
Abstract
Forkhead box O (FOXO) transcription factors have been implicated in regulating the metabolism, cellular proliferation, stress resistance, apoptosis, and longevity. Through the insulin receptor substrate → phosphoinositide 3-kinase → Akt signal cascade, FOXO integrates insulin action with the systemic nutrient and energy homeostasis. Activation of FOXO1 in liver induces gluconeogenesis via phosphoenolpyruvate carboxykinase (PEPCK)/glucose 6-phosphate pathway, and disrupts mitochondrial metabolism and lipid metabolism via heme oxygenase 1/sirtuin 1/Ppargc1α pathway. In skeletal muscle, FOXO1 activation underpins the carbohydrate/lipid switch during fasting state. Inhibition of FOXO1 under physiological conditions accounts for maintenance of skeletal muscle mass/function and adipose differentiation. In pancreatic β-cells, nuclear translocation of FOXO1 antagonizes pancreatic and duodenal homeobox 1 and attenuates β-cells proliferation and insulin secretion. Regardless, FOXO1 promotes the proliferation of β-cells through induction of Cyclin D1 in low nutrition, and elicits antioxidant mechanism to protect against β-cell failure during oxidative insults. In the brain, FOXO1 controls food intake through transcriptional regulation of the orexigenic neuropeptide Y, agouti-related protein, and carboxypeptidase E. In this article, we review the role of FOXO1 in the regulation of metabolism and energy expenditure based on recent findings from mouse models, and discuss the therapeutic value of targeting FOXO1 in metabolic diseases. Antioxid. Redox Signal. 14, 649–661.
Introduction
FOXO plays a critical role in metabolism and growth of mammals (72, 90). FOXO is expressed ubiquitously in mammalian tissues, especially adipose, brain, heart, liver, lung, ovary, pancreas, prostate, skeletal muscle, spleen, thymus, and testis (77, 90). It is regulated by several mechanisms, including Akt (or protein kinase B [PKB])-mediated serine phosphorylation. During insulin stimulation, the phosphoinositide 3-kinase (PI3K) → Akt cascade inhibits FOXO nuclear activity, which is characteristic of the postprandial state of nutrient excess. Whole-body glucose homeostasis is tuned by both endogenous glucose production and glucose uptake by peripheral tissues in response to insulin. FOXO1 is activated in the fasted state to promote gluconeogenesis in the liver, but in postprandial state hepatic FOXO1 is inhibited by insulin, which facilitates gene expression that drives the metabolism of glucose to acetate for oxidation or conversion into fatty acids (25, 117). FOXO-mediated gene expression has multiple effects upon lipid metabolism (18, 19, 53, 81). Insulin resistance leads to hyperactive FOXO transcriptional activity, and eventually induces hyperglycemia and dyslipidemia in mammals (12, 19, 38, 52, 118). In addition, activation of FOXO under insulin resistant conditions can result in muscle atrophy and impair adipose proliferation, which reduced the capacity of these tissues to respond to insulin stimulation (55, 78, 123). Genetic ablation of FOXO during insulin resistance is beneficial for metabolic homeostasis; however, under normal nutrition the deletion of hepatic FOXO has minimal effects (25, 82). These findings reveal FOXO transcription factors as potential therapeutic targets for metabolic syndromes associated with insulin resistance.
FOXO1 represents the predominant FOXO isoforms (35, 36, 90). In this review, we will review some recent research focusing mainly upon in mouse models. These studies reveal new strategies to understand the pathophysiology of metabolic diseases such as obesity, nonalcoholic fatty liver diseases, diabetes, and diabetic complications.
Regulation of FOXO
FOXO proteins contain 4 domains. Sequence alignment shows that these FOXO proteins have highly conserved regions, including the N-terminal region containing an Akt phosphorylation site, a highly conserved forkhead DNA binding domain (DBD), a nuclear localization signal located just downstream of DBD, a nuclear export sequence, and a C-terminal transactivation domain (92). The DBD domain contains 3 α-helices, 3 β-sheets, and 2 loops that are referred to as the wings. FOXO1, FOXO3, and FOXO6 have similar length of about 650 amino acid residues, whereas FOXO4 is shorter and contains about 500 amino acid residues. All the regulation mechanisms involve the retention of FOXO in the nucleus where it can promote or suppress the transcription of target genes containing a consensus DNA binding sequence—TTGTTTAC.
FOXO proteins are modified by several posttranslational modifications, including phosphorylation, acetylation, methylation, glycosylation, and ubiquinylation (Fig. 1) (reviewed in 92, 99, 119). The modifications affect protein–protein and protein–DNA interactions that eventually alter the DNA-binding characteristics of FOXO proteins and regulate their transcriptional activity (11, 92, 109, 110, 114, 119). For example, the serine-threonine kinase Akt can phosphorylate FOXO1, FOXO3a, and FOXO4 and exclude them from nucleus, which promotes cytoplasm ubiquitinylation and degradation. An exception to this nucleo-cytoplasmic trafficking is FOXO6 whose transcriptional activity is similarly blocked by Akt-phosphorylation but independent of shuttling to the cytosol (49, 112). Moreover, FOXO transcriptional activity is inhibited by inhibitor of NF-κB (nuclear factor κB) kinase and the serum/glucocorticoid-inducible protein kinase that cause phosphorylation and nuclear exclusion (76, 92); however, it can be enhanced by mammalian sterile 20-like kinase-1-induced phosphorylation (70) and AMP-activated protein kinase (34). Arginine methylation of FOXO blocks Akt-induced phosphorylation and inactivation (119). However, addition of O-linked β-N-acetyl-glucosamine to FOXO promotes expression of its target genes involved in stress resistance during hyperglycemia and cell stress (43, 44). Deacetylation of FOXO by sirtuin 1 (SirT1) was shown to suppress mammalian FOXO transcriptional factor (13, 86, 120), although some opposite evidence indicates that the lysine acetylation of FOXO increases FOXO phosphorylation and attenuate its ability to bind cognate DNA sequence (82a). Comprehensive review on the structure/posttranslational regulation of FOXO transcriptional factors can be found in this Forum issue of Antioxidants & Redox Signaling.
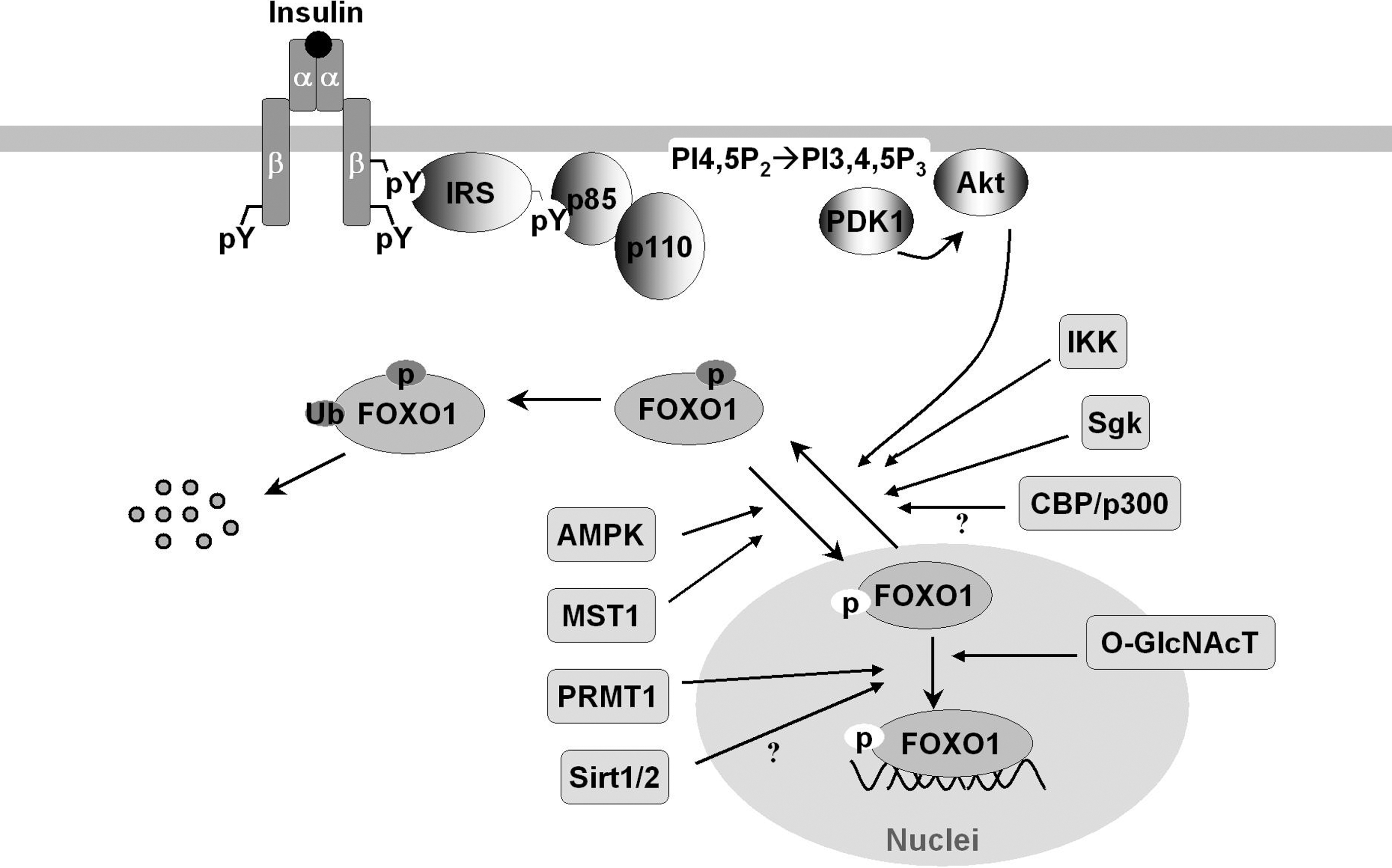
At the transcriptional level, expression of FOXO1 and FOXO3 can be induced by transcription factors E2F1 (91) and FOXC1 (10). Recent evidence shows that the transcription of FOXO is stimulated by FOXO3 in a positive feedback loop, but is repressed by growth factors such as platelet-derived growth factors and fibroblast growth factors (27). Ectopic FOXO3 activation blocked the proliferation of fibroblasts and induced expression of FOXO1 and FOXO4, mimicking the effects of growth factors. This regulatory mechanism may account at least in part for the coordinate upregulation of FOXO1, FOXO3, and FOXO4 in skeletal muscle from the mice treated by calorie restriction (20, 30). Moreover, the insulin receptor represses its own synthesis by a feedback mechanism directed by the transcription factor FOXO1 (97). These feedback loops, positive or negative, suggest that FOXO acts as a nutrient sensor to activate insulin (or growth factor) signaling that establishes an adaptive response to circulating factors.
FOXO1 in Regulation of Metabolism
FOXO1 is the best-studied member of FOXO subfamily. Loss and gain of FOXO1 function has been investigated in the tissues and cells of various genetically modified mice, including hepatocytes (18, 19, 25, 82), brain (95), muscle (20, 45, 55), adipose (17, 88), pancreas (2, 3, 84), and heart (99) (Table 1).
FOXO, forkhead box O; NGN3, neurogenin 3; PDX1, pancreatic and duodenal homeobox 1; PML, promyelocytic leukemia; PPARγ, peroxisome proliferator-activated receptor γ; SirT1, sirtuin 1.
FOXO1 in Liver
The liver is the primary organ for glucose and lipid metabolism (96). During fasting, glycogen stored in hepatocytes is hydrolyzed to release glucose (glycogenolysis). Upon glycogen depletion, gluconeogenesis ensures a sufficient glucose supply through transcriptional modifications controlled by CREB (cyclic AMP-responsive element binding protein)-regulated transcription co-activator 2 and FOXO1 (74). In the postprandial state, the liver stores the dietary carbohydrates through glycogen synthesis and de novo lipogenesis (18, 25, 81, 118). Under ordinary conditions, the fasting-feeding switch finely controls the hepatic glucose production/uptake and anabolic pathways of glucose disposal, which restricts postprandial in plasma glucose concentrations between 4 and 7 mM. The critical role of FOXO1 in hepatic glucose and lipid metabolism is well established with genetically modified mice (18, 25, 53, 81, 82). However, a critical role for hepatic FOXO1 is difficult to demonstrate definitively without additional efforts to stress the animal, including severe starvation or deletion of insulin signaling components. By deletion of hepatic insulin receptor substrates 1 and 2 [i.e., the liver-specific Irs1 and Irs2 double knockout (DKO)-mice], the mice lose the conserved response to insulin/feeding, and develop severe insulin resistance and glucose intolerance (25, 37). Microarray of the transcriptome in DKO-liver reveals markedly perturbed expression of growth and metabolic genes, including increased Ppargc1α (peroxisome proliferator-activated receptor gamma coactivator 1-α, also referred as Pgc1α) and Igfbp1, and decreased glucokinase, sterol-regulatory-element-binding protein 1c (SREBP-1c), Ghr, and Igf1 (25) (Fig. 2). Moreover, mitochondrial genes are dysregulated, which leads to abnormal mitochondrial morphology, function, and biogenesis in the DKO-liver (Table 2) (18). Signal transduction analysis indicates that FOXO1 is hyperactivated in these mice. Remarkably, expression of these genes is at least partially restored into a normal range upon deletion of FOXO1 (Fig. 3) (18, 25).
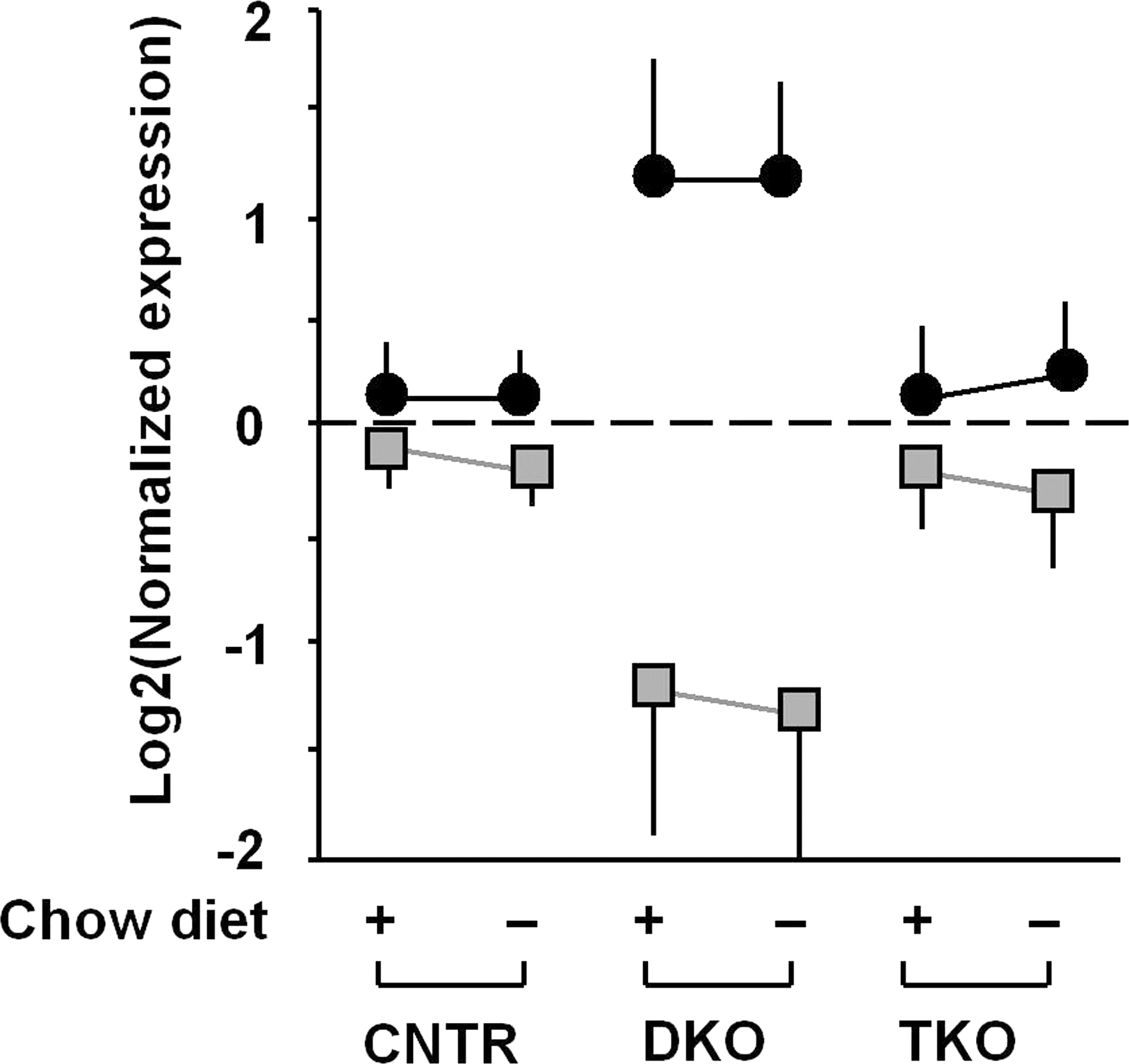
Adapted from Cheng et al. (18) with permission.
CNTR, Control; DKO, double knockout; Hmox1, heme oxygenase 1; TKO, triple knockout.
Chronic failure to control the hepatic glucose production leads to hyperglycemia and compensatory stimulation of insulin secretion by the pancreatic β-cells, which exacerbates peripheral insulin resistance in muscle and adipose (25, 117, 118). Hepatocyte-specific deletion of FOXO1 in DKO mice (triple knockout TKO) results in significant normalization of the gluconeogenic genes and partial restoration of the response to fasting and feeding, near-normal blood glucose and insulin concentrations (25). Single knockout of FOXO1 in mouse liver results in 40% reduction of glucose levels at birth and 30% reduction in adult mice after a 48 h fast (82). These changes are associated with impaired fasting- and cAMP-induced glycogenolysis and gluconeogenesis, because the complex between FOXO1 and the transcription coactivator Ppargc1α is absent, which compromises the cAMP response and gluconeogenesis (82). Similarly, FOXO1 deletion in liver curtails excessive glucose production caused by ablation of hepatic insulin receptor and prevents neonatal diabetes and hepatosteatosis in liver-specific insulin receptor knockout mice (82, 85). These findings corroborate the deleterious role of the constitutively active FOXO1 in hyperglycemia during severe hepatic insulin resistance, and that the insulin receptor substrates (Irs1/2) → PI3K → Akt → FOXO1 branch of insulin signaling underpins the hepatic insulin-regulated glucose homeostasis (Fig. 3) (25, 82). To this end, synthetic and optimized antisense oligonucleotides for specific inhibition of FOXO1 expression (FOXO1-antisense oligonucleotide therapy) has been used to improve both hepatic and peripheral insulin action that lowers plasma glucose concentration and the rate of basal endogenous glucose production in mice with diet-induced obesity (DIO) (101).
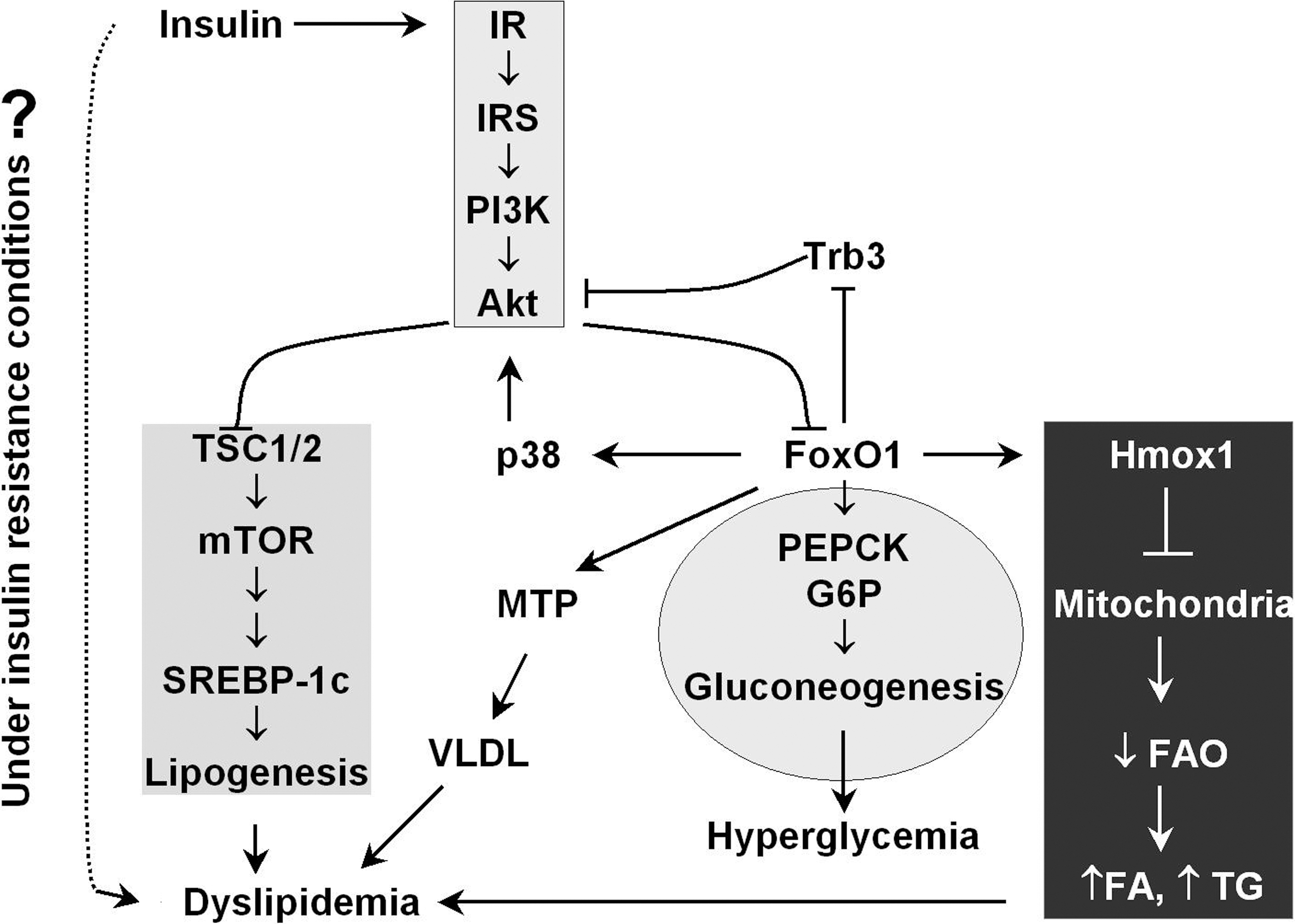
The mechanism by which insulin resistance fails to inhibit FOXO1 → PEPCK driven gluconeogenesis (hyperglycemia) while promoting SREBP-1c-mediated lipogenesis (dyslipidemia) remains unclear (Fig. 3) (12, 65, 118). In this respect, identification of the branch/divergent point that insulin signal control glucose production and lipogenesis has been the topic of intensive study. The results from mice that lack both major regulatory subunits of PI3 kinase in the liver (Pik3r1 and Pik3r2; L-p85DKO mice) suggest that Akt and atypical protein kinase C differentially define specific actions of insulin and PI3 kinase on hepatic glucose and lipid metabolism, respectively (107). In the obese, insulin-resistant mice induced by either leptin deficiency or high-fat diet, Akt is required for hepatic lipid accumulation (67). Leptin-deficient obesity mice lacking hepatic Akt exhibit a decrease in lipogenic gene expression and de novo lipogenesis, which prevents hepatic triglyceride accumulation. Whereas mice fed high-fat diet have reduced liver triglycerides in the absence of hepatic Akt, lipogenesis progresses normally presumably due to a compensatory adaptation to the uptake of dietary fat. These findings reveal Akt as a requisite component in insulin-dependent regulation of lipid metabolism during insulin resistance (67). Further corroborating evidence was reported most recently, showing that inhibition of PI3 kinase → Akt blocks both insulin-mediated PEPCK (gluconeogenesis) and SREBP-1c (lipogenesis) gene expression (71). Inhibition of mammalian target of rapamycin complex 1 (mTORC1), a downstream target of Akt can block insulin-induction of SREBP-1c but not insulin suppression of PEPCK, suggesting that insulin-mediated lipogenesis and gluconeogenesis diverge with Akt → mTORC1 driving the former and FOXO1 → PEPCK driving gluconeogenesis (Fig. 3); however, during severe insulin resistance FOXO appears to contribute to both processes (18, 19).
Hyperactivated FOXO during severe insulin resistance contributes to the accumulation of hepatic lipids (Fig. 3) (18, 19, 81). Adenoviral delivery of constitutively nuclear FOXO1 to mouse liver promotes hepatic triglyceride accumulation that can progress to steatosis (81). The lipid accumulation is associated with decreased fatty acid oxidation. Paradoxical stimulation of Akt signaling occurs owing to FOXO1-mediated repression of pseudokinase tribble 3 (a modulator of Akt activity) independent of DNA binding; however, suppression of FOXO1 with appropriate siRNA diminishes it (81). An accompanying study using constitutively nuclear FOXO1 (FOXO1-ADA mutant that is constitutively nuclear due to mutation of T24 and S316 to A and harbors a mutation of S253 to D) suggests that FOXO1 turns on a feed-forward loop, relayed by p38 and acting to amplify Akt activity (87). In addition, FOXO1 can induce expression of hepatic microsomal triglyceride transfer protein (MTP) that regulates the rate-limiting step of assembly of triglyceride-rich very-low-density lipoprotein (VLDL) (53). FOXO1 gain of function is associated with enhanced MTP expression, correlating with augmented hepatic VLDL production and elevated plasma triglyceride levels in FOXO1 transgenic mice. FOXO1 loss of function, caused by RNAi-mediated depletion of FOXO1 mRNA in liver, reduces hepatic MTP and VLDL production in diabetic, db/db and FOXO1 transgenic mice. The stimulating effect of FOXO1 on MTP expression and VLDL production is blunted by insulin in the control mice (53).
In addition to regulation of lipogenesis, recent observations indicate that activated FOXO1 impairs fatty acid oxidation. By this mechanism, dyslipidemia might arise at least in part from mitochondrial dysfunction under the insulin-resistant conditions (Fig. 3) (18, 19, 81). In mice lacking hepatic Irs1/Irs2, FOXO1 hyperactivation is accompanied by impaired mitochondrial oxidative metabolism and mild hepatic lipid accumulation (18, 19). By directly binding the promoter, the transcription factor FOXO1 induces heme oxygenase 1 that reduces the heme content/pool required for expression, stability, and function of electron transport chain (ETC) components (18, 21). The impaired ETC activity in these DKO hepatocytes fails to oxidize NADH to NAD+, thus decreasing the [NAD+]/[NADH] ratio due to NADH accumulation. These changes prevent 2 biological processes that are related to NADH metabolism and [NAD+]/[NADH] ratio (18, 19). First, the NAD+-dependent deacetylase SirT1 is inactivated, which blocks the mitochondrial biogenesis pathway mediated by Ppargc1α—the activation of Ppargc1α requires SirT1-catalyzed deacetylation. Second, NAD+ acts as the essential cofactor of acyl-CoA dehydrogenase in fatty acid oxidation. Reduction of [NAD+] due to ETC defects attenuates fatty acid oxidation, which promotes accumulation of fatty acid available for incorporated into triglyceride (46, 47, 100, 116). As such, pharmacological stimulation of NADH oxidation strongly promotes mitochondrial fatty acid oxidation and attenuates dyslipidemia and fatty liver in DIO and ob/ob mice (46). In line with the observation that adenoviral delivery of constitutively nuclear FOXO1 to mouse liver decreased fatty acid oxidation (18, 19, 81), targeting hepatic FOXO1 by anti-sense oligonucleotides or knockout lowers both hepatic triglyceride and diacylglycerol content, which is at least in part accounted for by the restoration of mitochondrial function (18, 19, 101).
Together, activated FOXO1 promotes hepatic glucose production through induction of the gluconeogenic enzymes, PEPCK and glucose 6-phosphate. FOXO1 may also participate in lipid metabolism by driving Akt-mediated lipogenesis through p38 and tribbles homolog 3. Under insulin-resistant conditions, the induction of heme oxygenase 1 that cause ETC defects results in impaired mitochondrial fatty acid oxidation and lipid accumulation. Hyperactive FOXO1 under insulin-resistant conditions causes hyperglycemia and dyslipidemia, the characteristics of diabetes and diabetic complications. Therefore, finely tuned FOXO1 activity is the premise of nutrient and metabolic homeostasis.
FOXO1 in Brain
FOXO1 is strongly expressed in the striatum and neuronal subsets of the hippocampus (dentate gyrus and the ventral/posterior part of the cornu ammonis regions) (41). In wild-type mice, hypothalamic FOXO1 expression is reduced by the anorexigenic hormones insulin and leptin (59). Deletion of Irs2 in the brain activates FOXO1 that accounts for extended life span of mice (106). After the observation that neuron-specific insulin receptor knockout mice developed diet-sensitive obesity with increases in body fat and plasma leptin levels (14), FOXO1 as the downstream target was recently found to play the key role in control food intake, energy disposal, and fuel metabolism (9, 48, 59, 60, 95). The adenoviral delivery of a constitutively active FOXO1 to the hypothalamus disables leptin to control food intake, and the mice showed an increased food intake and body weight (59, 60). Mechanistic studies indicate that FOXO1 stimulates the transcription of the orexigenic neuropeptide Y and agouti-related protein via PI3 kinase → Akt cascade, but suppresses the transcription of anorexigenic pro-opiomelanocortin (Pomc) by antagonizing the activity of signal transducer-activated transcript-3 (STAT3) (59, 60). Given that both insulin and leptin signaling pathways converge on PI3 kinase → 3-Phosphoinositide-dependent kinase 1 (PDK1) → Akt → FOXO1 in (Pomc) neurons, 2 independent groups confirmed that Pomc neuron-specific PDK1 knockout (PDK1ΔPomc) mice display hyperphagia, increased body weight, and impaired glucose metabolism caused by reduced hypothalamic Pomc expression (9, 48). Selective expression of a transactivation-defective FOXO1 mutant in Pomc cells ameliorates the energy imbalance in PDK1ΔPomc mice (9), whereas expression of a constitutively nuclear FOXO1 exacerbated the obesity of the PDK1ΔPomc mice due to excessive suppression of the Pomc gene (48).
Recently, ablation of Pomc-FOXO1 identified carboxypeptidase E (Cpe) as a critical downstream component in the dysregulation of food intake and energy expenditure (95). Cpe expression increases during the deletion of Pomc-FOXO1, which is concomitant with decreased food intake and normalized energy expenditure. Of note, Cpe expression is downregulated by DIO, but Pomc-FOXO1 ablation prevents Cpe downregulation and protects against weight gain. Moderate Cpe overexpression in the arcuate nucleus phenocopies features of the FOXO1 mutation (95). These findings suggest that targeting Pomc-FOXO1 can be a model for therapeutic intervention in obesity.
Thus, FOXO1 in brain appears to be a dysregulating factor of leptin/insulin-mediated food intake/energy expenditure. Persistently activated FOXO1 increases food intake and predisposes to obesity. Inhibition of FOXO1 dampens neuropeptide Y and agouti-related protein, but promotes Cpe expression, which ameliorates metabolic disorder and protects against weight gain.
FOXO1 in Skeletal Muscle
The skeletal muscle is one of the major peripheral tissues that are responsible for insulin-mediated fuel metabolism and energy expenditure. Skeletal muscle accounts for >30% of resting metabolic rate and 80% of whole-body glucose uptake (23). Expression of FOXO1 is increased in skeletal muscle by energy deprivation (such as fasting, calorie restriction, and severe diabetes), suggesting that FOXO1 may mediate the response of skeletal muscle to changes in energy metabolism (20, 29, 56). Indeed, FOXO1 suppresses SREBP-1c in skeletal muscle (54), and mice overexpressing FOXO1 lose their glycemic control and display a lower capacity for physical exercise due to severe muscle loss (55).
Skeletal muscle metabolism switches from oxidation of carbohydrates to fatty acids as the major energy source during fasting when the plasma glucose concentration is low. FOXO1 controls this switch by upregulating 3 enzymes: pyruvate dehydrogenase kinase-4 (PDK4) that shuts down glucose oxidation by targeting pyruvate dehydrogenase (PDH), lipoprotein lipase that hydrolyzes plasma triglycerides into fatty acids, and fatty acid translocase CD36 that facilitates fatty acid uptake into skeletal muscle (Fig. 4) (8, 29). PDK4 phosphorylates PDH and blocks PDH activity in catalyzing the conversion of pyruvate into acetyl-CoA, which can divert glucose flux to lactate and away from acetyl-CoA (8, 29). During starvation or glucocorticoid treatment, FOXO1 expression increases and directly induces PDK4 expression to inhibit glucose oxidation (29). In the mean time, FOXO1 overexpression in muscle C2C12 cells enhances lipoprotein lipase gene expression and increases the plasma membrane level of the fatty acid translocase CD36. These changes promote the membrane uptake of oleate and oleate oxidation (8, 56). FOXO1 also regulates triglyceride content via retinoid X receptor α (RXRα)/liver X receptor α (LXRα)/SREBP-1c pathway (54). FOXO1 suppresses RXRα/LXRα/-mediated SREBP-1c promoter activity: gene expression of both RXRγ and SREBP-1c is downregulated in skeletal muscle FOXO1 transgenic mice (Fig. 4). During nutritional changes caused by fasting and feeding, gene expression of RXRα and SREBP-1c in mouse skeletal muscle switches off and on, respectively, whereas expression of FOXO1 shows reverse correlation with SREBP-1c expression (54).
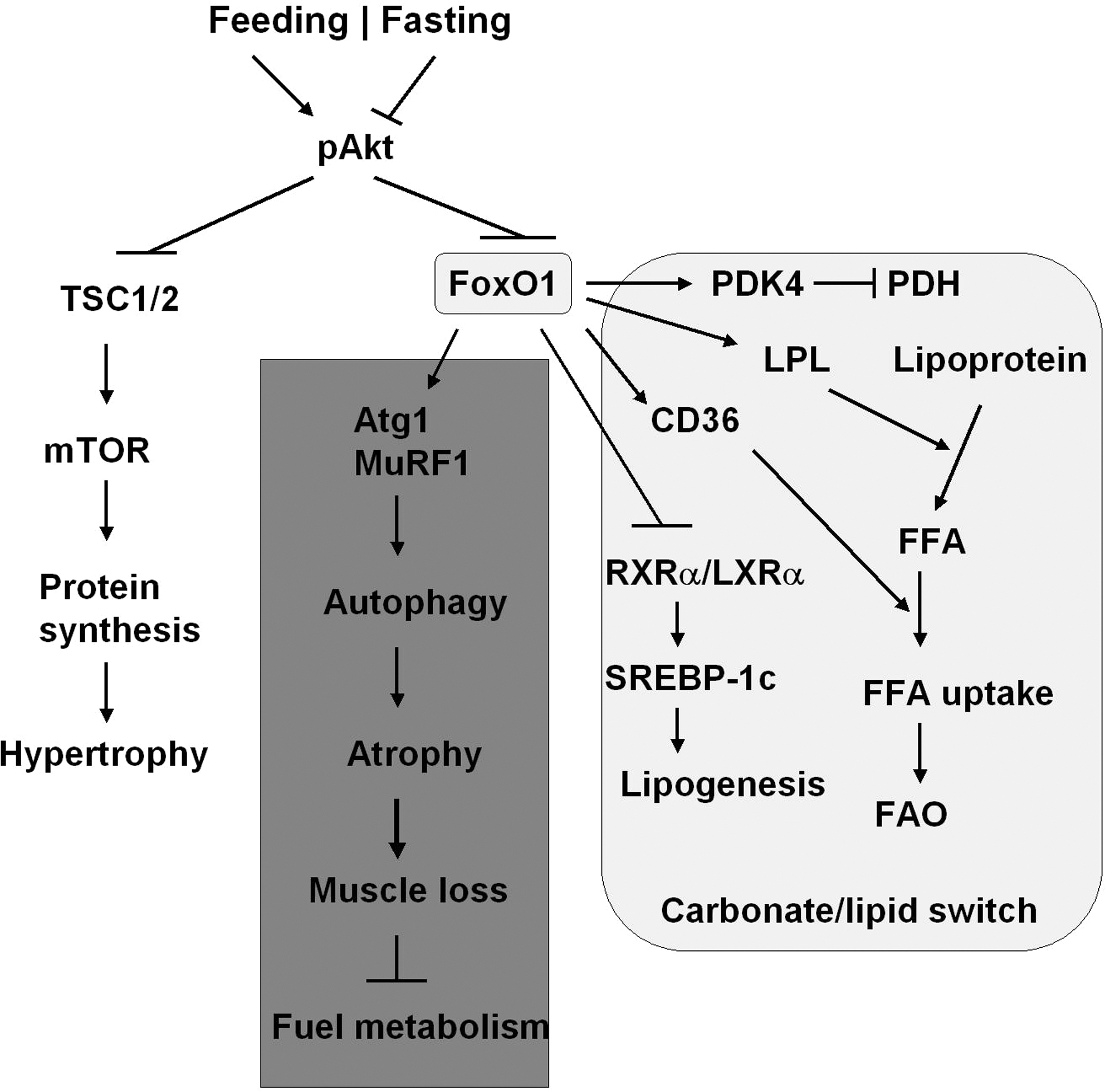
The muscle mass/fiber size is known to change with workload, activity, or pathological conditions, including diabetes mellitus. The maintenance of muscle mass is achieved by a dynamic balance of atrophy and hypertrophy (32, 102). Activation of FOXO1 or FOXO3 in the skeletal muscle, in fasting or diabetic conditions, can increase protein breakdown through ubiquitin-proteasome and autophagy-lysosome pathways, the 2 major mechanisms causing muscle atrophy (Fig. 4) (22, 78, 80, 103, 123). Overexpression of a constitutively active FOXO1 in C2C12 muscle cells promotes expression of atrogin 1 (F-box protein 32) and muscle-specific RING finger protein 1, the 2 ubiquitin ligases involved in skeletal muscle atrophy (105). Expression of a dominant-negative FOXO1 construct in myotubes or in rodent muscle decreases atrogin-1 expression and muscle atrophy (105). Transgenic FOXO1 in skeletal muscle increases expression of cathepsin L, an atrophy-related lysosomal protease, which is associated with reduced skeletal muscle mass and body weight (55). Moreover, the genes encoding structural proteins of type I muscles (slow twitch, red muscle) are downregulated concomitant with a decreased size of both type I and type II fibers. The coordinate regulation of cathepsin L and type I muscle genes may account at least in part for the loss of muscle mass and glycemic control owing to hyperactivated FOXO (55).
Hence, physiological switch of FOXO1 activity, that is, on in fasting and off in feeding sate, is required for the nutrient/energy homeostasis in the skeletal muscle through carbohydrate/lipid switch. Severe starvation may trigger FOXO1-mediated autophagy and atrophy that break down protein for energy supply, the mechanism that underlies the loss of muscle mass and glycemic control under insulin resistance.
FOXO1 in Adipose Tissue
Adipose tissue contributes to metabolic regulation in several ways: (a) to combust fuel and generate heat to maintain body temperature (mainly in brown adipose tissue) (72); (b) to store excess energy in the form of triglycerides and mobilize the stored lipids for energy supply during energy deprivation (mainly in white adipose tissue) (66); and (c) as an endocrine organ to secrete adipokines and cytokines (e.g., leptin and adiponectin) that control energy homeostasis in adipocytes and inform the central nervous system of nutrient homeostasis (31). The prerequisites for these functions are differentiation and maintenance of adipocytes, in which FOXO1 plays a key role by interacting with peroxisome proliferator-activated receptor γ (PPARγ), the transcription factor that promotes adipogenesis and fat storage (Fig. 5) (28, 88, 94, 115).
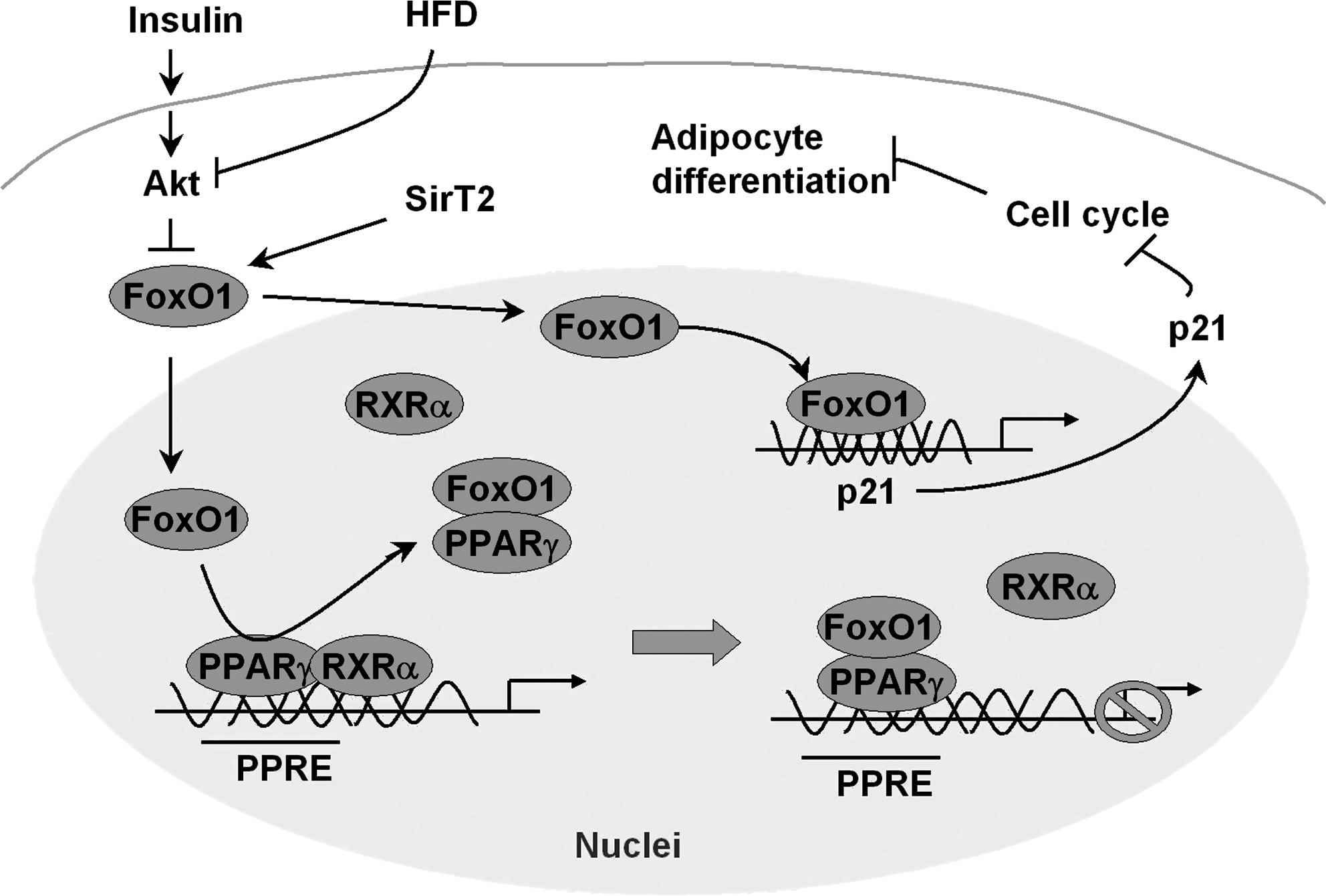
Adipogenesis, during which fibroblast-like preadipocytes differentiate into lipid-laden and insulin-responsive adipocytes, experiences several stages, including mesenchymal precursor, committed preadipocyte, growth-arrested preadipocyte, mitotic clonal expansion, terminal differentiation, and mature adipocyte (69). PPARγ exclusively regulates both the terminal differentiation and metabolism in mature adipocytes, by binding as a heterodimer with RXRα DNA at PPAR response elements (28). FOXO1 is a PPARγ-interacting protein that antagonizes PPARγ activity (6, 26, 28). This inhibition is achieved by disrupting the DNA binding activity of a PPARγ/RXRα heterodimeric complex (6, 26), and trans-repressing PPARγ transactivation via direct protein–protein interactions (Fig. 5) (28). FOXO1 represses transcription from either the PPARγ1or PPARγ2 promoter; however, this inhibition is attenuated by insulin treatment, suggesting that insulin-induced phosphorylation drives PPAR-γ activity by inhibiting FOXO1 (6). Mutation of insulin-induced phosphorylation sites in FOXO1 retains the repression of PPARγ1 promoter, and partly dampens the suppression of PPARγ2 promoter in either basal or insulin-stimulated cells (6).
Since acetyl-modification affects FOXO1 activity, the deacetylase SirT2 blocks adipocyte differentiation (50). SirT2-mediated deacetylation increases the association of FOXO1 with PPARγ, and knockdown of SirT2 restores PPARγ activity as observed with the use of constitutively acetylated FOXO1 (Fig. 5) (115). The trans-repression of FOXO1 is independent and dissectible from its trans-activation effect that is DBD dependent. The transrepression requires an evolutionally conserved 31 amino acid region containing the LXXLL-motif (28). Regardless, insulin prevents FOXO1–PPARγ interactions and diminishes the trans-repression of PPARγ genes by Akt-catalyzed phosphorylation and nuclear exclusion of FOXO1, enabling insulin signal to promote PPARγ-driven adipogenesis (28).
In line with the negative role for FOXO1 in adipocyte differentiation, FOXO1 might arrest the cell cycle that is required in the early stages of adipose conversion, via cell cycle inhibitor p21 (89). Constitutively active FOXO1 inhibits differentiation of the preadipocyte cell line 3T3-F442A cells, whereas haploinsufficiency of FOXO1 restores the size of white adipocytes in mice under a high-fat diet (89). Transgenic mice, which selectively overexpress the dominant-inhibitory FOXO1 in adipose tissue, exhibited improved glucose tolerance and insulin sensitivity, and increased energy expenditure (88). Overexpression of dominant-negative FOXO1 in white adipose tissue (WAT) increases fat mass, number of small adipocytes, and upregulates adiponectin and Glut 4. Overexpression of the same mutant FOXO1 in brown adipose tissue (BAT) increases oxygen consumption and upregulates the Ppargc1 and uncoupling protein 1 that promote mitochondrial metabolism (88). In particular, the effects of overexpression of mutant FOXO1 on glucose tolerance and insulin sensitivity under a high-fat diet are greater than under a normal diet (88). These findings support the paradigm of improving energy and nutrient homeostasis in adipose tissue through FOXO1 ablation that increases energy store in WAT and increases energy expenditure in BAT.
Therefore, FOXO1 can be an attractive target to improve the energy homeostasis in adipose tissue. Inhibition of FOXO1 activates PPARγ/RXRα but suppresses cell cycle inhibitor p21, which promotes adipocyte differentiation and maintenance. Targeting FOXO1 can eventually increase energy store in WAT and increases energy expenditure in BAT.
FOXO1 in Pancreatic β-Cells
Pancreatic β-cells respond to the elevation of circulating glucose and other nutrients, and secrete insulin that triggers the PI3K → Akt → FOXO1 signal cascade to adapt to changing metabolic demands and maintains nutrient homeostasis in both β-cells and peripheral tissues. During this process, β-cells metabolize glucose, increasing the [ATP]/[ADP] ratio to inhibit the K+-ATP channel and open the voltage-gated calcium channels that promote the secretion of insulin-containing granules (75). FOXO1 is the most predominantly expressed isoform in isolated mouse islets compared with FOXO3 and FOXO4 (62), and has been implicated in the regulation of proliferation (growth) and stress resistance of pancreatic β-cell (15, 33, 61).
The development and proliferation of β-cells require multiple transcription factors—pancreatic and duodenal homeobox 1 (PDX1), neurogenin 3, and cytokeratin 19—that induce expression of insulin, islet amyloid polypeptide, and Glut2, but suppresses glucagon expression (1, 3). In the human fetal pancreas FOXO1 has been found to negatively regulate β-cell differentiation by suppressing PDX1, neurogenin 3, and NKX61 (3). Disruption of PDX1 in mice leads to the development of type II diabetes due to impaired expression of both Glut2 and insulin (1). PDX1 transcription is regulated positively by FOXA2 but negatively by FOXO1 (62, 68). Because these 2 forkhead transcription factors share common DNA-binding sites in the Pdx1 promoter, FOXO1 competes with FOXA2 for binding to Pdx1 promoter and inhibits PDX1 transcription (62). Moreover, nuclear translocation of FOXO1 is prone to exclude PDX1 from the nuclei, or vice versa (3, 57, 62). This serves as a second mechanism that FOXO1 inhibits β-cell proliferation (Fig. 6). As such, Irs2 knockout mice that show highly nuclear distribution of active FOXO1 suffer from β-cell failure (62). Functional β-cells expressing Irs2 repopulated the pancreas, restoring sufficient β-cell function to compensate for insulin resistance in the obese mice (73). Remarkably, the ablation of one allele of FOXO1 restores β-cell proliferation (62), and the beneficial effect of one-allele-of-FOXO1 deletion was also observed in PDX1 knockout mice (β-cell-specific knockout for PDX1) (39). On the other hand, transgenic mice that selectively overexpress Akt in β-cells increase β-cell survival and cell size (24). Recently, downregulation of Akt activity was implicated in dexamethasone and prostaglandin E2-induced pancreatic β-cell dysfunction, in which c-jun-N-terminal kinase 1 activation leads to nuclear accumulation of FOXO1 and nucleo-cytoplasmic shuttling of PDX1 (57, 84, 122). This mechanism is implicated in fatty acid-induced β-cell apoptosis and endoplasmic reticulum stress-induced β-cell dysfunction (79). Inhibition of either c-jun-N-terminal kinase or FOXO1 protects pancreatic β-cells against dysfunction (57, 79, 84, 122).
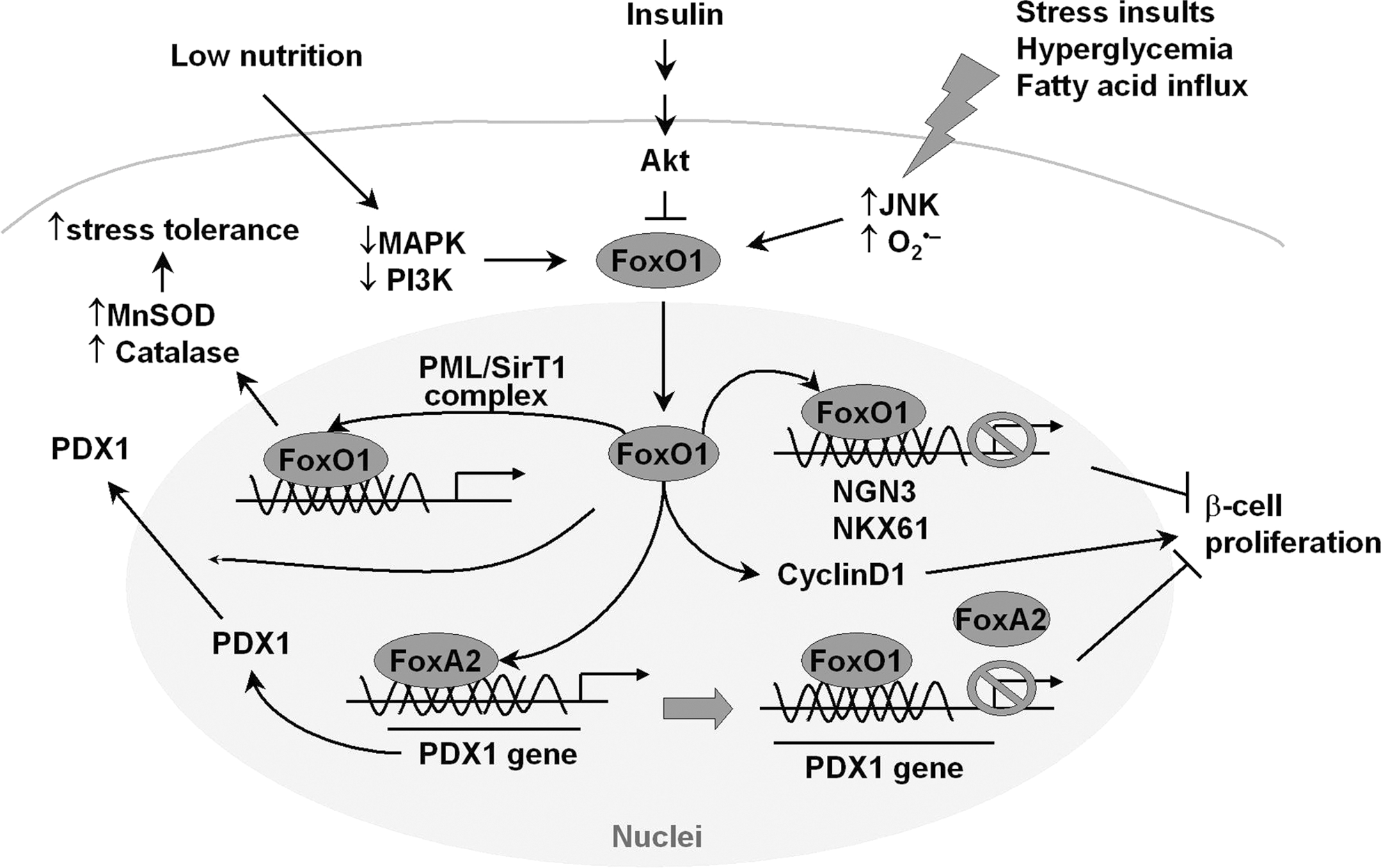
During stress conditions, however, nuclear translocation of FOXO1 protects β-cell against oxidative stress-induced failure (63) or promotes its proliferation in low nutrition (2) (Fig. 6). During hyperglycemia, glucose follows the enolization pathway to generate superoxide that induces oxidative insult because intracellular glucose is overloaded during glycolysis in the β-cell (98). In this case, FOXO1 enters the nuclei of β-cells and initiates NeuroD/MafA pathway to protect against pancreatic β-cell failure, by forming a complex with the promyelocytic leukemia protein and the NAD+-dependent deacetylase SirT1 (63). Most recently, the study of β-cells exposed to low nutrition reveals an additional beneficial role of FOXO1 activation: overexpression of FOXO1 promotes the proliferation of cultured pancreatic β-cells exposed to low nutrition, without obvious change in apoptosis compared with the control group (2). This study with specific inhibitors reveals that PI3 kinase and MAPS kinase signaling pathways are dampened, and that the induction of CyclinD1 expression by activated FOXO1 in low nutrition is responsible for the improved proliferation of β-cells (2).
Therefore, FOXO1 appears to play a dual role in regulating β-cell function: to increase stress resistance and to inhibit the proliferation of β-cells. This highlights the significance of a balanced FOXO1 activity in β-cell regulation under physiological conditions, and should be true in the regulation of other organs. Hyperactive or hypoactive FOXO1 could result in β-cell failure in either case.
Perspective and Conclusion
FOXO1 is a regulated transcription factor that is studied extensively in terms of its structure, activity regulation, and function in energy/nutrient homeostasis. Using genetically generated mouse models, FOXO1 has been shown to regulate general nutrient homeostasis; hepatic glucose production; insulin secretion in β-cells and β-cell growth; survival and function; and fat and muscle masses. Evidence from tissue-specific mouse models reveals an organ-dependent pattern of FOXO1 function, and suggests that FOXO1 could be a therapeutic target for metabolic diseases with insulin resistance, such as obesity, diabetes, and nonalcoholic fatty liver diseases.
However, a few outstanding questions remain to be addressed before therapeutic strategies can be developed for the metabolic diseases. First, at the molecular level, multiple posttranslational modifications, including phosphorylation, acetylation, methylation, glycosylation, and ubiquitinylation, modulate FOXO1 in response to nutrient or stress stimuli, which switch on or off FOXO1 activity. How these posttranslational modifications coordinate with each other to regulate FOXO1 activity remains elusive or controversial. Deacetylation of FOXO1 by SirT1 represses its transcriptional activity (13, 86, 120), but other evidence suggests that FOXO1 acetylation increases its phosphorylation and nuclear exclusion that promotes ubiquitination-mediated degradation (82a). Second, while FOXO1 appears to exert organ-dependent function in regulating metabolism, how FOXO1 achieves the cross talk between tissues is largely unsettled. For instance, knockout of insulin receptor in the pancreatic β-cell [β-cell-specific insulin receptor knockout (βIRKO) mice] conceivably activates FOXO1 and blunts PDK1 and insulin secretion, and the βIRKO mice did exhibit a selective loss of acute insulin release in response to glucose and a progressive impairment of glucose tolerance (64). However, β-cell and muscle insulin receptor double-knockout mice show an improvement rather than a deterioration of glucose tolerance when compared with βIRKO mice (83). Third, although ablation of FOXO1 appears to be beneficial to improve metabolism in many tissues, the physiological role of FOXO1 remains to be defined. For example, activation of FOXO (including FOXO1) has been shown to induce atrophy and heart dysfunction, and inhibition of transcription factors attenuates atrophy (99). Paradoxically, homozygous FOXO1 deletion is embryonic lethal as a consequence of incomplete vascular development (42). Moreover, FOXO1 does not necessarily act as a culprit of pathogenesis. Instead, FOXO1 can elicit signal cascade to increase stress tolerance in multiple organs (63, 113) or increase insulin sensitivity in adipose tissue (6, 7). In contrast to the observation of increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis (111), constitutive active FOXO1 was suggested to suppress lipogenesis in transgenic mice (121) and normalize fatty acid synthase expression in phosphatase and tensin homolog null cells (40). Therefore, the differential physiological roles of FOXO1 in metabolism, development, and stress resistance must be well established and taken into consideration before targeting FOXO1 for clinic therapy of metabolic diseases.
Footnotes
Acknowledgments
This project was supported by U.S. National Institutes of Health grants DK38712 and DK55326 (M.F.W.), and American Diabetes Association Mentor–based Postdoctoral Fellowship 7-08-MN-63 (M.F.W. and Z.C.). We apologize to the colleagues whose work is not specifically referenced owing to space limitations.