
Abstract
The ability of iron to accept or donate electrons, coupled with the ability of oxygen to act as an electron acceptor, renders both elements essential to normal cellular biology. However, these same chemical properties allow free iron in solution to generate toxic free radicals, particularly in combination with oxygen. Thus, closely interwoven homeostatic mechanisms have evolved to regulate both iron and oxygen concentrations at the systemic and the cellular levels. Systemically, iron levels are regulated through hepcidin-mediated uptake of iron in the duodenum, whereas intracellular free-iron levels are controlled through iron-regulatory proteins (IRPs). Cardiorespiratory changes increase systemic oxygen delivery, whereas at a cellular level, many responses to altered oxygen levels are coordinated by hypoxia-inducible factor (HIF). However, the mechanisms of iron homeostasis also are regulated by oxygen availability, with alterations in both hepcidin and IRP activity. In addition, many genes involved in iron homeostasis are direct targets of HIF. Furthermore, HIF activation is modulated by intracellular iron, through regulation of hydroxylase activity, which requires iron as a cofactor. In addition, HIF-2α translation is controlled by IRP activity, providing another level of interdependence between iron and oxygen homeostasis. Antioxid. Redox Signal. 12, 445–458.
Introduction
Systemic Iron Homeostasis
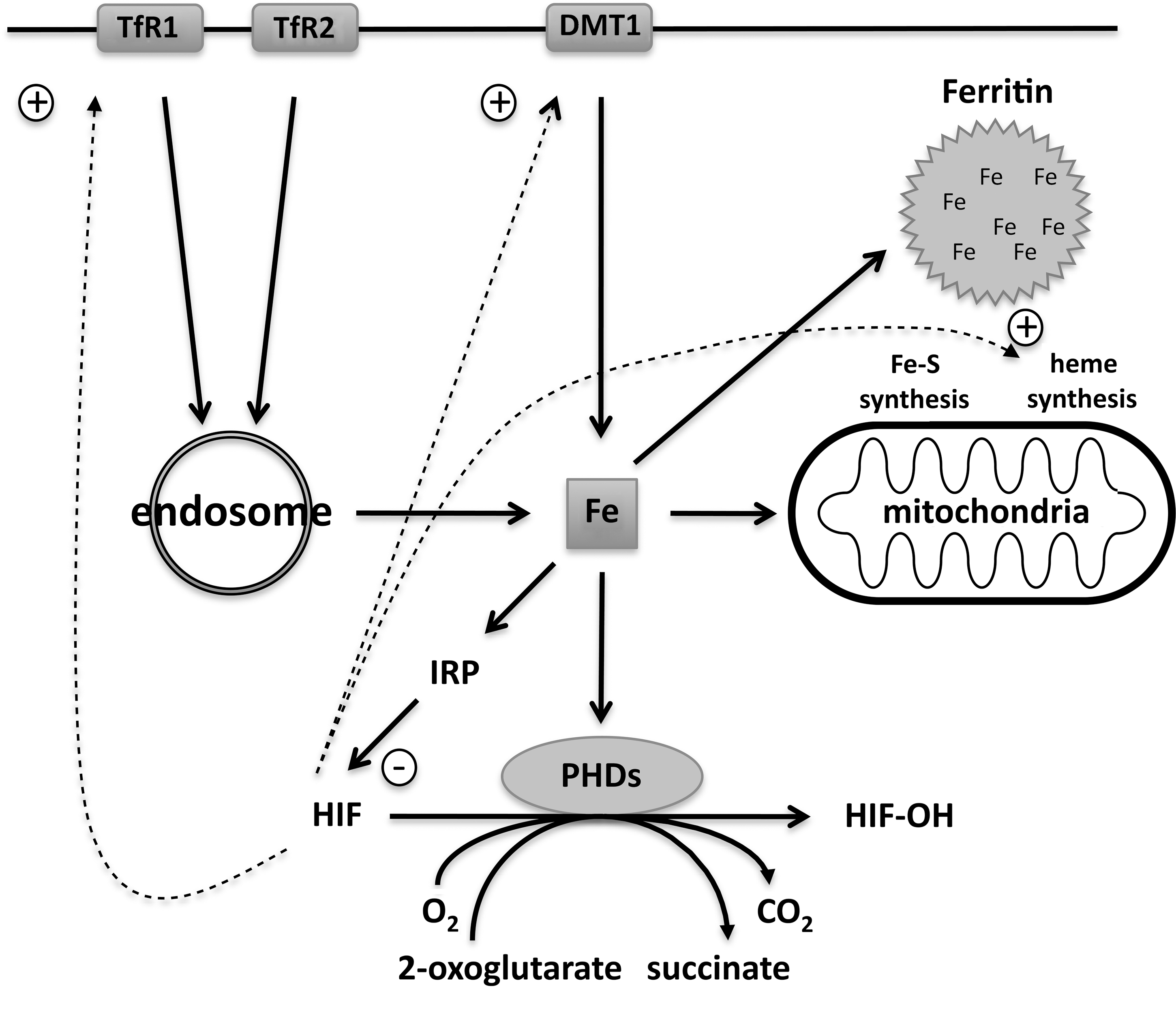
As a constituent of hemoproteins, iron–sulfur (Fe-S) proteins, and other functional groups, iron is important not only in the biochemical reduction of oxygen to release energy, but also in the transport of oxygen to the tissues. Indeed, nearly 80% of iron demand in humans is required for the daily production of haemoglobin to fill ∼200 billion new erythrocytes (52). Much of this iron is provided by recycling iron from old erythrocytes as they are destroyed, or by releasing iron stored in the liver, macrophages, or other tissues (Fig. 1). However, iron losses from sloughing of skin and mucosal surfaces, as well as blood loss, necessitate the absorption of iron from the diet. As no regulated excretory pathway exists for iron, systemic iron homeostasis is controlled through regulation of this dietary uptake. Iron is present in the diet mainly in its ferric (Fe3+) form or as heme. Ferric iron uptake into duodenal enterocytes involves first the reduction of iron to its ferrous (Fe2+) state by a ferric reductase (duodenal cytochrome b, DcytB) (89), and second, movement of iron across the cell membrane by a ferrous iron transporter, divalent metal transporter 1 (DMT1) (35, 45). These enterocytes are rapidly shed from the tips of the intestinal villi, so unless the iron is exported across the basolateral membrane into the plasma, it is not absorbed.
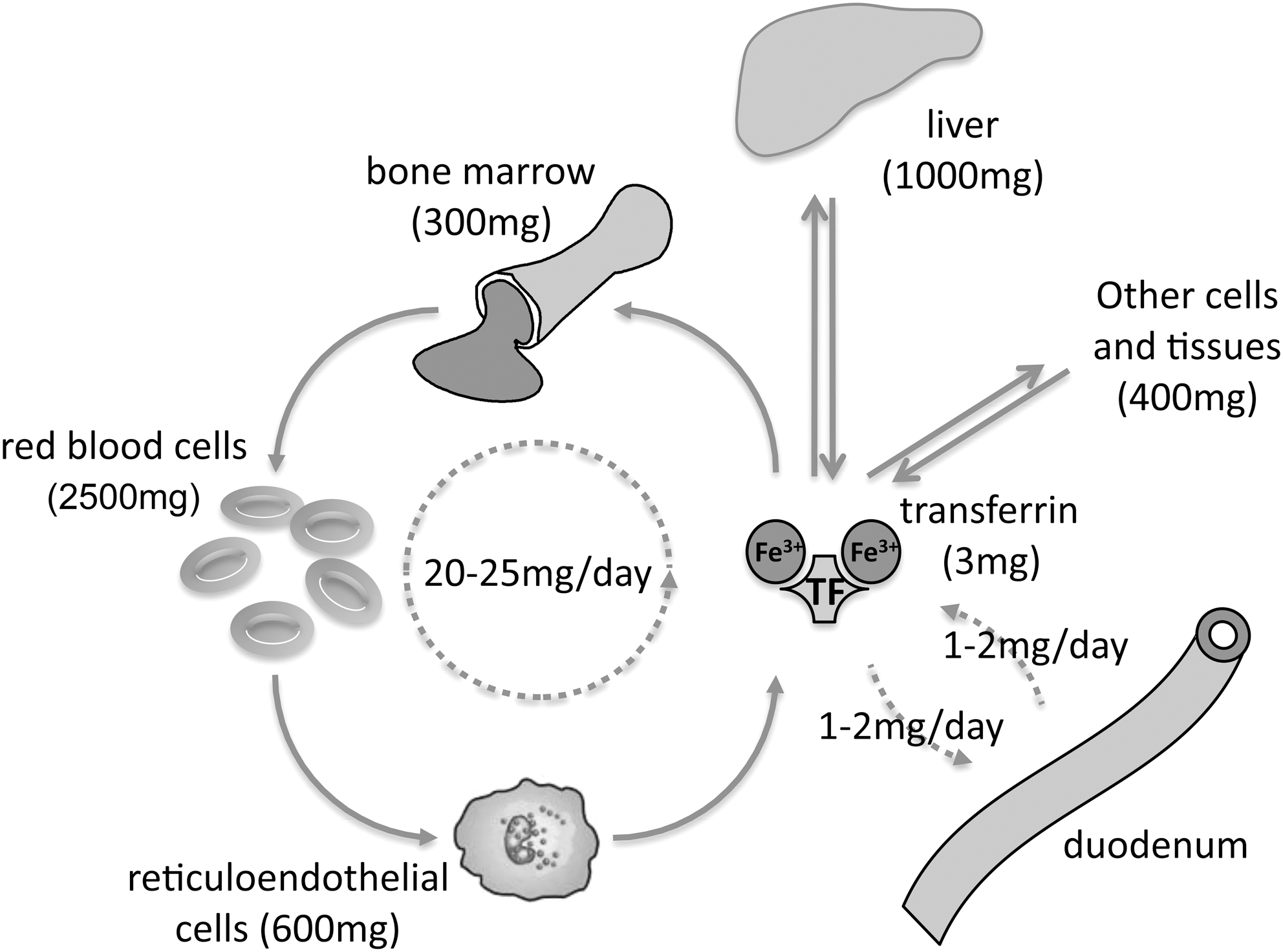
Serum iron is maintained by the regulated release of iron from these intestinal enterocytes, as well as recycling of the iron stored in macrophages and hepatocytes by the cell-membrane protein ferroportin (FPN) (1, 22, 90). Ferroportin is the sole known exporter of intracellular iron, so high levels of basolateral ferroportin expression on duodenal enterocytes will lead to maximal iron absorption (23). The iron-satiety peptide hormone hepcidin interacts with these ferroportin receptors, causing them to be endocytosed and degraded, thereby blocking the single pathway for uptake of dietary iron (101). In the oxygen-rich environment of blood, iron circulating in the plasma is largely bound to transferrin in a form that is nonreactive, but also difficult to extract (58). Several transferrin-dependent iron-uptake mechanisms mediate cellular uptake of this iron–protein complex from the plasma. The most studied of these is the transferrin receptor 1 (TfR1), which, after binding transferrin, internalizes to endosomes (58). Acidification of these endosomes releases iron. The ferric (Fe3+) iron released is reduced, by ferrireductase activity, and transported into the cytoplasm by DMT1 (34). Here, any iron that is not required for immediate synthesis of biologically active molecules is stored in ferritin. Each ferritin complex, consisting of 24 H- and L-subunits, is capable of sequestering up to 4,500 atoms of iron as chemically inert ferrihydrite (50). Only a small proportion of cellular iron remains available for ongoing biologic processes in the form of a labile iron pool.
Regulation of Hepcidin
The master regulator of systemic iron homeostasis is the liver-derived antimicrobial peptide hepcidin, which inhibits ferroportin-mediated intestinal iron absorption as well as iron release into the serum from macrophages and hepatocytes. The mature 25-amino acid peptide is cleaved from the prohormone by the proprotein convertase, furin (141). It has eight cysteine residues, forming four intramolecular disulfide bonds that are highly conserved among species, and is regulated primarily through transcriptional mechanisms. Changes in body iron stores, erythropoiesis, inflammation, and hypoxia all contribute to control hepatic hepcidin mRNA levels, often through complex interplay (52, 99). For example, hepcidin levels are increased in response to iron loading and are reduced during iron deficiency (39). When erythropoiesis is stimulated by blood loss or hemolysis, hepcidin expression is suppressed (38, 102). Inflammation and, in particular, the inflammatory mediator interleukin-6 (IL-6) leads to enhanced hepcidin through Janus kinase/signal transducers and activator of transcription (JAK/STAT) signaling, with concomitant hypoferremia (100). Hypoxia reduces hepcidin expression both in vivo and in tissue culture. Mice subjected to hypobaric hypoxia exhibited reduced hepcidin levels and increased iron uptake (102). In addition, hypoxia downregulates hepcidin mRNA in human cell culture (102, 108). However, the molecular mechanisms governing hepcidin regulation, and the way in which they interact, are as yet incompletely understood and the topic of much current research.
Nevertheless, insights have been gained through the study of mutations associated with inherited disorders of iron loading. HFE protein, mutated in the most common form of hereditary hemochromatosis, is a nonclassic MHC class I molecule that interacts, on the cell surface, with TfR1 (31, 32, 107). Binding of diferric transferrin blocks this interaction, allowing HFE to stimulate hepcidin expression in response to circulating levels of diferric transferrin. Furthermore, binding of diferric transferrin to TfR2 stabilizes the receptor (64, 114), leading to induction of hepcidin expression through activation of the extracellular signal-regulated kinase (ERK1/2) and p38 microtubule-associated protein kinase (MAPK) pathways (15). A recent study demonstrated the importance of HFE-TfR2 complexes to this hepcidin response (41).
BMPs [in particular, BMP6 (2, 93), as well as BMPs 2, 4, and 9] stimulate hepcidin transcription through the son of mothers against decapentaplegic (SMAD) intracellular pathway, and liver-specific knockout of SMAD4, leads to iron overload and low levels of hepcidin that cannot be stimulated by iron loading (21) (Fig. 2). Growth and differentiation factor 15 (GDF15), an iron- and oxygen-regulated (75) member of the TGF-β superfamily of proteins that includes all BMPs, exhibits increased expression and secretion during erythroblast maturation and inhibits hepcidin expression in vitro (140). Thus, GDF15 has been postulated as a mediator of both the iron stores and the erythropoietic regulation of hepcidin. However, no correlation was seen between hepcidin and GDF15 levels after erythropoietic stem cell transplants (66), or in hemodialysis patients treated with erythropoietin (6).
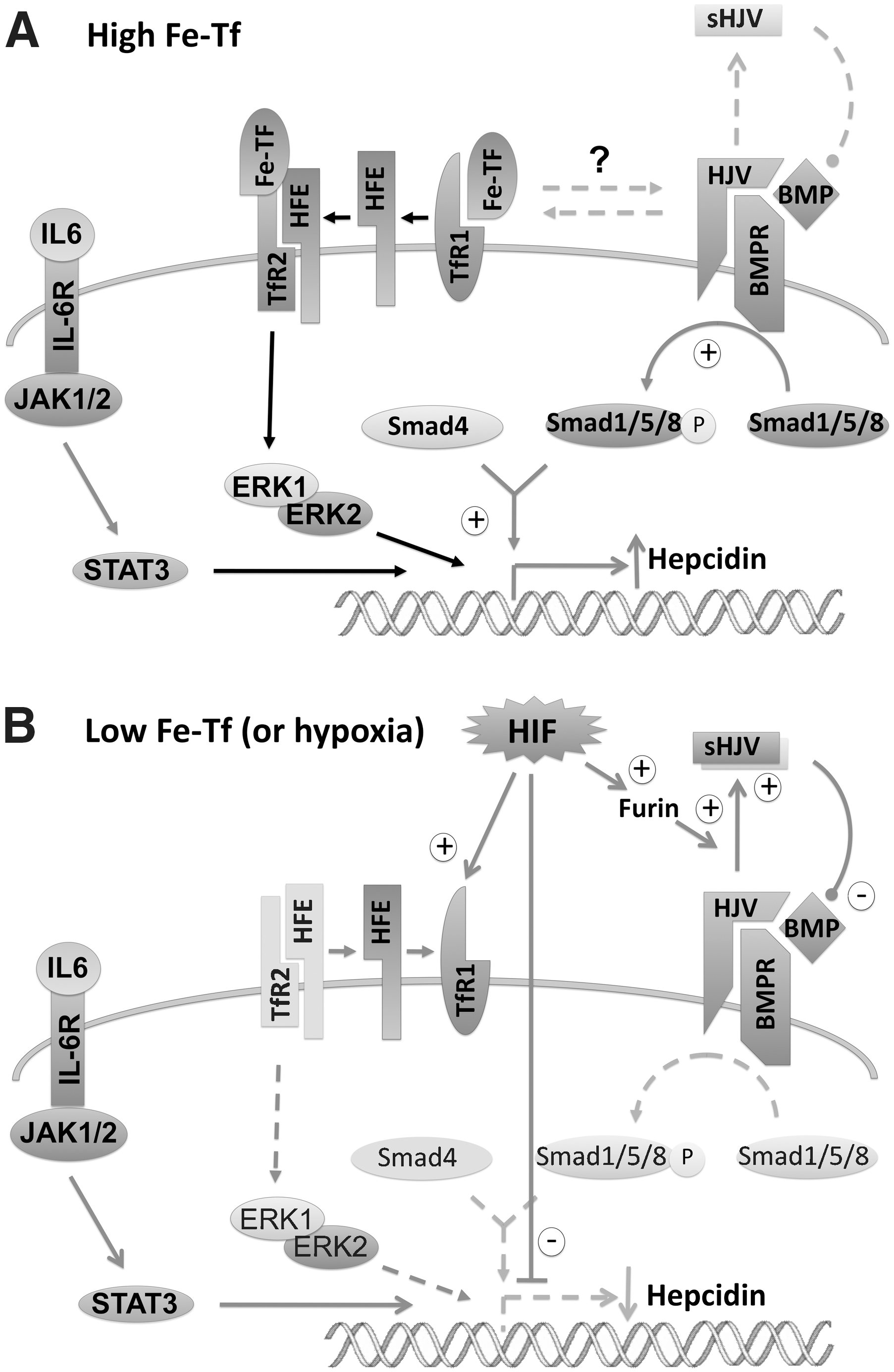
Hemojuvelin (HJV), a member of the repulsive guidance molecule (RGM) protein family functions as co-receptors for bone morphogenetic protein (BMP) signaling. The importance of hemojuvelin as well as HFE and TfR2 to hepcidin regulation is underscored by the lack of hepcidin expression in Hjv-knockout mice and the mutation of this gene observed in most cases of juvenile haemochromatosis (56, 106, 115). Hemojuvelin is expressed in two isoforms, a membrane-bound heterodimer and a secreted molecule (soluble HJV, sHJV) that is processed by furin (the same proprotein convertase that cleaves hepcidin to the mature form) (141). sHJV antagonizes the membrane-bound isoform, represses BMP signalling, and reduces hepcidin expression (7, 81, 82). Thus, furin has opposing actions on hepcidin.
Recently, another protease, matriptase-2, has been implicated in the regulation of hepcidin, mutation of which is associated with refractory iron-deficiency anaemia and inappropriately high hepcidin levels in humans and mice (25, 33, 36). Similar to furin, this membrane-bound serine protease is able to suppress hepcidin by cleaving membrane-bound hemojuvelin, although the fragments produced have not been shown to be active or specific (130). It is not known how these two proteins interact, or whether matriptase-2 is regulated in any way.
Cellular Iron Homeostasis
At the cellular level, most cells tightly regulate the intracellular labile iron pool through effective control of iron uptake (e.g., by adjusting the expression of TfR1) and of the sequestration of iron (e.g., by modulating levels of ferritin). Cytosolic iron concentrations are registered by two ubiquitously expressed, homologous members of the aconitase gene family, iron-regulatory proteins 1 and 2 (IRP1 and IRP2) (118). When labile iron levels decrease, the IRPs bind iron-response elements (IREs) within mRNAs that encode these genes. Binding to the 5'-untranslated region (5'-UTR) inhibits initiation of translation and downregulates expression of genes such as H- and L-ferritin, erythroid 5-aminolevulinic acid (the first enzyme of heme biosynthesis), mitochondrial aconitase, and ferroportin (22, 28, 51, 90) (Fig. 3). Conversely, binding of IRPs to IREs within the 3'-UTR protects mRNAs from endonucleolytic cleavage and degradation, thereby increasing expression of genes such as TfR1 (51). These processes both act to restore cytosolic iron levels.
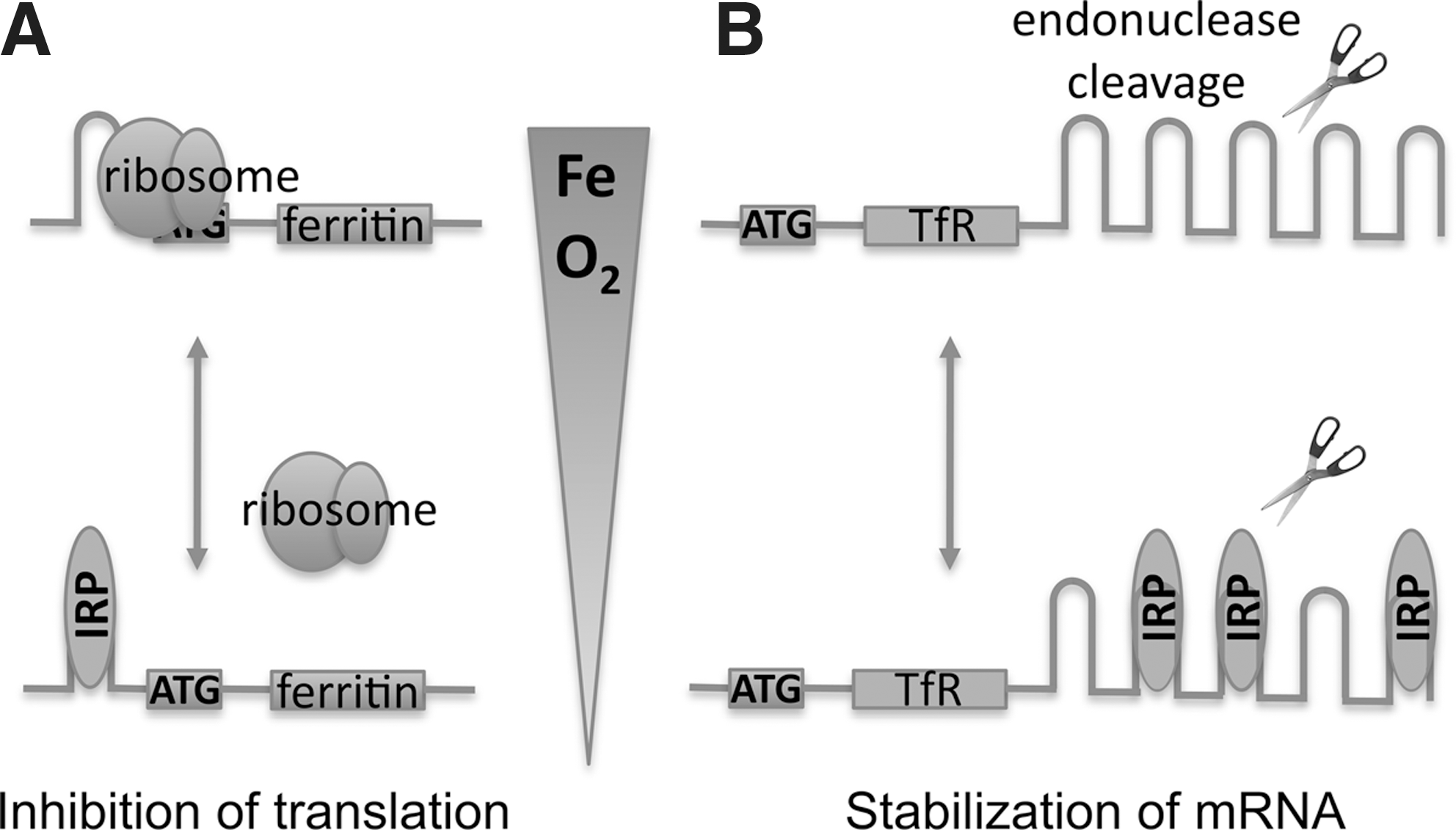
Embryos homozygous for deletions in both IRP1 and IRP2 die before implantation, emphasizing the importance of the IRP-IRE pathway to cellular function (132). Mice lacking both copies of IRP2 develop a microcytic anemia and neurodegeneration (particularly in the substantia nigra), a phenotype that is exaggerated in animals that also lack one copy of the IRP1 gene, underscoring that IRP1 and IRP2 have redundant functions (19, 131). This lack of IRP–IRE activity mimics iron overload, resulting in ferritin overexpression and reduced TfR1 levels, and likely leads to functional iron deficiency, despite accumulation of ferric iron sequestered by the ferritin. With their dependence on iron-facilitated aerobic ATP generation in mitochondria, neurons are thought to be particularly sensitive to this functional iron deficiency (118).
IRP1 exists in two structural forms, depending on the presence or absence of an iron–sulphur (Fe-S) cluster at its core. When present, the Fe-S cluster forms part of the enzymatic active site that converts citrate to isocitrate in the tricarboxylic acid cycle. When iron is restricted and Fe-S cluster assembly is impaired, the apoprotein takes on a more open structure and is able to bind the IRE stem–loop structure of regulated transcripts. However, the state of this Fe-S switch can also be influenced by degradation of the Fe-S cluster. For instance, the Fe-S moiety can be oxidized and destabilized by oxidative stress such as nitric oxide and peroxynitrite, as well as by oxygen (14, 48), thereby promoting its mRNA-binding function. However, whereas in tissue culture, IRP1 contributes to iron-regulatory activity (69), at physiologic oxygen levels, it is thought that most of the IRP1 exists as the holoprotein with aconitase rather than IRE-binding actions (118). Targeted deletions of IRP1 and IRP2 in mouse models have demonstrated that IRP2 is the main physiologic iron sensor (40, 77, 94), although synergistic effects also are seen (19, 131).
IRP2, which derived from a duplicated gene pair, does not bind Fe-S clusters and apparently lost its aconitase activity at some point in evolution (118). Instead, its IRE-binding activity is regulated at the level of protein abundance through iron-dependent degradation of IRP2 by the ubiquitin–proteasome pathway. Under conditions of low iron, IRP2 is stabilized and able to bind IREs on target mRNAs. This degradation is also facilitated by oxygen (47, 49), so that at the oxygen tensions present in most mammalian tissue, IRP2 is relatively stable and more abundant, whereas IRP1 is mainly in its aconitase form (118). Thus, low levels of oxygen promote IRP2–IRE binding but inhibit IRP1–IRE activity.
The mechanism by which IRP2 is regulated in response to iron and oxygen is incompletely understood. Relative to IRP1, IRP2 contains an extra cysteine-rich exon, although the contribution of this domain to iron-dependent oxidation, ubiquitination, and proteasomal degradation is debated (12, 46, 61, 62, 147). One hypothesis suggested that site-specific oxidation of three specific residues within this “iron-dependent degradation domain” targets the molecule for degradation by the proteasome (61, 62, 67, 158), although it also was proposed that this domain functions by binding to heme (44). Other workers have found that this domain is dispensable for iron-dependent degradation of IRP2 (12, 49, 147). Pharmacologic data using dimethyl oxalylglycine (DMOG), an inhibitor of 2-OG–dependent dioxygenases (63), has implicated this class of enzyme in the regulation of IRP2 (49, 147, 148), although the specific enzyme(s) involved have not yet been identified.
Oxygen Homeostasis
Oxygen is highly lipid soluble and so freely diffuses across cell membranes. The uptake of oxygen is therefore governed by matched ventilation and perfusion within the lung, the diffusion capacity of the alveolar wall, and the oxygen-carrying capacity of the blood, and is not reliant on specific uptake mechanisms.
Homeostatic responses to hypoxia (low levels of oxygen) involve changes at both the systemic and cellular levels. Systemically, respiratory, cardiovascular, and hematologic changes predominate. Most often studied after ascent to altitude, systemic hypoxia in humans induces both an acute increase in ventilatory rate, within minutes, as well as a later and more-sustained response that continues to increase for several days (135). This latter response, known as ventilatory acclimatization, is also accompanied by an increased sensitivity to further hypoxia (112, 121). Cardiovascular responses to acute high-altitude hypoxia also occur over a similar period, with progressive increases in both heart rate and cardiac output occurring over several days (143).
Unlike that in other vascular beds, hypoxia constricts rather than dilates the pulmonary vasculature. Similar to the effects on ventilation, this again shows a biphasic pattern, with an acute response that plateaus after 5 min, and a secondary pulmonary vascular acclimatization occurring between 45 min and 2 h, again accompanied by increased sensitivity to further hypoxic insult (24, 139). This pulmonary hypertensive response can be beneficial in matching pulmonary perfusion to ventilation, although its contribution to maintaining systemic oxygenation over the physiologic range remains unclear. However, global hypoxic pulmonary vasoconstriction, such as that seen in chronic hypoxic lung disease, leads to pulmonary vascular remodeling and pulmonary hypertension, worsening survival of these patients.
Perhaps the best-studied systemic effect of high-altitude hypoxia is the erythropoietic response (11, 65, 142). This lacks the acute effect seen within minutes. Instead, hypoxia induces a detectable increase in serum erythropoietin within 90 min that peaks by 2 days and thereafter gradually declines as hemoglobin increases (27). Occurring as part of a transcriptional response, study of the hypoxic control of erythropoietin expression has led to the discovery of a widespread network of oxygen-dependent gene regulation. These transcriptional effects are orchestrated by the transcription factor hypoxia-inducible factor (HIF). First identified as a hypoxia-inducible factor binding the hypoxia-response element (HRE) within the 3' enhancer of the erythropoietin gene (145), HIF is now known to be widely expressed in almost all cell types studied and to regulate the expression of many genes that are involved, both directly and indirectly, in restoring oxygen homeostasis and in reducing oxygen demand (88, 155).
These HIF targets include enzymes at key checkpoints in the glycolytic pathway, such as glucose-uptake transporter 1 (GLUT1), 6-phosphofructo-1-kinase L, and lactate dehydrogenase A (155). Hence, during conditions of oxygen deficiency under which oxidative phosphorylation by the tricarboxylic acid (TCA) cycle cannot proceed, HIF coordinately upregulates the less-efficient glycolytic pathway and facilitates conversion of the resultant pyruvate to lactate, for export to the liver (the Pasteur effect). Furthermore, in hypoxia, HIF directly upregulates pyruvate dehydrogenase kinase (PDK), which phosphorylates and inactivates the pyruvate dehydrogenase enzyme complex that converts pyruvate to acetyl-coenzyme A, thereby inhibiting pyruvate metabolism by the TCA cycle (70, 105). Also important in limiting oxygen demand are cell-based decisions involving cell proliferation and apoptosis with key HIF-mediated genes such as B-cell lymphoma-2 (Bcl-2) family members, and the cell-cycle regulators p21 and p27 (16).
Hypoxia and HIF in particular interact at many points with the angiogenic pathways and have a critical function in stimulating the growth of these new blood vessels. Both vascular endothelial growth factor (VEGF) and its receptor, Fms-related tyrosine kinase 1 (Flt-1), are transcriptionally activated by HIF (37, 43), and this alone is capable of initiating angiogenesis in quiescent vessels. However, for an efficient vasculature to be formed, a more coordinated response is required involving angiopoietin and its receptor Tie-2, fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF), as well as the balanced control of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases (TIMPs), all of which are under the transcriptional control of HIF (111). The combined effect is for new blood vessels to grow into areas of impaired oxygenation to restore adequate oxygen delivery. During localized hypoxia, vasomotor tone also is controlled, through HIF-mediated transcriptional regulation of factors such as endothelin 1, inducible nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), and heme oxygenase-1 (HO-1), endothelin-1, and atrial naturetic peptide (ANP), which improve blood flow (155).
Paradoxically, HIF also appears to play a role in the vasoconstriction response seen during hypoxic pulmonary acclimatization. Patients with Chuvash polycythemia have activation of the HIF pathway as a result of inactivating mutation in the von Hippel–Lindau protein (pVHL), which leads to constitutive stabilization of HIF (3). These patients exhibit both pulmonary hypertension and pulmonary vascular hypersensitivity to hypoxia, similar to patients with hypoxic lung disease (134). Similarly, these patients also have a reduced carbon dioxide set point for the respiratory controller and enhanced hypoxic ventilatory sensitivity, comparable to those of altitude-acclimatized individuals (134). This strongly suggests that HIF plays a role in both these responses.
HIF Target Genes Involved in Iron Metabolism
Tissue hypoxia, in the kidney, as a consequence of either systemic hypoxia or of anaemia, activates HIF, leading to increased erythropoietin production (127). Arguably, this is the most important gene in iron homeostasis, as even under basal conditions, erythropoiesis is the major user of iron and is capable of manyfold upregulation. Increased erythropoietin alone would rapidly lead to a state of iron deficiency, limiting further erythropoiesis. Iron deficiency is the most common limiting factor in red cell production. Therefore, efficient erythropoiesis requires not only stimulation of the bone marrow by erythropoietin, but also coordinated provision of iron. During hypoxia-mediated erythropoiesis, this increased iron provision is mediated in part through HIF-controlled upregulation both of its transporter molecule, transferrin (116), and of its cellular-uptake mechanism, the transferrin receptor 1 (TfR1) (85, 137) (Table 1). The hypoxic regulation of TfR1 by HIF was initially overlooked, as TfR1 expression is also enhanced by IRP/IRE interaction. Because only Fe3+ can be bound by transferrin, ceruloplasmin (also known as ferroxidase), which is required to oxidize Fe2+ to Fe3+, also is important in iron transport. This, too, is an HIF-target gene and likely further supports hypoxia-mediated iron transport (96). Furthermore, ferrochelatase, the enzyme that catalyzes insertion of ferrous iron into heme molecules, also is transcriptionally activated by HIF (84), thereby promoting synthesis of hemoglobin.
The major proportion of iron required for essential protein synthesis derives from the turnover and recycling of hemoproteins. Heme oxygenase, which catabolizes conversion of heme to biliverdin, with release of free iron, is also under HIF transcriptional control and facilitates recycling of iron during hypoxia (78).
However, long-term, hepcidin-regulated, ferroportin-mediated iron absorption is important for maintaining total body iron levels. Relative iron deficiency generated by erythropoietin-driven red cell production would act to suppress hepcidin production and to enhance ferroportin-mediated intestinal iron absorption. However, recent evidence suggests additional mechanisms by which HIF is able to regulate hepcidin more directly. HIF can bind hypoxia-response elements within the murine hepcidin promoter and downregulate hepcidin expression directly (108) (Fig. 2). Furthermore, the proprotein convertase, furin, that cleaves hemojuvelin, has been recognized as an HIF target gene with increased levels of furin in hypoxia leading to more sHJV, which also reduces the expression of hepcidin (91, 129). Conversely, activation of the HIF pathway does not alter hepcidin cleavage, suggesting that furin is not rate limiting in this conversion (141). Thus, it has been postulated that the overall effect of HIF-regulated furin will be reduction of hepcidin signalling (129). These novel links between the HIF pathway and hepcidin regulation provide additional mechanisms for coordinate upregulation of both erythropoietin and ferroportin, further supporting erythropoiesis by allowing the HIF pathway to enhance iron metabolism (Fig. 4).
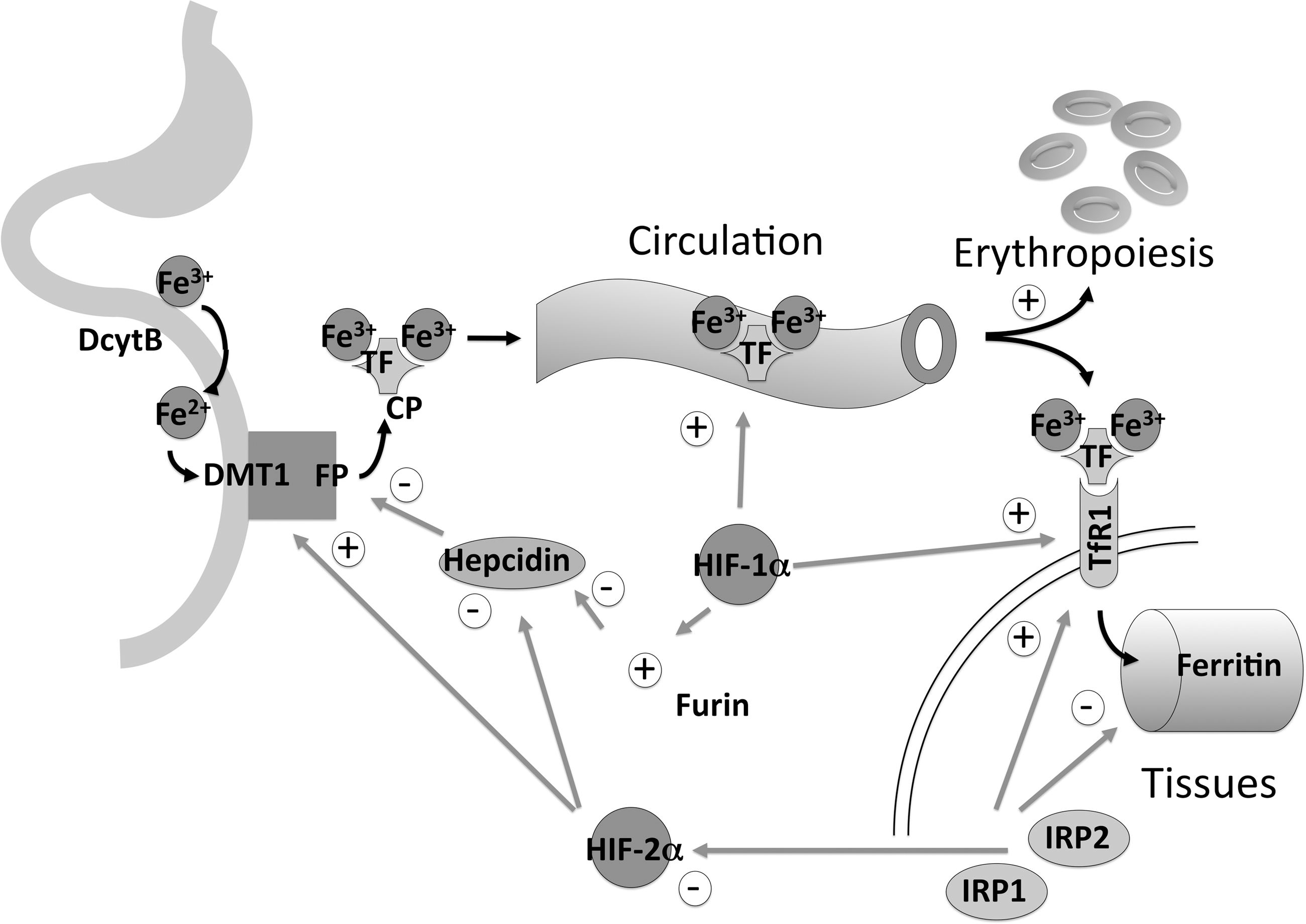
In addition, duodenal uptake from the intestinal lumen into the enterocyte involves reduction of Fe3+ to Fe2+ by the apical ferric reductase duodenal cytochrome b (DcytB), followed by uptake by the apical iron transporter divalent metal transporter-1 (DMT1). Both of the proteins have recently been demonstrated to be direct HIF-2α target genes (87, 128).
Differential Effects of HIF-1α and HIF-2α on Iron Regulation
Despite binding a common core consensus motif, HIF-1α and HIF-2α modulate the transcription of overlapping but distinct sets of target genes. Although HIF-1α was first identified as binding the erythropoietin enhancer, it transpires that erythropoietin-producing cells predominantly express HIF-2α (117, 156) and that this isoform is mainly responsible for hypoxic erythropoietin regulation (124, 125, 150).
The striking downregulation of hepcidin observed in conditional knockout of VHL in the liver, in which both HIF-1α and HIF-2α are stabilized, together with the minor effect of HIF-1α elimination on hepcidin expression, suggests that HIF-2α may play a dominant role in regulation of hepcidin by HIF (108), although direct proof is still lacking. Furthermore, both DcytB and DMT1, which are involved in apical uptake of iron into the enterocyte, are HIF-2α target genes (87, 128). This is consistent with a predominant role for HIF-2α in coordinated erythropoiesis. In addition, a phylogenetically conserved IRE has been identified in the 5'-UTR of the HIF-2α gene that limits erythropoietin translation during intracellular iron deficiency (120). This mechanism for feedback regulation between iron and oxygen metabolism may serve to match erythropoiesis better to iron availability.
Regulation of HIF by Hydroxylases
HIF is a heterodimeric transcription factor composed of two basic helix-loop-helix proteins, HIFα and HIFβ, members of the PAS family of proteins (PER, AHR, ARNT, and SIM family) (144). The HIFβ subunit is identical to a previously described constitutive nuclear protein, aryl hydrocarbon receptor nuclear translocator (ARNT), which has roles in other transcription pathways (113). By contrast, all three HIFα subunits (HIF-1α, HIF-2α, and HIF-3α) are novel proteins, the levels of which are highly induced by hypoxia.
The mechanisms regulating the abundance and activity of HIF-1α and HIF-2α, the two main isoforms, are largely concordant, and this is reflected in similar domain structures (123). HIF-1α and HIF-2α levels are both regulated by proteolytic degradation, dependent on two distinct oxygen-dependent degradation domains (NODDD and CODDD), located in the central region of the molecule (57, 103, 110, 119, 157). In addition, both isoforms possess two transactivation domains, required for recruiting cofactors that mediate transcription [such as the coactivator p300 (5)], an internal activation domain (NAD) that overlaps with the CODDD, and a carboxy-terminal activation domain (CAD). Both the proteolytic destruction and the transactivation domains are under oxygen-dependent regulation.
These same oxygen-dependent responses can be mimicked by iron chelators or cobaltous iron, indicating the involvement of a ferroprotein oxygen sensor. Initially, this was thought to be a heme protein. However, recent work has shown that HIF is regulated through posttranslational hydroxylation of specific prolyl and asparaginyl residues. Hydroxylation is catalyzed by specific oxygen-dependent enzymes that belong to the 2-oxoglutarate–dependent dioxygenase superfamily. These are nonheme, Fe2+-dependent enzymes, in which the iron is loosely coordinated by a two-histidine-one-carboxylate facial triad at the catalytic center, accounting for the ability of iron chelators to inhibit enzyme activity. During the enzymatic cycle, splitting of molecular oxygen is coupled both to hydroxylation of HIF-α and to oxidative decarboxylation of 2-oxoglutarate to succinate and carbon dioxide. The K M for oxygen (oxygen concentration at which enzymatic activity is half-maximal) is much higher than tissue oxygen concentrations, allowing enzymatic activity to respond in a graded fashion over the entire physiologic range. The reaction cycle proceeds by the formation of a highly reactive ferryl (FeIV = O) intermediate that oxidizes the HIF-α amino acid residue. In the absence of HIF-α substrate, uncoupled turnover leaves the iron center in an oxidized and inactive form. Ascorbate is required for full catalytic activity and likely functions to reduce the iron center in this event.
Genetic analysis in model organisms helped identify three closely related mammalian enzymes responsible for hydroxylation of prolyl residues within the NODDD and CODDD, termed PHD1, 2, and 3 (13, 30), and one asparaginyl hydroxylase responsible for modifying the CAD, named FIH (76). Inactivation of each PHD individually by using small interfering RNA has shown that loss of PHD2 alone is sufficient to increase HIF-1α levels in oxygenated cells, leading to the proposal that PHD2 is the most important isoform in oxygen sensing (10). Further evidence is lent to this hypothesis by the embryonic lethality seen in PHD2-knockout mice, whereas those lacking PHD1 and PHD3 survive relatively normally (138). However, these enzymes show differential patterns of organ expression, intracellular localization, inducibility by exogenous stimuli, and substrate selectivity, suggesting that each has a distinct, if overlapping, function (4, 17, 55, 80, 86, 92, 98). For instance, whereas PHD2 exerts its major effect on HIF-1α, under certain circumstances, PHD3 shows a greater bias toward regulation of HIF-2α through hydroxylation of the CODDD rather than the NODDD (4).
Enzymatic hydroxylation of residues within proteins proved to be a novel mechanism of signal transduction, although such modifications are seen with structural significance during the posttranslational processing of collagen molecules (97). The requirement for 2-oxoglutarate as a co-substrate and nonheme iron (Fe2+) as a cofactor, in addition to oxygen, raises important questions as to what extent HIF-hydroxylase activity is affected by physiologic or pathologic variation in iron or ascorbate availability or levels of TCA-cycle metabolites. It is tempting to see these enzymes not only as oxygen sensors, but also as iron, redox, and metabolic sensors, able to modulate the hypoxic response in a coordinated way in response to these influences.
Indeed, 2-oxoglutarate, a co-substrate of the reaction, is an intermediate metabolite of the TCA cycle. Furthermore, both succinate (a product of the hydroxylase reaction) and fumarate are also products of TCA-cycle enzymes and can both compete with 2-oxoglutarate to inhibit PHD enzyme activity (59, 126). It is possible, therefore, that in addition to sensing oxygen, these enzymes are in some way acting as metabolic sensors, able to detect activity or stress on oxygen-requiring TCA cycle metabolism. It is also interesting to postulate whether alteration in cytosolic aconitase activity in association with iron or oxygen levels can in some way alter metabolite levels to affect HIF hydroxylation.
The ability of iron chelators to stimulate erythropoietin production has long been recognized and is attributable to in vivo inhibition of HIF hydroxylases by these agents. Effects are seen after just a single dose of the poorly cell-penetrant chelator desferrioxamine, suggesting that only minor perturbation of iron homeostasis may be required (133, 146). Furthermore, alteration of iron status also has effects on the pulmonary vasculature. Desferrioxamine, administered to human volunteers, not only increases pulmonary vascular resistance, but also enhances its hypoxic reactivity (8, 133), whereas infusion of iron blunts hypoxic pulmonary vasoconstriction (133), suggesting that the response is only partially activated by physiologic levels of iron.
In vitro, iron chelation not only suppresses HIF hydroxylase activity (Fig. 5), but addition of iron or ascorbate enhances enzymatic action, suggesting that either iron or ascorbate or both are limiting (72, 104). In addition, differentiation of monocytic cell lines is associated with inhibition of HIF hydroxylase activity and stabilization of HIF-α, in a manner that is independent of oxygen, but dependent on intracellular levels of chelatable iron (71). In vivo, hepatic levels of HIF-1α were increased in normoxic mice fed an iron-deficient diet (108), and in a separate study, duodenal HIF-2α levels were increased in mice fed a similar diet (128). It therefore seems likely that the HIF hydroxylases are able to respond to changes in intracellular free iron across the physiologic range.
However, whether it is the total intracellular chelatable iron or the redox-specific form that is being sensed is unclear. The effects of ascorbate would suggest that intracellular free Fe2+ is important. Oxidative stress can lead to increase in HIF-α levels because of inhibition of HIF hydroxylases. This could be as a result of direct oxidative damage to the prolyl hydroxylases. However, JunD (jun D protooncogene) antagonized Ras (rat sarcoma oncogene homologue) mediated increase in reactive oxygen species, leads to oxidation of Fe2+ to Fe3+, and increased HIF-α levels that can be antagonized by exogenous Fe2+ or ascorbate (42), suggesting that Fe2+ levels are being sensed.
Other Hydroxylases
In addition, the question of hydroxylase activity against non-HIF substrates arises. PHD3 was originally cloned from rat smooth muscle cells as SM-20 and has additional roles in apoptosis of nerve growth factor–deprived sympathetic neurons (26). It is not known to what degree these actions of PHD3 are dependent on HIF, but importantly, these effects are specific to the PHD3 isoform, raising the possibility of an additional unique substrate (79). Furthermore, the hydroxylase FIH was recently shown to hydroxylate ankyrin repeat domains in inhibitor of NF-κB kinase (IκB) proteins (18). The human genome encodes other proteins predicted to encode 2-oxoglutarate–dependent dioxygenases, including those with known functions in collagen biosynthesis (collagen prolyl-4-hydroxylase), DNA repair (alkylation repair homologue-AlkB, a demethylase), and fatty acid metabolism (γ-butyrobetaine hydroxylase and phytanoyl CoA hydroxylase) (109). It therefore seems likely that protein hydroxylation will prove important in signaling pathways outside of HIF and may play as-yet-unrecognized roles in iron as well as oxygen sensing. For example, reports exist of hydroxylation of RNA polymerase II (74), and iron-, oxygen-, and 2-oxoglutarate-dependent degradation of the iron-regulatory protein IRP2 (49, 147).
Conclusions
Given the emerging complexity in both iron and oxygen sensing, together with the large degree of overlap already apparent between the homeostatic mechanisms regulating each, it is likely that yet more points of interaction remain to be discovered. Understanding the full extent of these interactions will likely lead to novel therapeutic strategies. For instance, activation of the HIF pathway will lead to a coordinate increase in erythropoietin, as well as delivery of iron to facilitate erythropoiesis. The possible involvement of as-yet-unidentified hydroxylases in iron metabolism further enhances the potential of manipulating this family of enzymes by using small-molecule inhibitors. Increasing numbers of such agents are being discovered (9, 54, 60, 73, 83, 95, 122, 149, 151 –154). Many were originally developed as inhibitors of procollagen prolyl hydroxylase in an effort to reduce tissue fibrosis (20, 53). However, these compounds highlight one problem with developing this class of drugs: the lack of specificity, with potential for “off-target” effects (29, 53). The possibility of unwanted actions is increased still further when the pleiotropic actions of HIF are considered. This is well illustrated by the ability of HIF to increase both erythropoiesis and angiogenesis, as well as the importance of HIF to tumorigenesis. Further understanding of the relative contribution of each individual HIF and prolyl hydroxylase isoform to these individual processes may aid the development of more-specific therapeutic approaches.
Conversely, the responsiveness of the HIF pathway to intracellular iron, the frequent observation of high levels of HIF in human cancer, and the importance of HIF to in vivo tumorigenesis raises important questions as to the contribution of altered intracellular iron to these processes. Furthermore, altered iron status could potentially contribute, through perturbation of HIF, to more global responses to hypoxia, such as pulmonary hypertension. Thus, manipulation of global or local iron metabolism in these circumstances could provide potentially novel therapeutic strategies.
Footnotes
Acknowledgment
This work is supported financially by the Wellcome Trust.