
Abstract
Like nitric oxide and carbon monoxide, hydrogen sulfide has historically been recognized as an industrial pollutant, but in recent years, it has been shown to be an important mediator of many physiological processes. Hydrogen sulfide contributes to the maintenance of gastrointestinal mucosal defense and repair. It also exerts many antiinflammatory effects, including inhibition of leukocyte adherence to the vascular endothelium and leukocyte migration to sites of inflammation. Conversely, inhibition of endogenous hydrogen sulfide synthesis leads to a loss of mucosal integrity and to an increase in mucosal inflammation. Hydrogen sulfide therefore appears to have overlapping actions with nitric oxide and prostaglandins in terms of modulating mucosal defense and resolution of inflammation. Recent evidence suggests that these properties of hydrogen sulfide can be exploited in the design of novel therapies for ulcerative and inflammatory diseases of the gastrointestinal tract. Antioxid. Redox Signal. 12, 0000–0000.
Introduction
In this article, the roles of H2S in modulating inflammation are discussed, as is the role of this mediator in maintaining mucosal integrity and in promoting the healing of injury. In each case, the effects of novel H2S donors in relevant animal models are reviewed. These studies suggest important roles for H2S in mediating mucosal defense, particularly in the stomach, in regulating inflammatory processes, and in promoting the resolution of inflammation and injury. H2S-releasing derivatives of nonsteroidal antiinflammatory drugs (NSAIDs) have been shown to be more effective than the parent NSAIDs but to cause markedly less gastric injury. An H2S-releasing derivative of mesalamine has been shown to be significantly more effective than the parent drug in rodent models of colitis. These H2S-releasing antiinflammatory drugs suppress leukocyte extravasation and expression of several proinflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and interferon-γ (IFN-γ).
Modulation of Inflammation
Leukocyte function
Several lines of evidence point to an important role of H2S in regulating some of the key events in the inflammatory cascade and in the resolution of inflammation. These actions of H2S (and of nitric oxide) contribute to the ability of these gaseous mediators to enhance gastrointestinal mucosal resistance to injury. The induction of adhesive interactions between circulating leukocytes and the vascular endothelium is the initial step in the process of extravasation of leukocytes and their migration to sites of injury or infection. An important role of H2S as a tonic regulator of this process was suggested by the observation that inhibition of H2S synthesis in rats resulted in a rapid increase in leukocyte adherence in mesenteric venules (60). When H2S synthesis is inhibited, an increase in expression of adhesion molecules on the endothelium (P-selectin, ICAM-1) and on leukocytes (LFA-1) can be detected (14). Leukocyte adherence stimulated either by aspirin or by a formylated tripeptide (fMLP) could be dose-dependently inhibited by exogenous H2S (i.e., through administration of an H2S donor). H2S donors also were effective in reducing leukocyte accumulation in a rat airpouch model in which inflammation was induced by injection of carrageenan (60). Conversely, administration of an inhibitor of endogenous H2S synthesis (BCA) significantly enhanced carrageenan-induced leukocyte infiltration (60). Modulation of leukocyte extravasation by H2S also was demonstrated in a rat model of endotoxin-induced lung and liver inflammation (25).
Altered leukocyte interactions with the endothelium also were observed in mice genetically deficient of one of the key enzymes for H2S synthesis (CBS). Mice completely deficient in this enzyme did not survive, but those that were heterozygous for CBS exhibited increased vascular permeability, reduced leukocyte-rolling velocity, and increased leukocyte adherence to the vascular endothelium (21).
Resolution of an inflammatory process is complex, involving the removal of infiltrating leukocytes and the repair of tissue injury (39). Several groups of mediators have been identified that play key roles in the resolution of inflammation, and H2S appears to be one such mediator. Its role in promoting the resolution of gastrointestinal injury and inflammation is discussed later. One of the mechanisms through which H2S may contribute to resolution of inflammation is by inducing neutrophils to undergo apoptosis (29), a necessary precursor to their engulfment by macrophages (39).
The vasodilator action of H2S has been suggested to occur, at least in part, through activation of ATP-sensitive K+ channels (K+ ATP). Some of the antiinflammatory actions of H2S also may be mediated through activation of K+ ATP channels. Thus, the inhibitor effects of H2S donors on leukocyte adherence stimulated by aspirin were blocked by glibenclamide, an antagonist of K+ ATP channels (14).
The observation that administration of an inhibitor of endogenous H2S synthesis resulted in significant adhesion of leukocytes (primarily neutrophils) to the vascular endothelium in mesenteric venules of rats (60) suggested that H2S tonically downregulates leukocyte adhesion and emigration. Recent studies in the rat provide evidence that supports this hypothesis. Treatment of rats twice daily for a week with β-cyano-alanine (BCA), a reversible inhibitor of CSE, resulted in a significant increase in myeloperoxidase activity in the colon (51). Myeloperoxidase is an enzyme found in cells of myeloid origin and, in particularly high concentrations, in neutrophils. It has been used extensively as a biochemical marker of neutrophil infiltration (8). Similar results were obtained in rats treated with an inhibitor of CBS [O-(carboxymethyl) hydroxylamine hemihydrochloride] (51).
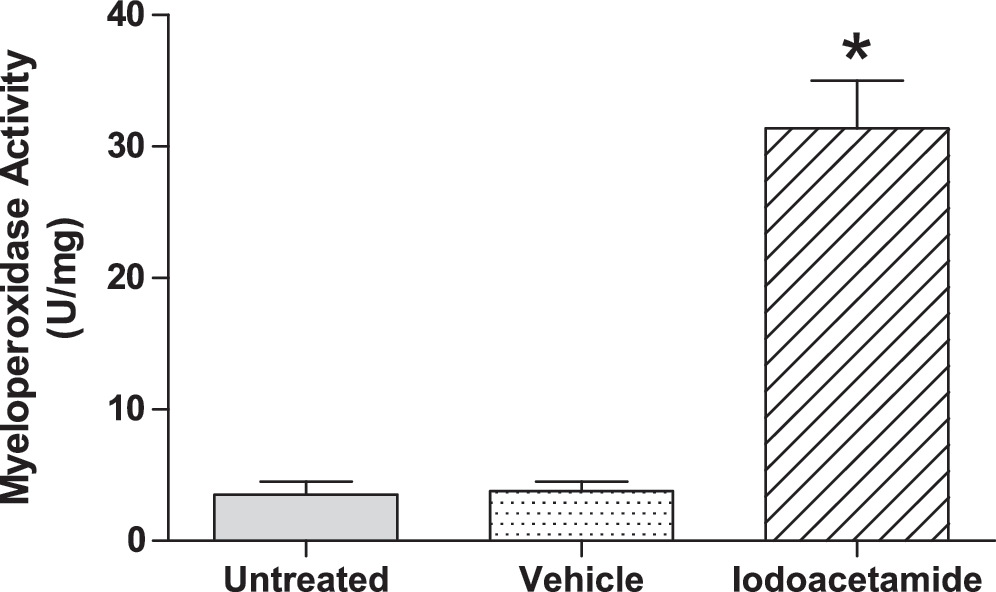
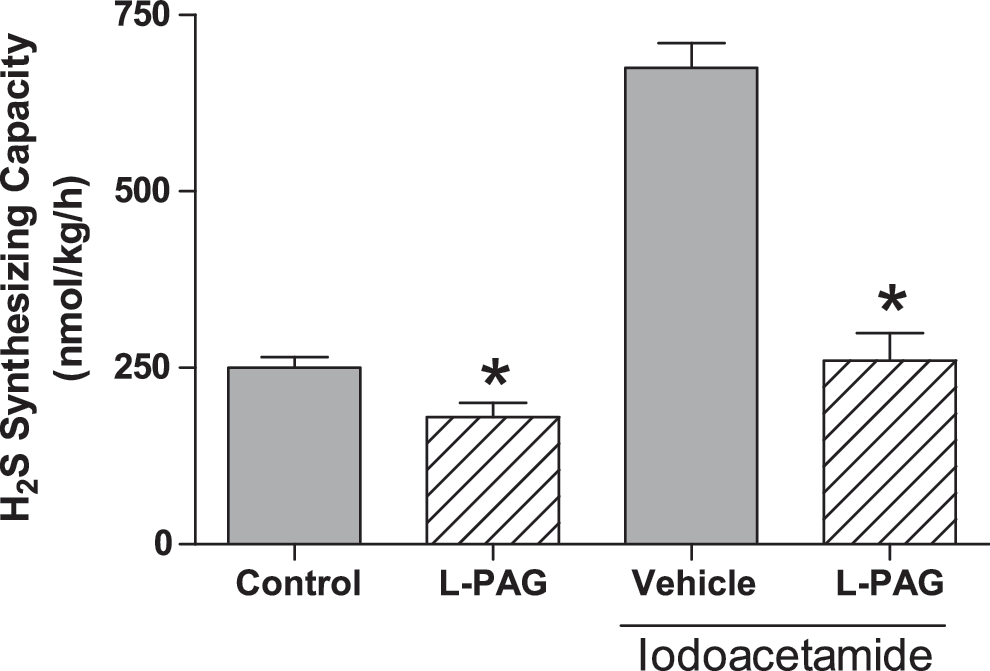
Iodoacetamide is an agent frequently used to induce inflammation in the gastrointestinal tract. When given orally, it induces gastritis (4), whereas intracolonic administration results in the development of colitis (41). These effects have been ascribed to the ability of iodoacetamide to reduce mucosal levels of sulfhydryls, such as glutathione. However, iodoacetamide binds covalently to cysteine, thereby limiting its availability for conversion to H2S. Iodoacetamide has been used as an inhibitor of H2S synthesis (5). Given the observations described earlier with respect to elevated mucosal myeloperoxidase levels (i.e., increased leukocyte infiltration) after inhibition of H2S synthesis, it is possible that the mucosal inflammation induced by iodoacetamide is also a response to diminished mucosal H2S synthesis. Interestingly, suppression of H2S synthesis by iodoacetamide may result in a compensatory increase in the expression of CSE. We observed that treatment of rats for 5 days with iodoacetamide resulted in substantial mucosal inflammation (Fig. 1), as had been observed previously (4). When the ability of the mucosa to generate H2S from exogenous
Edema formation
Plasma exudation is a hallmark feature of inflammation that also appears to be modulated by H2S, although conflicting results have been reported in this regard. For example, intraperitoneal administration of H2S donors significantly and dose-dependently reduced paw edema induced by intraplantar injection of carrageenan (54). These effects of H2S appeared to be mediated through activation of K+ ATP channels, as the antiedema effects of H2S donors were inhibited by glibenclamide and mimicked by pinacidil (an activator of K+ ATP channels) (60). Interestingly, carrageenan-induced paw edema was exacerbated by BCA, an inhibitor of H2S synthesis, consistent with the hypothesis that H2S tonically downregulates inflammation. In studies by two separate laboratories, oral administration of an H2S-releasing diclofenac derivative also was found to reduce carrageenan-induced paw edema in the rat, significantly more potently than did diclofenac (40, 46).
In contrast to these studies, Bhatia et al. (7) reported that injection of carrageenan into the footpad of rats resulted in a significant increase in the capacity of the tissue to convert
Visceral pain
Distrutti et al. (12) used the colorectal distention model in rats to assess the potential role of H2S as a modulator of visceral sensation. Rats were treated with vehicle and then subjected to graded distentions of a balloon catheter inserted into the distal colon of the rat. After a recovery period, the experiment was repeated, but the rats were treated with an H2S donor (NaHS),
A more marked antinociceptive effect of H2S was observed when experiments similar to those described earlier were performed in rats that were recovering from a bout of colitis induced by trinitrobenzene sulfonic acid (12). Such studies are relevant to the human conditions of inflammatory bowel disease and irritable bowel syndrome, in which patients experience severe pain and an increased sensitivity to colorectal distention (6). In the rat model, allodynia (i.e., perception of pain from a stimulus that is usually not perceived as painful) was observed in the rats with colitis, and enhanced hyperalgesia occurred, as compared with that observed in healthy rats (Fig. 3). Treatment with NaHS produced a reversal of the allodynia and a marked reduction of hyperalgesia.
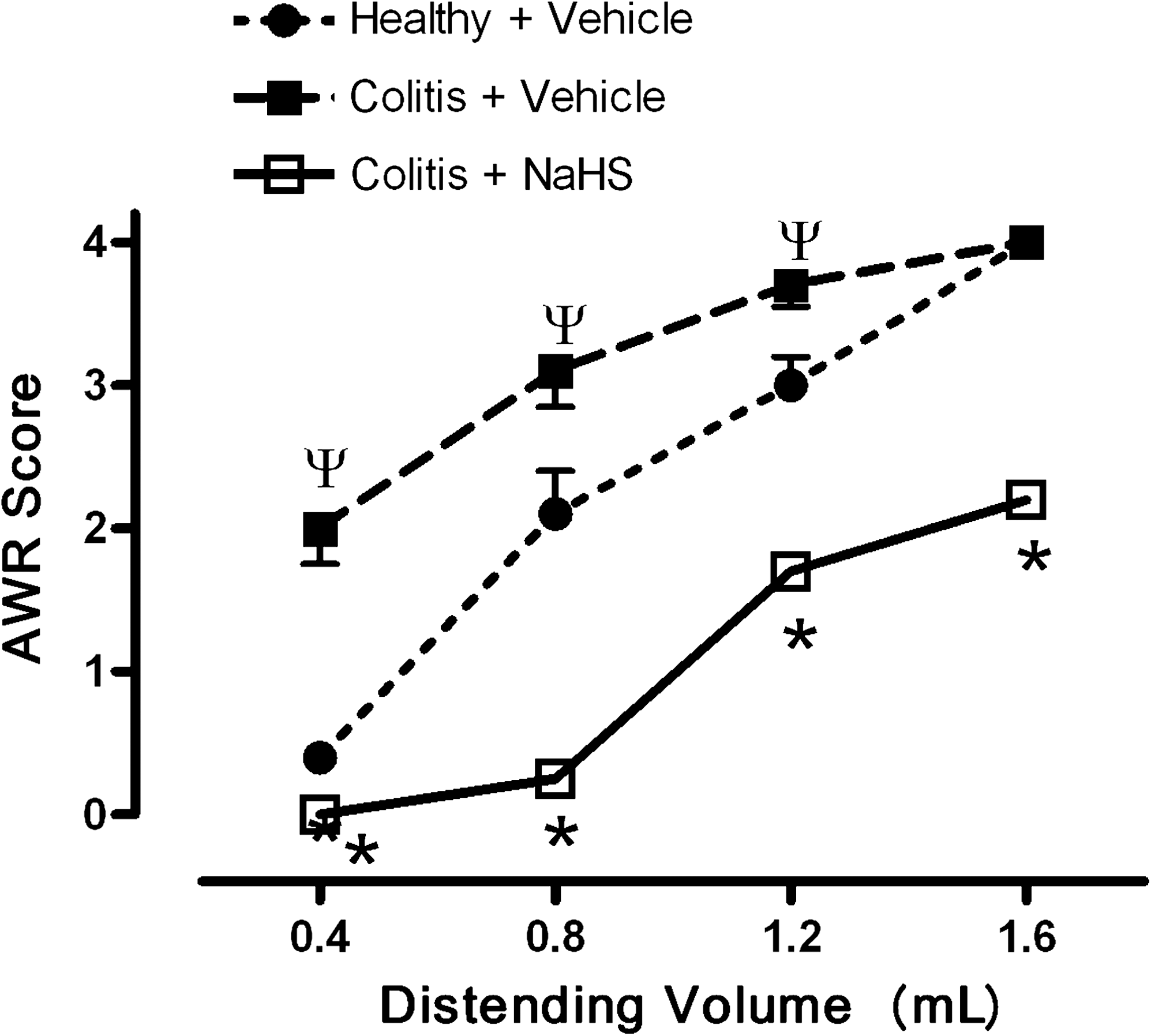
Distrutti et al. (11) provided further evidence for a role of H2S in modulating visceral pain in a study in which a novel drug in development for the treatment of inflammatory bowel disease was tested. Mesalamine (5-aminosalicylic acid) is the first-line therapy for mild-to-moderate colitis and Crohn disease, but it is a weak drug, with doses of up to 6 g/day being required to induce remission. ATB-429 is H2S-releasing derivative of mesalamine, which has been shown to be significantly more effective in accelerating resolution of colitis in mouse and rat models (16). In the colorectal distention model, both with healthy rats and with rats recovering from a bout of colitis, treatment with ATB-429 produced a significantly greater effect than did mesalamine in terms of reducing visceral pain perception.
H2S has been reported to activate transient receptor potential vanilloid-1 (TRPV1), which might be expected to be pronociceptive rather than antinociceptive. Matsunami et al. (31) recently reported pronociceptive effects of intracolonic administration of 0.5 and 5 nmoles of NaHS to mice, but no effect of lower or higher concentrations. Unfortunately, no other H2S donor was studied. The pronociceptive effects of NaHS were not due to actions on K+ ATP channels, but were blocked by a T-type calcium channel blocker.
Some of the issues relevant to the controversy regarding H2S as a pro- versus antinociceptive in the GI tract were recently reviewed (37). Evidence suggests that H2S can be pro- or antinociceptive, or neither, in tissues other than those of the GI tract (1, 10), and it seems likely that the particular effect produced by an H2S donor is context-specific and may also be variable with the concentration of H2S that is generated and the kinetics of release of H2S.
Proinflammatory mediator expression
Considerable evidence exists from both in vitro and in vivo studies that H2S can modulate the expression of a number of other inflammatory mediators, and this may contribute to the modulation of inflammation and injury by H2S. Oh et al. (33) observed that H2S inhibited endotoxin-induced iNOS expression and NO production in cultured macrophages, and further demonstrated that this was a consequence of inhibition of the activation of the transcription factor NF-κB. An inhibitory effect of H2S on endotoxin-induced upregulation of iNOS expression, NO production, and TNF-α expression was similarly observed in cultured microglia (20) (Table 1). At least some of these effects appeared to be due to inhibition of p38 MAP kinase phosphorylation (20). Effects on H2S on p38 MAP kinase also were reported in studies of neutrophils (35). H2S also dose-dependently reduced heme oxygenase-1 expression, through an ERK-dependent mechanism (33). Although not directly studied, an effect on heme oxygenase expression could affect the production of another gaseous mediator, carbon monoxide.
This is a list of some of the possible mechanisms through which hydrogen sulfide can exert antiinflammatory actions.
Modulation of proinflammatory mediator expression also was demonstrated in several in vivo models, including models of GI injury and inflammation. For example, mucosal expression of mRNA for iNOS, the chemokine RANTES, and several proinflammatory cytokines (TNF-α, IFN-γ, IL-1, IL-2, IL-12 p40) was markedly upregulated in a mouse model of colitis (16). Expression of each of the aforementioned was significantly reduced in mice that had been treated for several days with ATB-429, an H2S-releasing derivative of mesalamine. In contrast, neither ATB-429 nor mesalamine reduced expression of the antiinflammatory cytokine, IL-10 (16). Similarly, H2S donors (given intracolonically) reduced TNF-α mRNA and protein levels in the colon in a rat model of colitis, in association with a significant reduction in the severity of the colitis (51). In an in vitro organ-culture study of colonic tissue from patients with ulcerative colitis, Bai et al. (3) demonstrated that diallyl trisulfide, which releases H2S, decreased TNF-α expression.
As discussed in more detail later, the modulation of proinflammatory mediator expression also can be observed in models of NSAID-induced gastric injury. NSAIDs trigger an upregulation of TNF-α expression in the gastric mucosa of rats (2, 14, 36). When H2S donors were administered before the NSAIDs, the upregulation of TNF-α expression was inhibited (14). An H2S-releasing NSAID derivative did not trigger the increase in expression of TNF-α in the gastric mucosa that was observed when the parent NSAID was administered (46). Moreover, the H2S-releasing NSAID inhibited endotoxin-induced NF-κB activation and the accompanying increase in plasma TNF-α and nitrate/nitrite (25). Interestingly, and consistent with observations in experimental colitis (16), this compound augmented the increase in plasma IL-10 that was induced by endotoxin (25).
Resistance to Injury
The resistance of the GI mucosa to injury induced by luminal agents, including endogenous [acid, bile, digestive enzymes] and exogenous [ethanol, nonsteroidal antiinflammatory drugs (NSAIDs)] is dependent on a complex network of factors collectively referred to as “mucosal defense” (55). Mucosal defense is modulated by a number of soluble mediators, most notably, prostaglandins and nitric oxide. Recent studies suggest that H2S can be added to this list of mediators that contribute significantly to mucosal defense. Moreover, as has been accomplished with prostaglandins and NO, it may be possible to exploit the GI-protective effects of H2S in the development of novel therapeutic agents.
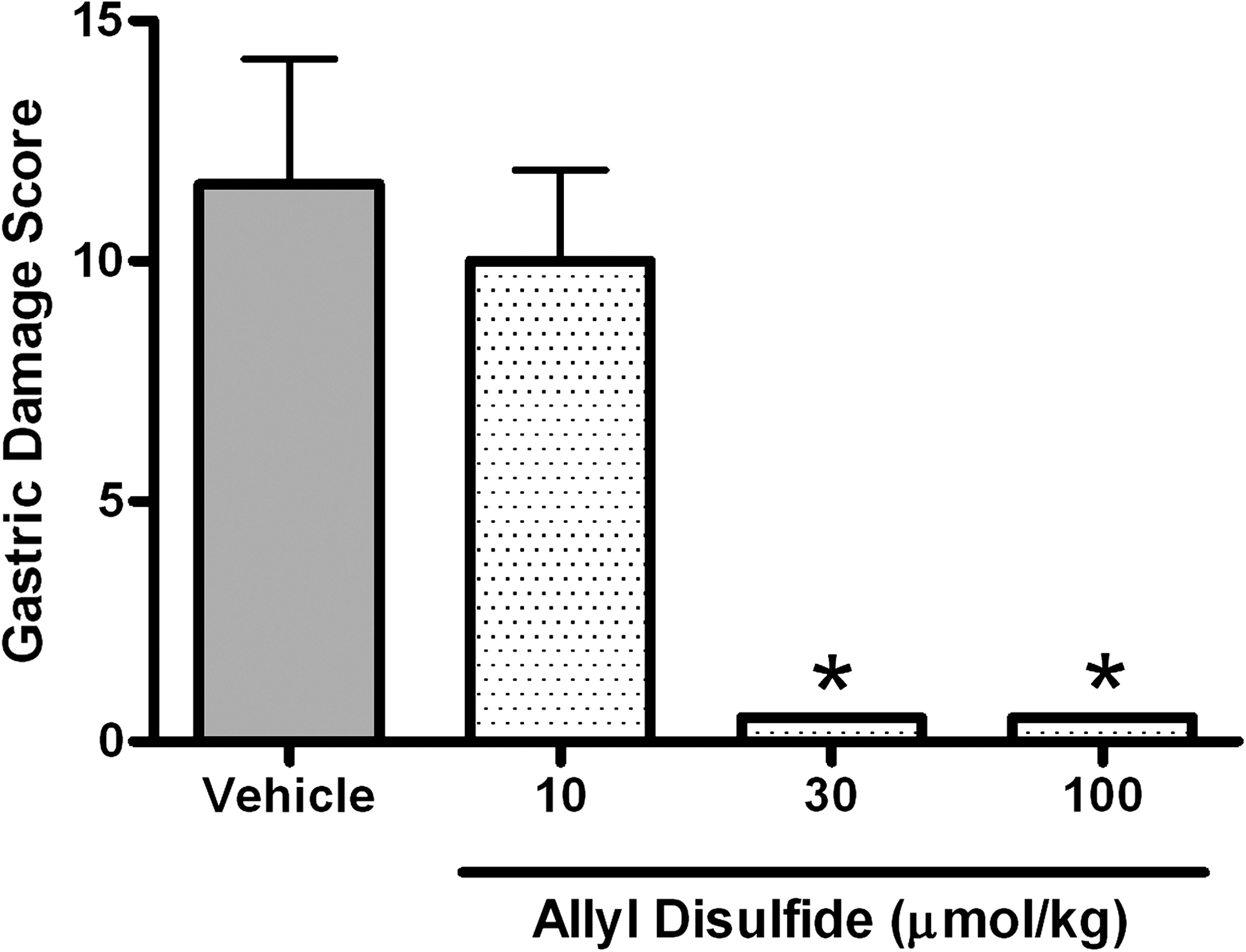
NSAIDs are among the major causes of GI ulceration and bleeding. The damaging effects of NSAIDs in the GI tract, particularly in the stomach, are directly linked to their ability to inhibit mucosal prostaglandin synthesis (55). Suppression of prostaglandin synthesis impairs mucosal defense (reduced blood flow, reduced bicarbonate secretion), results in TNF-α release, and triggers adherence of leukocytes (primarily neutrophils) to the vascular endothelium in the GI microcirculation (55). This latter event is critical in the development of erosions in the stomach (52). Blockade of NSAID-induced leukocyte adherence by immune-depleting circulating neutrophils (48), by administration of antibodies directed against key adhesion molecules (45, 49), or by co-administration of NO (54) prevented damage developing in the stomach. These observations provided the basis for the development of NO-releasing NSAIDs, also called “COX-inhibiting nitric oxide donors” (44, 50). Given that H2S shares with NO the ability to suppress leukocyte adherence, including that induced by aspirin (60), and, like NO, is a vasodilator (14, 56), we hypothesized that H2S may also play an important role in mucosal defense and may be able to prevent NSAID-induced gastric damage (53). We observed that suppression of endogenous H2S synthesis increased the susceptibility to NSAID-induced gastric damage in the rat (14). H2S donors, conversely, reduced the severity of NSAID-induce damage. Figure 4 shows the effects of allyl disulfide, a garlic-derived H2S donor, on gastric damage in the rat induced by oral administration of naproxen. H2S donors also were shown to prevent NSAID-induced decreases in gastric blood flow and the increase in leukocyte adherence (14). The latter was likely attributable to the observed reduction in the expression of key adhesion molecules (LFA-1, ICAM-1), as well as a significant reduction in NSAID-induced TNF-α synthesis (14).
Based on the observations that concomitant administration of an H2S donor and an NSAID largely abrogated the gastric-damaging effects of the latter, attempts have been made to develop NSAID derivatives that liberate H2S in vivo. Thus, H2S-releasing derivatives of diclofenac (ATB-337) and indomethacin were shown to spare the gastric mucosa of the rat from damage, even though they suppressed gastric prostaglandin synthesis to the same extent as did the parent drug (46, 53). ATB-337 also produced markedly less intestinal damage and bleeding (i.e., reduced the decrease in hematocrit) as compared with diclofenac. In terms of antiinflammatory activity, ATB-337 (which is also referred to as “S-diclofenac” in some publications; 25) exhibits somewhat better effects than equimolar doses of diclofenac. For example, ATB-337 was more effective in reducing edema than was the parent drug (25, 46).
The ability of the H2S-releasing NSAID to spare the gastric mucosa from injury despite suppression of mucosal prostaglandin synthesis may be attributable to the suppression of leukocyte adherence by this agent. ATB-337 did not stimulate leukocyte adherence, as was observed with diclofenac, and did not cause the elevation of expression of LFA-1 or ICAM-1 that was observed with diclofenac (46). This effect of the H2S-releasing NSAID derivative also was evident in the studies of Li et al. (25), who observed a significant reduction of LPS-induced accumulation of neutrophils in the lung and liver in animals treated with S-diclofenac.
Resolution of Injury and Inflammation
Gastric ulcer healing
Gastric and duodenal ulceration has decreased in incidence markedly since the discovery that most such ulcers are associated with Helicobacter pylori infection. However, NSAID-induced ulcers remain a clinical concern, particularly in patients requiring antiinflammatory therapy. NSAIDs interfere with the healing of ulcers, most likely related to suppression of COX-2–derived prostaglandin synthesis (55). Previous studies demonstrated key roles for endogenous NO in healing of experimental gastric ulcers in rodents (13, 24, 27). Given the similar actions of NO and H2S with respect to mucosal defense, we explored the possibility that H2S may influence gastric ulcer healing in a rat model. Ulcers were induced by brief serosal application of acetic acid (13). Over the 2- to 3-day period after this treatment, an ulcer forms on the mucosal surface of the stomach and penetrates into the submucosal layer. NSAIDs and selective COX-2 inhibitors have been shown to significantly delay ulcer healing in this model (13, 28), consistent with what occurs in humans.
Two of the key enzymes for H2S synthesis (CSE and CBS) were significantly upregulated at both 3 and 6 days after serosal application of acetic acid (47). In parallel, a marked upregulation of H2S synthesis by gastric tissue was found. When rats with ulcers were treated twice daily with vehicle for 7 days, the ulcers healed to about half the size they were before treatment. However, if the rats were treated with an H2S donor (Lawesson's Reagent or 4-hydroxybutyrate) in the same manner, a significant enhancement of ulcer healing was observed (30 μmol/kg produced this effect, but 10 μmol/kg did not) (Fig. 5). Combined treatment with an H2S donor (Lawesson's Reagent) and an NO donor (glyceryltrinitrate) produced greater healing that was seen with either of these donors alone, but no synergistic effect was evident. Interestingly, treatment with the K+ ATP agonist, pinacidil, or the K+ ATP antagonist, glibenclamide, did not affect ulcer healing, suggesting that actions on this channel were not responsible for the beneficial effects of H2S (47).
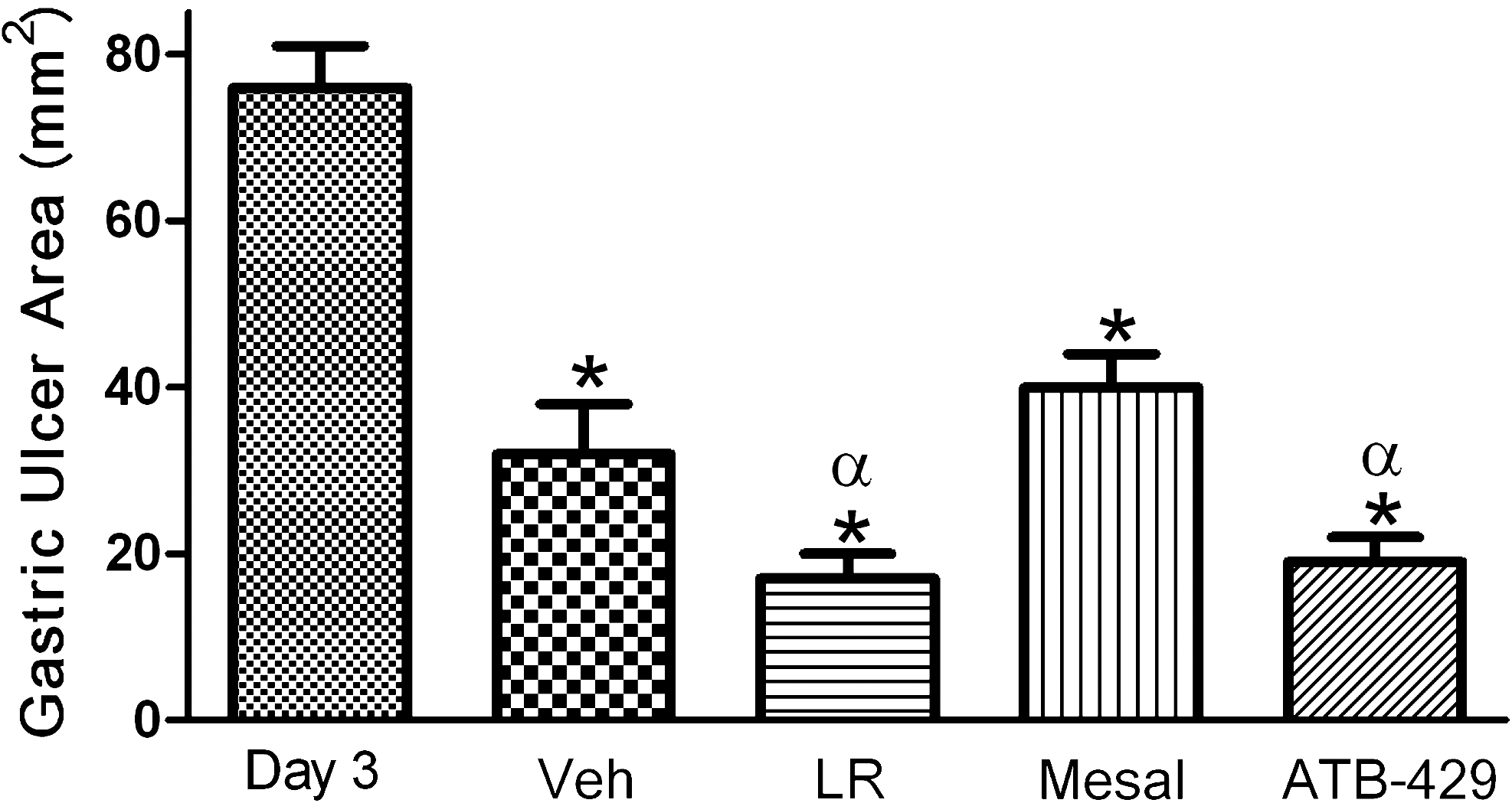
Ulcer healing also was significantly enhanced, in a dose-dependent manner, by twice-daily administration of
As mentioned earlier, the ability of NSAIDs to delay healing of ulcers is a significant clinical concern. With the rat ulcer model, experiments were performed to determine whether co-administration of an H2S donor with an NSAID would abrogate any detrimental effect of the NSAID on ulcer healing (47). Twice-daily treatment of rats with diclofenac, a commonly used NSAID, resulted in a significant impairment of ulcer healing (i.e., significant healing was not observed, as compared with rats treated with vehicle) (Table 2). However, in rats co-treated with diclofenac and Lawesson's Reagent, healing of ulcers was restored.
This is a list of some of the possible mechanisms through which hydrogen sulfide can increase the resistance of the gastrointestinal mucosa to injury. In some cases, the mechanism has been demonstrated only in nongastrointestinal cells or tissues.
The mechanism of action of H2S in promoting the healing of ulcers is not clear. Suppression of gastric acid secretion can result in more rapid ulcer healing, but the H2S donors were found to have no effect on intragastric pH or on volume of gastric acid secreted. It is possible that the H2S donors promoted angiogenesis in the ulcer bed, which could contribute to ulcer healing, but this has not been investigated. H2S has been shown to promote angiogenesis (9).
Colitis
Ulcerative colitis and Crohn disease are serious chronic conditions associated with severe pain, diarrhea, and bleeding. Treatment of these diseases is a significant challenge and is only modestly effective, except in the mildest cases. Several inflammatory mediators, cytokines, and chemokines have been implicated in the pathogenesis of colitis. Of these, TNF-α has been the best exploited from a therapeutic standpoint, through the use of monoclonal antibodies directed against this cytokine. However, these therapies are expensive and are not without risk. The most commonly used medication for treatment of colitis is mesalamine (5-aminosalicylic acid), which is effective in inducing remission in mild-to-moderate cases.
Given the observations that H2S can attenuate some elements of inflammation (leukocyte adherence, edema formation, etc.) and can accelerate healing of injured GI tissue, we explored the possible role of this mediator in a rat model of colitis. Intracolonic administration of trinitrobenzene sulfonic acid (TNBS) results in severe colitis bearing a histologic similarity to Crohn disease (32). Within a few hours, a marked increase in H2S synthesis can be detected, which increases further (to ∼100-fold over levels in the normal colon) in parallel with increased infiltration of granulocytes into the damaged tissue (51). Over the weeks that follow, the severity of the colitis diminishes, and in parallel, a decrease in colonic H2S synthesis is observed. The colonic H2S synthesis could be inhibited by >70–95% by co-incubation of the colonic tissue with inhibitors of CSE and CBS (51).
Whether the elevated H2S synthesis by the inflamed colon contributes to the tissue injury or to the resolution of that injury was investigated by treating rats with a number of inhibitors of H2S synthesis (51), including an inhibitor of CBS (CHH), a reversible inhibitor of CSE (BCA), and an irreversible inhibitor of CSE (PAG). Twice-daily treatment with these inhibitors resulted in significant mortality, with deaths occurring fastest with the CBS inhibitor, and the least with the reversible inhibitor of CSE. In the rats that survived treatment with BCA, a marked exacerbation of the severity of colitis was observed. Treatment of healthy rats with the same inhibitors also resulted in significant mortality, particularly with the CBS inhibitor. In surviving rats, treatment with the CSE and CBS inhibitors resulted in atrophy in the intestine and a significant increase in tissue myeloperoxidase activity (indicative of elevated granulocyte infiltration). Thus, these results suggest that H2S produced in the context of colitis is exerting a predominantly beneficial effect (inhibition of its synthesis leads to exacerbated colitis). Moreover, in healthy animals, H2S synthesis appears to act as a tonic downregulator of granulocyte infiltration into intestinal tissue.
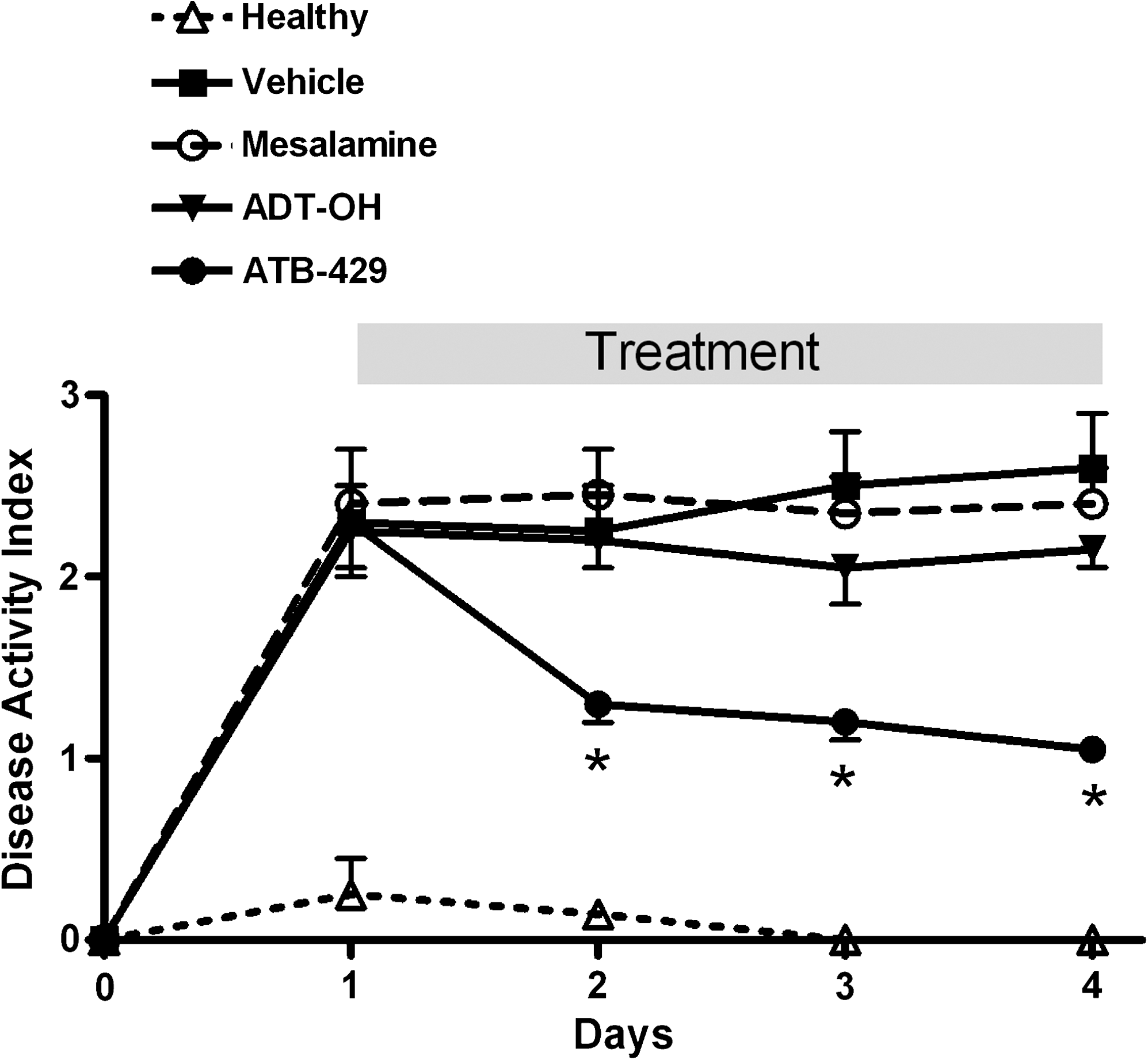
These conclusions were further supported by studies of H2S donors. Twice-daily treatment of colitic rats, by enema, with Lawesson's Reagent or with NaHS resulted in a significant attenuation of the severity of colitis (51). Moreover, a significant reduction of expression of TNF-α was observed in the rats treated with the H2S donors. Moreover, studies of an H2S-releasing derivative of mesalamine (ATB-429) further supported the notion that H2S exerts beneficial effects in the context of colitis. In TNBS-induced colitis in mice, treatment twice daily for a week with ATB-429 resulted in significantly more-rapid resolution of colitis than did treatment with mesalamine (16) (Fig. 6). ATB-429 also was more potent than mesalamine. Although mesalamine had little or no effect on colonic expression of TNF-α, IL-1β, and IFN-γ, treatment with ATB-429 resulted in significant inhibition of expression of each of these proinflammatory cytokines. As well as reducing the severity of inflammation, ATB-429 was shown to accelerate mucosal healing. When tested in the acetic acid–induced gastric ulcer model, described earlier, ATB-429 significantly enhanced healing as compared with vehicle or with an equimolar dose of mesalamine (47) (Fig. 6).
Future Perspectives
Over the past decade, our knowledge of the physiological and pathophysiological roles of H2S has grown considerably, and this is true for studies of the digestive system as much as for any other system in the body. As was the case in the early 1990s with respect to studies of NO, efforts to understand better the importance of H2S are limited by the paucity of highly selective pharmacologic and molecular tools. As such tools are developed, and this is already occurring with respect to H2S donors, investigators will be able to acquire a better understanding of the roles of this molecule in various processes. With more-selective inhibitors and with better systems for measuring H2S production in vivo, some of the controversies regarding the effects of H2S (such as whether it is anti- or pronociceptive) may be resolved. Whereas studies of cardiovascular function have been performed in mice genetically deficient in either CSE or CBS (21, 59), no studies have been published to date on the GI phenotypes of these mice.
Many unanswered questions remain regarding H2S synthesis and catabolism in the digestive system. Although more is being learned about the enzyme systems responsible for H2S production (17, 26, 30), little is known regarding the specific cellular sources of this mediator. It also is not clear to what extent luminal bacteria contribute to the production of H2S from samples of intestine.
The development of H2S-releasing antiinflammatory drugs appears to be a promising approach to the exploitation of the potent and broad effects of this molecule, at least based on preclinical studies that have been published. H2S exerts actions in the digestive system that are very similar to those of NO and, to some extent, prostaglandins. Thus, administration of H2S donors may be of particular benefit in circumstances in which production of one or both of these other “mucosal-protective” mediators is impaired.
Footnotes
Acknowledgments
Dr. Wallace holds the Farncombe Family Chair in Digestive Health Research at McMaster University, and his research is supported by grants from the Canadian Institutes of Health Research, the Crohn's and Colitis Foundation of Canada, and the Ontario Premier's Summit Award.
Author Disclosure Statement
Dr. Wallace holds shares in Antibe Therapeutics, Inc., and Ginova Pharm, Ltd., companies that are developing hydrogen-sulfide–releasing drugs for various indications.