
Abstract
Heme oxygenases (HOs) are the rate-limiting enzymes in the catabolism of heme into biliverdin, free iron, and carbon monoxide. Two genetically distinct isoforms of HO have been characterized: an inducible form, HO-1, and a constitutively expressed form, HO-2. HO-1 is a kind of stress protein, and thus regarded as a sensitive and reliable indicator of cellular oxidative stress. The HO system acts as potent antioxidants, protects endothelial cells from apoptosis, is involved in regulating vascular tone, attenuates inflammatory response in the vessel wall, and participates in angiogenesis and vasculogenesis. Endothelial integrity and activity are thought to occupy the central position in the pathogenesis of cardiovascular diseases. Cardiovascular disease risk conditions converge in the contribution to oxidative stress. The oxidative stress leads to endothelial and vascular smooth muscle cell dysfunction with increases in vessel tone, cell growth, and gene expression that create a pro-thrombotic/pro-inflammatory environment. Subsequent formation, progression, and obstruction of atherosclerotic plaque may result in myocardial infarction, stroke, and cardiovascular death. This background provides the rationale for exploring the potential therapeutic role for HO system in the amelioration of vascular inflammation and prevention of adverse cardiovascular outcomes. Antioxid. Redox Signal. 14, 137–167.
I. Introduction
II. HO Expression
A. Heme oxygenase-1
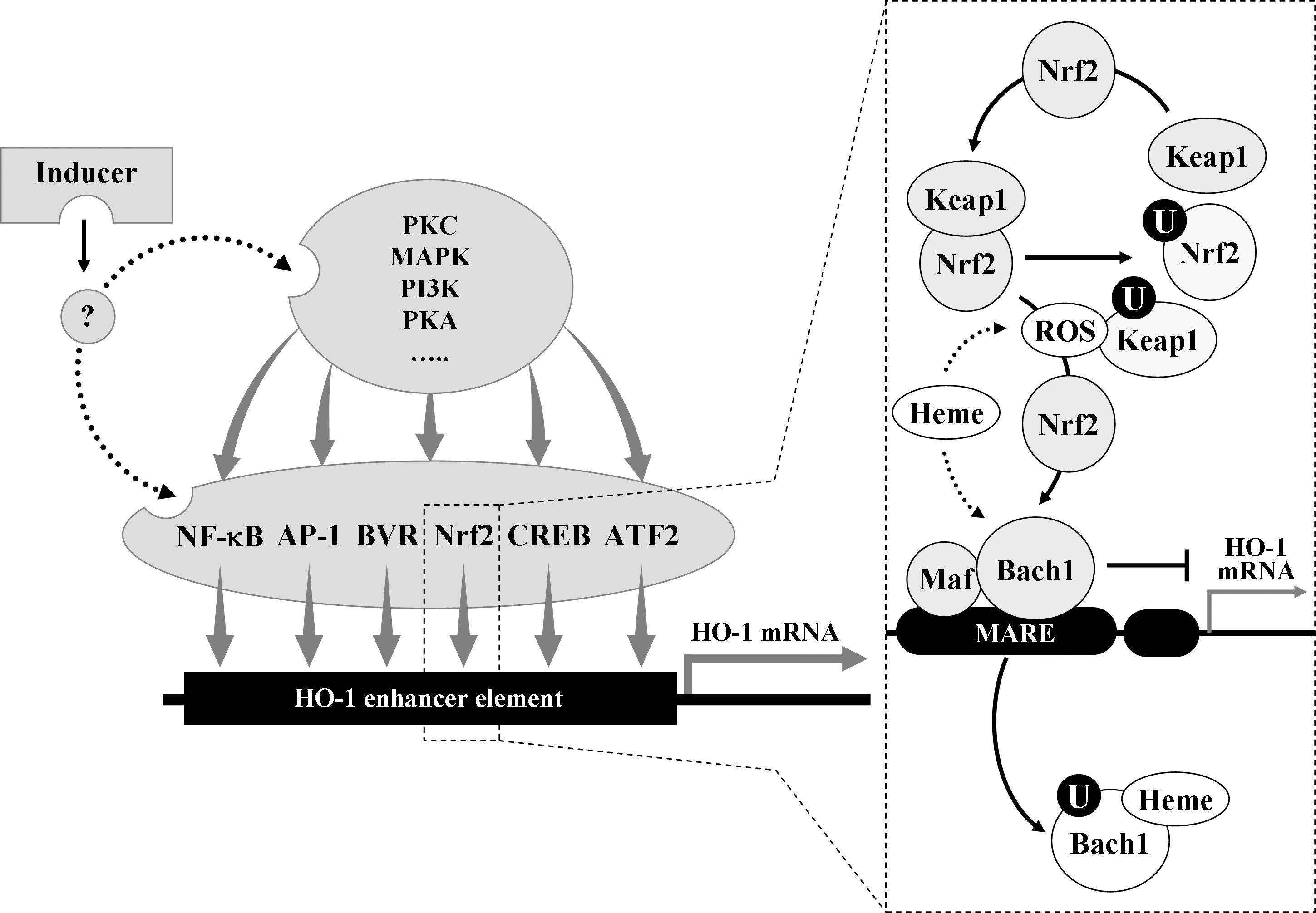
HO-1 expression is induced transcriptionally by a large number of pharmacological agents as well as a variety of circumstances, such as heat shock and other forms of extracellular and intracellular stresses (218). HO-1 expression in human cells is repressed under thermal stress or hypoxia, or by the treatment with interferon-γ [reviewed in (233)], which may represent a regulatory mechanism that prevents the noxious induction of HO-1. Because the modulation of HO-1 activity is of potential therapeutic value (142, 241), the detailed understanding of the mechanisms responsible for the transcriptional activation of the ho-1 gene (Hmox-1) is imperative. A number of signaling molecules and transcription factors have been indentified to be involved in regulating HO-1 expression [reviewed in (270)]. These molecules include mitogen-activated protein kinases (MAPKs), nuclear factor E2-related factor 2 (Nrf2), Bach1 (bric-à-brac, tramtrack and broad complex and cap ’n’ collar homology 1), protein kinase C, protein kinase A, phosphatidyl inositol 3-kinase/Akt, activator protein-1 (AP-1), nuclear factor-κB (NF-κB), cyclic adenosine monophosphate-responsive element-binding protein, BVR, and activating transcription factor 2. It is most likely that HO-1 expression may result from the complex cooperative interactions between these molecules (Fig. 1). Because a number of investigations have implicated the involvement of MAPK pathways in the regulation of HO-1 expression and have focused on characterizing the roles of Nrf2/Bach1 system in Hmox-1 activation in diverse cell types in response to various inducing conditions, MAPKs and Nrf2/Bach1 system, together with brief comments on AP-1 (which has previously been proposed to mediate Hmox-1 activation) and BVR (which has recently been recognized as a new transcription factor for Hmox-1 activation), are discussed below.
1. Mitogen-activated protein kinases
The MAPKs are a family of serine-threonine protein kinases that regulate many cellular events, including responses to environmental stimuli (199). The MAPK superfamily encompasses three important signaling pathways: the extracellular regulated kinases (ERK) pathway, the c-Jun N-terminal kinases or stress-activated kinases (JNK) pathway, and the p38 MAPKs (p38 pathway). A variety of structurally and functionally diverse agents that induce HO-1 expression also in parallel can activate MAPK cascades in multiple cell types (126, 137, 195), implicating the involvement of MAPK pathway in the regulation of HO-1 expression. Simvastatin, a low-density lipoprotein (LDL) cholesterol-lowering drug, stimulates HO-1 expression in vascular smooth muscle cells (VSMCs) through p38 MAPK pathway (140). Atrial natriuretic peptide, a cardiovascular hormone, induces HO-1 expression in endothelial cells (ECs) through ERK pathway (89). Salicylate, the active metabolite of aspirin, induces expression of HO-1 in ECs via JNK pathway (88). The inhibition of ERK, JNK, or p38 MAPK activation individually is insufficient to completely abolish HO-1 expression by oxidized LDL (Ox-LDL); however, inhibition of these MAPK pathways in combination results in a significantly greater attenuation of HO-1 expression (12), suggesting that these MAPK pathways act in concert to regulate HO-1 expression. The terminal, activated MAPKs phosphorylate their downstream targets. In the case of Hmox-1 transcriptional events, downstream targets of MAPKs may be the transcription factors that directly regulate ho-1 gene activation and/or components that are indirectly involved in activation of the transcription factors.
2. Nrf2 and Bach1
Nrf2, which belongs to the cap ’n’ collar family of transcription factors, controls a critical cellular defense response by coordinated upregulation of many of its target genes, culminating in a cell survival response. Deletion mutagenesis and transfection studies have identified an element designated as the antioxidant response element (ARE) in the promoter regions of Nrf2 downstream genes (215). Molecular cloning and bioinformatics have shown that the promoter of Hmox-1 contains numerous sequence-related cis-elements, a number of which are clustered around −4.0 and −9.0 kb (215). In the mouse, the regions of Hmox-1 promoter around −4.0 and −9.0 kb have been called enhancer 1 and enhancer 2, respectively, and contain multiple stress response elements (StREs) that are likened to Musculo-aponeurotic fibrosarcoma (Maf) protein recognition elements [reviewed in (111)]. The majority of the StREs in Hmox-1 contain the core ARE sequence. Because Hmox-1 has been found to contain functional AREs in the promoter region, HO-1 expression is considered highly likely to be regulated directly by Nrf2.
Under basal conditions, Nrf2 is rapidly ubiquitinated by forming a complex with the Kelch-like ECH-associated protein 1 (Keap1) and degraded by the 26S proteassome [reviewed in (126)]. Keap1 serves as a bridge between Nrf2 and the Cullin-3-dependent E3 ubiquitin ligase complex, leading to ubiquitination of multiple lysine residues located in Nrf2 domain. Keap1 targets Nrf2 for ubiquitin-dependent degradation and, hence, represses Nrf2-dependent gene expression (126). It has been initially proposed that Keap1-dependent degradation of Nrf2 occurs in the cytoplasm (111). Recently, it has also been proposed that Nrf2, even under normal condition, is localized in the nucleus where Nrf2 is targeted for degradation by Keap1 [reviewed in (186)]. It is most likely that oxidative stress may somehow prevent Keap1-dependent degradation of Nrf2 in either the cytoplasm or the nucleus, perhaps resulting in Nrf2 nuclear accumulation. Accumulated or stabilized Nrf2 may bind to ARE sequences to form complexes with the small Maf proteins (111, 126), leading to initiation of Hmox-1 transcription. Thus, Keap1 has been shown to be the main cytosolic inhibitor of Nrf2.
The Bach1 protein is a potent repressor of HO-1 expression. It also binds to StREs (i.e., AREs) in the promoter of Hmox-1 as a heterodimer with small Maf proteins and competes against Nrf2 for binding to the ARE sequences [reviewed in (96)]. In response to oxidative stimuli, Bach1 is exported from the nucleus, ubiquitinated and degraded, allowing the formation of Nrf2/small Maf protein complexes (96). This hypothesis is supported by the findings that Bach1-deficient cells express constitutive high levels of HO-1 mRNA (190), which is not observed in cells lacking both Nrf2 and Bach1. However, it is most likely that the Bach1/Nrf2 transcriptional system may interact functionally with other transcription factors to regulate Hmox-1 transcription.
3. Activator protein-1
AP-1 is a dimeric combination of basic leucine zipper proteins of the Jun and Fos family, Jun dimerization partners, and the closely related activating transcription factor subfamilies [reviewed in (7)]. Due to sequence similarity of the Maf protein recognition elements with the consensus AP-1 binding site, AP-1 factors were proposed to regulate Hmox-1 activation in response to multiple stimuli. A study has demonstrated that the phorbol ester, a classical activator of AP-1 factors, induced HO-1 expression in mice through AP-1 binding to the 5′-flanking region of the target gene (8). Other studies have also demonstrated that expression of HO-1 by sodium arsenite (74) and bacterial lipopolysaccharide (LPS) (42) is mediated by AP-1 activation. Because Maf proteins can form part of the AP-1 complex, it is likely that the dissociation of Bach1 from Maf proteins would enhance the binding of other positive transcriptional regulators of Hmox-1, such as Nrf2.
4. Biliverdin reductase
BVR enables continuous protection of cells against oxidative stress by its conversion of BV to BR, but this is not a sole function of BVR (82, 141, 151). BVR has been shown to have a wide spectrum of its potential functions in cell signaling pathways. BVR has two DNA binding sites known as AP-1 recognition sequences (82). Thus, it may bind to AP-1 recognition sequences of DNA to play a role in the AP-1 pathways of cellular signaling. Because AP-1 binds to multiple copies of consensus sequence in the ho-1 promoter (7), BVR could also bind to DNA of ho-1 promoter to activate Hmox-1 transcription (82). In addition, BVR binds to cyclic adenosine monophosphate-responsive element-binding protein recognition sequences of ho-1 promoter [reviewed in (125)], which may also activate Hmox-1 transcription. In human BVR-infected cells, levels of HO-1 mRNA and protein were increased, while blockage of BVR synthesis by small interfering RNA (siRNA) attenuated chemical-mediated increase in HO-1 expression (164).
B. Heme oxygenase-2
Under physiological conditions, HO-2 expression is found in various tissues, particularly in testis and brain, whereas HO-1 expression is relatively low, with the exception of spleen [reviewed in (218)]. HO-2 expression can be transcriptionally modulated by corticosteroids [reviewed in (154)] but may not by the environmental stress that can remarkably induce HO-1 expression at transcriptional level (218). It is most likely that relatively constant expression levels of HO-2 may be suitable for its regulatory role in heme homeostasis. However, HO-2 activity may be dynamically regulated via posttranslational mechanisms that involve protein phosphorylation (29). Unlike HO-1, HO-2 contains three cysteine residues, each of which is present as a dipeptide of cysteine and proline and may function as the heme-binding site (233). It has been, therefore, postulated that HO-2 may play a regulatory role by sequestering heme to maintain the intracellular heme level (233). It has also been postulated that HO-2 may function as an oxygen sensor (129). In addition, mice deficient in HO-2 have revealed several important functions of this enzyme. For example, HO-2 deletion causes EC activation marked by oxidative stress, inflammation, and angiogenesis (23), which underscores the importance of HO-2 in the regulation of EC homeostasis. HO-2 deficiency disables execution of the acute inflammatory and reparative response after epithelial injury and leads to an exaggerated inflammatory response in antigen-induced peritonitis (231), implicating a role for HO-2 in the regulation of the inflammatory and reparative response to injury. HO-2 deletion has been associated with impaired HO-1 induction (238), which may explain the reason why HO-2-null mice could not compensate for the loss of HO-2 by increasing HO-1 expression. Thus, it is most likely that HO-2 may be critical for HO-1 expression.
III. HO By-Products
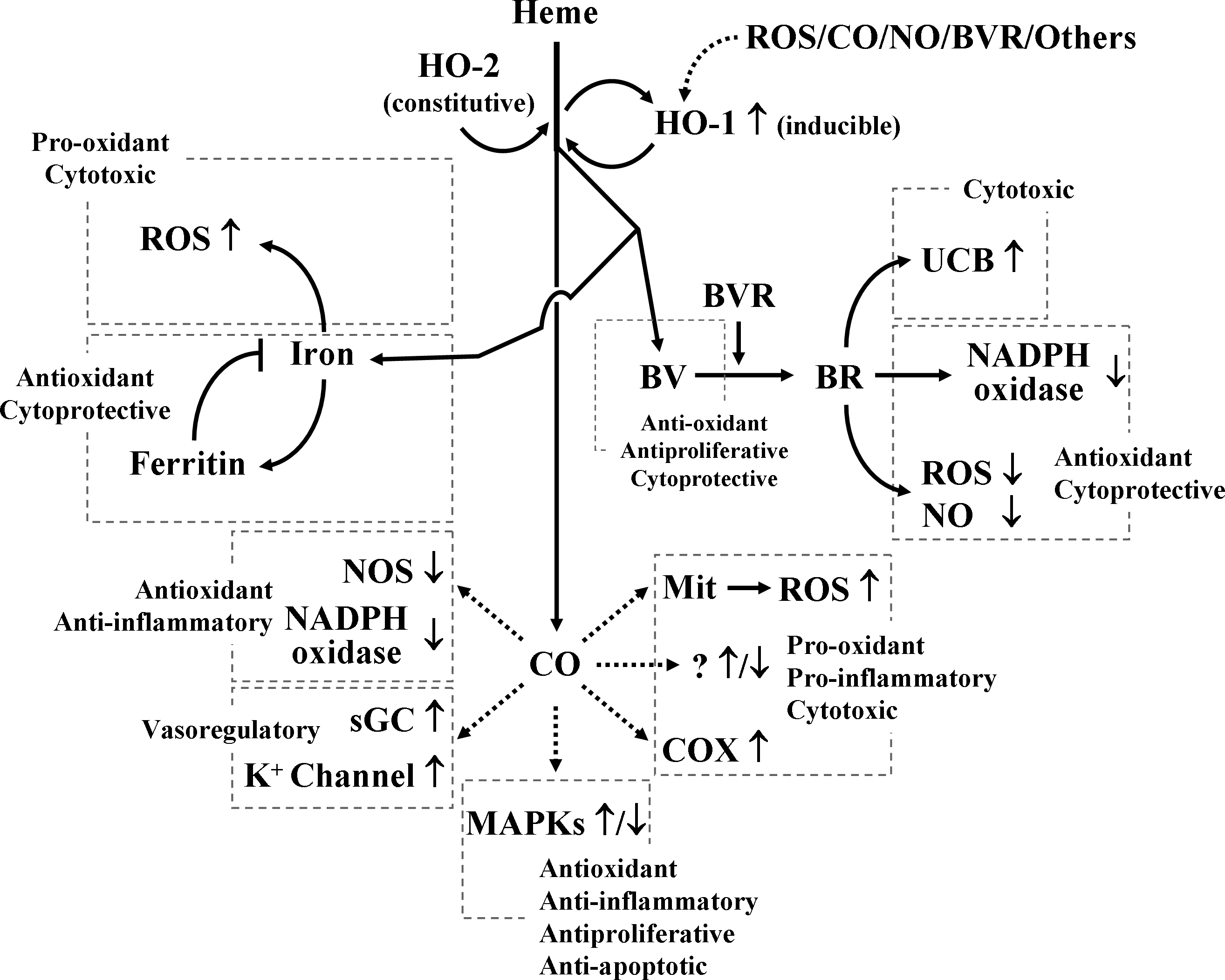
The protective effects of HO-1 have been confirmed in a number of experimental models, but the mechanisms of its multiple actions have not been completely elucidated. Heme, even though the heme is an essential prosthetic group of various enzymes in the biological system, it is inherently dangerous particularly when released from intracellular heme-containing proteins. The extreme hydrophobicity of free heme permits it to intercalate into cell membranes, resulting in increased cellular susceptibility to oxidant-mediated damages as well as generation of reactive oxygen species (ROS) (103). Once heme is in the cell membrane, hydrogen peroxide from sources such as activated leukocytes can split the heme ring and release free redox-active iron, which can catalytically amplify the production of ROS inside the cell (21, 27, 103). Thus, degradation of free heme by HO-1 appears to first aid in tissue protection; however, recent evidence suggests that one or more of the by-products of its heme catabolism (e.g., CO, ferrous iron and BV/BR) mediate the protective effects of HO-1 (Fig. 2).
A. Carbon monoxide
CO is classically thought of as a toxic molecule and cellular asphyxiate, but recent studies have revealed that CO has profound effects on the intracellular signaling processes, culminating in antiinflammatory, antiproliferative, and antiapoptotic effects (198, 199). These effects of CO may involve three general mechanisms; (a) CO can activate soluble guanylyl cyclase (sGC), with the resultant production of cyclic guanine monophosphate (cGMP), (b) CO can modulate MAPK pathway, and (c) CO can bind to a range of intracellular heme proteins, thereby resulting in the formation of metal-CO complexes that may generate diverse biological effects, including inhibition or activation of heme-containing enzymes. CO, like nitric oxide (NO), binds to the heme moiety of sGC, leading to the stimulation of sGC and subsequent elevation of cGMP levels that causes protective vascular relaxation (199). Besides binding to sGC, CO may bind to the heme moiety of other hemoproteins thus affecting their enzymatic activity; there is the experimental evidence about the stimulatory role of CO on cyclooxygenase (COX) activity [reviewed in (158)]. Hemin, the substrate of HO stimulated COX to produce prostaglandin E2 in rat hypothalamic explants and in primary cultures of rat hypothalamic astrocytes (156, 159). This effect was inhibited by the HO inhibitor tin-mesoporphyrin-IX and abolished by the CO scavenger hemoglobin (156, 159). Moreover, treatment of rat hypothalamic explants with CO-saturated medium resulted in significant increases in prostaglandin E2 release (156). All these data provided experiment evidence supporting that CO could stimulate COX activity in the rat hypothalamus.
CO induces a general downregulation of pro-inflammatory cytokine production through the MAPK-dependent pathway, leading to antiinflammatory tissue protection (218). Antiapoptotic and antiproliferative actions of CO also involve the modulation of MAPK pathways (169, 218). It has been shown that the effects of CO on MAPK signaling pathway may depend on cell types and stimuli. For examples, the antiapoptotic effect of CO was exerted via the p38 MAPK signaling pathway in human ECs (31), whereas CO-mediated antiapoptotic effect via ERK signaling pathway in rat hepatocytes (56). Whereas CO inhibited the proliferation of pancreatic stellate cell by activating p38 MAPK (226), CO suppressed the proliferation of human airway SMCs by inhibiting ERK activation (240). The mechanisms by which CO can modulate MAPK pathways are not clear. CO slows the rate of electron transport, enabling electrons to accumulate at complex III in mitochondria, and this promotes the formation of ubi-semiquinone from which electrons can be donated to produce superoxide and its conversion to H2O2 by manganese superoxide dismutase (17). It is most likely that the mitochondrium-derived H2O2 may activate the MAPK and Akt signaling pathways [reviewed in (27)].
Besides its general protective mechanisms, CO may have an ability to activate other protective pathways. CO expose induced expression of heat shock protein (HSP) 70, which plays vital roles in its protective effects against cytokine-induced EC apoptosis (131). Treatment of PC12 cells with a CO donor upregulated expression of the catalytic subunit of glutamate-cysteine ligase, the rate-limiting enzyme in glutathione biosynthesis, and this was associated with the protective effects of CO against NO-induced cell death (54).
CO does not always exert cytoprotective activity; CO also produces noxious effects in certain organs (e.g., the brain) (233). CO increases the formation of pro-inflammatory prostaglandins by activating COX in rat hypothalamic explants and in primary culture of rat hypothalamic astrocytes (see references in 153, 154, 233), suggesting that CO stimulates pro-inflammatory responses at least in the brain. Moreover, CO reduces cellular levels of antioxidants (e.g., glutathione) by increasing mitochondrial ROS formation through the binding of CO to cytochromes residing in complex IV (i.e., cytochrome a 3) [reviewed in (27)], indicating that CO may cause oxidative tissue damages. Whether the effects of CO would be protective or not, in all probability, may depend on several factors, including the type of cells, the concentration of CO produced or administered, and the tissue-specific signaling transduction pathway(s) that may be involved in its biological activity, and this has been well reviewed by Mancuso and Barone (154).
B. Ferrous iron and ferritin
Ferrous iron, an extremely pro-oxidative molecule, is released during the breakdown of free heme by HO-1, but this molecule is rapidly removed by ferritin, a ubiquitously existing intracellular protein that is able to effectively sequester intracellular iron and, hence, limits the pro-oxidant capacity of ferrous iron. It is most likely that increased ferritin expression in conjunction with HO-1 expression may contribute to additional protection afforded by HO-1 (16, 199, 218).
C. BV and BR
BV and BR have been shown to provide vascular protection by preserving EC integrity, preventing EC death, enhancing vascular reactivity, and inhibiting restenosis (218, 252). In the cellular system, BV resulting from the HO-1 catabolism of free heme is rapidly converted into BR. BV and BR are now recognized as being potent antioxidants manufactured by the body (147). Indeed, BV and BR can directly scavenge ROS and interact with the free radical NO and the oxidant peroxinitrite (155, 157, 159). BR also exerts a potent suppression of the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that generates the free radical superoxide (62, 86). The mechanisms by which BR can inhibit NADPH oxidase activity are not clear, but independent of its ROS-scavenging effect (62). Moreover, general antiinflammatory effects of BV and BR based on their antioxidant potentials have been described (94, 199).
IV. Effect of HO-1 on Vascular Inflammation
HO-1 serves as a protective gene by virtue of its antiinflammatory, antiapoptotic, and antiproliferative actions, as being variously manifested in endothelial, epithelial, smooth muscle, and other cell types (31, 138, 207, 263). HO-1 is also involved in blood vessel relaxation regulating vascular tone and participates in blood vessel formation by means of angiogenesis and vasculogenesis. Current accumulating data have highlighted the critical importance of HO-1 in ameliorating vascular inflammation in various diseases (11, 22, 116, 177, 178, 246, 257). The scope of this section will be to address the recent discoveries on the role of HO-1 in vascular inflammation, with special emphasis on its antiinflammatory mechanisms implicated in various mediators and cells, especially macrophages and ECs.
A. Cytokines, chemokines, and mediators
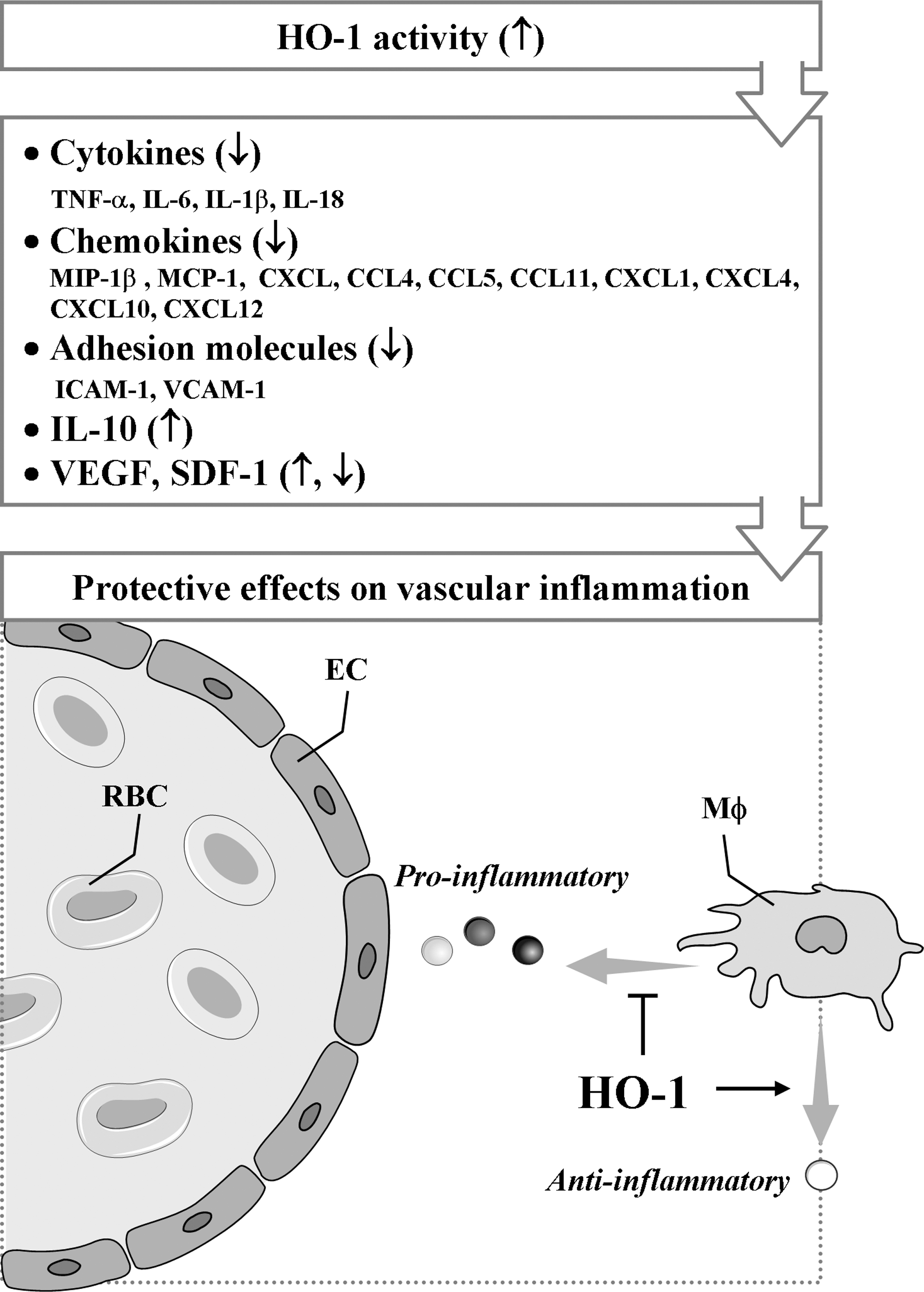
Various cytokines are implicated in vascular inflammation (259). HO-1 and its metabolites are well known to attenuate the secretion of pro-inflammatory cytokines and to augment the production of antiinflammatory cytokines (Fig. 3). Actually, HO-1 reduces the production of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, whereas it enhances IL-10 expression in vitro (183, 197). In addition, HO-1 upregulation blocks IL-18 signaling and reduces IL-18-dependent vascular injury and inflammation (297). IL-18 contributes to both systemic and acute inflammation by inducing expression of TNF-α and other pro-inflammatory cytokines and activation of NF-κB. A study has demonstrated that HO-1-dependent blockade of NF-κB, which can be induced by IL-18, is associated with reduction in EC death (297).
CO has been shown to be responsible for the inhibition of pro-inflammatory cytokine production, including TNF-α, IL-1β, IL-6, and macrophage inflammatory protein (MIP)-1β, in macrophages stimulated with LPS (193). The effects of CO on its regulation of pro-inflammatory and antiinflammatory cytokine production, at least, in macrophages are exerted mainly via the p38 MAPK signal pathway [reviewed in (130)]. In addition, upregulation of IL-10 is a further mechanism responsible for the antiinflammatory actions of CO (193). IL-10 can downregulate the pro-inflammatory response only when HO-1 expression or activity is upregulated in response to IL-10 (139). When HO activity is blocked pharmacologically, the antiinflammatory effect of IL-10 is lost (139). The ability of IL-10 to prevent lethality from endotoxic shock (139) and to attenuate inflammation in aortic allografts (49) is also abolished when HO activity is inhibited pharmacologically. After HO-1 is activated by IL-10, HO-1 itself, along with the products of heme degradation (e.g., CO), can mediate the IL-10 effect (49, 139). Hence, these observations indicate that IL-10 can upregulate HO-1 expression and its activity but also is augmented by HO-1, thereby exerting its antiinflammatory properties. In addition, several studies have suggested that expression of HO-1 is essential for the function of a variety of endogenous molecules and pharmacologic agents used therapeutically to suppress inflammation (1, 15).
CCL2, also known as a monocyte chemotatic protein 1 (MCP-1), was the first chemokine shown to affect atherosclerosis. CCL2 and its receptor are most prominently involved in monocyte recruitment from bone marrow into the arterial wall (230, 266, 285). Overexpression of HO-1 or administration of CO suppresses the production of the chemokines such as MCP-1 or MIP-1β (CCL3). Another HO-1 product, BR has been shown to inhibit MCP-1 expression in human ECs (127), but the underlying mechanism(s) remains to be established. As for vascular pathologies, a study has shown that elevation of IL-8 level in plasma can be a biomarker predicting early recurrence of ischemia, myocardial infarction, and sudden cardiac death after percutaneous coronary interventional procedures (211). Blocking of IL-8 signaling substantially attenuates polymorphonuclear neutrophil infiltration and vascular and tissue injury in the postischemic heart (30). Induction of HO-1 expression/activity has been reported to be associated with reduction in IL-8 production in microvascular endothelium in vitro and in vivo (188), but it is not clear that HO-1 expression could modulate IL-8 production. Other chemokines, such as CCL4, CCL5, and CCL11 are implicated in vascular inflammation; however, their relationship with HO-1 remains to be clarified.
It has been shown that HO-1-mediated resolution of inflammation may originate from the modulation of adhesion molecule expression. Studies have shown that overexpression of HO-1 attenuates expression of adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin, in vitro and in vivo, whereas selective inhibition of HO-1 activity aggravates expression of those adhesion molecules (104, 268, 272, 273). Confirming these observations, Rucker et al. (217) have shown the decrease in ICAM-1 expression in an animal model of arterial injury when HO-1 was upregulated before injury. Sun et al. (248) have also reported in vitro data showing that induction of HO-1 inhibits TNF-α-induced monocyte adhesion to human ECs by suppressing expression of ICAM-1. In addition, HO activity decreases expression of VCAM-1 and ICAM-1 by depleting heme and generating antioxidants (i.e., BV/BR) and another gaseous mediator (i.e., CO), which shares many properties with NO (24, 63).
The role of HO-1 in angiogenesis seems to vary depending on the underlying conditions. Bussolati and coworkers (34, 35) have demonstrated that vascular endothelial growth factor (VEGF)-induced angiogenesis requires HO-1 activity, whereas inflammation-induced blood vessel formation is attenuated by HO-1 overexpression. Thus, a concept that during chronic inflammation, HO-1 may inhibit leukocytic infiltration and facilitate tissue repair by promoting VEGF-driven noninflammatory angiogenesis has been accepted (35). Recently, Deshane et al. (67) have also demonstrated that stromal cell-derived factor-1 (SDF-1) promotes angiogenesis and the function of endothelial precursor cells (EPCs) through a mechanism dependent on HO-1 expression. Although the molecular mechanisms underlying the induction of VEGF and SDF-1 by HO-1 or vice versa remain unclear, it is envisioned that HO-1 and angiogenic factors can activate a positive-feedback circuit to amplify neovascularization in adult tissues. Consistent with these observations, HO-1-mediated cardioprotection after ischemic injury can be offered by promoting neovascularization through inducing expression of VEGF and SDF-1 and the recruitment of circulating progenitor/stem cells (145).
B. Macrophages
Macrophages seem not only to promote inflammation, but also to downregulate it (77, 283, 288) (Fig. 4). Macrophages produce pro-inflammatory cytokines, participate in lipid retention and vascular cell remodeling, and express pattern-recognition receptors (PPRs) during atherosclerosis (90). During apoptosis, however, macrophages can suppress inflammatory responses through phagocytosis of apoptotic debris (214, 216). In addition, macrophages act as the first-line defense against invading pathogens through undergoing immediate oxidative burst to overproduce superoxide anion radicals, delaying overproduction of NO, and thereby leading to generation of peroxynitrite. While the peroxynitrite can kill invading pathogens, this oxidant can also kill macrophages and surrounding host tissues (243). However, macrophages and host cells can protect themselves from the toxicity of various assailants, including peroxynitrite, by enhancing expression of HO-1 and other protective enzymes. Under vascular inflammatory conditions, HO-1 is highly expressed in all the main cell types, including ECs, macrophages, and SMCs, but HO-1 is virtually absent in neighboring unaffected vascular tissue (113, 274).
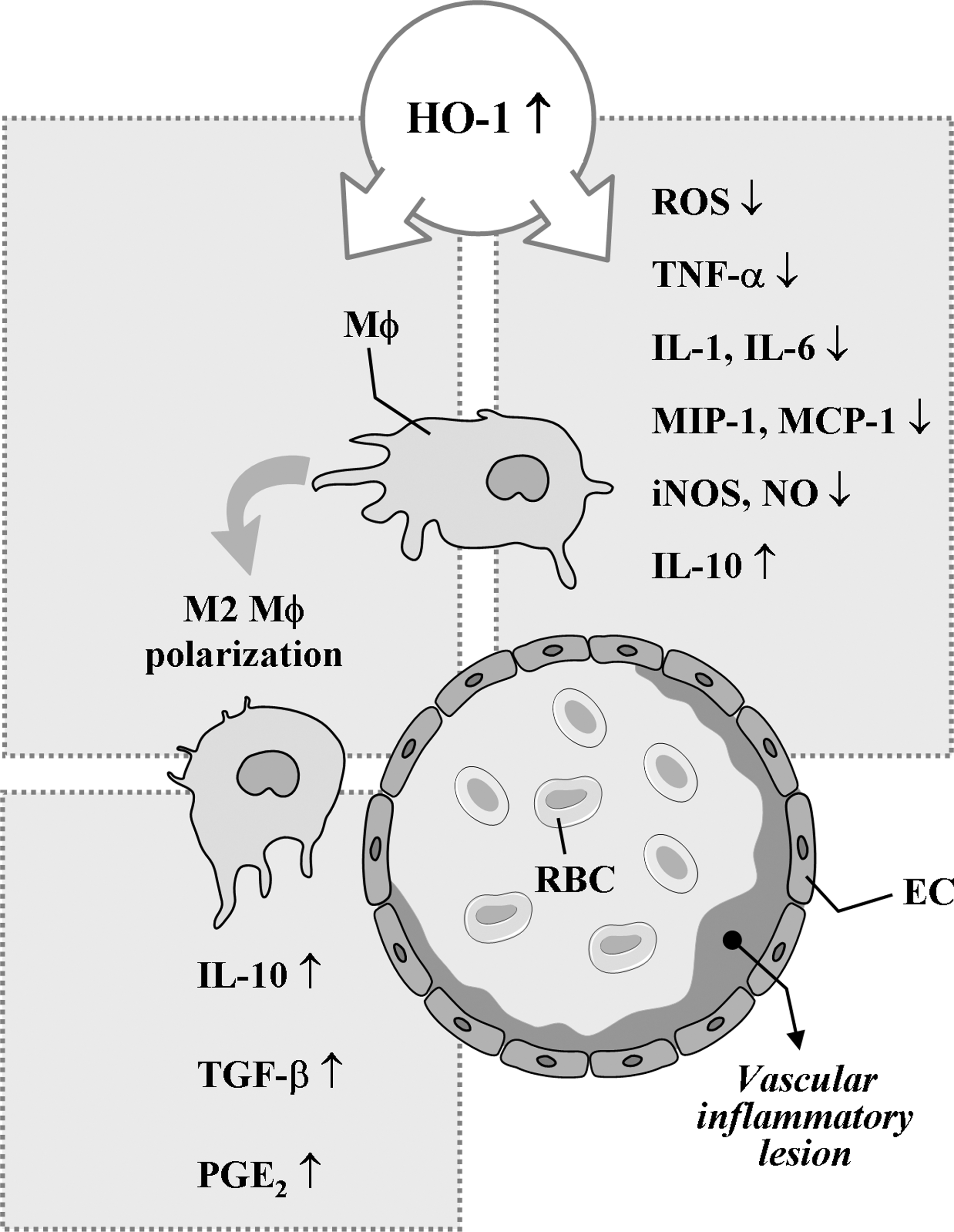
HO-1 can inhibit NO production through suppression of inducible NO synthase (iNOS) expression and HO-1 by-product, BR, has been shown to effectively counteract NO-induced stress through its ability to bind and inactivate NO (37, 39), indicating the possible presence of functional interaction between NO/iNOS and CO/HO-1 systems in regulating inflammation. There is an excellent study supporting the negative feedback regulation of the pro-inflammatory NO/iNOS system by the antiinflammatory CO/HO-1 system (13). Using iNOS-deficient macrophages, Ashino et al. (13) showed that LPS failed to induce HO-1 expression, suggesting that NO/iNOS is implicated in HO-1 expression. In other experimental sets, they investigated iNOS and HO-1 expression by LPS using Nrf2-deficient macrophages. Whereas LPS failed to induce HO-1 expression, LPS strongly induced iNOS expression in Nrf2-deficient macrophages, supporting that CO/HO-1 system can inhibit iNOS expression.
Recent studies have demonstrated that the major site of HO-1 expression is mainly monocytes/macrophages in the inflammatory and ischemic injured areas (48, 99, 140, 149). As expected, HO-1 expression in macrophages is one of the antiinflammatory mechanisms of macrophage. Orozco et al. (192) have shown that HO-1 expression in macrophages constitutes an important component of its antiatherogenic effects. Supporting these in vivo data, macrophages stimulated with LPS produce several pro-inflammatory cytokines, including TNF-α (26). If macrophages overexpress HO-1 or are exposed to CO in vitro before stimulation with LPS, the pro-inflammatory response (i.e., TNF-α production) is markedly inhibited, whereas the antiinflammatory response (i.e., IL-10 production) is enhanced (163). In addition, chalcone potently induces HO-1 expression in murine macrophages, leading to reduced LPS-mediated NO and TNF-α production (5).
A study using a CO-releasing molecule (CORM) has demonstrated that overproduction of CO allows the survival of LPS-stimulated macrophages (a) by eliminating the free heme to prevent Fenton reaction, (b) by limiting the availability of free heme required for induction of NO producing heme enzyme (i.e., iNOS), and (c) by limiting additional production of superoxide and NO via CO-derived inhibition of the activities of heme enzymes like NADPH oxidase and iNOS, allowing the LPS-activated macrophages to return back to the normal quiet state (243). Further, in LPS-stimulated murine macrophages, hydrogen sulfide (H2S), a putative vasodilator, can inhibit NO production and NF-κB activation through a mechanism that involves the action of HO-1/CO, displaying antiinflammatory effects (189). However, H2S can reduce NO reactivity by interacting with it and forming an S-nitrosothiol compound (282).
Macrophages are classically activated by microbial cell wall components and/or interferon-γ. The resulting phenotype is known as M1 macrophage, which is characterized by the production of pro-inflammatory mediators such as NO, superoxide, TNF-α, IL-1β, and IL-6 (93, 95). Polarization toward the alternatively activated phenotype (i.e., M2 macrophage) is achieved by, for example, glucocorticoids, IL-4, IL-13, or IL-10 (94, 160). M1 and M2 macrophages play opposite roles during inflammation, but both are present in vascular inflammatory diseases, such as atherosclerosis. M2 macrophages suppress the release of pro-inflammatory mediators and provoke the formation of antiinflammatory mediators, such as IL-10, transforming growth factor-β, or prostaglandin E2 (79, 87, 271). In addition, the antiinflammatory mechanisms of macrophages are at least in part attributed to defective LPS-induced NF-κB activation (61). A recent work by Weis and colleagues (281) has demonstrated that the establishment of this antiinflammatory phenotype of macrophages is in part dependent on the induction of HO-1 by apoptotic cell-derived sphingosine-1-phosphate. Taken together, these findings suggest that HO-1 expression in macrophage and the downstream effectors could be a promising target for the therapeutic approach for various vascular pathologies.
C. Endothelial cells
Although the molecular basis of the antiinflammatory effects of HO-1 in ECs remains to be fully elucidated, it has been reported that HO-1 overexpression in human ECs reduces TNF-α-induced E-selectin and VCAM-1 expression but not ICAM-1 expression and inhibits monocyte chemotaxis (34, 237) (Fig. 5). In addition, substantial expression of HO-1 induced by hypoxia inducible factor (HIF)-1α attenuates IL-8 secretion in microvascular endothelium in vitro and in vivo (104). The products of HO, such as BV, BR, and CO, have also cytoprotective effects for ECs against various stresses (210, 287). BR inhibits activation of ECs by suppressing expression of E-selectin, VCAM-1, MIP-1, and monocyte/macrophage-colony stimulation factor (127, 202, 237). Notably, CORMs have been shown to have antiinflammatory effects on vascular ECs. CORMs inhibit EC death via suppression of IL-18-mediated NF-κB activation and phosphatase and tensin homolog induction and reversion of IL-18-mediated suppression of Akt activity (265), suggesting that CO donors have the therapeutic potential to block IL-18 signaling and reduce IL-18-dependent vascular injury and inflammation. CORM-3 modulates polymorphonuclear leukocyte migration across the vascular endothelium by reducing levels of cell surface-bound elastase (165), and also inhibits TNF-α-induced expression of VCAM-1 and E-selectin in human umbilical vein ECs (239). CORM-2 attenuates ICAM-1 expression induced by high glucose in ECs (187).
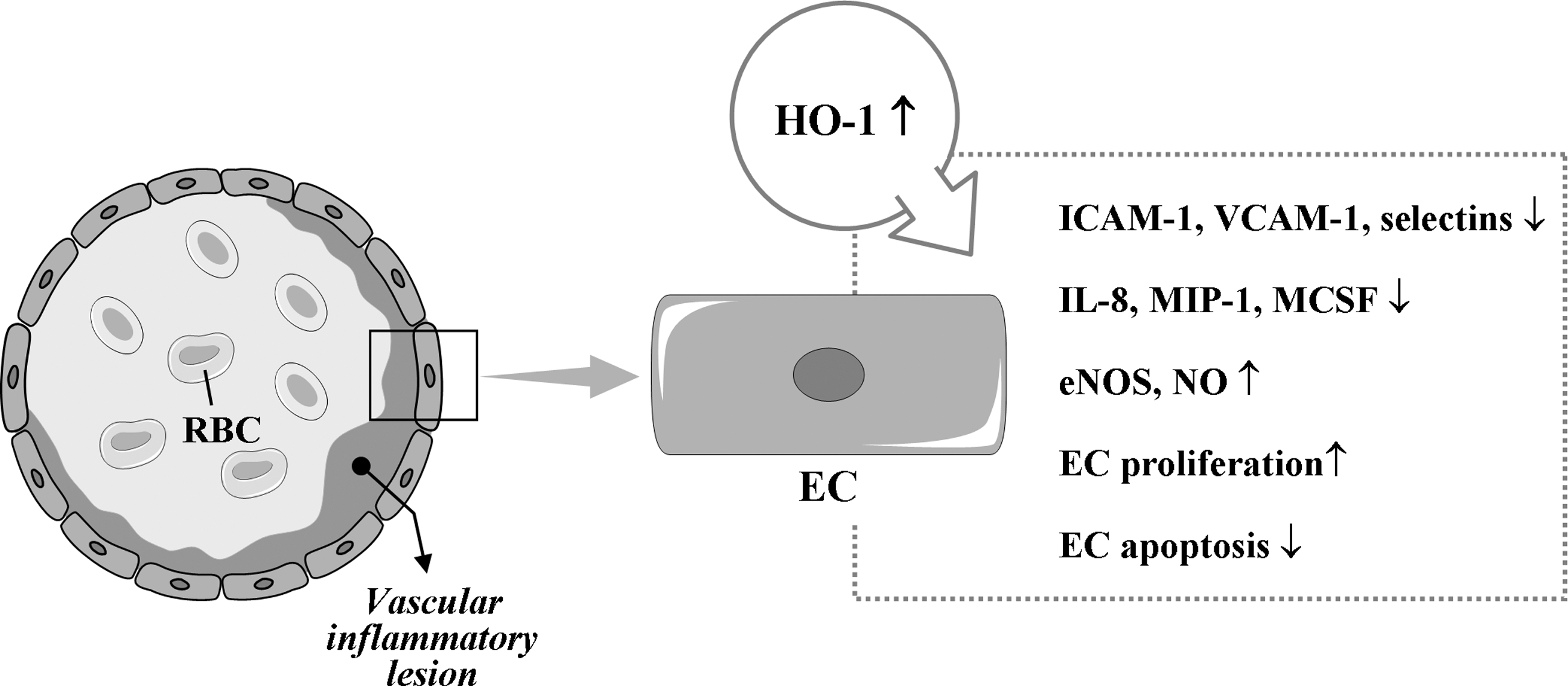
Kim et al. (133) have demonstrated that HO-1 expression by exogenous CO administration in ECs. HO-1 activity, in turn, protected the cells from endoplasmic reticulum (ER) stressors. The novelty of this study is that protein kinase R-like ER kinase-dependent activation of Nrf2 by CO is critical for cell survival signal during ER stress. They suggested a positive feedback loop of the signaling pathway in HO-1/CO axis, in which protein kinase R-like ER kinase-Nrf2 signaling played a critical role in endothelium.
Recently, Kawamura et al. (127) have revealed that BR restores endothelial NO synthase (eNOS) expression and improves endothelium-dependent vascular relaxation responses in atherosclerosis. On the other hand, in diabetic mice, HO-1 induction decreases EC sloughing and fragmentation, partly by a mechanism involving increased eNOS and decreased iNOS expression (137). Conversely, it has been evidenced that NO-producing cells possess regulatory pathways where protective mechanisms can operate to control pro-inflammatory responses via its induction of cytoprotective enzymes, such as HO-1, and thus limit the destructive potential (200). Durante et al. (74) have reported that NO, either exogenously administered or endogenously generated from cytokine-treated cells, selectively induced HO-1 expression and CO release in VSMCs. The mechanism responsible for the induction of HO-1 expression by NO included the activation of Nrf2 that was independent of the MAPK pathways but is dependent on oxidative stress (74). Moreover, the induction of HO-1 by NO functioned in an autocrine manner to limit SMC apoptosis (74). To date, the interrelationship between HO-1 and NO seems to be dependent on type of inflammation/injury and tissues and is not completely elucidated in vascular inflammation.
HO-1 has been demonstrated to stimulate cell cycle progression and proliferation in vascular endothelium (66, 143). The mechanism by which HO-1 is able to stimulate the growth of vascular endothelium is not known; however, the ability of HO-1 to stimulate the synthesis of VEGF from vascular cells may contribute to its proliferative action (71). Additionally, HO-1 can directly affect cell viability by blocking programmed cell death. Soares et al. (236) first demonstrated that the overexpression of HO-1 prevents apoptosis of ECs. Recent in vitro data have revealed that HO-1 induction reverses IL-18-mediated EC death, reducing IL-18-dependent vascular injury and inflammation (269). Further, HO-1-derived CO may contribute to the reendothelialization of the vessel wall at sites of vascular injury by stimulating EC growth and by protecting ECs from apoptosis (269). In contrast, a pro-apoptotic effect of CO in ECs has also been reported (261). The reasons for these divergent outcomes are not known but may reflect differences in the dose and duration of CO exposure and/or the vascular source of ECs. CO abrogates apoptosis by activating a discrete signaling pathway, the p38 MAPK pathway, in vascular ECs (31). Hence, the protective effects of HO-1 on vascular inflammation, to some degree, seem to closely implicate with the EC responses through various mechanisms.
V. Control of Vascular Diseases by HO-1/CO
The HO system present in organisms from bacteria to eukaryotes is the main enzyme that can degrade heme, playing a critical role in heme and iron homeostasis. There is the possibility that heme could be degraded through HO-independent pathways. In fact, NADPH-cytochrome p450 reductase has been demonstrated to destroy heme into dipyrrolic propentdyopents and other products (224, 296). The majority of heme is present in hemoglobin, whereas other heme proteins include myoglobin, mitochondrial and microsomal cytochromes, and various catalytic enzymes such as NOSs, catalase, and respiratory burst oxidase (107, 208). These heme proteins play a critical role in many physiological processes, including oxygen transport, mitochondrial respiration, and signal transduction (208). However, free heme is cytotoxic at 1 M concentrations in the presence of ROS (18). In the presence of H2O2, the heme ring is split open releasing free iron witch amplifies ROS production via Fenton chemistry (17 –19). The cell either dies or survives by induction of cytoprotective pathways such as HO-1 and ferritin.
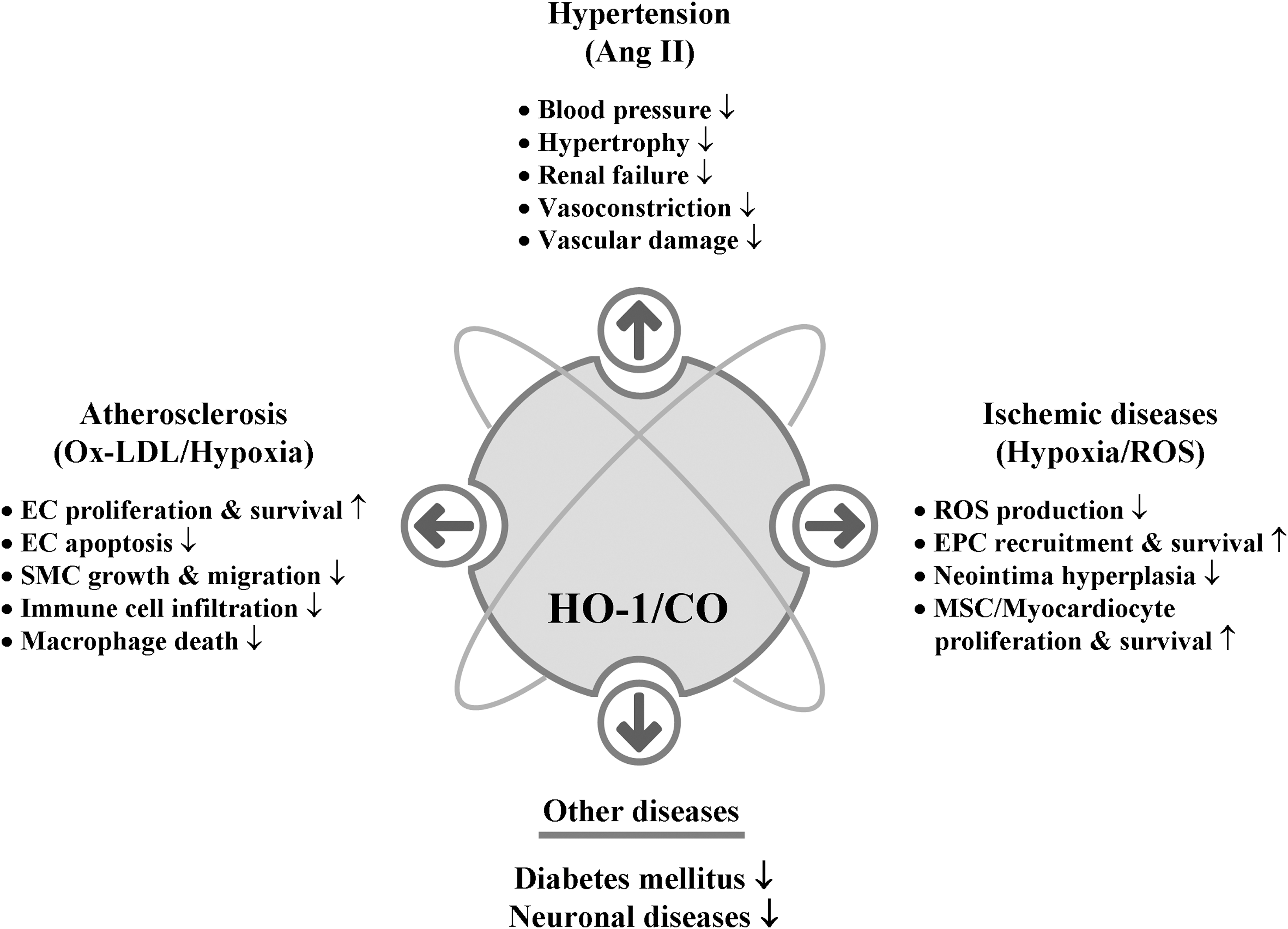
We know that CO, which is generated endogenously during cellular metabolism where heme amount is high (152), is indispensible for the homeostasis of living organisms. Still prolonged exposure to high concentrations of CO seems deleterious especially as seen in chronic disease states such as asthma and diabetes (205, 291), and it is apparent that a physiological dose of CO [reviewed in (233)], exerts beneficial roles such as vasodilatory, antiapoptotic, and antiinflammatory effects. CO now emerges as a key signaling molecule that regulates numerous vascular processes such as blood pressure (BP), vessel tone, smooth muscle proliferation, platelet aggregation, neurotransmission, and stress response, suggesting a crucial role of CO in maintaining physiological homeostasis. Restoration of physiologic CO levels exerts a beneficial effect in many disease settings, such as I/R injury, atherosclerosis, septic shock, hypertension, and metabolic syndrome (73, 170).
A. Ischemic diseases
Evidence suggests that the HO-1/CO system plays an important role against coronary artery I/R injury (Fig. 6). A study demonstrated that the lack of HO-1 makes reendothelialization at the site of injury difficult because of the reduction of EPC recruitment and differentiation compared with wild-type EPCs (67). EPC number also correlates with the extent of ischemia in stroke or myocardial infarction (68, 150). Ischemia results in an increase of growth factors such as SDF-1 and VEGF that lead to increased EPC number and formation of new blood vessels in the injured tissue (64), leading to increased neovascularization for postischemic repair. It has been shown that recruitment of EPCs from bone marrow to periphery is dependent on the establishment of the SDF-1 gradient in a model of heart (14) and hind limb ischemia (64). This gradient is caused both by increased SDF-1 in ischemic tissues (44), and the decrease in SDF-1 in the bone marrow (64, 264), pointing to the mechanisms operating between the peripheral tissues and bone marrow. Although initial human trials have shown only modest short-term benefits in the setting of coronary artery disease (CAD) (215), the therapeutic efficacy of EPC has drawn much attention to improve neovascularization and subsequent recovery of ischemic tissues.
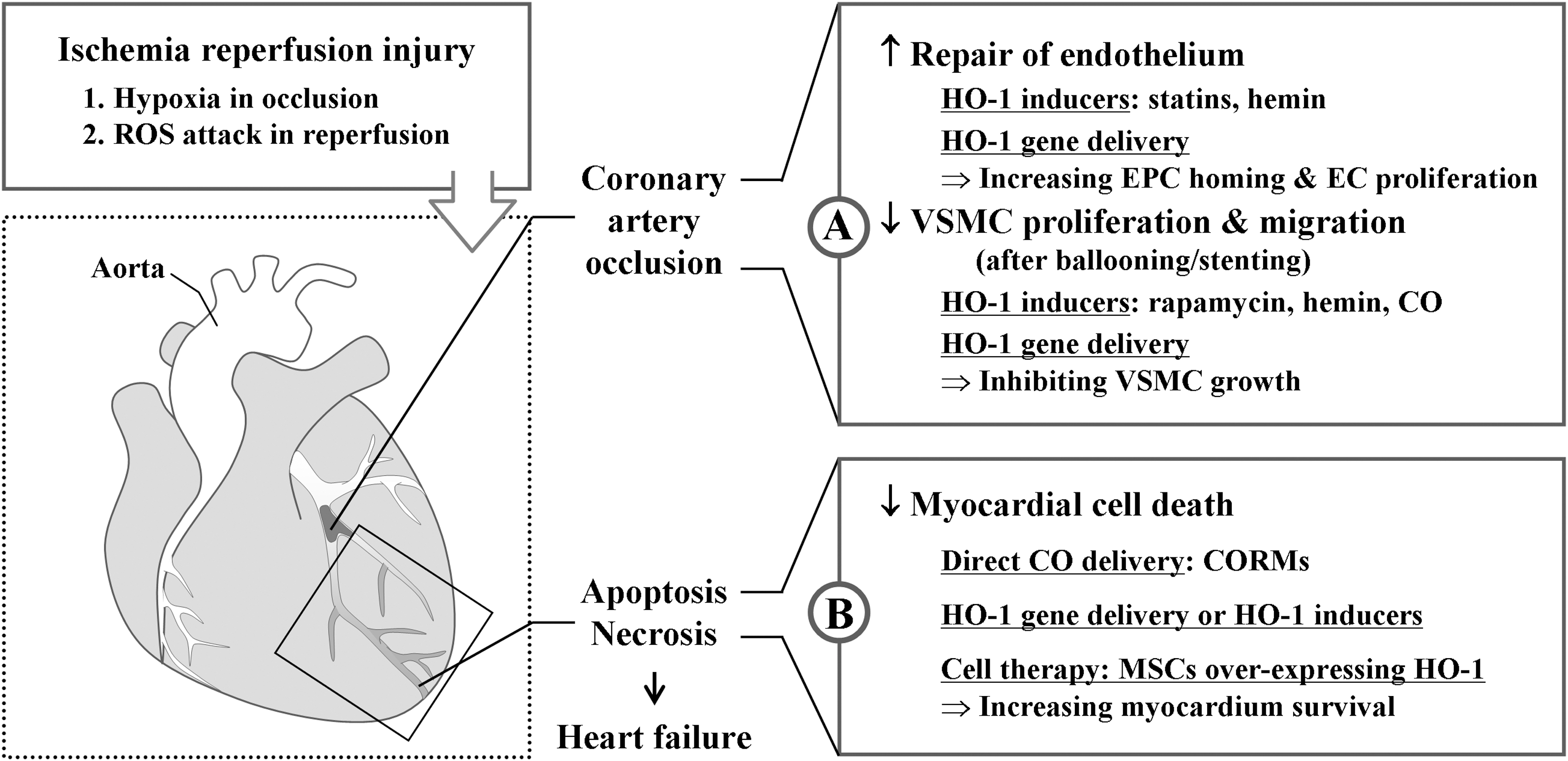
Importantly, regulatory mechanism of cell cycle by HO-1 appears different between ECs and SMCs. In SMCs, HO-1 expression decreased cell cycle progression, whereas in ECs, HO-1 expression increased cell cycle progression (2, 143). The mechanisms underlying the HO-1 cell-specific effect on cell cycle progression within the vascular wall are yet to be explored. Nevertheless, such a difference in regulation of cell cycle by HO-1 led to the inhibition of neointimal hyperplasia (135). Since restenosis caused by VSMC proliferation occurs within 1–3 months of stent implantation, favorable and differential effects of HO-1/CO on ECs and VSMCs make them attractive therapeutics to prevent restenosis after percutaneous coronary intervention.
Although there is no clinical evidence about a therapeutic role of CO, this gas might be therapeutically used for protecting cells from reoxygenation injury. The importance of CO in vascular biology leads many investigators to study the mechanisms of the action of CO and to find CORMs (206). CORM-3 has been shown to generate vasodilation of aorta ex vivo and reduce BP in vivo (46, 85, 173). Administration of CORM-3 reduces infarct size in vivo when given at the time of reperfusion (101). Intriguingly, a study showed that combined treatment with CO and BV increased survival up to 80% with a significant decrease of myocardial injury and improved cardiac function, whereas single treatment with either CO or BV did not alter the survival of heart grafts in a rat model (147).
BR may also display cytoprotective properties in the cardiovascular system. BR at physiological levels can provide cardioprotection by suppressing oxidation of lipid membranes and preventing EC death caused by hydrogen peroxide [reviewed in (245)]. Administration of BR or its precursor BV to rodents can suppress I/R injury (219, 256). It has been demonstrated that low serum BR levels correlate with an increased risk for CAD and oxidative metabolites of BR are detected in atherosclerotic lesions (92, 127, 255). However, there is no clinical evidence supporting the hypothesis that the protection provided by BR or BV in the ischemic myocardium could be clinically significant.
HO-1 may also have therapeutic benefits during chronic heart failure as well. Upregulation of HO-1 during heart failure mitigated pathologic left ventricular remodeling and reduced myocardial hypertrophy, oxidative stress, and inflammation (222, 223, 298). Further, HO-1 induction increases adult cardiomyocyte tolerance by reducing apoptosis to ischemia after in vivo transplantation (258), indicating the role of HO-1 in repairing infarcted myocardium. Tang et al. (258) have also shown that HO-1 transduction in bone marrow-derived mesenchymal stem cells (MSCs) improved cell survival, attenuated left ventricular remodeling, and improved functional recovery after myocardial infarction in hearts transplanted with MSCs. In this case, preconditioning with HO-1 acts to retain functional viability in vivo in adult cardiomyocyte cellular grafts after implantation. Thus, autologous atrial cardiomyocytes or MSCs could be useful cell sources and HO-1 can be used to improve the function of cardiomyocytes and MSCs to treat infarction.
Understanding the inherited factors such as candidate gene expression or finding novel genes that influence susceptibility for developing diseases may lead to find better comprehensive therapies (110). A very important case with HO-1 deficiency in human revealed that low level of BR and extremely high level of heme in serum by lack of heme catabolism (287), which caused severe malfunctions and advanced plaques development with damaged ECs, providing a direct evidence for crucial role of HO-1 in pathophysiology of cardiovascular disease. HO-1 expression in monocytes and lymphocytes from patients with CAD is significantly higher than in patients without CAD (50, 144). The HO-1 expression levels showed significant differences in order: the highest in acute myocardial infarction, followed by unstable angina pectoris, and finally by stable angina pectoris.
Nevertheless, the ultimate goal in the treatment of cardiovascular disease is the timely delivery of the best therapeutic agents to protect the heart from the deleterious effects of prolonged ischemia or the effects of repeated challenges of ischemia. Numerous studies using animal models have been developed and proved the role of HO-1/CO in I/R injury. HO-1 null mice during hypoxia show enhanced ventricular dilatation, infarction, and thrombosis (294). In addition, isolated hearts from heterozygote HO-1-knockout mice demonstrate an increased susceptibility to I/R injury compared to hearts from controls (295). A strategy for tissue protection has been developed using an adeno-associated vector system under the control of the erythropoietin hypoxia response elements for ischemia-regulated expression of the therapeutic human ho-1 gene (196). A single administration of this vector several weeks in advance of I/R injury produced a rapid and timely induction of human HO-1 during ischemia, which resulted in a dramatic reduction in tissue damage. In addition, HO-1 overexpression prevented long-term pathologic tissue remodeling and normalized tissue function (196).
HO-1 inducers and ho-1 gene delivery do not seem to induce organ-specific gene expression. As expected, biochemical inducers administered intraperitoneally or subcutaneously induced HO-1 in most organs and intracardiac injection of the ho-1 gene altered other organs, including lungs, kidneys, liver, and brain (94, 220). Thus, a key to future therapeutic application of HO-1 for humans is to properly target and regulate HO-1 expression at a specific site to allow maximal cytoprotection and minimum adverse effects at the site of the lesion.
B. Hypertension
HO and CO participate in the homeostatic control of cardiovascular functions, including the regulation of BP (185). Studies regarding HO-1/CO effects on hypertension can be approached in two ways. One is whether the increase of HO-1 is a consequence of increased BP. The other is whether the administration of exogenous CO or the induction of HO-1 by chemical inducers or gene delivery reduces hypertension.
Angiotensin II (Ang II) is a peptide that is known to increase BP when present continuously in the bloodstream. Ang II, depending on the kind of tissue, can stimulate cell proliferation, hypertrophy, apoptosis, and ROS production, and also cause oxidative DNA damage (Fig. 7). Ang II-induced hypertension is prevented by losartan or hydralazine accompanied by reduction of HO-1 expression, which is diminished by HO inhibitors (115). The interplay between Ang II and HO-1 was observed both in vitro in VSMCs (115) and ECs (2) and in vivo in vessels (115), hearts (83), and kidneys (6). Unexpectedly, weaker HO-1 induction inhibited Ang II-induced hypertrophy (108), whereas stronger HO-1 induction had no effect on hypertrophy (83).
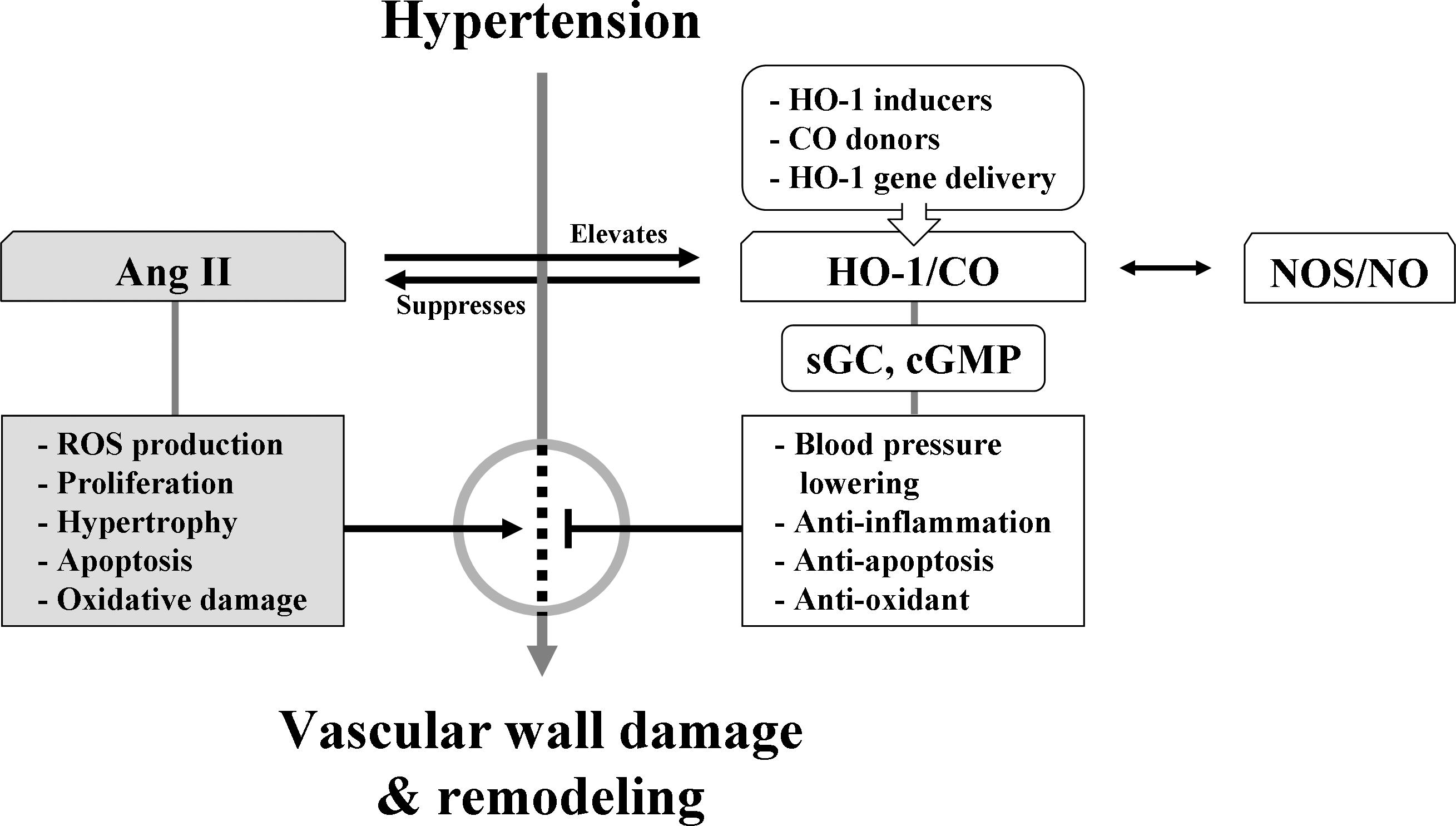
Most of our understanding of the metabolism of CO in hypertension is from studies in spontaneously hypertensive rats (SHRs), in which age is an important determinant factor for the manifestation of the hypertension (183). Using a hypertensive rat model with chronic Ang II infusion, Ishizaka et al. (115) reported that pressure overload upregulated aortic HO-1 expression and activity. Through its antioxidant and antiinflammatory properties, increased aortic HO-1 may act favorably against the tissue damage elicited by Ang II and pressure overload. Results of using another hypertensive rat model with chronic norepinephrine infusion demonstrated that HO-1 is upregulated in myofibroblasts and infiltrated inflammatory cells of the heart of Ang II-induced hypertensive rats and inhibits BP elevation (118). Upregulation of HO-1 expression by hemin or stannous chloride has been reported to be successful in decreasing BP (118). The BP-lowering effect was mediated by CO but not by other HO-1 by-products (118). BP and the level of HO-1/CO are correlated with reciprocal influence, implying that an impaired HO/CO system might constitute one of the pathogenic mechanisms of hypertension.
Administration of ZnPP, a HO inhibitor, increased total peripheral resistance and mean BP in Sprague-Dawley rats (117). Other studies reported that daily injection of ZnPP for 4 days resulted in a striking increase in BP in prehypertensive (4-week-old) and young (8-week-old) SHRs, whereas ZnPP did not affect BP in adult Wistar-Kyoto rats (20 weeks) or age-matched SHRs (184, 185). Other nonspecific effects of metalloporphyrins include the inhibition of muscle relaxation and suppression of cyclic adenosine monophosphate and cGMP. These inhibitory effects may be consequential to the interaction of metalloporphyrins with membrane receptors or their downstream signal transduction pathways.
One of the ultimate goals of the use of HO-1 inducers is to increase CO delivery to the injured tissues. When hypertension was induced by NOS inhibitor in Lewis rats, BP was significantly decreased by a CO donor (173). In addition, when adult SHRs were administrated with HO substrates, BP was decreased within an hour and sustained for 1–2 h. Since this time cannot be enough to induce HO-1, BP reduction could be attributable to CO's effect, probably by HO-2 activity.
Delivery of the ho-1 gene by retroviral vectors is capable of long-term expression but also have some limitations, such as poor efficiency in infecting nondividing cells and unknown immunological response (228). Most HO inducers (e.g., heme analogs or heme derivatives) are administered intraperitoneally, whereas the ho-1 gene is usually delivered by intracardiac injection. When the ho-1 gene delivery was accomplished with a single injection of ho-1 retroviral vectors, this single gene delivery has proven to be sufficient to upregulate expression of HO-1 and decrease BP at different ages in the following weeks and months in SHRs (220). Thus, development of proper HO-1/CO delivery strategies may help to augment the antihypertensive effect and possibly to decrease BP to normotensive values.
Hypertension is characterized by increased vascular contractility, concomitant increase in oxidative stress (286), and enhanced vascular inflammation and vascular remodeling (Fig. 7). An upregulated HO-1/CO system would not only normalize the endogenous CO concentration, but also increase the production of BV and BR, two potent antioxidants. Antiinflammatory and antioxidant protection of cardiovascular tissues by CO will also protect the primary and secondary damage inflicted on tissues by hypertension.
C. Atherosclerosis
Well-known risk factors for the development of atherosclerosis are smoking, hypertension, hyperlipidemia, and diabetes. With the risk factors, vascular cells in the vessel wall can produce ROS, which can initiate endothelial dysfunction by altering the cellular redox state and subsequently induces vascular inflammation (168). Therefore, understanding the adaptive and/or protective responses of the vasculature to oxidative stress are important in preventing atherogenesis (25). During the atherosclerotic process (Fig. 8), one of the most protective proteins is the enzyme HO-1. HO-1 is upregulated at transcriptional level by Ox-LDL (112), high BP (115), and shear stress in blood vessels (51) and a multitude of systemic inflammatory processes (218). A report demonstrated that HO-1 expression and its by-product, BR, were present only in ECs from advanced atherosclerotic lesions but not in cells from early lesions or normal arteries (171). The finding that the higher level of HO-1 in ECs with a greater disease was observed might indicate that HO-1 expression could be a defense mechanism.
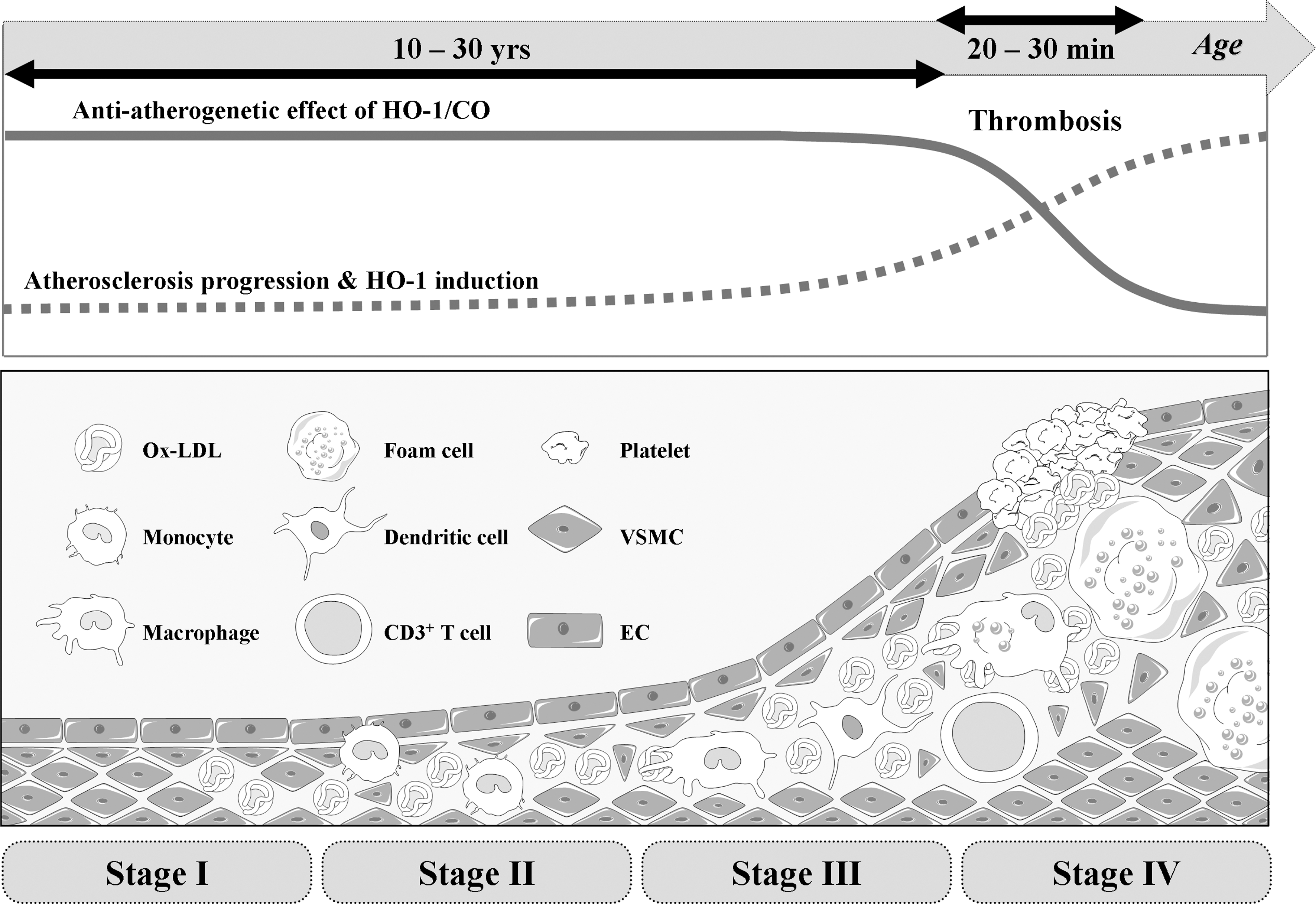
HO-1 is expressed in vascular ECs and macrophages in the early stages of atherosclerotic lesion formation and in foam cells and VSMCs residing in the necrotic core of advanced lesions (113, 114, 274). HO-1 in vascular ECs, VSMCs, and macrophages is markedly upregulated by Ox-LDL, but not by native LDL in ECs and VSMCs (112, 274, 290). In atherosclerotic lesions, HO-1 has been found to be colocalized with oxidized phospholipids, supporting that HO-1 is induced by oxidized phospholipids in vivo. HO-1 contributes to the balance of pro-oxidant and antioxidant factors in the vascular wall through multiple mechanisms.
Most importantly, studies with a 6-year-old boy who had HO-1 deficiency revealed that intravascular hemolysis and EC injury were prominent in association with hyperlipidemia (128, 287). This might be caused by deposition of iron derived from methemoglobin in plasma. In turn, Ox-LDL with iron might increase susceptibility of ECs to oxidative insults, eventually leading to the development of fatty streaks and fibrous plaques in the aorta (128, 287). These studies strongly suggested that HO-1 plays a crucial role in protecting vessels from oxidative insults in human. Additionally, importance of HO-1 in human vascular diseases came from promoter polymorphism analysis in studying the 5′-flanking sequence of the human ho-1 gene (52, 53, 76, 78, 124). A cohort study by Exner et al. (78) revealed that patients with short (25 guanine-thymine dinucleotide [GT]) dinucleotide repeats in the ho-1 promoter region on either allele had often significantly less developed restenosis compare to patients with longer (≥25 GT) dinucleotide repeats. Another study that assessed microsatellite polymorphisms in the ho-1 gene promoter showed that patients with type 2 diabetes carrying longer (≥25 GT) repeats had higher oxidative stress and increased susceptibility to the development of atherosclerosis and CAD (53). They both found that shorter dinucleotide repeats was associated with higher transcriptional activity and thus higher expression levels of HO-1, indicating a protective role of HO-1 in vascular diseases, such as atherosclerosis. Another report, a single nucleotide polymorphism in the ho-1 promoter, suggests that the AA genotype had significantly higher basal promoter activity that was independent of the length of (GT)n repeats (191). These studies claim the potential role of ho-1 gene regulation in atherosclerotic disease processes.
Many investigators have developed the strategy of delivering the ho-1 gene with a vector and generating transgenic mice to study the long-term and specific protective effects of HO-1 against atherosclerosis and vascular injury. Studies have demonstrated that ECs that were transduced by retroviral vector containing HO-1 in rat model of diabetes resisted to ROS attack (3, 213). Delivery of the ho-1 gene in rat carotid artery after balloon injury reduced neointimal hyperplasia by inhibiting VSMC proliferation in media and neointima layer (267). Transduction of adenoviral vector containing HO-1 to pig femoral artery directly reduced vasoconstriction through sGC and cGMP and inhibited vascular cell proliferation through p21 induction (69).
Overexpression of HO-1 was able to decrease atherosclerotic lesion formation by Western diet in apolipoprotein-E (apoE)−/− mice (122). Using apoE−/−HO-1−/− mice, Yet et al. (293) demonstrated that HO-1 played a protective role in atherosclerotic lesion formation. When apoE−/− mice were fed with a Western diet for 8 weeks, atherosclerotic lesions developed and apoE−/−HO-1−/− mice had larger and more advanced lesions than apoE−/− mice despite similarly elevated total plasma cholesterol. The lesions in apoE−/−HO-1−/− mice were complicated with fibrous caps, comparable to plaques seen in apoE−/− mice on a Western diet for longer periods (12 weeks). These results provide strong evidence for a beneficial effect of HO-1 on experimental atherosclerosis. Also, vein grafts from HO-1−/− mice showed a robust neointimal hyperplasia derived from VSMCs as compared with that from wild-type mice and VSMCs from HO-1−/− mice were more susceptible to H2O2-induced cell death than those from wild-type mice (293). Nrf2−/− mice showed a decreased susceptibility to apoE-mediated atherosclerotic plaque formation (249), which is surprising in light of the crucial role of Nrf2 in HO-1 expression. Although Nrf2 stimulates antiatherosclerotic HO-1 expression, this transcription factor can also stimulate pro-atherosclerotic CD36 expression (249), which may explain why Nrf2 disruption in mice attenuates apoE-mediated atherosclerosis.
Together, the above results strongly support that HO-1 plays a protective role in experimental atherosclerotic vascular disease. This is very important because when evaluating the potential for HO-1 as a therapeutic target for atherosclerosis, we should also assess the ability of HO-1 to ameliorate vascular complications, not only in atherosclerosis but also after coronary artery bypass surgery or percutaneous transluminal angioplasty/stenting, namely, neointimal hyperplasia or restenosis (Fig. 9).
As described, statins, inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A reductase, exert pleiotropic activities (144): one of which is HO-1 induction and subsequent reduction of ROS formation (98). Lee et al. (140) first demonstrated that simvastatin at micromolar concentration induced HO-1 expression in rat and human VSMCs as well as in VSMCs in the tunica media after intraperitoneal injection in mice, but not in ECs or macrophages (149). Ali et al. (9) have shown that atorvastatin and simvastatin at low micromolar concentration upregulated HO-1 expression in ECs with potential link between HO-1 and Kruppel-like factor 2 by laminar flow. Heeba et al. (105) have identified a novel mechanism related to induction of HO-1 by statins, which is mediated through the NO pathway and could explain some of effects of these drugs. Activation of the HO-1 pathway by statins inhibited inflammatory disorders and protected the endothelium from oxidative damage by improving eNOS functional activity and decreasing lipid peroxide formation. Habeos et al. (102) have provided evidence supporting that some of the beneficial-reported effects of statins may be attributed to the activation of Keap1/Nrf2 signaling and the concomitant increase of HO-1.
Although the effect of statins on HO-1 induction seems to be significant in vitro, the pharmacological relevance remains to be confirmed, because the physiological concentrations of statins used in clinical trials were not likely to induce HO-1. Rapamycin has been reported to block cell cycle progression in VSMCs; therefore, rapamycin- or its derivative-coated coronary stents are most popularly used to treat obstructive coronary artery lesions (166). Rapamycin has been shown to induce HO-1 expression and suppress growth factor-dependent VSMC growth (270), and a question is raised whether HO-1 expression could be involved in the reduction of restenosis rate by rapamycin.
Combination therapy of statins with antihypertensive and immunosuppressive rapamycin may also be considered to induce HO-1. Another drug, probucol, is likely to have the beneficial effect on atherosclerosis and restenosis not only by inhibition of lipid oxidation but also by induction of HO-1 (65, 284). Recent study suggests that lipid-lowering fibrates and insulin-sensitizing thiazolidinediones can be used as potential therapeutics since these drugs induce HO-1 expression via activation of peroxisome proliferator-activated receptor-α (136). Besides the aforementioned drugs, naturally occurring polyphenols are also able to induce HO-1 expression (242, 252), suggesting that a considerable part of the protective effects of fruit consumption against cardiovascular diseases could be mediated through HO-1 pathway.
Paradoxically, HO-1 inducers could be both stimulators and inhibitors of a particular disease process. For example, pro-athrogenic stimuli (e.g., TNF-α, LPS, and hypoxia) or antiatherogenic stimuli (e.g., IL-10) have been reported to induce HO-1 (123, 139, 260). One thing is clear that preemptive HO-1/CO shows protective roles in any type of vascular diseases, and therefore it can be useful as therapeutic means (15). Regardless of their capabilities, most of the HO-1 inducers, such as hemin or tin compound, appear to show cellular and tissue toxicity, such as severe nephrotoxicity. In addition to direct gene delivery, therefore, drugs that are clinically used to treat cardiovascular diseases should be carefully studied.
D. Diabetes mellitus
Diabetes mellitus (DM) is characterized by hyperglycemia, insulin resistance, and a relative impairment in insulin secretion. Insulin resistance, a key factor in the pathogenesis of DM, is frequently accompanied by hypertension, high serum LDL, low serum high-density lipoprotein, and high serum triglyceride levels, which promote the development of atherosclerotic cardiovascular disease (4). Recent studies suggest that upregulation of HO-1 system may be used for the prevention of DM. An enduring antidiabetic effect of the HO-1 inducer, hemin, on Zucker diabetic-fatty rat, a model of insulin-resistant DM, has been evaluated (181). Hemin treatment improved glucose tolerance, reduced insulin intolerance, and lowered insulin resistance. The comparative effects of the HO-1 inducer, hemin, and the HO blocker, chromium mesoporphyrin, on insulin sensitivity/glucose metabolism have been examined, and an antidiabettic effect of hemin has been further confirmed (179). Hemin therapy lowered BP and increased plasma insulin and the insulin-sensitizing protein adiponectin with slight but significant reduction of glycemia, while chromium mesoporphyrin abolished the hemin effects. Hemin also enhanced insulin sensitivity and improved glucose metabolism in insulin-resistant Goto-Kakizaki rats (180) and in adult SHRs (182), suggesting the hypothesis that upregulation of HO-1 may reverse the detrimental effects of diabetes. This hypothesis has been supported by a finding that diminished upregulation of visceral adipose HO-1 correlated with waist-to-hip ratio and insulin resistance in human obesity (232). A putative mechanism underlying the antidiabetic effects of HO-1 may include the antioxidant and antiinflammatory actions of HO-1 (4).
E. Therapeutic potential of HO-1/CO in vascular diseases
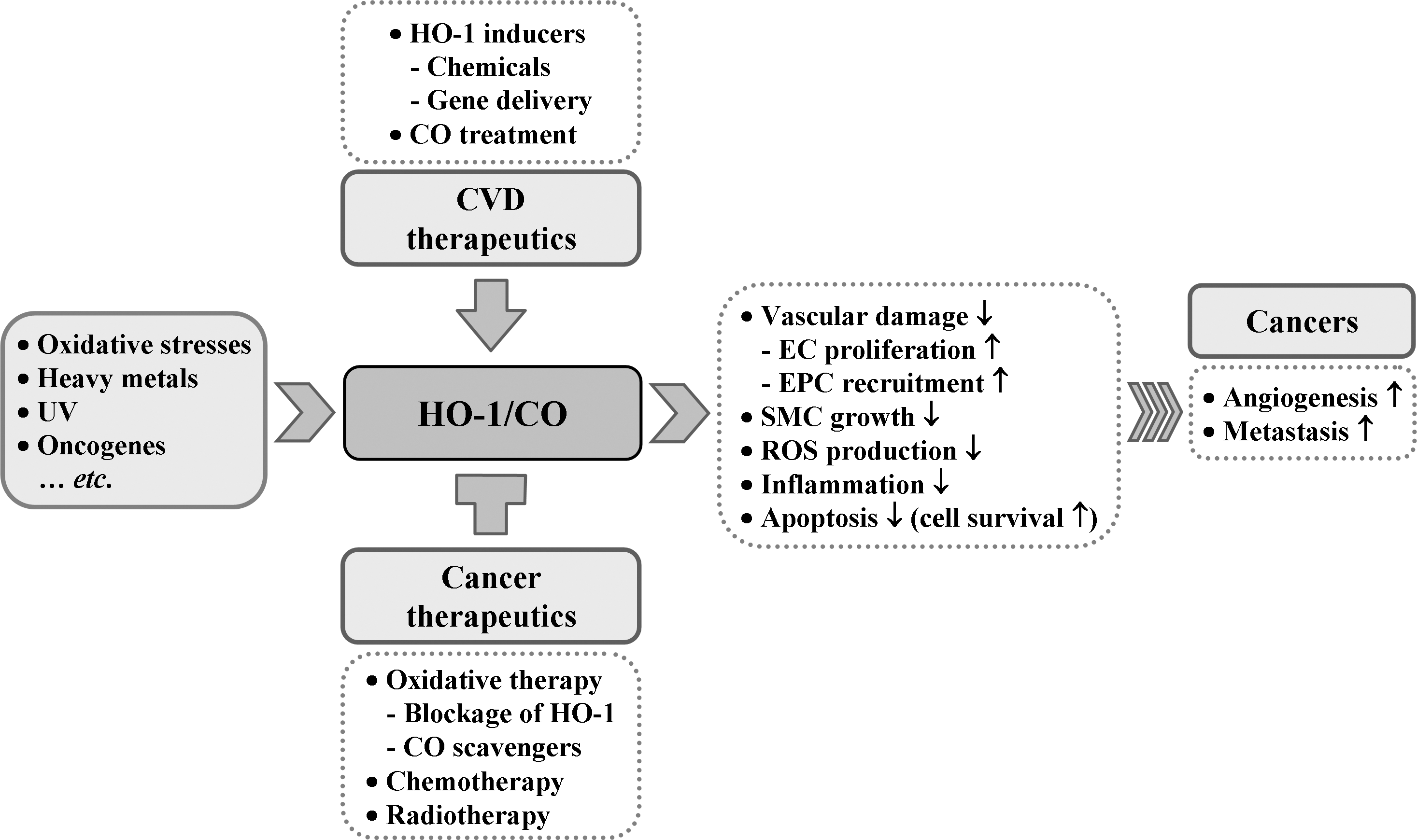
Given that cardiovascular disease risk conditions converge in the contribution to oxidative stress and oxidative stress leads to endothelial dysfunction, this background may provide the rationale for seeking pharmacological modulation of oxidative stress pathways in the treatment of cardiovascular diseases. According to Calabrese et al. (40), the ability of a cell to counteract stressful conditions requires the activation of survival pathways as well as the production of molecules endowed with antioxidant activity, which is under control of such protective gene products as HSP32, HSP70, thioredoxin, and sirtuin, all of which are encoded by so-called vitagens (36, 38). HO-1, also referred to as HSP32, is naturally induced by cellular stresses, which leads to the production of molecules with antioxidant, antiapoptotic, and immunomodulatory properties (154). Although the administration of exogenous CO alone may have potent cytoprotective effects in cell culture systems or in animal models, the coordinated response of all products may be necessary for the best outcome; because BR or BV are known to be strong antioxidants (154). Many clinically used drugs have been shown to induce HO-1 in various cell types and in animal disease models. HO-1/CO efficiently functions to recover the damaged tissues in I/R injury, hypertension, and atherosclerosis mainly by improving EPC/EC functions and inhibiting proliferation of SMCs (Fig. 10). As our understanding of the cellular and molecular events of HO-1/CO regarding cardiovascular diseases and other diseases continues to expand, we may be able to find more treatment targets for the preservation of tissues from oxidant injury and ultimately archive better outcome of patients.
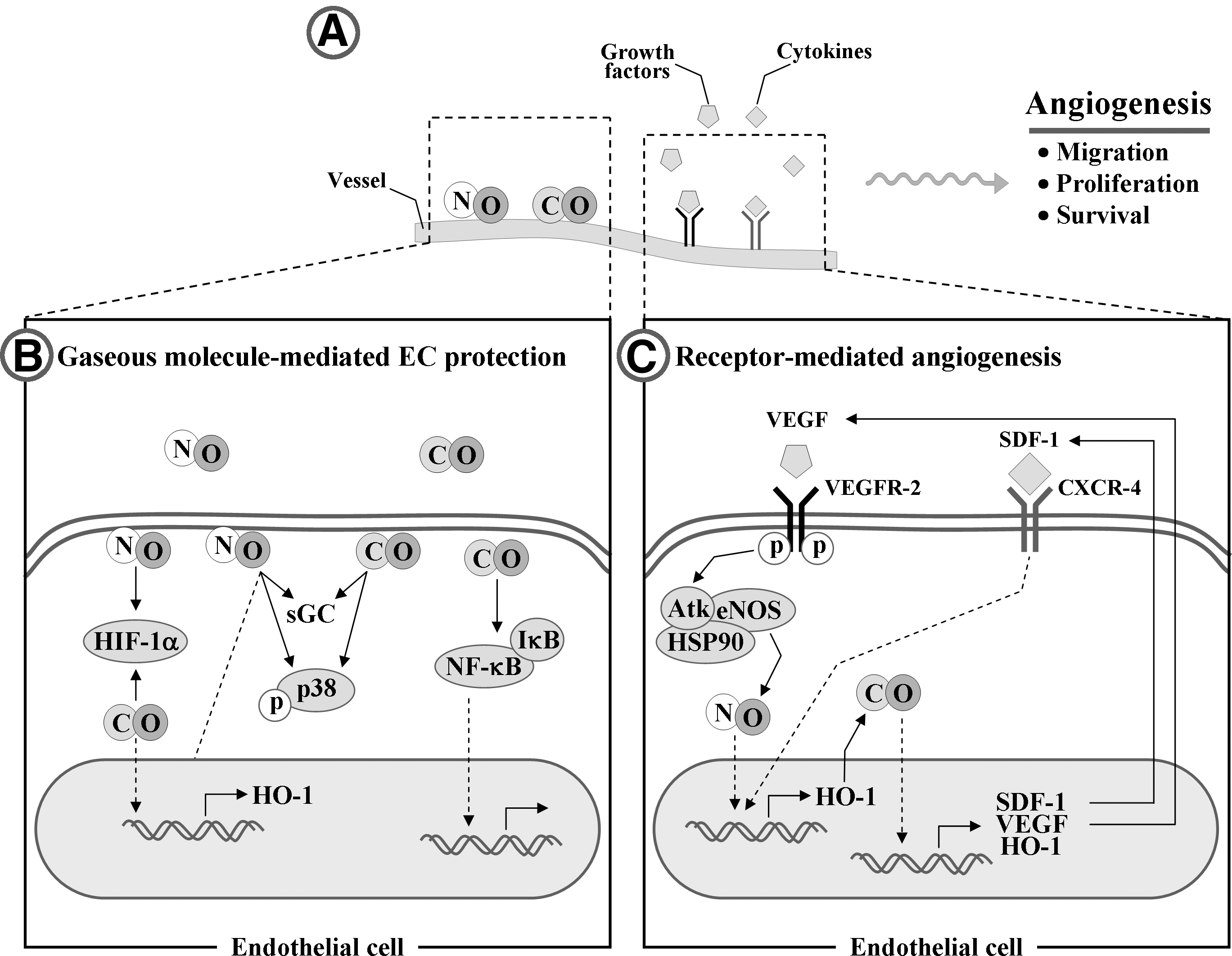
VI. Effect of HO-1 on Angiogenesis
The development of the vascular system begins with an assembly from bone marrow-derived precursor cells that shape the first primitive plexus of vessels, a process called vasculogenesis. Subsequently, new vessels are established from preexisting vessels along with their migration and proliferation for the generation of tube-like structures, a process called angiogenesis. As the new vessels are formed, the endothelial layer endothelium forms an important interface between tissue and blood to control the transportation of nutrients and O2 from the blood to all cells. ECs adapt to oxygen tension using various O2-sensing mechanisms, such as HO-2 and eNOS (45). The O2-sensing mechanisms regulate several stages of vessel formation, ranging from EC fate decision to vasculogenesis and angiogenesis.
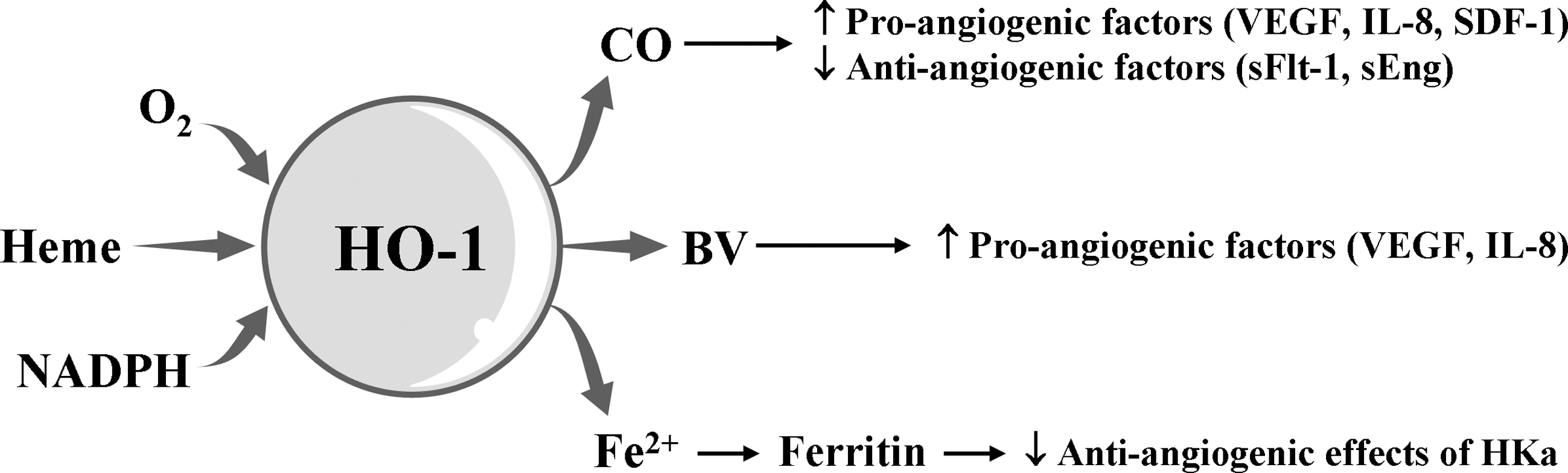
HO-1 induction could be a critical event for angiogenesis. CO synthesized by the catalytic reaction of HO-1 induces the production of angiogenic mediators, such as VEGF, IL-8, and SDF-1 and decreases the antiangiogenic mediators such as soluble VEGF receptor 1 and soluble endoglin, consequently resulting in the promotion of EC proliferation, migration, and antiapoptotic response (60, 70) (Fig. 10). BV stimulates the induction of pro-angiogenic factors, such as VEGF and IL-8 in human keratinocytes cells (148). Ferritin, Fe2+ sequestering protein, binds to HKa (cleaved high molecular weight kininogen) and antagonizes the antiangiogenic effects of HKa (58) (Fig. 11).
Genetic and gene transfer studies have shed light on the distinct role of HO-1 in vascular functions such as angiogenesis and endothelial dysfunction. Genetic overexpression of HO-1 enhances VEGF synthesis and augments formation of vascular capillaries, improving the blood flow in ischemic tissues, and this effect is completely abolished by treating the animals with the HO activity inhibitor, ZnPP (66, 250). Recent study demonstrated that VEGF stimulates HO-1 induction in ECs and that this induction is required for VEGF-dependent angiogenesis because the inhibition of HO-1 activity by SnPP or ZnPP abrogates the formation of blood vessels in a Matrigel assay in vivo (35). Further, Deshane et al. (67) demonstrated that aortic rings isolated from HO-1-deficient mice are unable to form capillary sprouts ex vivo in response to the chemokine SDF-1, which plays a role in the recruitment of EPCs to home to sites of injury (289) and facilitate repair and that this defect is reversed by treatment with CORM (67). This study further confirmed the functional significance of HO-1 in angiogenesis in HO-1−/− mouse models of Matrigel plug and wound healing. Deficiency of HO-1 is closely associated with impairment of neovascularization and wound healing in injured tissue. These findings demonstrate an important role for HO-1 in VEGF- and SDF-1-mediated angiogenesis for vascular remodeling and repair.
Angiogenesis can occur both prenatally as well as postnatally. Mice lacking the functional HO-1 gene demonstrate the low survival percentage (20% of expected HO-1-deficient mice) of embryos, probably by suppressing embryonic angiogenesis (209), whereas the functional loss of VEGF induces the impaired angiogenesis during embryonic development and is lethal in the mouse embryos between days 8.5 and 9.5 (81). In addition, VEGF treatment increases angiogenesis, simultaneously with HO-1 expression during chick embryo development, and VEGF-stimulated angiogenesis in chick embryos is markedly attenuated by the HO inhibitor zinc mesoporphyrin (80). These suggest that cross-talk between VEGF and HO-1 plays an important role in prenatal angiogenesis and that VEGF possesses more potential angiogenic activity than HO-1. On the other hand, inhibition of HO-1 activity by SnPP or genetic knockdown of HO-1 also leads to the suppression of neovascularization in wounded tissues and the retardation of wound closure in an adult animal model (97), indicating that HO-1 is also involved in postnatal neovascularization. HO-1-deficient human patients showed that both intravascular hemolysis and EC dysfunction are prominent, which are similar clinical symptoms to those of HO-1 knockout mice (128, 287). These findings demonstrate that the HO-1 pathway participates to the biological process of both prenatal and postnatal angiogenesis.
In addition to the adoptive function of ECs for hypoxia by inducing angiogenesis to ensure a continuous supply of O2, ECs also consume O2 to generate the signaling molecules like ROS and NO. Low levels of NO are beneficial for cardiovascular functions, such as vasodilation, inhibition of platelet aggregation, and angiogenesis (241). Like NO, HO-1-derived CO may also possess signaling properties for vascular protection, antiinflammation, and vascular remodeling, presumably by inhibiting the release of pro-inflammatory cytokines in LPS-stimulated macrophages through augmentation of the interaction between caveolin-1 and toll-like receptor-4 (278), platelet aggregation through the activation of the enzyme guanylyl cyclase and subsequent generation of cGMP (221), and cytotoxic ROS production through downregulation of NADPH oxidase activity (277). Thus, the inducible effect of HO-1/CO on vascular inflammation and function plays a quite important role in the pathophysiological condition.
The definitive link of HO-1 to pathological angiogenesis has been suggested by recent studies. HO-1/CO can be associated with several vascular disorders such as cancer and vascular restenosis (70). The persistent angiogenesis has been known to be a hallmark of pathologic status, such as inflammation and tumors. Several human tumors express high levels of HO-1, which are closely linked to tumor angiogenesis, supplying necessary nutrients and O2 for tumor growth and providing a route for tumor invasion and metastasis (279). HO-1/CO-enhanced VEGF production may lead to the formation of leaky and immature vessels in pathological tumor angiogenesis. SDF-1, which can be induced by HO-1 expression (145), also promotes tumor cell growth, migration, and invasion by inducing angiogenesis (100), and SDF-1-induced neovascularization was strongly reduced in aortic rings from HO-1−/− mice compared with those of wild-type animals (67). Therefore, HO-1 can be involved in pathological tumor angiogenesis, and regulation of HO-1 expression and activity may be potential therapeutic strategy for cancer treatment.
In a study using HO-2-null mice, Seta and colleagues have demonstrated that a constitutive isoform of HO-2 is also important in inflammation and angiogenesis (231). HO-2-deficient mice are characterized by increased production of pro-angiogenic chemokines (e.g., MIP-2 and MCP-1) and elevated angiogenesis in corneal wound region. The interesting feature of HO-2-null mice is the downregulation of HO-1 expression, which is associated with the exaggerated production of pro-angiogenic chemokines and subsequent induction of inflammatory corneal angiogenesis. Another constitutive isoform, HO-3, is a nonfunctional enzyme and remains incompletely characterized (162).
A. Cross-talk between the HO-1/CO and NOS/NO pathways
CO and NO, produced by the catalytic reactions by HO and NOS, share similar properties: (a) they are retrograde messengers which are highly diffusible; (b) they act as neurotransmitters by elevation of GMP production; (c) they affect adjacent cells without acting through a surface receptor; (d) they are extremely short-lived. Thus, CO and NO can serve as transcellular messengers. Because of the concept that the endogenously derived gas NO could exert a significant biological function in vascular physiology and pathology, CO has gained much attention as a molecule with many similar chemical and biological features.
Expression of iNOS under prooxidant conditions and the coordinate induction of HO-1 in the tissues support the concept that HO-1 could sense NO and thus be protective against ROS- and NO-mediated insults. The sequence of iNOS and HO-1 expression is supported by the following findings: (a) NO and NO-related species induce HO-1 expression in aortic vascular cells; (b) cells pretreated with NO-releasing molecules acquire increased resistance to H2O2-mediated cytotoxicity at the time when HO-1 is maximally activated; (c) in the same study, the presence of NOS inhibitors suppressed both NO production and HO-1 expression (41).
The protective functions of NO are suggested to be mediated through transcriptional activation of the Keap1/Nrf2/ARE pathway, which is responsible for the induction of >100 cytoprotective genes, including the HO-1 gene. Activation of the Keap1/Nrf2/ARE pathway leads to rapid adaptation to a variety of stress conditions and promotes cell survival (172).
Endothelial-derived CO and NO determine the supply of oxygen to tissues through angiogenesis. HO-derived CO contributes to the angiogenesis not only by increasing the synthesis of VEGF, HO-1 and SDF-1, but also by potentiating their synergistic effects on ECs (121, 146) (Fig. 12). The effect of CO on VEGF expression is shown in an animal model by Marti and Risau (161). They demonstrated that animals kept for 6 h in an atmosphere containing 0.1% CO exhibit significant induction of VEGF in various organs. In addition, genetic overexpression of HO-1 leads to the stimulation of VEGF expression in ECs (121). VEGF is also able to stimulate HO-1 expression (35) and eNOS-derived NO production (204) in ECs, consequently inducing angiogenesis (Fig. 12C). Because expression of HO-1 can protect EPCs from oxidative injury and stimulate EPC homing to injured regions for promoting angiogenesis, such a positive feedback can be of beneficial importance in EPC function. In addition, recent in vivo study shows that the cytoprotective effect of VEGF on hyperoxic lung injury is mediated by upregulation of HO-1 expression (235). In concert with CO, NO also induces angiogenesis by upregulating pro-angiogenic mediators, such as HO-1, VEGF, and IL-8 (120, 174). Treatment of ECs with an NO donor enhances the protein levels of HO-1, VEGF, and IL-8 (201), and, conversely, VEGF increases eNOS-dependent NO production in ECs (204), suggesting that NO-HO-1/CO-VEGF axis/circuit is important regulating pathway for angiogenesis.
HO-1/CO is closely linked to the pro-angiogenic effects of SDF-1 (Fig. 12C). ho-1 gene transfer highly appears to increase neovascularization through the recruitment of circulating progenitor/stem cells and the high expression of angiogenic factors such as VEGF and SDF-1 (146). Treatment with CO (250 ppm) enhances reendothelialization after vascular injury by increasing the circulating SDF-1 in a mouse model (146). Further, SDF-1 promotes angiogenesis in EPCs and aortic ECs isolated from wild-type, but not from HO-1−/− mice, and the impaired capillary sprouts in HO-1-deficient mice are restored by exposure to CO (67), indicating that vascular remodeling by SDF-1 requires HO-1 induction and subsequent CO production. In addition, local gene transfer of SDF-1 enhances ischemia-induced vasculogenesis and angiogenesis in vivo through the elevation of VEGF expression and NO production (106), suggesting that the VEGF/eNOS pathway is critically involved in SDF-1-induced vascular remodeling. These findings indicate that a positive link among HO-1/CO, eNOS, VEGF, and SDF-1 can provide new avenues for ischemia-induced neovascularization in adults, as well as support a vital role of HO-1 and its reaction byproduct, CO, in the vascular repair through enhancing EPC mobilization.
It can be interesting to investigate whether HO-1/CO is involved in eNOS/NO-dependent angiogenic events or vice versa. NO upregulates HO-1 with production of CO (84). The high levels of CO inhibit NOS activity and NO generation, whereas the low concentration of CO induces NO release from either eNOS stimulation or a large intracellular pool of NO (254, 262) (Fig. 13). Therefore, the relationship between the HO-1/CO and eNOS/NO pathways is still complex, dynamic, and adaptable, although the effect of HO-1/CO pathway on vascular system can be cross-talk with the eNOS/NO pathway, which controls expression of angiogenic factors, including VEGF.
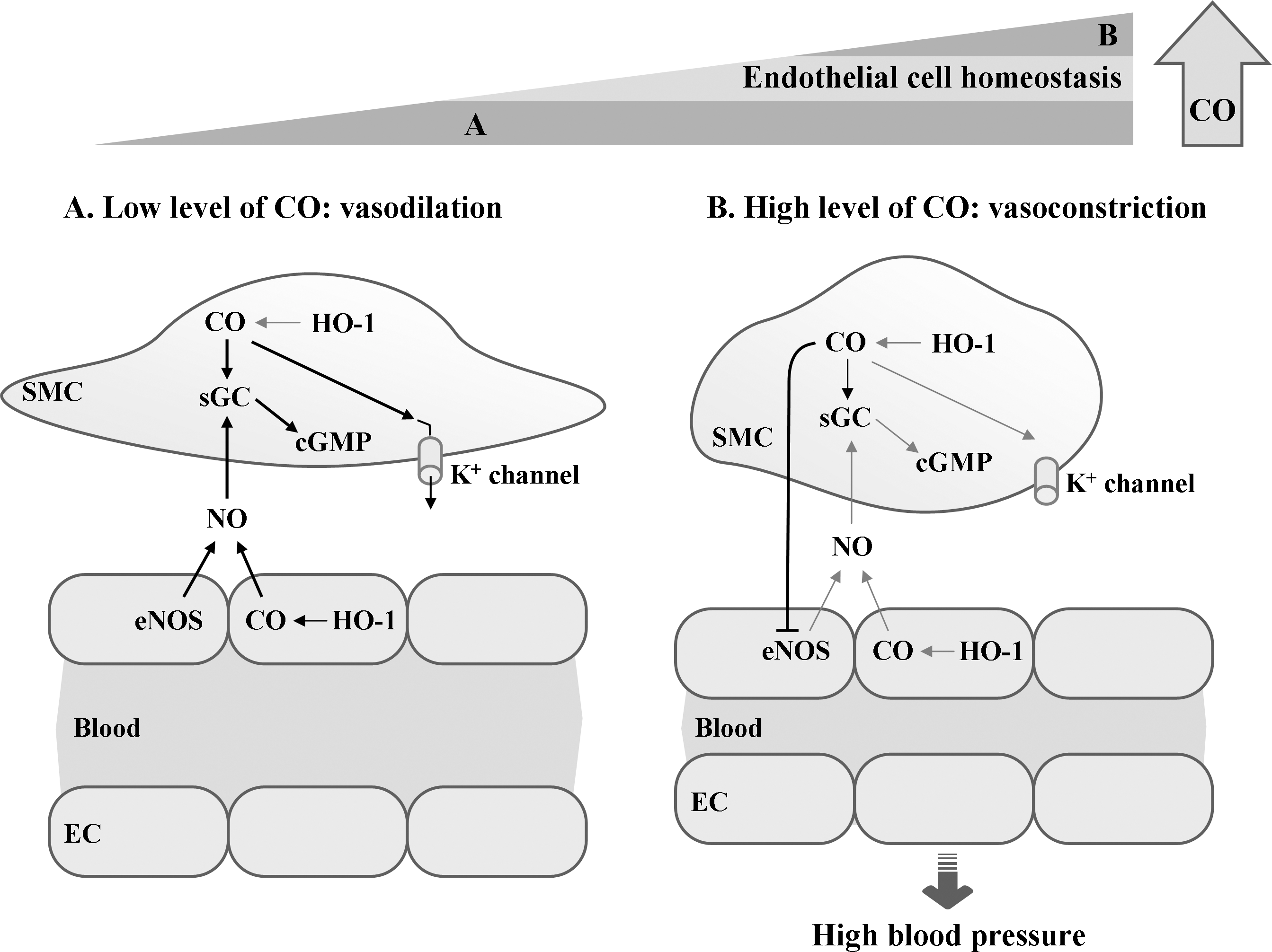
B. Effect of HO-1/CO pathway on vascular homeostasis
CO or NO production by ECs not only enhances O2 delivery by stimulating angiogenesis but also influences O2 consumption during mitochondrial respiration by acting as a reversible inhibitor of cytochrome c oxidase in the electron transfer chain and as a guardian of cellular energy homeostasis (119). Like evidence of NO action in ECs (134, 280), the gaseous molecule CO produced from HO-1 exerts both paracrine and autocrine effects on various vascular cells, leading to the regulation of vascular tone, endothelial function, the proliferation of smooth muscle cells, and vascular inflammation (27, 206).
To maintain normal vascular system against diverse forms of stimuli, living cells should communicate with adjacent cells. Gaseous retrograde messengers, CO and NO, are highly diffusible, thereby transmitting signals from ECs to smooth muscle cells or vice versa. Both CO and NO may cause relaxation of the smooth muscle cells of vessel walls, allowing the vessels to dilate (Fig. 13). Both gaseous molecules bind to iron in the heme moiety of sGC (247, 251), a stimulator of the cGMP synthesis. Several reports show that vasodilatory properties of HO-1-derived CO are clearly linked to the production of cGMP and the activation of calcium-activated K+ channels (167, 275) (Fig. 13). The activation of K+ channels leads to membrane hyperpolarization, which in turn inhibits voltage-gated Ca2+ channels, causing SMC relaxation (276). HO-derived CO may facilitate the vasodilatory action of NO by stimulating cGMP levels in VSMCs (84, 244) (Fig. 13A). High levels of CO, however, can act as a negative regulator of eNOS activity by interacting with its catalytic heme moiety in ECs, leading to the suppression of NO production and the elevation of vasoconstriction (85, 262) (Fig. 13B). Therefore, vasodilatory property of HO-1-derived CO is not only directly dependent on cGMP production but also coordinately connected with EC-derived NO.
EC injury or apoptosis can occur, leading to the vascular dysfunction, when ECs are exposed to pro-inflammatory stimuli, nutrient deprivation, or DNA-damaging agents. Cytoprotective functions of HO-1 may be attributed to heme turnover and the concerted action of its enzymatic reaction products, such as CO, BV/BR, and iron. Each of these metabolites has their own set of physiological properties that may contribute to protection of ECs against apoptosis. Expression of HO-1 or administration of exogenous CO prevents ECs from TNF-α-initiated EC apoptosis through the activation of the p38 MAPK pathway (31). The antiapoptotic effects of CO (15 ppm) in anoxia/reoxygenation-exposed rat pulmonary artery ECs is dependent on the activation of several signaling pathways, such as phosphatidyl inositol 3-kinase and p38 MAPK pathways, and associated with decreased Fas expression and caspase-3 activity (299). HO-1/CO has been shown to block IL-18-mediated human cardiac EC death by reversing IL-18-mediated p38 MAPK and NF-κB activation (297). Although a majority of in vitro studies show the protective effects of CO on vascular endothelium, exposure of bovine pulmonary artery ECs with CO (100 ppm) induces a pro-apoptotic effect (261), implicating that the cytoprotective effects of CO can be influenced by a dose of CO and cell types. Inhibition of BR level by siRNA targeting for BVR increased the ROS production, leading to cell death in HeLa and primary neuronal cells (20). BR can protect ECs against oxidative or nitrosative stress via ROS scavenging (72). Administration of BV to various injured models, such as transplantation, I/R, and vascular balloon injury, attenuates apoptotic cell death [reviewed in (170, 176)]. Free iron produced by HO-1 induces the synthesis of ferritin (292), which also confers cytoprotective effect (18).
Further, Wang et al. showed that CO attenuates hyperoxia-induced EC apoptosis by inhibiting both extrinsic (Fas-dependent) and intrinsic (mitochondria-dependent) pathways (277). They showed that CO inhibits hyperoxia-induced extrinsic apoptosis signaling initiated by death-inducing signal complex trafficking from the Golgi apparatus to the plasma membrane and downstream activation of apical caspase-8 through the suppression of NADPH oxidase activity for ROS production (175), which is involved in intrinsic apoptosis signaling. CO also inhibits hyperoxia-induced EC death by regulating the Bcl-2-related proteins involved in inhibiting both intrinsic and extrinsic apoptotic signaling. Indeed, CO inhibits the activation of Bid and expression and mitochondrial translocation of Bax, whereas it increases Bcl-XL/Bax interaction and Bad phosphorylation, resulting in the suppression of cytochrome c release and caspase-9/-3 activation. These results indicate that HO-1/CO prevents apoptotic EC death by regulating the activation of both extrinsic and intrinsic apoptosis signal pathways.
VSMCs closely contact with vascular ECs and play a primary role in vascular tone. HO-1 expressed in the vasculature and its catalytic products CO and BV are involved in the control of SMC proliferation (207, 253). Inhibition of SMC proliferation by CO (250 ppm) is mediated by G0/G1 arrest via upregulation of G1-cyclin-dependent protein kinase inhibitor p21 and downregulation of cyclin D1 expression (240) (Fig. 13B). CO can inhibit platelet-derived growth factor–induced SMC proliferation by blocking the mitochondrial ROS-ERK-cyclin D1 pathway and the NADPH oxidase-cyclin D1 pathway (254). HO-1 activators and BR decrease platelet-derived growth factor–induced human SMC proliferation by reducing ROS production and ERK phosphorylation (253). Since exaggerated SMC proliferation is correlated with vascular inflammatory diseases, such as atherosclerosis and restenosis, expression of HO-1 and its metabolic products may play a vital role in vascular therapy.
Increases in HO-1 expression and/or HO-1 metabolic products suppress a variety of inflammatory responses in the vascular endothelium. In quiescent state, the vascular endothelium regulates vascular tone and inflammation to maintain proper vascular homeostasis (57). However, in inflammatory conditions, ECs become highly vasoconstrictive, prothrombotic, and pro-adhesive (57). Upregulated adhesion molecules in ECs, such as selectins (VCAM-1 and ICAM-1) by inflammatory stimuli induce the recruitment of inflammatory cells to the endothelium, ultimately inducing vascular dysfunction such as atherosclerosis and restenosis (28). ECs isolated from HO-1-deficient mice show higher levels of intracellular free iron, ROS and TNF-α-induced VCAM-1, ICAM-1, and E-selectin as compared with ECs isolated from wild-type mice (227). Induction of HO-1 expression inhibits hypoxia/reoxygenation-induced stasis by suppressing NF-κB-dependent expression of ICAM-1 and VCAM-1 in the skin of sickle mice (22). Similarly to the effect of HO-1 expression, exogenous CO attenuates an increase in ICAM-1 expression and polymorphonuclear leukocytes adhesion to LPS-stimulated human ECs. CO released from systemically administered chemical CO donors provides antiinflammatory effects by interfering with NF-κB activation and subsequent downregulation of pro-adhesive vascular EC phenotype in the liver of septic mice (43). This effect is involved in inhibition of the transcription factor NF-κB by blocking NF-κB RelA phosphorylation at serine 276, which is critically involved in expression of adhesion molecules such as ICAM-1 and VCAM-1, leading to the promotion of vascular inflammation and diseases (297).
C. Regulation of VEGF expression by HO-1/CO
VEGF is known as an essential mediator not only for developmental and pathological angiogenesis but also for metabolism. So far, the VEGF family is composed of at least seven members: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, PlGF, and snake venom-derived VEGF. VEGF-A is regarded as a key regulator of angiogenesis since both VEGF-A−/− and VEGF-A+/− mice die in the embryonic stage showing defects in angiogenesis (59, 234). VEGF-A expression is mainly upregulated through the stabilization of HIF by hypoxia, cytokines, growth factors, NO, HO-1/CO, ROS, and others. HIFs are potent regulators of VEGF-A, VEGF receptor-1, and VEGF receptor-2 expression by various stimulants, including hypoxia (212, 229). Particularly, the biological gaseous molecules, CO and NO, have been shown to stabilize HIF-1α to elicit angiogenesis-associated gene expression, such as VEGF.
CO acts as an inhibitor of electron-transport complex IV (33) and results in a significant and transient burst of ROS arising from the mitochondria (55, 300), which can be involved in HIF-1 stability (45). Exposure to CO promoted the activation of HIF-1α by ROS production from mitochondria in macrophages, consequently protecting against lung I/R injury in an animal model (55). However, precise mechanisms by which CO promotes HIF-1α stability and VEGF expression in ECs remain incompletely clarified. The contribution of CO to the regulation of HIF-1α is little complicated since CO is reported to stabilize HIF-1α in normoxia whereas they promote HIF-1α degradation under hypoxia (109). These findings indicate that the effect of CO on HIF-1α activity is dependent on the oxygen availability.
A body of evidence shows that VEGF-induced angiogenesis is initiated through the induction of HO-1 [reviewed in (72)], which in turn promotes the angiogenic processes, including vascular permeability, vasodilation, EC survival, proliferation, migration, and angiogenic sprouting (47). VEGF transgenic mice induce a more than 10-fold accumulation of HO-1 mRNA in lung tissues and protect mice from lethal hypoxia (232). This protective effect is suppressed by the inhibition of HO-1 expression using siRNA and its activity using chemical inhibitors, indicating that HO-1 is an important contributor to biological functions of VEGF. Further evidence that the HO-1/CO pathway plays an important role in angiogenesis has been also confirmed by in vivo Matrigel plus assay (35). Administration with VEGF significantly increases angiogenesis in a mouse model, and this angiogenic effect was abrogated by the inhibition of HO-1 activity. The angiogenic effect of fibroblast growth factor was not affected by HO-1 inhibitors. These results suggest that VEGF and fibroblast growth factor induce angiogenesis through distinct pathways for HO-1 induction (93). The signal pathway of HO-1 induction in ECs by VEGF is likely to be associated with an increase in NO production by increasing Akt-dependent eNOS phosphorylation and association of eNOS with HSP90 (32, 91), leading to angiogenesis (Fig. 13C).
HO-2 deletion in mice affects the phenotype of ECs (23). The alteration in the VEGF pathway, including changes in expression of VEGF receptors (VEGFRs) and VEGFs, was observed in ECs from HO-2-deficient mice. Compared with wild-type cells, HO-2−/− ECs displayed an increase in the mRNA levels of pro-inflammatory VEGFR-1, whereas the mRNA levels of pathologic VEGFR-2 and −3 were markedly decreased. It was found that expression of VEGF-A, the primary ligand for VEGFR-1, was largely unchanged, whereas expression of VEGF-C and -D, ligands for VEGFR-2 and −3, was significantly reduced. This finding suggests that HO-2 activity may regulate the VEGFR-2/VEGF-B and VEGFR-3/VEGF-C pathways in ECs.
The above-menthioned observations suggest that regulation of HO-1 induction and activity by gene delivery or chemical inhibitors controls angiogenesis and may be useful for therapeutic strategies for angiogenesis-associated diseases, including I/R injury and tumor growth and metastasis.
D. Restenosis and vasculopathy
Restenosis, neointimal hyperplasia, and vasculopathy are vascular disorders after percutaneous angioplasty, coronary artery bypass grafting, lower extremity vein bypass grafting, and transplantation (225). These vascular proliferative disorders remain common problems resulting in significant long-term morbidity and mortality. Their pathogenesis is multifactorial; the most common initiating event appears to be endothelial disruption and subsequent overproliferation of the underlying smooth muscle cells (Fig. 14A).
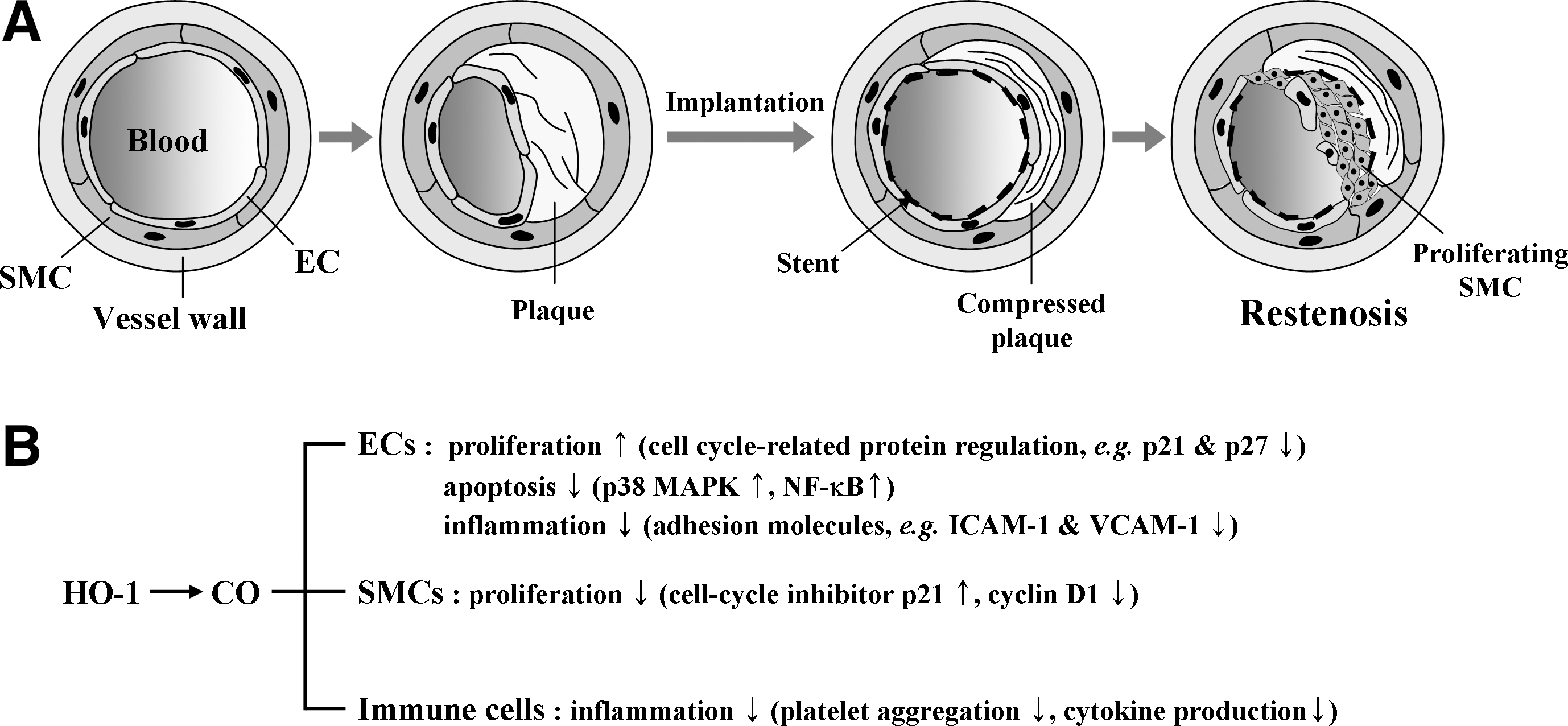
Many studies have demonstrated that HO-1/CO is involved in regulating the pathogenic process of vascular proliferative disorders (Fig. 14B). HO-1 gene transfer into the vessel wall or CO delivery resulted in a significant reduction in intimal hyperplasia in a rat model of allogenic aorta transplantation compared to controls by decreasing activated leukocytes, adhesion molecule expression, and VSMC accumulation in the intima (47). However, HO-1 gene transfer shows a strong inhibitory effect on leukocyte infiltration and expression of adhesion molecules and cytokines compared with CO delivery, whereas CO reveals more inhibitory effect on VSMC accumulation in the intima than HO-1 gene delivery. This study indicates that the beneficial effects of HO-1 gene expression on the reducing graft arteriosclerosis are associated with CO production, but possibly also with other end products (iron and BR), from heme degradation by HO-1. However, this distinct mechanism should be further investigated.
The differential induction of HO-1, which is certainly correlated with differential production of CO from heme by the catalytic reaction of HO-1, may account for the inhibition of VSMC proliferation, constrictive neointimal hyperplasia, and recurrent lumen narrowing. Indeed, direct exposure to CO suppresses restenosis after carotid balloon injury in rats and mice, and this protective effect is closely associated with a strong inhibition of vascular inflammation and VSMC proliferation (132, 195). This effect was elicited by the generation of cGMP, the activation of p38 MAPK, and expression of the cell cycle inhibitor p21 (195). Probucol, a drug used to prevent restenosis, reveals its beneficial effect by inhibiting the proliferation of VSMCs via induction of HO-1 (65). These findings strongly suggest that end products, mainly CO, from heme degradation by HO serve as key modulators for vascular inflammation, EC function, and VSMC proliferation, which are pathogenic factors for human vascular proliferative diseases such as restenosis, neointimal hyperplasia, and vasculopathy.
VII. Conclusion
Optimal vascular health depends on a delicate balance in the vascular wall of pro-oxidative and antioxidant cellular mechanisms. Presence of more than one major cardiovascular risk factor creates an environment in which the principal oxidative systems such as NADH/NADPH oxidase, xanthine oxidase, and the uncoupling of eNOS are activated in the vascular wall in addition to increased production and decreased dismutation of superoxide. As a result, functional and structural alterations develop in the vascular cells. Moreover, inflammation can potentiate the deleterious effects of oxidative stress on the vasculature. These contribute to the development of clinical sequences such as atherosclerosis, myocardial infarction, stroke, congestive heart failure, and cognitive dysfunction related to vascular damage (Fig. 15). However, the HO system could attenuate/block the progression of vascular diseases via their antioxidant, antiinflammatory, and antiproliferative effects.
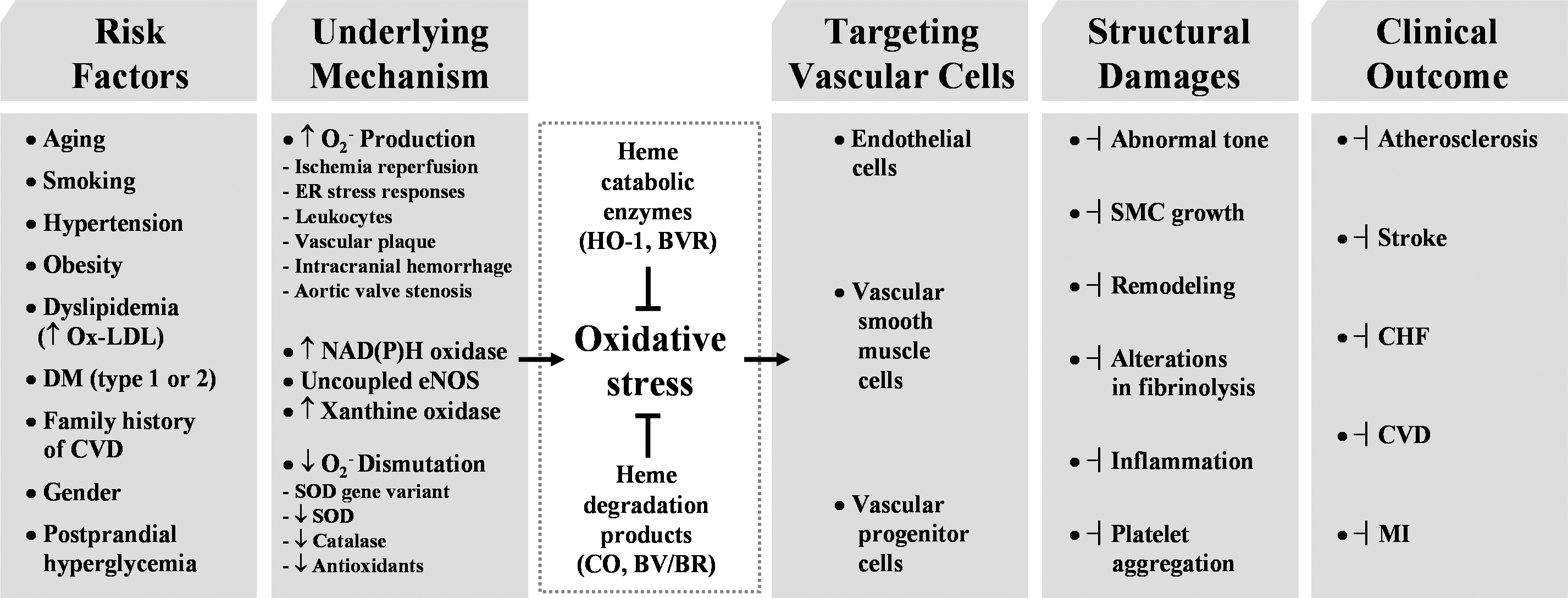
Laminar shear stress (LSS) may protect vascular endothelium against atherogenesis. LSS increases NO biosynthesis, prolongs EC survival, and generates an anticoagulant, antiadhesive cell surface [reviewed in (212)]. In contrast, disturbed blood flow, with low shear reversing or oscillatory flow patterns, may promote atherogenesis; ECs exposed to disturbed blood flow exhibit reduced eNOS levels, increased apoptosis, oxidative stress, permeability to LDL, and leukocyte adhesion (203). Recently, Ali et al. (10) have presented evidence that LSS increases endothelial responsiveness to statins and that HO-1 induction represents an important component of the vasculoprotective profile of these drugs. They found that treatment of mice with atorvastatin induced HO-1 expression in the aortic endothelium and that this occurred preferentially at sites exposed to LSS. The mechanisms underlying a synergistic relationship between LSS and statins include maximal Akt phosphorylation, dependence upon eNOS, Kruppel-like factor 2, and Nrf2 activation. This study may have important implications for the efficacy of statins in patients with CAD, and also emphasize the need for novel therapies, such as those targeting HO-1/CO system, to optimize vasculoprotection.
The strategies to target HO system as described in this review may offer promising therapeutic approaches to clinicians for the effective management of a number of vascular disease states that have, in the past, proved difficult to treat. Also, this review has laid a foundation to prepare the researcher for translation of these current discoveries into new therapeutic modalities to combat vascular diseases.
Footnotes
Acknowledgment
This work has been supported by a National Research Foundation Grant funded by the Korean Government (MOEHRD) (BRL-2010-0087350).
Abbreviations Used
Reviewing Editors: John Belcher, Francois Boucher, Vittorio Calabrese, Damian Calay, De-Xing Hou, Giovanni Li Volti, Mahin Maines, Cesare Mancuso, Giovanni Mann, and Helena Parfenova