown in Figures 8A and B, HeLa cells overexpressing PON2 had significantly lower superoxide and significantly higher ATP levels than control cells.
Discussion
The PON gene family consists of three members, PON1, PON2, and PON3. All PON proteins have been implicated in the pathogenesis of inflammatory diseases, including atherosclerosis, Alzheimer's, Parkinson's, diabetes, and cancer. We have shown that stably transfected cells overexpressing PON2 exhibit significantly lower levels of intracellular oxidative stress when exposed to H2O2 (23). In addition, studies in mice suggest that PON2 protects against atherosclerosis (21) by modulating intracellular oxidative stress. We have previously reported that PON2 is localized to mitochondrial membranes and may play a role in mitochondrial oxidative stress (3); however, the mechanism(s) how PON2 modulates oxidative stress is still elusive and has been the focus of a great deal of research in recent years.
Accumulating evidence from in vitro, in vivo, and epidemiological studies suggests an important role for oxidative stress in the pathogenesis of atherosclerosis. Mitochondria are the major source of free radical-related oxidative stress. Decreased activity of mitochondrial ETC complexes activity results in increased production of ROS (2, 15), which play an important role in the development of many inflammatory diseases, including atherosclerosis. Our results suggest that PON2 deficiency under atherogenic conditions disrupts the activities of ETC complexes I + III, resulting in the production of superoxide that leads to enhanced mitochondrial oxidative stress. Further, mitochondrial function is impaired in PON2-def peritoneal macrophages, based on multiple indications, including oxygen consumption, ATP synthesis, and superoxide production, which are all direct measures of the efficacy of respiratory complex activity. In addition, mitochondrial superoxide was significantly elevated in the arotas of PON2- def mice. All these parameters indicate mitochondrial dysfunction in PON2-def in a manner that aggravates atherosclerosis lesions.
We initially identified Rieske iron sulfur protein as an interacting partner of PON2 by yeast two-hybrid screen (data not shown). This protein is localized to the mitochondria, as a subunit of respiratory complex III, where superoxide is primarily generated via the Q cycle (13, 24, 30). In the current investigation, we found that PON2 indeed localizes in the mitochondria, preferentially localizing to the IMM. Consistent with our study, McDonald et al. (16) isolated the IMM and identified 154 previously unreported proteins that included PON2 (but not PON1 nor PON3). The IMM plays a vital role in the regulation of energy metabolism, oxidative stress, and apoptosis. Since the IMM harbors the respiratory complexes of the ETC, we subsequently examined whether PON2 interacted with any of the respiratory complexes and found that, among the four respiratory complexes, PON2 was found to be potentially associated with complex III. It is well established that within the IMM, superoxide is continuously generated and released into the matrix at complex I and released both into the matrix and intermembrane space at complex III (15). The steady state concentration of superoxide in the mitochondrial matrix has been shown to be approximately 10-fold higher than that in the cytosolic and nuclear spaces (2, 15). All other ROS such as peroxynitrate, hydrogen peroxide, and hydroxy radicals are formed from superoxide. Therefore, as major sources of ROS production, the IMM could be a major target of ROS attack. Accordingly, the effects of ROS would be expected to be greatest at the level of IMM constituents, including the complexes of the respiratory chain and phospholipid(s) constituents particularly rich in unsaturated fatty acids, including cardiolipin (10). Although mitochondria possess antioxidant enzymes, they are not positioned to protect the immediate surroundings of the respiratory complexes. For example, Mn superoxide dismutase (SOD) and glutathione peroxidase are located on the matrix side, whereas CuSOD resides both in cytoplasm and in the inner mitochondrial membrane space (15). Perhaps PON2 on the inner membrane serves to reduce and inactivate some of the oxidized lipids as soon as they are formed, and at their source.
While the immunoprecipitation data demonstrate association of PON2 with complex III of the IMM (Fig. 5), it is feasible that PON2 is binding directly to the fatty acid tail of CoQ10 that associates with complex III. It has been reported that 32% of the CoQ10 in the mitochondria is associated with membrane proteins (12) and that species with longer life-span contain higher amounts of protein-bound CoQ10. X-ray crystallography studies suggest that fatty acid tail side chains are likely to fit into the substrate pocket of PON enzymes (8). CoQ10 has a fatty acid tail, is synthesized in the IMM, and plays a role in the bioenergetics pathway. Ubisemiquinone is a free radical that results from the process of dehydrogenation of hydroquinone to quinone. This unstable ubisemiquinone (involved in Q cycle) can donate a single electron to molecular oxygen to produce superoxide and reduce the ETC activity (13, 24, 30). Our lipid protein binding studies suggest that CoQ10 binds rPON2 with an affinity of KD of 4.4 × 10−8 M. Indeed, PON2-def mice show enhanced mitochondrial superoxide compare to control with impaired mitochondrial dysfunction. It has been widely accepted that steady-state concentration of ubisemiquinone was increased in the IMM by antimycin in a manner that produces superoxide, and our in vitro studies further suggest that overexpression of PON2 reduces the superoxide level induced by antimycin. It is therefore plausible that PON2 maintains the respiratory chain by promoting the sequestration of the unstable reactive intermediate ubisemiquinone, thereby preventing the superoxide production. Supporting our hypothesis, previously, it has been shown that mitochondrial superoxide is inversely related to the amount of CoQ10 bound to membrane proteins (12).
It is well established that oligomycin inhibits ATP synthase by blocking its proton channel (Fo subunit, which is required for oxidative phosphorylation of adenosine diphosphate to ATP) and leads to an increase in the proton gradient, which in turn reduces the respiratory activity and oxidative phosphorylation leading to mitochondrial dysfunction (11). Interestingly, PON2 overexpression restores the ATP level, which further supports our hypothesis that PON2 protects the mitochondria from adverse conditions.
Despite the role in mitochondrial function, we did not see a severe phenotype in PON2-def mice as was reported for the MnSOD knockout mouse model. It should be noted that in the MnSOD knockout mouse model, superoxide can not be converted to hydrogen peroxide and the accumulating mitochondrial superoxide promotes the formation of peroxynitrite and hydroxyl radicals that result in a severe phenotype, which occurs only in the homozygous mice and not heterozygous mice. In contrast, in PON2-def model, we speculate that the downstream pathways needed to convert superoxide into water including MnSOD, Glutathione Peroxidase, and Catalase are intact. Moreover, PON2-def mouse is not a total knockout. We have previously reported that the PON2-def mouse is deficient in PON2 and that tissue expression of PON2 is roughly 5–10% that of wild-type mice (21). Finally, it has been reported previously that mitochondrial dysfunction does not always correlate with severity in phenotype (1, 6, 27, 31).
Accumulation of unfolded proteins in the endoplasmic reticulum causes oxidative stress via the unfolded protein response (UPR) (28). Horke et al. (9) reported that overexpression of PON2 protects vascular cells from UPR-induced oxidative stress and apoptosis. Recent work from various laboratories suggests that under pathophysiological conditions there is a crosstalk between endoplasmic reticulum and mitochondria via both calcium-dependent and -independent pathways, which ultimately affects the function of mitochondria (28). Studies are underway in our laboratory to address and understand the role of PON2 in this respect.
In conclusion, our data suggest that PON2 is an IMM protein that plays an important role in the redox mechanisms of the respiratory chain. We report here for the first time that PON2-deficiency causes mitochondrial dysfunction, which may, in part, be responsible for the aggravated atherosclerosis observed in PON2-def mice.
Footnotes
Acknowledgments
We thank Dr. Paavo Korge, Dr. Robert I. Lehrer, and Dr. Satoshi Imaizumi for valuable discussions. We also thank Feng Su, David Meriwether III, and Alan Wagner for their expert technical assistance, and Aditya Gangopadhyay for his work on the Yeast Two Hybrid screen.
Author Disclosure Statement
The authors have no conflicts to disclose.