
Abstract
The diverse physiological actions of the “biologic gases,” O2, CO, NO, and H2S, have attracted much interest. Initially viewed as toxic substances, CO, NO, and H2S play important roles as signaling molecules. The multiplicity of gas actions and gas targets and the difficulty in measuring local gas concentrations obscures detailed mechanisms whereby gases exert their actions, and many questions remain unanswered. It is now readily apparent, however, that heme-based proteins play central roles in gas-generation/reception mechanisms and provide a point where multiple gases can interact. In this review, we consider a number of key issues related to “gas biology,” including the effective tissue concentrations of these gases and the importance and significance of the physical proximity of gas-producing and gas-receptor/sensors. We also take an integrated approach to the interaction of gases by considering the physiological significance of CO, NO, and H2S on mitochondrial cytochrome c oxidase, a key target and central mediator of mitochondrial respiration. Additionally, we consider the effects of biologic gases on mitochondrial biogenesis and “suspended animation.” By evaluating gas-mediated control functions from both in vitro and in vivo perspectives, we hope to elaborate on the complex multiple interactions of O2, NO, CO, and H2S. Antioxid. Redox Signal. 13, 157–192.
I. Introduction
Molecular oxygen (O2) is produced solely by plants and cyanobacteria and is a product of oxygenic photosynthesis, whereas CO, NO, and H2S are enzymatically produced in mammals. O2 is often viewed as a “physiological” ligand of the key heme proteins hemoglobin (Hb) and mitochondrial cytochrome c oxidase (COX). Conversely, the other three gases are often viewed as “toxic” ligands. This characterization has arisen because CO, NO, and H2S are all capable of binding to either hemoglobin or mitochondrial cytochrome c oxidase, where they can either block O2 transport to the tissue or inhibit energy production, respectively. [The result being that the tissue may experience either a hypoxic hypoxia or cytopathic hypoxia state (87)]. Alternatively, a growing body of evidence now indicates that these same gases also function in a regulatory capacity, controlling important physiologic functions, including vascular tone, host defense against pathogens, neuromodulation, apoptosis, and energy metabolism. Many excellent reviews are available on signal transduction by various gases, and the reader is referred to a number of articles [e.g., CO (140, 260, 373), NO (33, 128, 206), H2S (144, 181, 187, 237, 304, 340), and interactions of CO, NO, and H2S (67, 94, 147, 180, 225)].
A central question in the field of “gas biology” is how do these gases interact with one another when transducing signals and modulating cell function? There are several reasons why this is a difficult question to answer. In the first place, the effects of gaseous molecules are often not dependent on one specific receptor; accordingly, they can produce myriad effects virtually simultaneously. Additionally, gases exert biologic activity through interactions with macromolecules in ways that are fundamentally different from other signaling molecules, such as hormones or peptides.
This review focuses primarily on gaseous interactions involving coordinate bonding of gases to prosthetic heme complexes in gas sensors/receptor proteins. Additionally, key questions regarding the interactions of gases are considered for CO, NO, H2S, and O2. We focus on soluble guanylate cyclase as a primary gas target.
How do gases interact with one another to control cell and ultimately organ function? This question can be broken down into several related parts. First, where, when, and how are gases generated? Second, what and where are the molecular targets and sensors of these gases? Third, what effector systems are associated with these gases? And finally, how do multiple gas-generating systems and gas-reception systems interact with one another?
In this review, we address these questions, discuss controversial areas, and review the physiological significance of CO, NO, and H2S on mitochondrial signaling and their relation to O2 metabolism.
II. Overview: Heme Proteins as the Key to the Generation, Signal Transduction, and Interaction of Gases
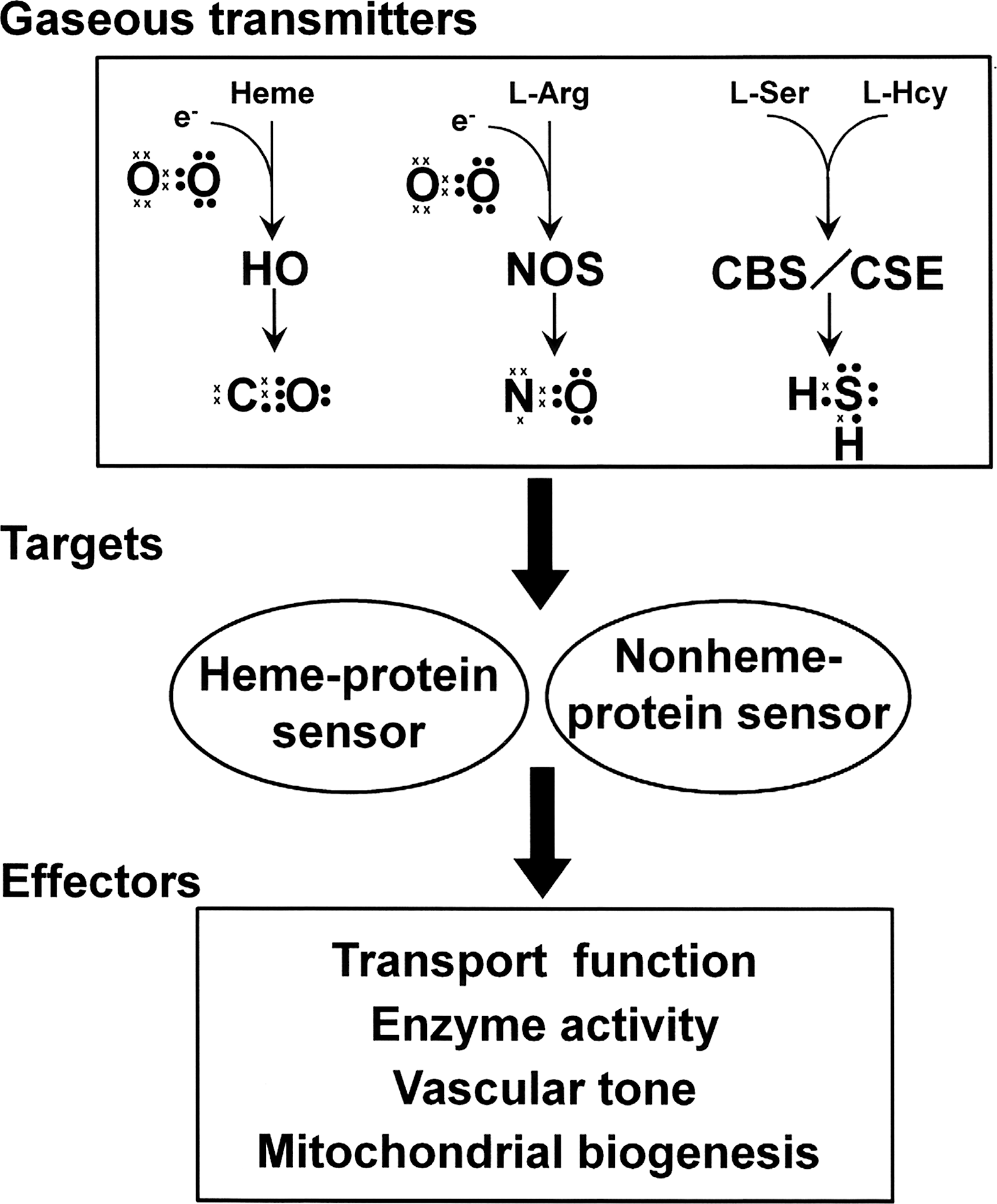
We begin this review by pointing out four main functions of heme proteins (see Table 1 for a list of main functions) because these metal-containing protein molecules are key molecular entities in gas transport, gas generation and gas sensing, as well as important sites of multiple gas interactions.
The first function of heme proteins to be considered is that of gas transport, typically recognized as O2 transport by myoglobin and hemoglobin. These iron-containing heme proteins have a broad range of ligands, including CO (9), NO (364), and H2S (86, 166, 242). The reversible nature of coordinate bonding between a gaseous ligand and the heme allows a gas to be bound and released and forms the basis for competition with another gas.
A second function of heme proteins is to transfer electrons, something typically recognized in mitochondrial cytochrome c. This heme protein mediates single-electron transfer between integral membrane complexes in the mitochondrial respiratory chain of eukaryotes. The transfer of an electron takes place at the iron of the prosthetic heme where the iron switches between two oxidation states: Fe2+ and Fe3+ (151).
A third function of heme proteins is to facilitate reduction–oxidation (redox) reactions that occur at catalytic sites of specific enzymes. In this case, enzyme reactions mediated by these heme proteins start with the reaction of O2 and an electron or H2O2 at the iron of a prosthetic heme. During these reactions, the heme iron is activated by O2 to form high-valence states (195, 257). Oxygenases are a subclass of enzymes that catalyze the addition of O2 to a substrate. Heme oxygenase (HO), the CO-producing enzyme, and nitric oxide synthase (NOS) belong to this category.
A fourth function of heme proteins is that of gas sensor. Unlike the other three heme-protein functions, in which the heme is located at the functional site, in gas-sensing proteins, the prosthetic heme is not found at the functional site, but rather, it is located at a regulatory site. In this case, the heme group conveys a signal to the functional site of the protein. Cystathionine β-synthase (CBS, an H2S-producing enzyme) and soluble guanylate cyclase (sGC) are examples of enzymes that belong to this category.
III. Gas Metabolism
A. O2
Oxygenic photosynthesis is the biologic process occurring in plants and cyanobacteria that generates O2 from water (2H2O → 4H+ + 4e- + O2). Although O2 is essential for mammals to generate energy (ATP) for cell survival and to break down larger substances into simpler components, our body cannot produce its own O2. Most of the O2 entering the human body is consumed during oxidative phosphorylation by the terminal enzyme of the electron-transport chain, COX, in a four-electron reduction of O2 to yield H2O (4Cytochromes c 2++ 4H+ + O2 → 4Cytochromes c 3+ + 2H2O).
B. CO
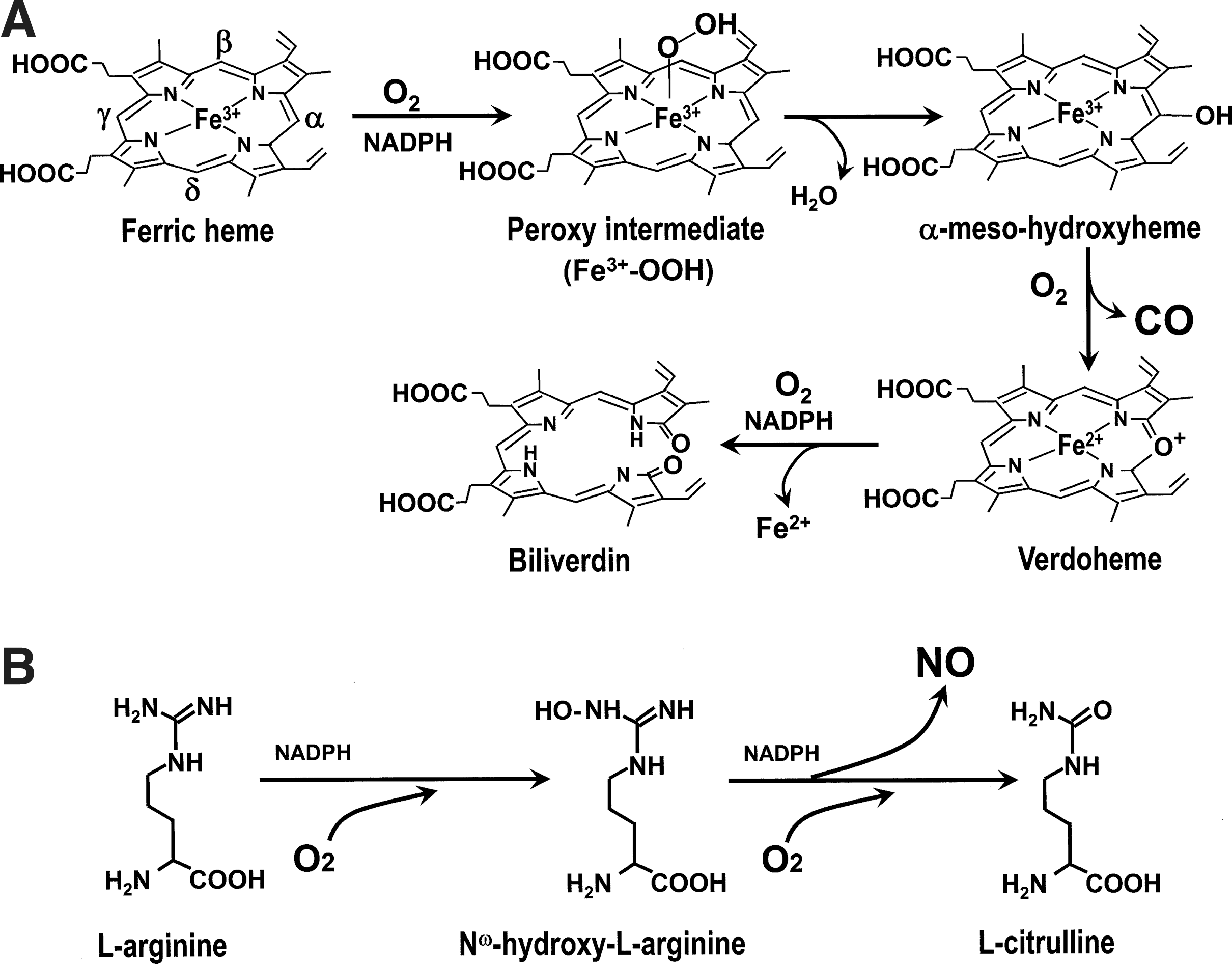
In the human body, the predominant endogenous source of CO is from oxidative degradation of heme (iron protoporphyrin IX) by heme oxygenase (HO, EC 1.14.99.3). Although most heme is derived from senescing red blood cells and ineffective erythropoiesis, a small fraction comes from the degradation of other heme proteins, such as myoglobin, catalase, peroxidases, and cytochromes (23). Under pathophysiologic conditions, additional sources of non-heme CO are thought to be lipid peroxidation (331) and the metabolic activity of intestinal bacteria (82). CO is generated by HO, which catalyzes three successive monooxygenation steps to convert heme to biliverdin, Fe2+, and CO in the presence of reducing equivalents (Fig. 2A) (270, 324).
(
HO is a unique heme protein, in that the heme serves as both a substrate and a catalytic center of this reaction. Reducing equivalents are supplied by cytochrome P450 reductase, which provides an electron, derived from NADPH, to the ferric heme. Overall, the reaction requires three O2 molecules. CO is transported by the red blood cell, where it is bound to ferrous heme of hemoglobin (COHb). CO is subsequently eliminated through the lung, when it is displaced by O2 as the red cell transits along a capillary in the alveolar membrane. Although the binding of CO to hemoglobin is strong (i.e., high affinity), O2 can outcompete CO (186) under the atmospheric O2 conditions that exist in the lung. The reported values of COHb half-life in the circulation range from 22 to 360 min in humans (280, 287).
C. NO
NO is synthesized from
D. H2S
H2S is a gas, which is very soluble in water. Although somewhat ambiguous, the term “H2S” in this article refers mostly to combinations of the inorganic sulfides as undissociated hydrogen sulfide (H2S), hydrosulfide anion (HS-), and the sulfide anion (S2-) in water, unless otherwise specified. Which of these species exerts biologic action(s) is not currently known (180, 352). The biochemical pathways and the mechanisms whereby endogenous H2S are generated are not well understood and are currently the subject of active research. Readers are referred to excellent articles by Kamoun (144), Kimura (156), and Li et al. (180) for more-comprehensive reviews on this subject. In brief, as shown in Fig. 3, the multiple reactions associated with three main enzymes, cystathionine β-synthase (CBS, EC 4.2.1.22) (57, 199, 283, 284), cystathionine γ-lyase (CSE, EC 4.4.1.1) (361), and 3-mercaptopyruvate sulfurtransferase (MPST, EC 2.8.1.2) (279) are responsible for its production in mammals (144, 156).
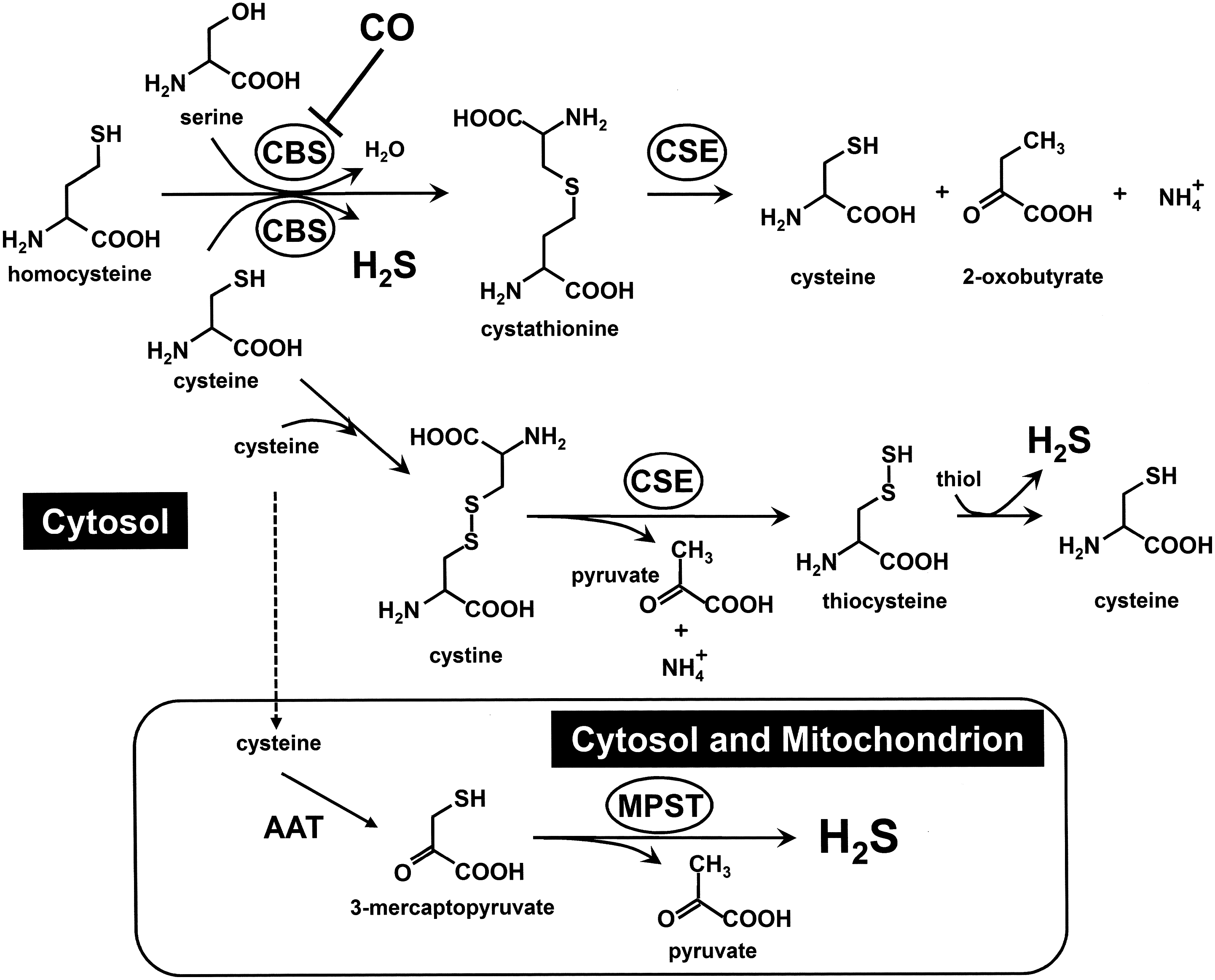
The first enzyme, CBS, uses pyridoxal phosphate (PLP) as a cofactor (152). CBS catalyzes the production of H2S from cysteine by β-elimination (
As seen in Fig. 4, in animal tissues, the sulfur atom exists in several oxidation states [i.e., −2; H2S and organic thiols such as cysteine, −1; polysulfide, 0; elemental sulfur such as protein-bound-S0, +2; thiosulfate, +4; sulfite, and +6; sulfate (185, 323)]. On generation, H2S can be stored in three different forms: (a) stable, (b) acid-labile, and (c) bound-sulfane sulfur [the term “sulfane” designates a compound containing a sulfur-bonded sulfur (144); e.g., polysulfides, polythionates, thiosulfate, thiosulfonates, and elemental sulfur. Compounds containing reduced divalent (−2) or oxidized hexavalent (+6) forms are defined as “stable” because the sulfur atoms are not liberated by simple chemical treatment with acid or dithiothreitol. The stable form includes organic sulfur present in the divalent state, including cysteine and methionine. By contrast, other forms of sulfur compounds are defined as “labile,” because H2S is liberated by simple chemical treatment (323). The ion-sulfur (FeS) complex is an example of the acid-labile form, which releases H2S under acidic conditions, whereas polysulfide (R-Sn-R where n ≥3) is an example of a bound-sulfane sulfur form, which releases H2S under reducing conditions (156, 323).
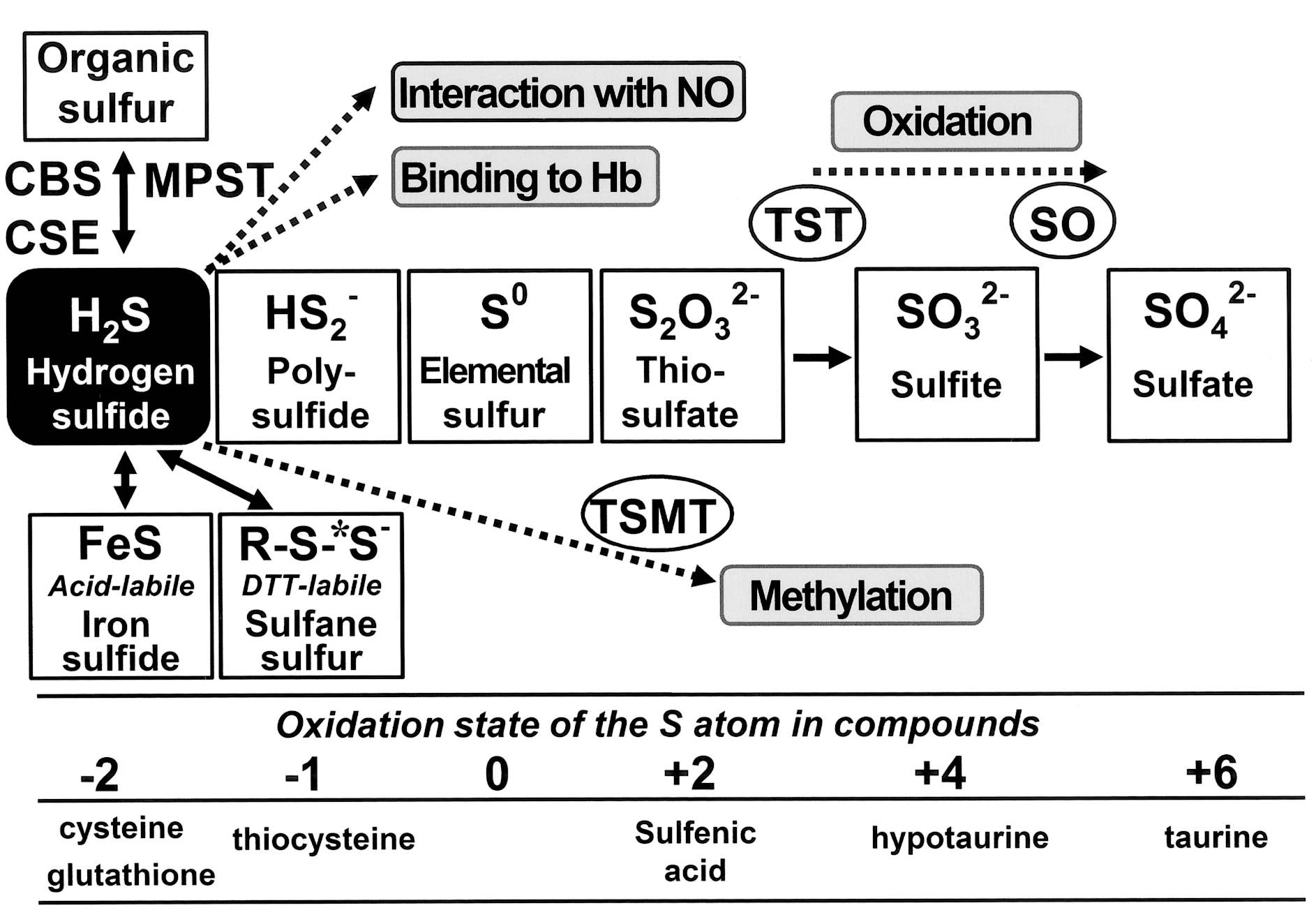
The nonenzymatic production of H2S from the organic polysulfides found in garlic has been suggested as a potential source in the circulation (22, 271), suggesting that stored forms of H2S are physiologically significant. The existence of labile-sulfur fractions not only elaborates the biology of H2S, but also complicates detection of endogenous levels of H2S in the tissue. This issue is discussed later (Section IV.B.3).
What are the mechanisms of H2S catabolism in the body? In a pioneering study, Haggard (110) reached the conclusion that sulfide is rapidly metabolized, based on his demonstration that a quick bolus injection of Na2S in the dog was lethal, whereas a slow rate of injection caused no harm. However, the catabolic pathway(s) whereby endogenous H2S is metabolized appear to be more ambiguous than those of CO or NO (181). Although several systems for breaking down, scavenging, and sequestering H2S have been identified, most data were obtained by using exogenous H2S (187).
The first pathway of H2S catabolism is its oxidation. Studies using an organ-perfusion model indicated that H2S was oxidized in mitochondria to thiosulfate (S2O3 2-), sulfite (SO3 2-), and sulfate (SO4 2-) (20, 144, 181, 247), and then excreted in the urine (63). Sulfate (SO4 2-) is the major end product of H2S (181). The initial oxidation to thiosulfate is most likely a nonenzymatic step. Conversions of thiosulfate to sulfite and sulfite to sulfate are catalyzed by thiosulfate:cyanide sulfurtransferase (TST) (241) and sulfite oxidase (SO), respectively. The second pathway of H2S metabolism is the thiol S-methyltransferase (TSMT)-mediated methylation of H2S to yield monomethylsulfide and dimethylsulfide (97). The third pathway involves the binding of H2S to methemoglobin to form sulfhemoglobin. Additionally, H2S can react with NO (350). It has also been reported that H2S can diffuse across the alveolar membrane (83, 132, 209).
IV. Determinants of the Effective Gas Concentrations at the Target
A. General considerations
To understand how gases are functioning in physiological processes, it is important to know how much gas is actually being delivered to a given target. Three major determinants of the effective gas concentration are (a) the physiochemical properties of the gas itself (258); (b) the properties of the local environment, including the surrounding media through which a gas travels including viscosity, temperature, and tissue composition (117, 167); and (c) scavenging systems, including chemical reactions that consume the gas. Here we attempt to compare and contrast factors affecting gas-transport efficiency, while referring the reader to excellent books and reviews on the transport properties of O2 (167, 346), CO (332), and NO (38, 39, 56). Although physical (203, 216), biochemical and physiological (144) properties of H2S have been well studied, experimental and mathematical modeling of H2S transport properties in vivo is limited.
Biologic membranes create a diffusion barrier. In general, gaseous molecules have a high solubility in nonpolar solvents, such as the lipid bilayer. The dipole moment indicates the polarity of the gas molecule. The larger this value, the more polar the gas (i.e., the less permeable through hydrophobic membranes). As seen in Table 2, H2S stands out among the four gases under consideration in this review. It has the largest dipole moment (H2S >> NO > CO > O2), suggesting that it has the lowest permeation through lipid bilayers, and it displays the greatest water solubility (H2S >> NO >O2 > CO). Unlike CO and NO, H2S possesses an acidic proton with a pK a of 6.8, making H2S the anionic conjugate base, HS−, the predominant form at the physiologic pH 7.4 (202). Different fractions of H2S species exist under different conditions. For example, in water at 25°C, pH 8.1, the following composition has been reported: H2S (7.05%), HS− (92.25%), and S2- (2.9 × 10−5%) (203); whereas, in physiologic solution (160), a composition of H2S (∼30%) and HS− (70%) has been reported. Because of its ionization, it can be speculated that H2S may have a reduced ability to permeate the lipid bilayer compared with either O2 or CO. NO (free radical) also changes its appearance; the ease of oxidation to the nitrosonium ion (NO+), the probability of reduction to the nitroxide ion (NO−), and the attack by O2 leading to formation of NO2 (197). It remains difficult, however, to determine the fractional contributions of each H2S and NO species involved in physiologic processes.
Numbers in parentheses denote references.
From the diffusion coefficient (D), one can calculate how far a gas molecule travels in a given time and can estimate the time for a certain diffusion process. For O2 to travel a 10-μm distance in water at 20°C, it takes 0.021 seconds; whereas, for H2S that has a smaller D, it takes a slightly longer time, 0.028 s. It would be of great interest to compare the D of O2, CO, NO, and H2S in heme protein–containing solutions (the values listed in Table 2 were obtained in water). Although no such information can be found in the literature, Longmuir and Roughton (186) did compare the D of CO and molecular nitrogen (N2), which have similar molecular weights (i.e., 28.0) and physical properties. Measured values of D N2 (2.8 × 10−6 cm/s) and D CO (3.2 × 10−6 cm/s) at 20°C in 40% hemoglobin in water were similar. To our knowledge, no information exists on the diffusion of H2S through biologic tissues, as has been done with O2 (167).
Furthermore, the reported values of the redox potentials of NO are +0.71 volts in acidic solution (2NO + 2H+ +2e− → H2N2O2) and +0.18 volts in basic solution (2NO +2 e− → N2O2 2-) (102). This indicates that NO is readily reducible or oxidizable, allowing it to function as either an oxidizing or a reducing agent (367). By contrast, H2S tends to be regarded as solely a reducing agent (180, 185). However, the biologic chemistry of H2S has not fully been investigated.
NO has a higher affinity than either O2 or CO for metal ions (367). Because NO has an unpaired electron, it can readily accept or donate an electron to a metal ion, as described earlier. Although H2S has been recognized to react with metals such as silver and zinc, no report exists comparing the interaction energy of NO and H2S with a certain metal. The interaction with a metal depends on the first ionization energy of the lone pair electron in the gas ligand and the electron affinity of the metal (100). It can be speculated that H2S exhibits a slightly weaker affinity to a metal as compared with that of NO (H2S, 10.46 eV, vs. NO, 9.26 eV), but a stronger metal affinity than either O2 or CO.
B. Membrane permeability of gases
An important question has recently been raised regarding the membrane permeability of gases. Do gases cross biologic membranes by simple diffusion alone, or might they be transported? Factors influencing the permeability of small neutral molecules such as gases are (a) how easily the gas dissolves into the membrane's hydrocarbon, and (b) the rate at which the gas diffuses through the hydrocarbon. Based on this “solubility-diffusion” model (88, 158), it is generally assumed that biologic membranes are no barrier to small gases, which can readily cross membranes without specific transporters. However, some experimental data do not support this model. For example, the apical membrane of gastric gland cells was found to have no demonstrable permeability to either H+, CO2, HCO3 −, NH3, or NH4 + (333). Investigators suggest that the lipid bilayer can offer resistance to simple diffusion, based on data showing that changes in lipid composition alter gas permeability (119), whereas other investigators have challenged the solubility-diffusion model (69, 93), suggesting that gases are transported across biologic membranes by proteins.
Recently, NO membrane permeability was suggested to be facilitated by the aquaporin-1 (AQP1) protein (115). Additionally, when AQP1 was purified from human red blood cells and reconstituted into proteoliposomes, it was found to increase the permeability of both water and CO2. As both effects were abolished with HgCl2, the authors concluded that AQP1 served as a protein-mediated channel that increased CO2 permeability (245). Although the notion that AQP-1 mediates gas permeability is supported by various studies (69, 81, 342), the experimental system has been criticized (204, 253) and the concept refuted. Moreover, in a study using transgenic mice lacking AQP1, CO2 permeability in red blood cells was not reduced in the absence of AQP1 (84, 360). To date, no report suggests facilitated transport of either O2, CO, or H2S.
V. Mechanisms of Gas Sensing and Gas Actions
A. Heme-protein sensors
1. General aspects of heme-protein sensors: biochemical characteristics and structural–functional relations
Heme-based sensor proteins are key regulators of cellular responses to changes in O2, CO, NO, and H2S levels. These gas sensors act as signal transducers by coupling a “regulatory” heme-binding site to a “functional” signal-transmitter site. Four different types of heme-binding domains are known: (a) globin-coupled sensors, (b) heme-binding PAS domains, (c) CooA, and (d) heme-NO-binding (HNOB). Coupled transmitter domain sites include cyclase, histidine protein kinases, and phosphodiesterases, as well as transcription factors with the basic helix–loop–helix motif.
What are the molecular mechanisms whereby heme-based gas-sensing molecules transduce signals? Understanding the structural–functional relations of the coordinated complex at the site of a prosthetic heme of these sensor proteins can help provide answers. Here we first describe key biochemical features of heme-protein sensors that should be taken into account when discussing the concept of gas sensing and signaling. This includes the relations between (a) oxidative states of the central iron of the prosthetic heme and the binding affinity of gases, (b) ligand binding and base affinity, (c) conformational changes within the protein arising from ligand binding, and (d) structural changes and protein functions.
The oxidative state of the heme iron is an important determinant of ligand discrimination. The iron atom in the prosthetic heme can exist as either an Fe2+ (ferrous) or an Fe3+(ferric) oxidation state that, as described earlier, has the ability to form six coordinate bonds. As shown in Fig. 5, each of the lone pairs on the nitrogen atom of the porphyrin ring in hemoglobin can form a coordinate bond with either an Fe2+ or an Fe3+ ion, holding it at the center of the porphyrin ring. This leaves two more coordinate positions, one above and one below the plane of the porphyrin ring. The globin attaches to one of these positions by using a lone pair of the nitrogen molecule of histidine known as proximal histidine. The other position is the point at which a gas forms the sixth coordinate bond.
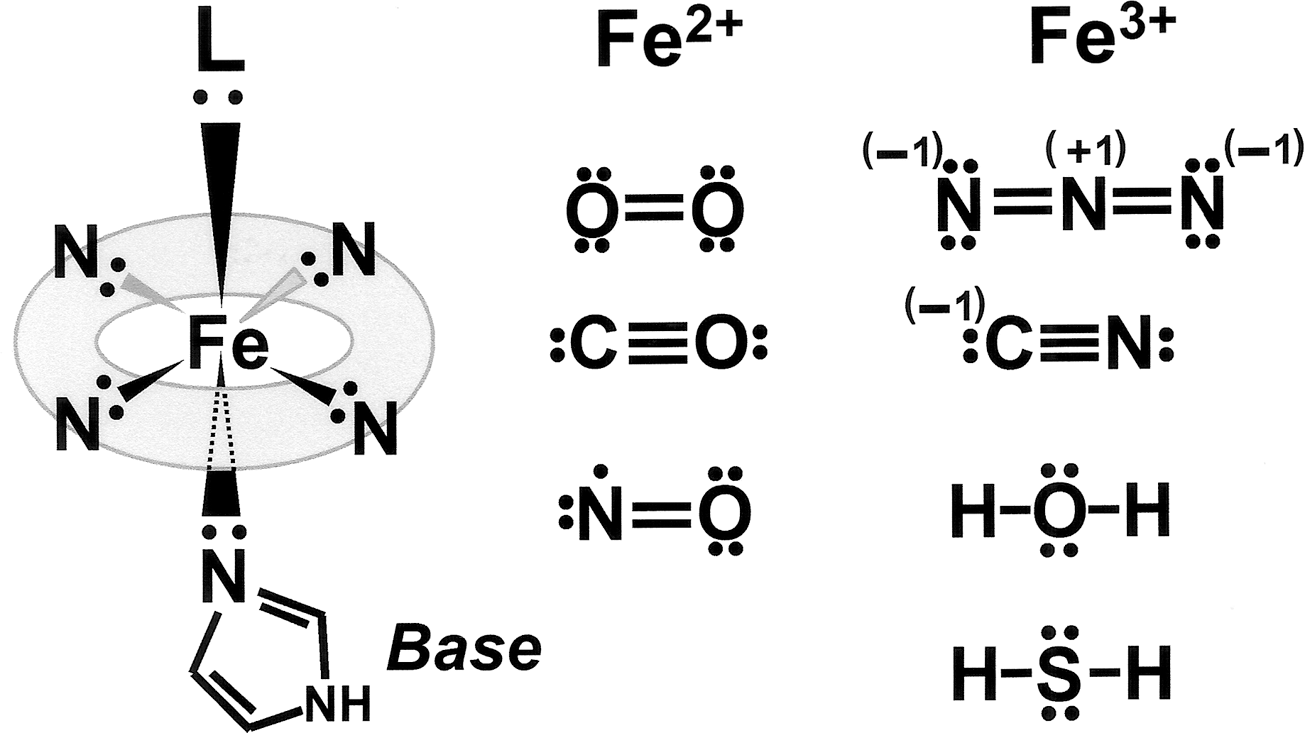
The electronic structure of the iron atom in its ground state can be expressed by specifying the number of electrons in each orbital as follows: 1s22s22p63s23p63d64s2. When iron is oxidized to Fe2+, it loses two electrons from its 4s orbital, changing its electronic state to 1s22s22p63s23p63d6. When it is further oxidized to Fe3+, an additional electron is lost from the 3d orbital, resulting in a 1s22s22p63s23p63d5 electronic configuration. In an octahedral complex with six ligands, the energy level of the iron 3d orbitals splits into two, giving low energy t2g (dxy, dxz, dyz) and high energy eg (dx2-y2, dz2) orbitals. The former are essentially non-bonding, while the latter are weakly anti-bonding with respect to the coordination bonding with the ligands. The bonding molecular orbitals in the complex are close in energy to those of the atomic (or molecular) orbitals of the isolated ligands that compose a lone pair, although the iron 4s and 4p orbitals also contribute significantly to the stabilization of the coordination bonds (17, 194).
Electronic configurations of O2, CO, NO, and H2S are an important determinant for gas binding. All of O2, CO, NO, and H2S have lone pairs of electrons in their outer energy levels, making them active electron-lone-pair donors (Fig. 5). The ferrous oxidation state (Fe2+) of hemoglobin preferentially binds neutral ligands such as O2 (the physiologic ligand), CO (a typical toxic ligand), and NO (9). Conversely, the ferric oxidation state (Fe3+) preferentially binds H2S and water (138), and anions such as CN−, N3−, and OH− (Fig. 5) (367).
Why does the Fe3+ of the prosthetic heme of myoglobin or hemoglobin prefer an anion to a neutral ligand? Under the condition of low electron density of the metal (M)-d-orbitals, donation of an electron from the ligand (L) to M (“π-donation”) tends to stabilize the complex by forming “π-bonding” (194). Because an Fe3+-heme possesses one less electron than an Fe2+-heme, the electron density of its d-orbitals is lower, making it more readily accepting of an electron from L. This is one explanation for why Fe3+ prefers more-electronegative ligands to neutral ones (194). Furthermore, the charge-transfer property from L to M depends on the ionization energy of the lone-pair electron in L and the electron affinity of M (100). In this regard, the first ionization energy among different lone-pair–donating ligands (the energy required to remove the first electron in the molecule) appears to become an important parameter. Here the smaller the ionization energy, the more readily the transfer of electronic charge occurs. As shown in Table 2, H2S (10.46 eV) displays a smaller ionization energy compared with CO (14.01 eV), making H2S a better ligand for an Fe3+-heme complex than CO (194).
Why then does the neutral ligand CO bind to Fe2+-heme, but not to Fe3+-heme? The bonding of CO to M is thought to have two steps. The first step is the donation of electron density from CO to M, “π-donation”, whereas the second step is the back donation from the dπ-orbital of M to an empty π* antibonding orbital of CO, called “π-back donation” (197). Such a “π-back donation” occurs more readily for the Fe2+-heme than that for the Fe3+-heme because the difference in energy between the d-orbital energy of an Fe2+ and the π*-orbital energy of CO is relatively small (194). Consequently, CO can interact with an Fe2+-heme, but not with an Fe3+-heme.
This description makes us realize the importance of the metal center redox state. The Fe2+ of hemoglobin can be rapidly oxidized to the Fe3+ of methemoglobin in the presence of O2. However, in our body, the endogenous source of the reducing equivalent modulates the heme oxidative state. For hemoglobin, the electron donor is methemoglobin reductase, and for HO, it is cytochrome P450 reductase: however, the electron donor for CBS is unknown. Nitric oxide poses a more-complicated story because the relative distributions of the Fe oxidative states are not well understood (295).
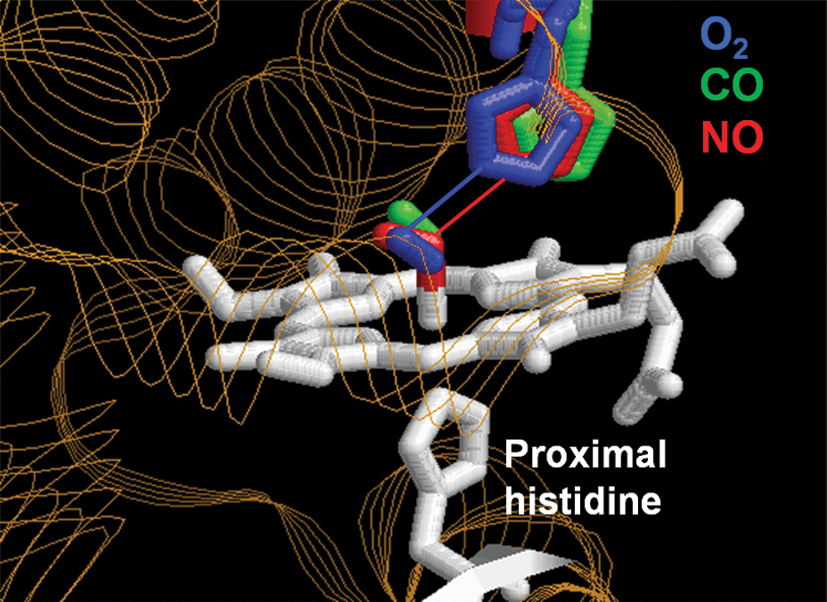
Although myoglobin is not considered a gas sensor per se, it does bind O2, CO, and NO. Figure 6 shows the structures of native sperm-whale myoglobin whose ferrous heme has been ligated by O2, CO, or NO, respectively (35). Each structure was determined by x-ray crystallography and then superposed. As seen, ligand-binding by the different gases causes distinct positional changes of the distal histidine group. A change of this kind is considered to be the first step in the signal-transduction mechanism of heme-protein gas sensors.
How then is this first conformational change, induced by ligand binding, coupled to structural changes at distant sites within the heme-protein gas sensor? And furthermore, what is the mechanism whereby NO acts differently than CO? Hemoglobin can be used as a model to describe such a mechanism. Figure 7 shows a scheme comparing binding affinities (iron-to-gas bond) and base affinities (iron-to-imidazol) of CO and NO. Although both CO and NO are strong heme-ligands, NO has a much higher affinity for ferrous heme (nitrosylheme) than does CO. Conversely, the base affinity of nitrosylheme is lower than that for CO-heme.
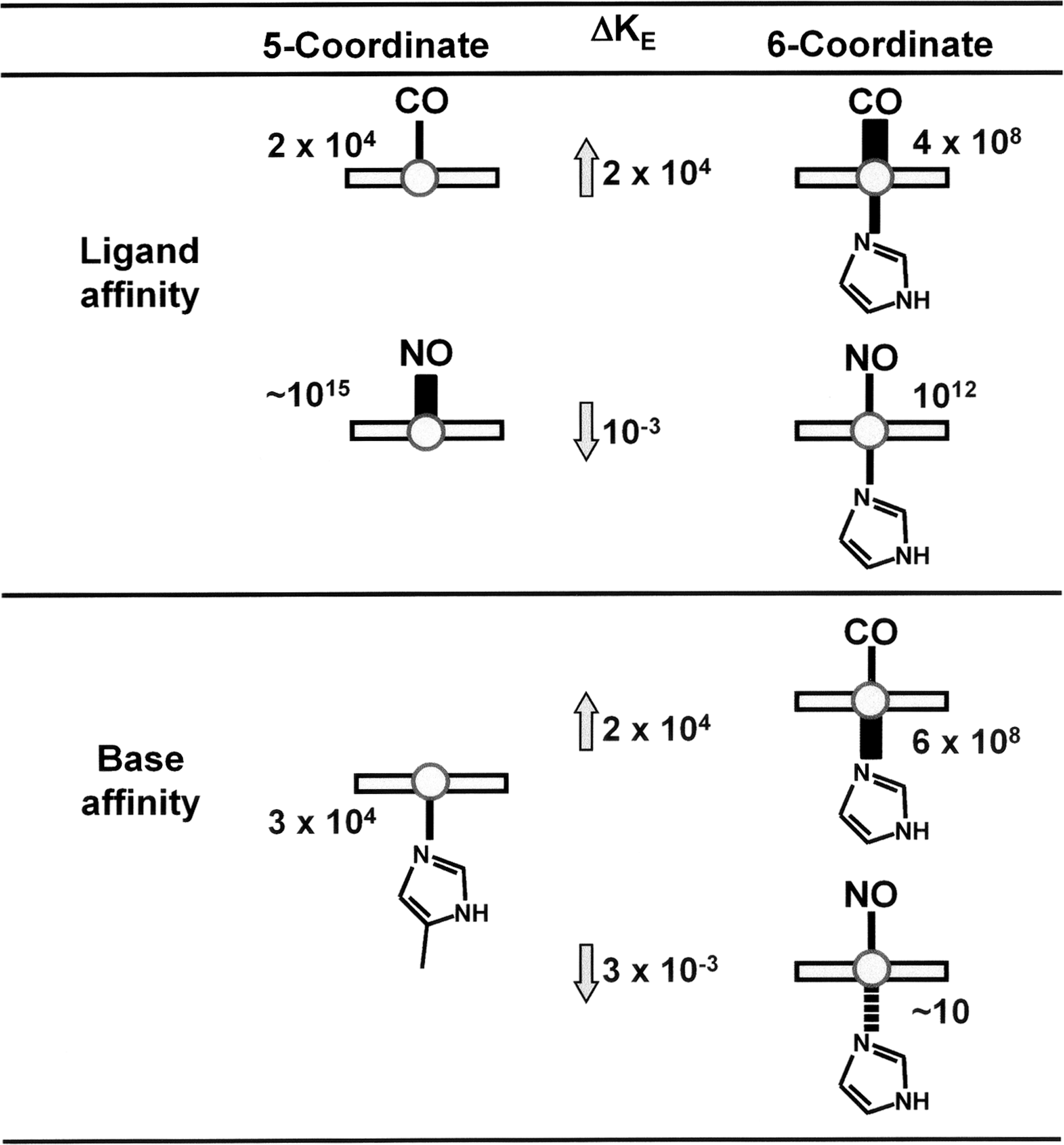
CO binds preferentially to the six-coordinated structure with an equilibrium constant (K E) of 4 × 108 M −1, compared with 2 × 104 M −1 for the five-coordinated structure, a 4 orders-of-magnitude difference. The opposite is true for the binding of NO, which prefers the five-coordinated structure to the 6-coodinated structure by a 3 orders-of-magnitude difference (255, 256, 266, 320). The consequence of this differential binding is that the binding of CO strengthens the Fe-axial bond to the imidazol base of proximal histidine; whereas that of NO markedly weakens the same bond (Fig. 7). Under some conditions, the proximal iron-histidine bond is severed, forming a 5-coodinated nitrosylheme complex. This has been observed in both α-NOHb in the presence of inositol hexaphosphate (364) and in the heme-regulatory subunit of sGC (101, 161).
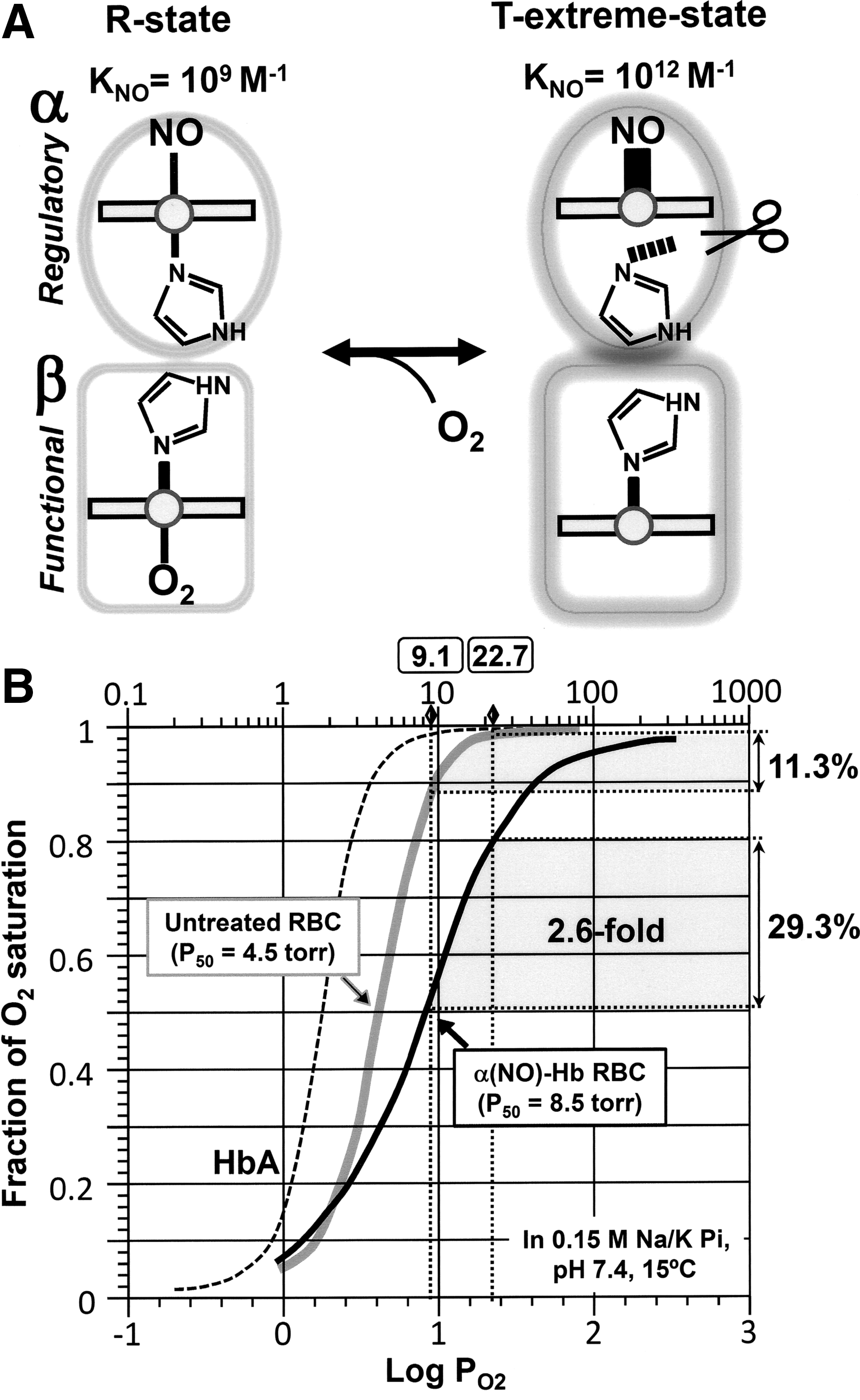
Iron in the six-coordinated nitrosylheme is situated in the plane of the porphyrin ring; whereas, iron in the five-coordinated structure is displaced out of the plane, causing a shift of the iron toward the NO ligand. This shift is thought to induce further changes in protein structure (161, 364, 367). By using α-nitosyl hemoglobin (α-NOHb) as a model, in which the two α- subunits are bound by NO, we relate a structural change induced by NO into a functional change of hemoglobin, namely the O2 binding characteristics and O2-delivery performance of α-NOHb. Yonetani et al. (364) showed that α-NOHb modulates the quaternary structure of hemoglobin between the R (relaxed) and T (tense) forms. The T-state displays a dramatic decrease in affinity for O2 in the β-subunits, attributable to the quaternary conformational change caused by NO-induced cleavage of the axial bond in the α-subunits. Conversely, O2 binding to the β-subunits shifts the quaternary conformation toward the R-state, causing a decrease in the affinity of NO for the α-subunits (Fig. 8A). Indeed, the O2 saturation curve of human erythrocytes containing α-NOHb (α-NO-RBC) is right-shifted compared with that of the NO-free erythrocytes (Fig. 8B) (298, 322). In this manner, α-NO-Hb is suggested to augment O2 delivery in the peripheral tissues.
2. Specific heme-protein sensors
a. NO sensor
The soluble guanylate cyclase enzyme (sGC, EC 4.6.1.2), which catalyzes the conversion of GTP to the second-messenger cGMP, is a heme-based NO sensor and was the first definitive receptor of NO to be identified in mammals (73, 188, 243, 293, 319). sGC is a heterodimer consisting of α- and β-subunits (72). The N-terminal regulatory domain of each subunit has a
The catalytic activity of sGC is controlled by both NO and CO (Fig. 9). NO binding can increase the catalytic activity of the enzyme by several hundred-fold (293); whereas, the stimulatory effect of CO is far less potent, increasing enzyme activity by only three- to four-fold (142, 154, 161, 233, 292). However, in the presence of the allosteric activator YC-1 (3-(5'-hydroxymethyl-2'-furyl)-1-benzyl indazole) (189), CO synergistically activates the enzyme to the same level as NO (294). For detailed kinetics, ligand-binding characteristics, and structure–function relations of this enzyme, readers are referred to excellent reviews by Marletta and his co-workers (49, 73) and others (211, 233, 243).
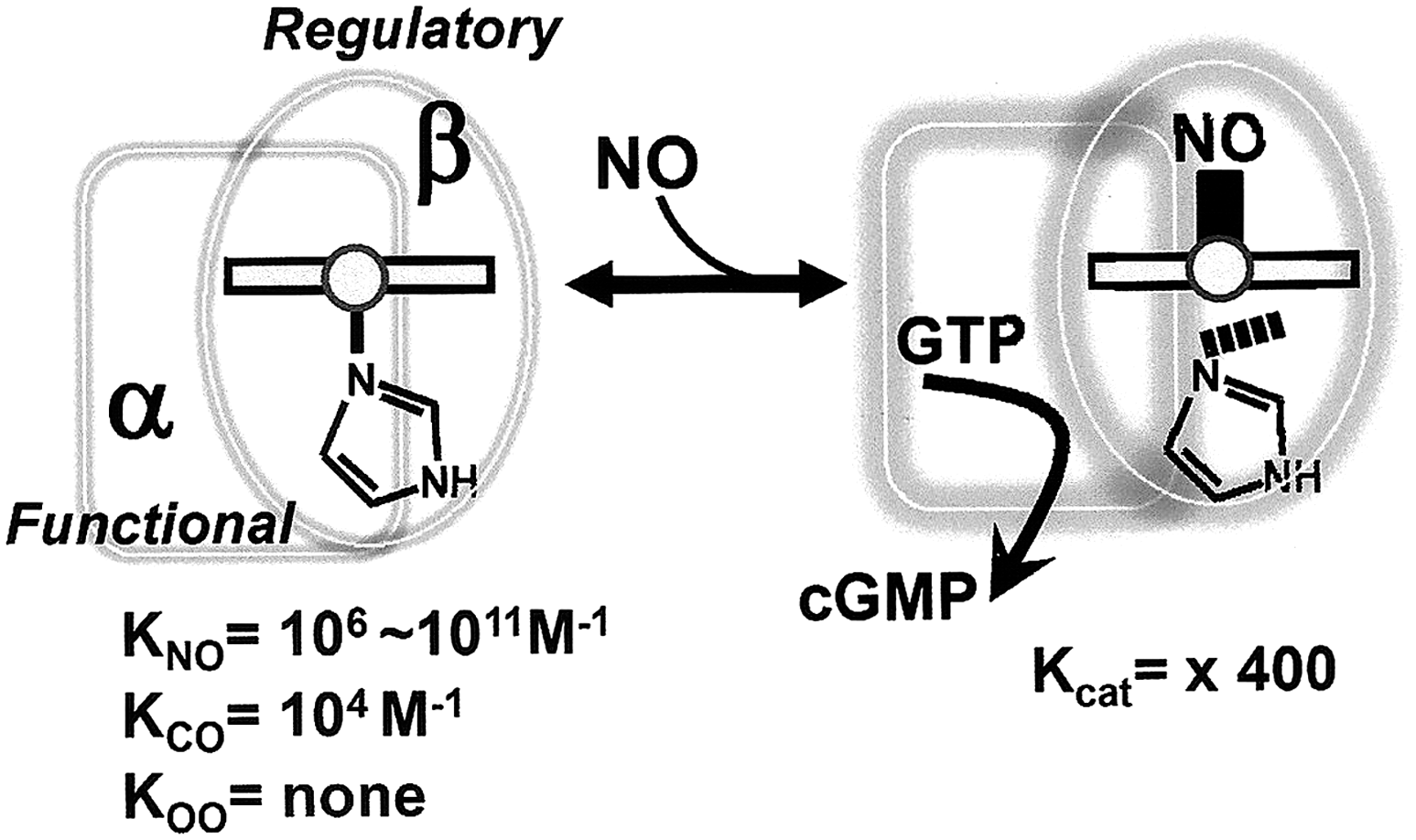
How are CO and NO different from each other with respect to their sGC-binding properties? Sharma and Magde (274) summarized important kinetic data for CO and NO binding to sGC, and the following values are taken from their table. CO rate constants for association and dissociation are 1.2 × 105 M−1s−1 and 28 s−1 at 23°C, respectively (154), whereas Stone and Marletta (291) reported values for CO on and off rates as 3.6 × 104 M−1s−1 and 3.5 s−1 at 10°C, respectively. By contrast, these values for NO are 1.4 × 108 M−1s−1 and 8 × 10−4 s−1, respectively. This means that the association of NO to the heme is faster than that of CO, whereas dissociation of NO from the heme is slower than that of CO.
How is the binding of NO translated into an increase in the enzyme activity? Although no three-dimensional structural information of sGC is yet available, kinetics and equilibrium binding studies of NO to sGC have made it possible to propose a complicated multistep mechanism of sGC activation that involves at least two NO-binding sites (49). It was once thought that sGC followed a simple two-step activation process whereby NO bound to heme, forming a sGC-NO complex (six-coordinated histidine-Fe-NO), which subsequently changed to form a fully active five-coordinated histidine-Fe-NO complex. However, this model was revised by Zhao et al. (371), who showed that full activation required two NO-dependent kinetic processes. By monitoring NO binding and catalytic activity simultaneously, Russwurm et al. (259) showed that when 80% of heme is saturated, as in the Fe-NO complex, sGC displayed only 10% of maximal activity, supporting two NO-bound states of the sGC. Poulos (243) suggests in his review that sGC exists in a mixture of two species of five-coordinate Fe-NO complexes displaying identical spectra, in which one has low or no activity and the other has full activity. By contrast with NO, CO ligation results in the formation of a six-coordinated Fe2+-CO complex (161). Stone and Marletta (291) suggest that the binding of CO to sGC is a simple one-step process, in which the off-rate of CO from the hexacoordinate complex is much faster than typically found in heme proteins. The differences in sGC ligand binding described earlier become important when the interactions of CO and NO are considered in vivo.
b. CO sensor
Are specific CO-specific sensors not affected by NO under physiologic conditions? The transcriptional activator CooA in the photosynthetic bacteria Rhodospirillum rubrum is the first example of a heme protein in which CO plays a physiological role. Here only the CO-bound form of CooA binds to its target DNA and acts as a transcriptional activator (11, 127, 275). In mammals, the heme protein neuronal PAS domain protein 2 (NPAS2) was reported to be a specific CO sensor (78, 251). It was identified as a member of the bHLH family of transcription factors expressed in the forebrain. The resonance Raman spectra indicated that CO coordinated to the heme iron histidine on the proximal side, whereas NO did not bind to the heme group (318). Here, CO is suggested to regulate the formation of a complex between NPAS2 and another bHLH transcription factor, BMAL1, in a process that regulates the circadian rhythms (78).
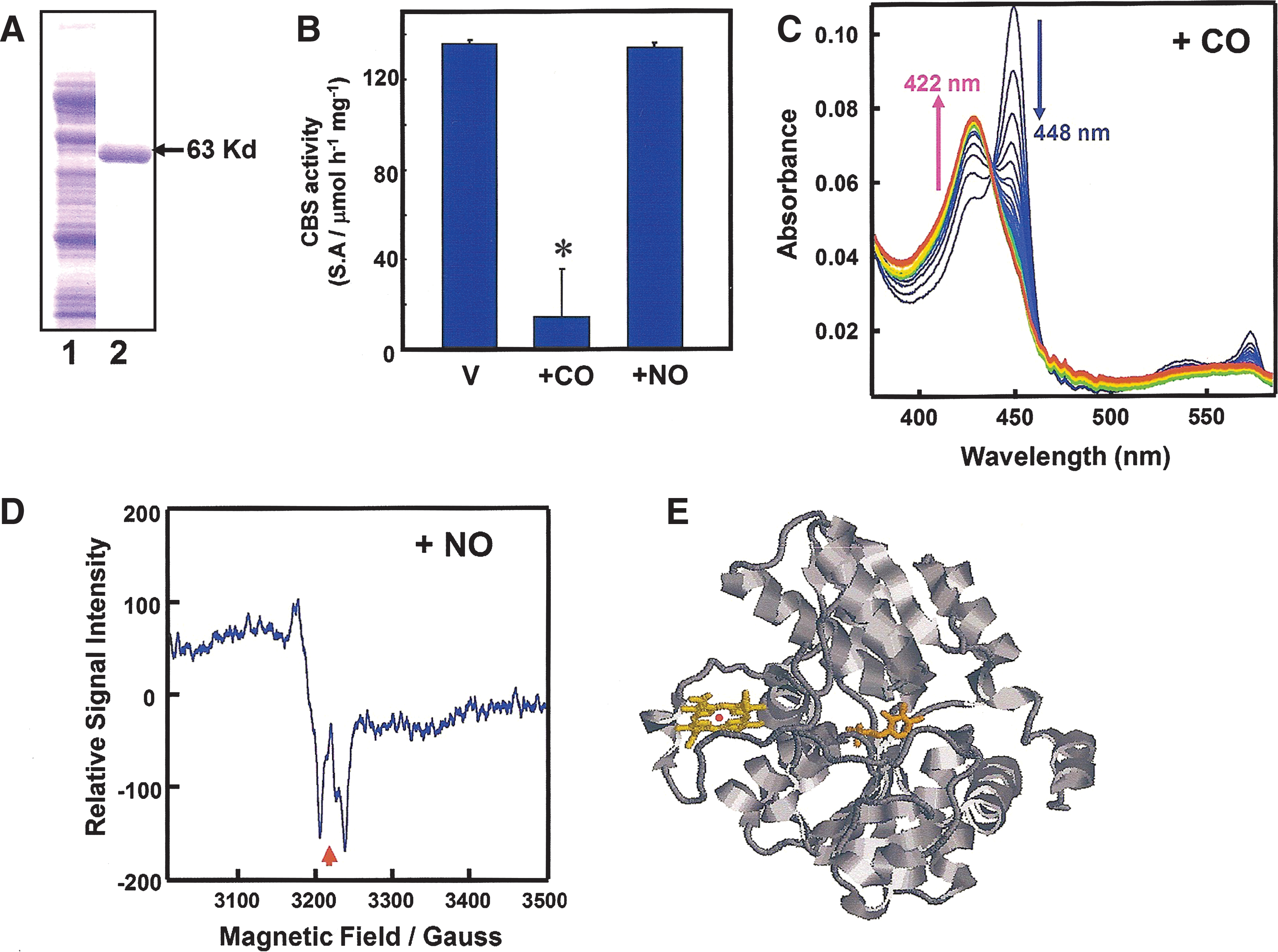
Cytochrome P450 enzymes were once considered putative CO-sensitive signal transducers (118, 312, 313). However, the ferrous heme of these enzymes has been found to be sensitive to both CO and NO, ruling them out as specific CO sensors. CBS, the “pseudo-cytochrome P450,” conversely, was found to be a bona fide CO-specific sensor. In vitro studies using recombinant CBS have shown that CO acts as an apparent competitive inhibitor of CBS, with the K i value of ∼5 μM, much smaller than that for NO (200 μM) (312). The result is striking because such a low K i for CO suggests that CBS acts as a specific CO sensor in vivo under physiologic conditions. CO inhibits recombinant rat CBS by stabilizing the six-coordinated structure of the heme. By comparison, NO binds to heme, but stabilizes the five-coordinated structure.
CBS was first identified as H-450, in which the addition of CO to its reduced form produced a new spectral species that resembled that of the reduced CO-complex of a denatured form of cytochrome P-450 (226). Among heme proteins, CBS is unique, in that it catalyzes a PLP-dependent reaction (152). The prosthetic heme of this enzyme is coordinated to histidine and cysteine as axial ligands in human and rodents. Because a distance of ∼20Å exists between the heme and the PLP cofactor, a direct catalytic role of the heme is excluded (Fig. 10) (199). Although the crystallographic structure of CO-ligated forms has yet to be determined, perturbation of the heme environment by CO, but not by NO, is believed to be communicated to the active site with concomitant inhibition of enzyme activity. Thus, it can be postulated that CBS functions as a CO-sensing heme protein.
VI. Interactions of Multiple Gas-Transducing Systems
A. Multiplicity of actions: a hallmark of gas behavior
HO, NOS, and CBS are heme proteins that generate CO, NO, and H2S, respectively. Although these enzymes have adapted ways to recognize specific substrates and cofactors and protect themselves from being bound to nonphysiologic ligands of similar polarity and shape, interactions among gas ligands do occur. At least nine different interactions between a gas and a generating system can be considered (CO/HO, CO/NOS, CO/CBS, etc.), without taking into account the binding effects of O2. Here, we mostly limit consideration to acute interactions of CO and NO at the level of their biosynthetic enzymes, NOS and HO, and sGC, a primary heme protein target/receptor for these gases.
1. Does CO inhibit NOS and does NO inhibit HO?
CO has been suggested to inhibit NOS activity by coordinating to the NOS prosthetic heme group. However, such inhibition seemingly requires a high concentration of CO. For example, by using purified murine macrophage iNOS, White and Marletta (349) found that a mixture of 80%CO and 20% O2 (a near-saturated CO solution that resulted in direct CO-binding to the NOS prosthetic heme) inhibited NOS activity by 73 to 79%. Similar results were obtained independently (196, 198, 265) by using recombinant neuronal NOS (nNOS). However, an important question is what is the actual physiologic inhibitory concentration of CO? Scheele et al. (265) note that the CO affinity for NOS is relatively weak; it is 300 times less than the CO affinity for human Mb, and they suggest that 1 mM CO, a near-saturated concentration, would be required to inhibit NOS. It would appear, however, that such inhibition by CO is unlikely in vivo, unless such a high concentration can be achieved locally. Although local CO concentration cannot be currently measured and is unknown, such a possibility should be pursued.
Whereas CO does not appear to inhibit NOS unless it is at high concentrations, based on in vitro experiments, NO has been reported to bind and inhibit HO (338). The ligation of NO to the Fe2+ (ferrous) heme of recombinant human HO-2 was demonstrated by resonance Raman and EPR spectroscopy (134), and stopped-flow studies revealed that NO binds 500-fold more tightly to Fe3+-human HO-1 than to the Fe3+-myoglobin (338). In terms of the inhibitory effect, by using the NO donor, NOC9, Wang et al. (338) reported the IC50 for human HO-1 as 0.08 mM.
2. Does CO autoinhibit HO and does NO autoinhibit NOS?
Can a gas self-inhibit the activity of its own synthesizing enzyme? When exogenous gases were applied to their respective enzymes in vitro, the answer was “yes” for both CO and NO. The HO reaction was substantially attenuated under a gas phase of 20% CO and 80% O2 (366). By contrast, however, endogenously produced CO did not appear to inhibit the same reaction (191). It is therefore worth noting the mechanism by which HO can escape from CO autoinhibition (170, 299, 300). A notable feature of the HO reaction resides in the oxidation of verdoheme to biliverdin. This reaction proceeds without interference by CO, which is produced in the second step of the overall HO reaction as α-meso-hydroxyheme is converted to verdoheme (Fig. 2A).
The endogenously produced CO does not seem to interfere with the third step (single-turnover reaction) of the overall HO reaction (155). The affinity of CO for verdoheme is low compared with that for heme. As CO is being generated during the second step of the HO reaction, it is temporarily trapped within a special space in the heme pocket of the enzyme and then released after the generation of biliverdin. It is through this mechanism that CO autoinhibition of HO is avoided (299). However, excess exogenous CO does inhibit this third reaction step. Under a gas phase of 20% CO and 80% O2 (366), the HO reaction is substantially attenuated, specifically at the verdoheme stage. These findings suggest that the active site of HO may be equipped with structural features that allow the HO to escape autoinhibition by local CO during the reaction, but not by external CO. Somewhat consistent with this concept is x-ray crystallography data, which show that the heme-HO complex has a structure that increases the affinity for O2, but decreases the affinity for CO (300).
Similar to the inhibition of HO by exogenous CO, exogenous NO (105, 254) has been shown to inhibit NOS activity at concentrations between 0.1 and 10 μM (1, 105, 192). Griscavage et al. (105) suggested that enzymatically generated NO could autoregulate NOS, with direct consequences on NOS activity and local changes in NO concentration. The mechanism of NO autoregulation may be somewhat different, however, from that of CO autoregulation, as NO binds to both Fe2+ and Fe3+ oxidation states in heme proteins, whereas CO binds solely to the Fe2+ oxidation state of NOS heme proteins.
Stuehr et al. (295) comprehensively summarized a novel catalytic model that emphasized the importance of heme oxidation states during NO biosynthesis, suggesting a possible role for NO as an intrinsic regulator. During NO synthesis, the iron of the NOS prosthetic heme alternates between the Fe2+ and Fe3+ oxidation states (192). Under an anaerobic atmosphere, the heme iron of nNOS was shown to bind NO as a sixth ligand in both the Fe2+ and Fe3+ oxidation states, generating stable NOS heme iron–NO complexes (339). During NO synthesis, NO binds to the Fe3+-heme before exiting the NOS, and the reduction of the Fe3+-heme is the rate-limiting step for the overall enzyme reaction (264).
3. CO Attenuation of NO-mediated sGC activation
As discussed previously, a principal means by which CO and NO transmit signals is through binding to the heme moiety at the active site of sGC. What happens if both CO and NO simultaneously diffuse to the sGC? To answer this question, an experiment was conducted with purified bovine sGC to determine whether CO modulates NO-dependent activation of sGC (1.7 nM in a reaction mixture) (142). At in vitro concentrations of 10 to 30 μM, CO exhibited dual effects on the activation of purified bovine sGC induced by the NO donor S-nitroso-N-acetylpenicillamine (SNAP). In the presence of SNAP at <100 nM, CO elicited a modest activation of sGC. However, in the presence of SNAP at greater concentrations, the application of CO modestly but significantly attenuated sGC activation. These results suggested that CO serves as a partial antagonist for sGC, limiting the dynamic range of the NO-dependent activation of the enzyme. Many biochemical investigations have provided good evidence for the acute interactions of CO and NO at the level of the generating enzymes. To use these findings to discuss more-complicated systems in vivo, we must consider the following factors of gas generation: (a) concentrations of gases in tissues, cells, or even compartments in the cells; (b) spatial localization of gases; and (c) temporal regulation of gas generation.
B. Quantitative arguments
Organs and cells are specialized in their ability to produce different gases at different rates, and it is this specialization that controls cellular function. Many studies have extrapolated from the protein expression of gas-producing enzymes to the amount of gas generated. However, such extrapolation is far from ideal because it is the availability of substrates and trace elements that controls enzyme activities and is more likely to determine the rate of gas formation in a tissue than simple protein expression.
Unlike NO, which is synthesized from
1. Tissue concentrations of CO
CO has attracted much interest since being implicated as a gaseous messenger for various biologic systems (230, 261, 296, 328). To understand the molecular basis and mechanisms whereby CO mediates cellular functions, it is important to determine the local concentration of CO in target tissues. Analytic procedures used to measure endogenous CO concentrations include gas chromatography–gas-reduction detection (330), gas chromatography–mass spectroscopic detection (14), laser sensor–infrared absorption (165, 208), and UV-visible spectrophotometric measurement of the CO–hemoglobin or the CO–myoglobin complex (2). Although recent quantum cascade laser technology (165, 208) enabled us to perform the real-time detection of biogenic CO generation, the technology to resolve both spatial and temporal dynamics of CO is not yet available. Readers are referred to comprehensive articles by Vreman et al. (332) and Marks et al (191).
One of the first attempts to determine the level of endogenous CO was made by using an isolated perfused liver preparation. Concentrations of CO in the effluent were determined spectrophotometrically by measuring the formation of the ferrous–CO complex of myoglobin (296, 297). The steady-state generation of CO was calculated to be 0.7 nmol/min per gram of liver. When the differences in local flow rates between ex vivo and in vivo systems are considered, it appears that local concentrations of CO in and around sinusoidal vessels are approximately 1 μM (296).
Recently, the development of a gas chromatography/reduction gas detector (GC/RGD) system allowed measurement of CO with a sensitivity of 1 pmole and a linear range up to 120 pmoles (330). By using this technique, the CO tissue concentration in rat liver was found to be 4 pmol CO/mg FW (fresh weight), which was similar to that measured by Suematsu et al. (296). In the rat brain, Vreman et al. (359) found the CO content to be 2 pmol CO/mg FW. By using GC, Ishikawa et al. (135) detected 1 μM CO in rat cerebrospinal fluid (CSF). In the same CSF, the concentration of bilirubin-IXα, an end product from heme degradation through the HO reaction, was determined to be 0.8 μM, suggesting that the stoichiometry of CO and bilirubin-IXα is close to 1 to 1. Taken together, these data suggest that CO concentrations in the liver and brain are in a micromolar range. By comparison, this value is one or two orders higher than the concentration of NO.
2. Tissue concentrations of NO
It is a difficult task to measure endogenous NO gas in vivo because of its low concentration and its short half-life. Analytic procedures to measure endogenous NO concentrations include electrochemical detection by using microelectrodes (16, 321) and chemical detection by using fluorescent indicators (163). NO microelectrodes can achieve a low detection limit [e.g., 6 nM (16)] and fast response time with high temporal resolution, but they cannot provide information on the spatial distribution of the gas within the tissue of interest. Conversely, fluorescent probes can provide information on spatial distribution, but cannot provide real-time information on NO flux. This is because fluorescent probes irreversibly react with NO. In vivo studies indicate that the NO concentration in tissues is likely to be in the 0.1 to 100 nM range (21, 38, 41), whereas values of perivascular NO concentrations from the resistance arterioles have been reported to be 500 to 600 nM (29, 321).
Numerous studies have tried to answer the following question: How much NO is required to activate sGC in vivo? The answer depends on (a) how much NO is actually produced, and (b) how much NO is delivered and coordinated to the prosthetic heme group of sGC, as an axial ligand. In the literature, the low-end concentration of NO required for half-maximal activation of purified sGC is 1.7 nM (103). By contrast, Stone and Marletta (293) reported an NO concentration of 250 nM at 10°C in vitro. Theoretically, the physiologic half-maximal activation of sGC at 37–38°C should be even higher, when one considers that NO reacts with not only with sGC, but also with other molecules, such as superoxide, thiol groups, hemoglobin, and cytochrome c oxidase, thereby reducing the effective concentration of the gas at the site of sGC (37, 196, 234).
In vascular smooth muscle cells (VSMCs), NO binding to the heme group of sGC occurs within milliseconds to a few seconds, although subsequent release of cGMP is much slower (49). Half-maximal activation of sGC requires 23 to 250 nM NO. The deactivation of sGC, with a half-life of 1 to 2 min, is at least an order of magnitude slower than its activation (64). This might imply that, even after the NO in the local environment dissipates, the sGC remains activated. This adds another level of complexity to the question of how much NO is needed to relax the smooth muscle in situ.
The difficulty in measuring the tissue concentration of NO has accelerated the field of mathematical modeling of NO delivery. Many mathematical models predicted a concentration range between 100 and 250 nM (38, 56, 327). Direct enzymatic production of NO in tissue depends on the availability of substrates and cofactors and the expression of the NO synthases (nNOS, iNOS, and eNOS (207)). Until recently, most NO available for vascular walls was believed to be derived from eNOS in endothelial cells. However, Kashiwagi et al. (148) found that nNOS-containing nerve fibers, which innervate arterioles and nerve terminals, are major sources of arteriolar NO, indicating an additional source in the vicinity of arteriolar walls (Fig, 11). Others also reported the presence of nNOS in perivascular nerve fibers (64, 219). These findings are reinforced further by the theoretic study in which consideration of the perivascular source of NO gives rise to a more realistic prediction of the NO-concentration profile in and around an arteriole, making it closer to the measured values (149). Although determination of tissue concentrations of NO is not easily achieved, these investigations may allow us to speculate that its concentration is in a nanomolar range, much less than that of CO. Possible consequences of the difference in CO and NO gas tissue concentrations are discussed later.
3. Tissue concentrations of H2S
Notwithstanding the numerous reports of potent actions of H2S in many organs, the cellular and molecular sources of this gas and the mechanisms of its release are still far from clear. To unravel these mechanisms, spatiotemporal determination of the H2S concentration must be acquired. However, among CO, NO, and H2S, the determination of H2S concentration in biologic samples, let alone the spatial determination of gas generation, appears to be the most challenging case. As mentioned in Section III.D. besides existing as free H2S, the gas is reversibly converted into different molecular entities of its related species. Processes of this conversion are sensitive not only to the natural biologic stimuli but also to the experimental conditions, making it difficult to determine actual local concentrations of H2S. Technical uncertainties of this kind challenge unraveling the mechanisms whereby H2S evokes biologic events.
Current methods to analyze H2S concentrations in biologic samples fall into two categories. One is designed to measure solely the “free” H2S, involving no derivatization process. The polarographic H2S sensor (79) is one such method. The other is designed to measure “labile” H2S, involving chemical treatment of samples with either acid or reducing agents to liberate H2S from sulfur compounds of cellular-H2S pools such as Fe–S complex (acid-labile) and bound-sulfane sulfur (dithiothreitol-labile). Colorimetric assays using methylene blue (108), gas chromatography–mass spectrometry (126), and high-performance liquid chromatography are often used to measure labile sulfur [see reviews by Tangerman (310) and Ubuka (323)].
Reported values of labile H2S in plasma and blood varied mostly between 20 and 300 μM (351, 352). In contrast, with a polarographic H2S sensor with the detection limit of near 10 nM (79, 160), no detectable H2S was found in either blood or plasma without any derivatization (352). Readers are referred to comprehensive summaries by Whitfield et al. (352) and Whiteman and Moore (351), in which values and the analytic methods used are comprehensively surveyed.
To measure free H2S in the rat brain homogenate, Ishigami et al. (133) developed a method using silver particles to trap free H2S, with the detection limit of near 9 μM; however, as in blood and plasma, no detectable H2S was found. In other studies, free H2S in the mouse brain was reported as 14 nM (96), whereas labile sulfur varied between 30 and 70 μM (323, 344). Investigators (351) interpreted these results to mean that H2S was stored in plasma rather than as free H2S. It is further suggested that H2S release from labile sulfur might take place in response to physiologic stimuli (156).
By using brain tissues from rodents, Ishigami et al. (133) reported the intriguing result that alkalization of the homogenates caused a release of H2S from bound sulfur. Moreover, in murine primary astrocytes, an increase in extracellular K+ concentration causes an alkalization of intracellular pH ([pH]i). An increase in [pH]
C. Functional arguments
1. Anatomical proximity of gas-producing and gas-reception sites
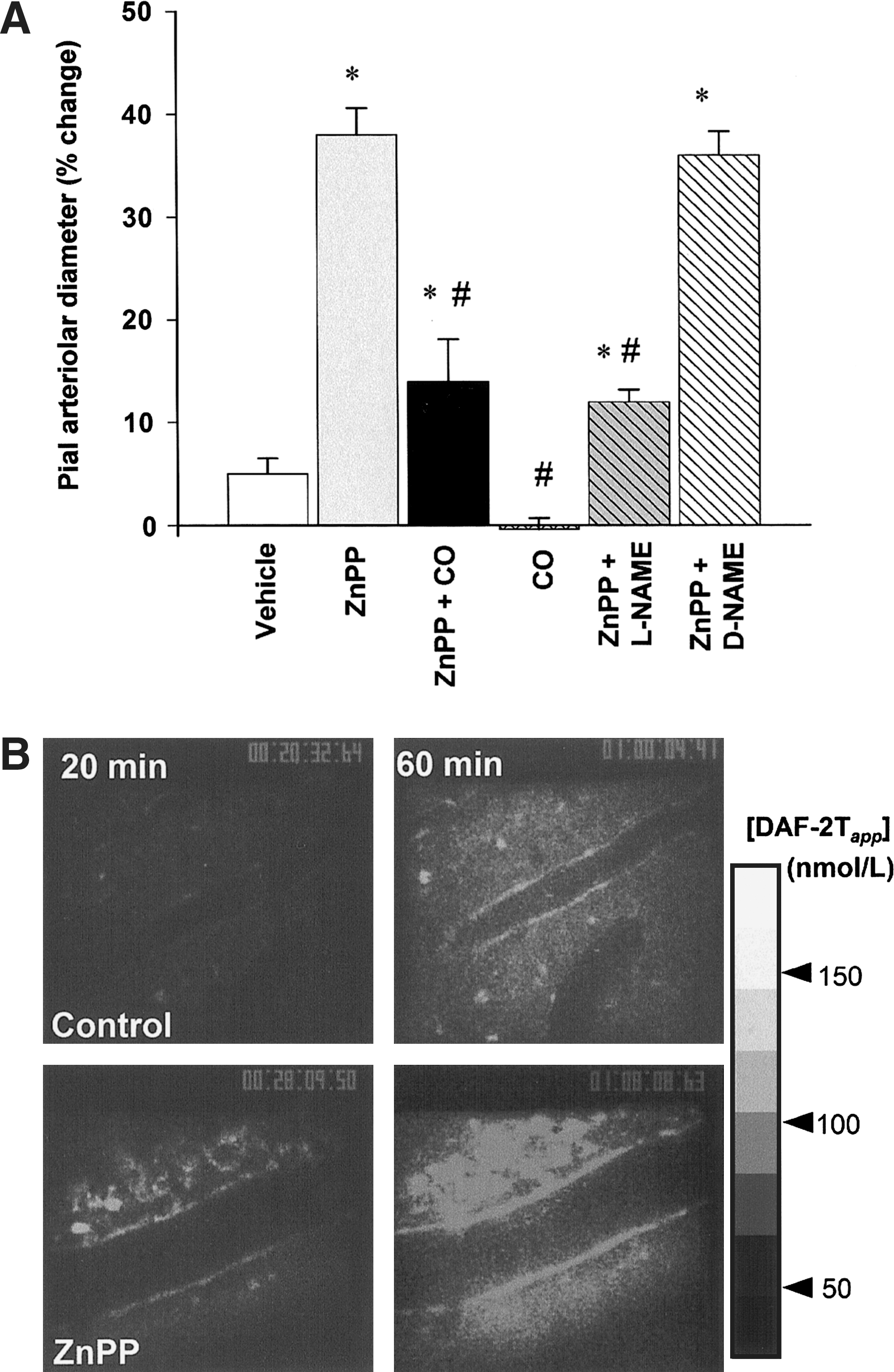
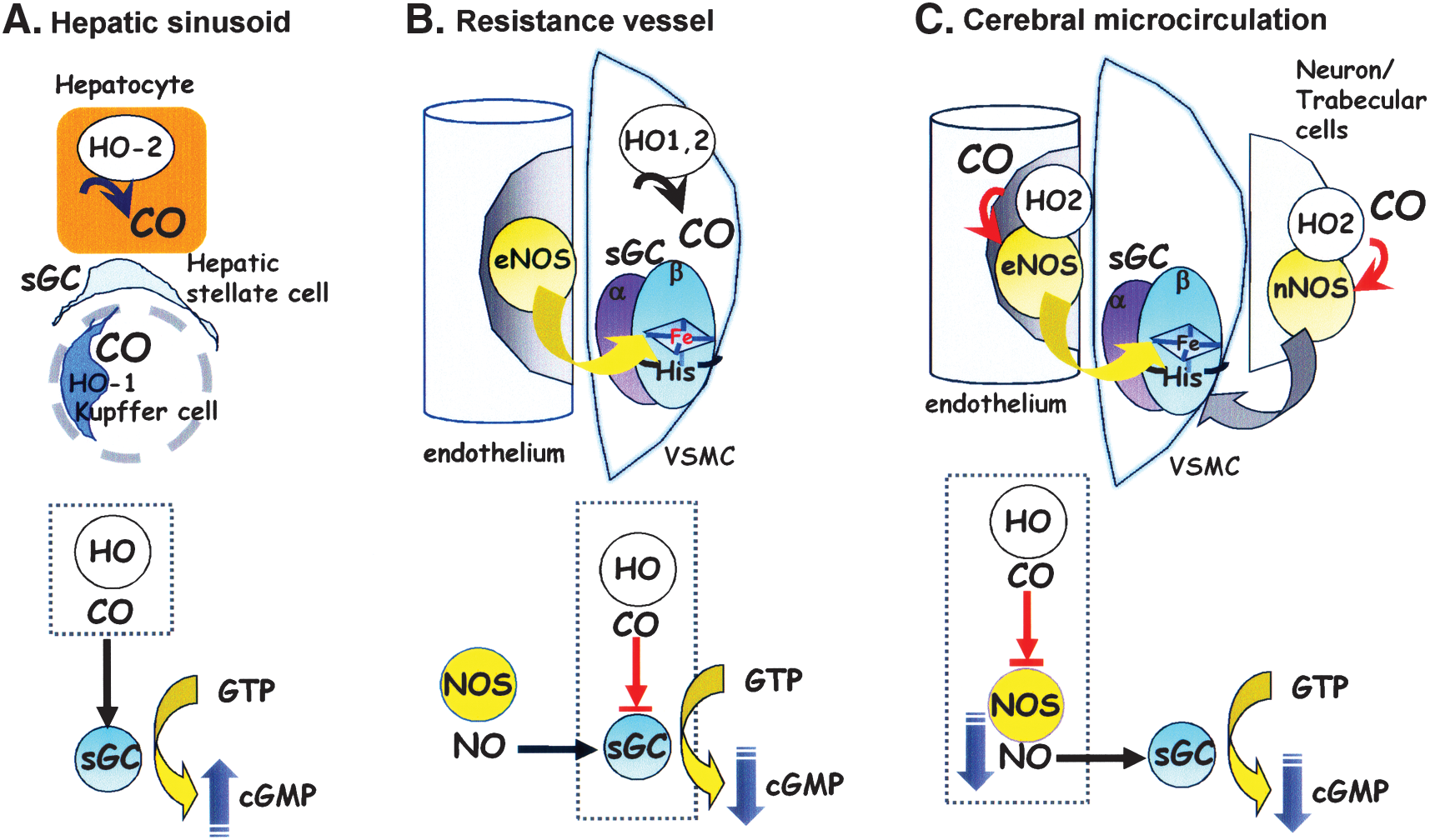
Experimental evidence suggests that CO modulates the generation of NO, and, by so doing, regulates vascular tone. Like NO, CO has vasodilatory properties in the liver where endogenous NO production appears low (296). In the brain, which produces relatively high levels of NO, how these gaseous monoxides interact with each other to control cerebrovascular tone is unclear. Ishikawa et al. (135) found that CO derived from HO acted as a tonic regulator against NO-dependent vasodilation in the adult rat brain. The authors showed that suppressing endogenous CO caused an increase in arteriolar diameter that was accompanied by an increase in local NO generation, demonstrating a causal relation between CO and the rate of NO production (Fig. 11). Such an inhibitory function of CO on NO generation appeared to be mediated by the ability of CO to bind the prosthetic heme of NOS. Additionally, immunohistochemical analyses of the rat brain showed that HO-2, the constitutively expressed form of HO, was present in neurons and arachnoid trabecular cells expressing nNOS and the vascular endothelium expressing eNOS, suggesting a colocalization between CO- and NO-generating sites. These results demonstrate the importance of spatial relations among the gas-producing enzymes and their reception systems.
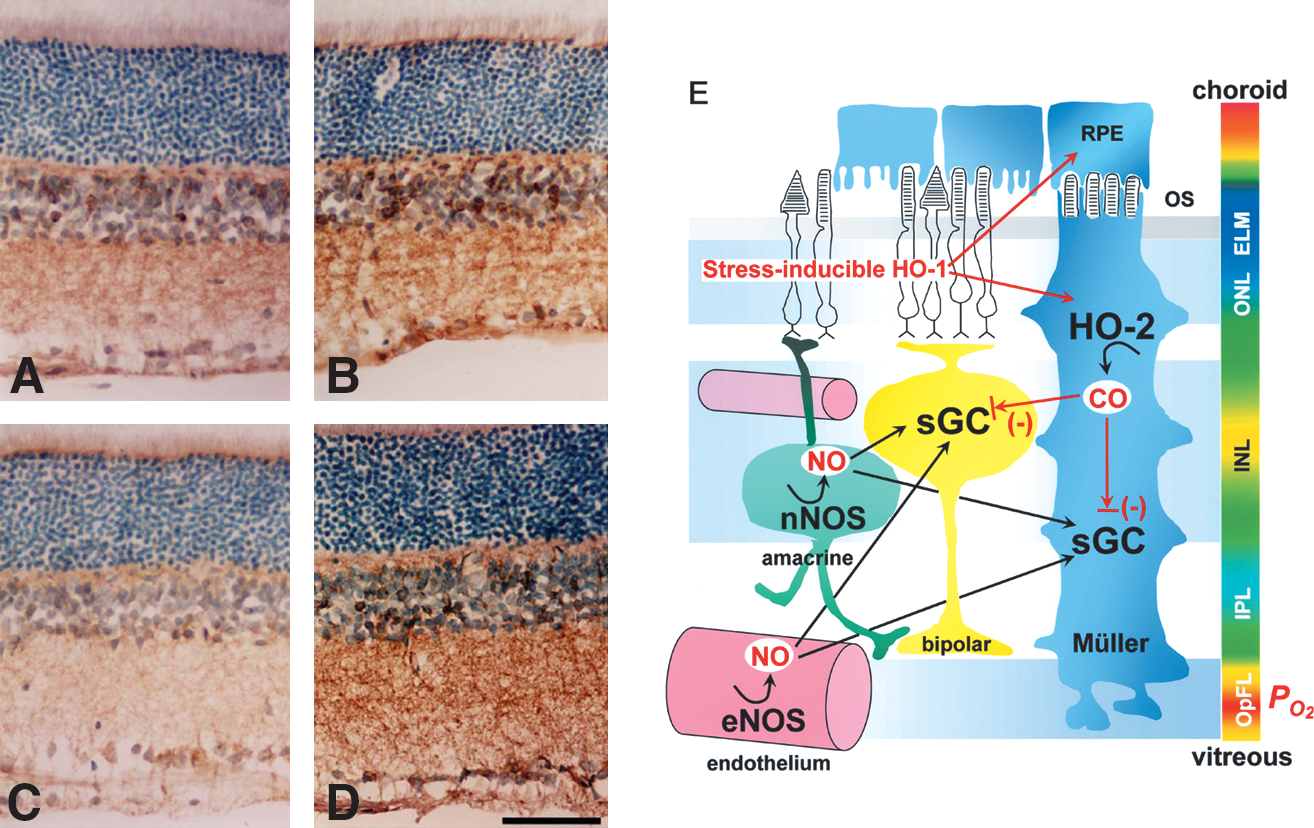
Taken together with other studies, it can be argued that CO regulates vascular tone in at least three distinct ways that depend on the microanatomic arrangements of the vasoactive gas-generation and -receptor systems. These arrangements are summarized as cartoons in Fig. 12. First in the liver, CO modestly stimulates sGC in hepatic stellate cells, thereby reducing the tonic contractile tension of sinusoids. For this action to take place, the local amount of NO must be low, as found in the liver microcirculation, in which constitutive NO appears to be negated by basal superoxide (99, 169, 296, 297). Second, in resistance arterioles, where sufficient amounts of NO are found, CO could target sGC and modulate NO-mediated vasodilatation. The observation that transgenic mice overexpressing cell-specific HO-1 in VSMCs exhibit systemic hypertension (130) supports this concept. Additionally, we found that endogenous CO produced by HO-2 in Müller glial cells plays a role in refining the NO-mediated activation of sGC in the retina (Fig. 13) (142). Finally, in the cerebral microcirculation, CO interferes with NOS activity and subsequently reduces NO generation, thereby limiting vasodilation. It appears that the mechanisms by which the gases exert their actions depend in part on the colocalization of the NOS and HO enzymes with sGC. These findings demonstrate the importance of spatial relations among the gas-producing enzymes and their reception systems in evaluating the functional roles of gases.
Several mechanisms exist by which the HO-CO system might modulate NO-dependent biologic events. First, oxidative degradation of heme by HO can downregulate catalytic activities of heme proteins, including NOS (305). A second possibility is related to substrate availability. Because both HO and NOS enzymes use NADPH as a reducing equivalent and molecular O2 as co-substrates, competition for these substances may mediate enzyme activity under normal conditions. Because NADPH is an intracellular substance that is not transported between cells, for HO and NOS to be in competition for these substrates, they must reside in the same cell. The finding that these enzymes do colocalize in the same cell supports the possibility that substrate competition may modulate enzyme activity. A third mechanism is related to the key observation made by White et al. (349), in which NOS activity is directly inhibited by CO in vitro, suggesting that the site of CO action is on the prosthetic heme of the NOS enzyme. As mentioned earlier, this inhibition requires a high concentration of CO, as much as 1 mM (265). By contrast, however, the reported values of tissue CO concentration are only on the order of ∼1 μM, significantly lower. One possibility to explain this discrepancy is that the in vivo mechanism involves local concentrations of CO, which may be much higher than global averages would suggest. If the turnover of HO-2 were fast enough, then it is possible that a large concentration of CO may exist locally. Provided that both HO-2 and NOS are expressed in the same cell, it is conceivable that NOS activity could be regulated by HO-2 in an autocrine manner. Although a gas is often thought to diffuse easily to a distant location in the tissue, the effective distance from the gas-generating site to its target could be much shorter than expected.
2. Is CO vasodilatory or vasoconstrictive?
The observation that CO acts as a tonic inhibitor of NO-mediated vasodilatation appears to contradict the results of other studies reporting that CO acts as a vasodilator (137, 176, 177, 297, 341). However, several possibilities exist that may help to explain this discrepancy. First, the principal target molecules for CO could be different in different experimental systems; thus, it is reasonable to expect different cellular responses based on different receptor systems. Leffler et al. (137, 164, 176) showed that superfusion of either CO (10−11 to 10−9 M) or heme-
Second, expression levels of CO- and NO-producing enzymes and those of target proteins could differ widely in different developmental stages within one animal model or between different species. By using cultured cerebellar granule cells of neonatal rats, Ingi et al. (131) demonstrated that production of CO decreased with the maturation of cells, whereas that of NO increased. More importantly, in mature cultures in which NO production reached a high level, HO inhibition potentiated the NO-mediated cGMP increase. These data were interpreted to mean that endogenous CO acted as an antagonist of NO-dependent cGMP production. Such results are consistent with previous studies from our laboratory in the rat retina (142). These investigators concluded that the role of CO varies depending on whether NO is present. Furthermore, studies revealed that the expression levels of sGC and nNOS change dramatically during postnatal development of the rat cerebral cortex (77).
A key factor that determines whether CO acts as either a constrictor or as a dilator could lie with the bioavailability of NO in each organ. In the brain, appreciable amounts of NO are present in both the vessel wall and the perivascular space (40, 58), whereas, in the liver, NO released from the sinusoidal endothelium appears to be extremely low, in part due to reaction with superoxide produced by Kupffer cells (296, 297).
What is the physiologic implication of having differential CO effects, particularly in the brain? We postulate that the effect of CO in regulating vascular tone is multifaceted where low tissue availability of NO renders CO a vasodilator, whereas high tissue NO availability renders CO a constrictor (141). Under resting conditions in the rat brain, endogenously produced CO could keep blood vessels from unnecessary dilation by suppressing local NOS activity, which may, in turn, contribute to the maintenance of normal intracranial pressure. Under traumatic brain injuries, such as cerebral ischemia and subarachnoid hemorrhage, CO can be overproduced, at least at some time point, because of an increase in free heme on the hydrolysis of heme proteins (55) or by an induction of HO-1 (303) or both. In addition, substantial alterations in NO and H2S might occur in relation to a reduced supply of O2, substrate availability, or altered redox states of gas-producing enzymes. Thus, aberrant actions of CO, together with NO, H2S, and O2 under these pathologic circumstances, deserve further investigation.
To summarize, although NO acts as a principal vasodilator, it may be important physiologically to have a modulating mechanism whereby NO alone does not dominate the control of cerebral vascular tone. CO generated by HO-2 might play a role in counteracting unnecessary overproduction of NO. This would imply a critical role for endogenous CO in maintaining resting cerebrovascular tone.
3. CBS as a CO sensor in vivo.
Several lines of evidence support the concept that CBS acts as an in vivo CO sensor. First, studies using recombinant CBS have shown that CO inhibits CBS with a K
i value of
Shintani et al. (281) demonstrated that an increase in hepatic CO content caused a global decrease in transsulfuration metabolites such as cystathionine, cysteine, and hypotaurine, suggesting that CO inhibits the transsulfuration pathway. Furthermore, CO-overproducing livers showed a decrease in labile H2S amount, whereas the livers of heterozygous CBS-knockout mice showed no decrease, suggesting that CO inhibits the activity of CBS in vivo. Furthermore, an administration of a stress-inducible level of CO (as 20 μmol/kg of CO-releasing molecule) caused a decrease in hepatic H2S content to stimulate HCO3 --dependent biliary choleresis. Such a CO-sensitive metabolic adaptation may play a role in quality control of bile excretion under disease conditions (281).
A remaining question is what is the physicochemical mechanism whereby CO interferes with the activity of CBS. Specifically, how is the iron center of the heme in this enzyme reduced to the Fe2+ oxidation state, and what kind of reducing agent accounts for this reduction in vivo. In the case of hemoglobin in the erythrocyte, methemoglobin reductase prevents the iron center of the heme from being oxidized. What might be playing the equivalent role in the case of CBS? At present, the regulation of CBS oxidative states in vivo is unknown and remains an important issue to be investigated.
4. HO-2 as an O2 sensor: a possible role in hypoxia-induced vasodilation
It was reported that HO-2 enzyme activity was activated by phosphorylation of serine 79 (28), implying the existence of an endogenous control mechanism in the central nervous system. Because HO-2 has been proposed to be an O2 sensor in chemosensing tissues, such as carotid body glomus cells (244, 353), it is reasonable that HO-2 plays a role in local hypoxia-induced vasodilatation in the brain (65, 85, 175, 238, 286). In this hypoxic situation, the action of CO could be mediated in a tonic manner in which no on/off switch is required. If the rate of CO production by HO-2 were more sensitive to a decrease in O2 than the rate of NO production by NOS isozymes (i.e., the O2 affinity of an enzyme-intermediate-product complex at a rate-limiting step for the HO reaction is weaker than that for NOS reaction), then NOS would still be able to operate at the concentration of intracellular O2 at which HO-2 can no longer produce CO efficiently. It could then be argued that hypoxia-induced vasodilation is mediated by augmented NO formation because of the reduced capacity of CO to inhibit NOS activity.
Here, it is worth noting that an overall K m value for O2, which is usually an important determinant of enzyme function during hypoxia, does not provide a key to the HO reaction because it consists of three oxygenation steps (Fig. 2). To gain insight into the O2 requirements of HO, one must find out which of the three oxygenation steps limits the rate of activity and the O2 equilibrium constant for each step. In vitro studies demonstrated that the rate-determining step of the heme degradation is the conversion of verdoheme to the ferric biliverdin complex (184, 195). However, although the rate-determining step involves binding of O2 to verdoheme, which is much slower than the binding of O2 to the heme complex, we can only speculate on the nature of the mechanisms in vivo, as the HO-2 kinetic parameters are extremely difficult to determine in vivo.
5. Cross-talk between H2S and NO
Besides the modulation of enzymatic activity by gaseous molecules, chemical reactions should be considered. Whiteman et al. (350) proposed that a reaction between H2S and NO produces S-nitrothiols (RSNO), by showing that in vitro incubation of sodium hydrosulfide (NaHS, H2S donor) with NO leads to the formation of an RSNO. Conversely, H2S was found to reduce GSNO and release NO (314). This led to the development of a novel method for measuring RSNO by using H2S (314).
Although H2S is generally considered to act as a vasodilator, through its action on ATP-dependent K+ channels located on vascular smooth cell membrane (370), studies using the aortic ring model showed a contractile response to H2S (6, 345). This provided evidence that the opposite response to H2S was due to the reaction of NO with H2S. These findings add a new mechanism to the maintenance of cellular redox states [see Whiteman and Moore (351) for a comprehensive summary of experimental evidence showing cross-talk between H2S and NO].
D. Non-heme protein O2 sensors/HIF/PHD
Hypoxia-inducible factor (HIF) is a heterodimeric complex consisting of α- and β-subunits, which belong to a family of helix–loop–helix Per/Arnt/Sim (bHLH-PAS)-domain proteins. At least three distinct α-subunit isoforms (HIF-1α, HIF-2 α and HIF-3 α) and a single β-subunit (HIF-1β) are known. Protein stability is regulated by the degradation of the α subunit. Under normoxic conditions, HIF prolyl hydroxylases (PHD1, PHD2, and PHD3) hydroxylate two proline residues (Pro-402 and Pro-564 in HIF-1α) in the oxygen-degradation domain (ODD) of the HIF-1/2 α-subunit by using O2 as a substrate. Modified ODDs allow binding of von Hippel-Lindau (VHL) ubiquitin ligase, leading to polyubiquitination and proteasomal degradation (150). Thus, HIF-1α protein is barely detectable under normoxia. Under hypoxic conditions, HIF-1α subunits can escape from hydroxylation because of decreased PHD activity (HIF-1α stabilization) and translocate to the nucleus, where they form heterodimers with the HIF-1β subunit and bind to the hypoxia-responsive element of target genes.
Hypoxia-inducible factors (HIF-1 and 2) are responsible for the upregulation of genes involved in hypoxic adaptation, including those of glycolysis, erythropoiesis, and angiogenesis, all of which are essential for survival under anaerobic conditions (273). In this sense, O2 sensing is the physiologic response in which activated HIFs modulate cellular functions by regulating gene expression (HIF pathway).
Under normoxia, both exogenously administered and endogenously produced NO stabilizes HIF-1α in a dose- and a time-dependent manner via distinct mechanisms from the classic cGMP-mediated pathway (36). This HIF-1α stabilization is due to decreased ubiquitin-dependent HIF-1α degradation. Under normoxia, NO inhibits PHD activity, most likely through direct interaction between NO and iron in ligand-like manner. The NO donor SNAP (S-nitroso-N-acetylpenicillamine) activated the HIF pathway under normoxia. However, SNAP does not inhibit hydroxylation of HIF-1α by PHD2, but causes S-nitrosation of cysteine 520 in HIF-1α ODD (236). This inhibits recruitment of VHL to hydroxylated HIF-1α, thereby inhibiting HIF-1α degradation.
In contrast, NO inhibits HIF-1α stabilization and hence activation under hypoxia. Several mechanisms have been reported for this inhibition. First, physiologic levels of NO can inhibit COX under hypoxia, which reduces the O2 consumption at mitochondria, leading to redistribution of O2 from mitochondria to other part of the cells. This increases the O2 availability for other O2-consuming enzymes, such as PHDs (36). Second, under hypoxia, NO is converted to peroxynitrite on interaction with mitochondria-driven superoxide, which leads to mitochondrial damage.
VII. Physiological Significance of CO, NO, and H2S on Mitochondrial Signaling and Their Relation to O2 Metabolism
CO, NO, and H2S are known to inhibit O2 consumption by inhibiting COX, the terminal electron acceptor of the electron-transport chain (ETC). Chemical mechanisms of this inhibition by three gases are analyzed and well-compared by Cooper and Brown in their recent review (67). COX possesses four redox-active metal centers (CuA, heme a, heme a 3, and CuB) all of which can be targeted by gases. The three gases, CO, NO, and H2S, can all bind to the iron center of the prosthetic heme in the COX enzyme complex. During electron transport, CuA accepts the first electron from ferrocytochrome c. The electron is transported to heme a, from which it passes to the binuclear heme a 3/CuB center, where the reduction of O2 takes place (12). CO is a competitive inhibitor that binds only to the reduced binuclear heme a 3/CuB center of COX. NO, however, can bind to both the reduced and oxidized states of the heme a 3/CuB complex, and its inhibition can be either competitive or uncompetitive. H2S, which binds to both the oxidized enzyme and the turnover intermediate, is a noncompetitive inhibitor. COX is thus a target and central mediator of mitochondrial respiration, not only through its natural ligand, O2, but also through the binding of CO, NO, and H2S. Here, we review four physiological processes affected by gaseous interactions in mitochondria: (a) mitochondrial redox signaling (MRS), (b) O2 sensing and hypoxia, (c) mitochondrial biogenesis, and (d) cytoprotection.
A. Mitochondrial redox signaling
The inhibition of COX by CO, NO, and H2S suppresses oxidative phosphorylation (OXPHOS) and reduces ATP production. Simultaneously, this downregulation of OXPHOS changes the redox state of the ETC and produces reactive O2 species (ROS). In some cases, ROS function as signaling molecules, thereby controlling cell functions. This process is known as “mitochondrial redox signaling (MRS).” In OXPHOS, reducing equivalents produced in TCA cycle send electrons into the ETC from complexes I and II. These electrons are passed to complex III and finally transferred to O2 at complex IV (COX) (143). However, a small fraction of the electrons leak from the ETC, even under normal conditions. These electrons react with O2 and produce superoxide anion (O2 −). It is worth noting that ∼1% of the O2 consumed in the cell is used for this reaction (31, 249).
B. Relations between CO, NO, H2S, COX, and hypoxia
CO binds to the reduced form of COX with a K i value of 0.3 μM (67). Its binding to COX is reversible and competitive with O2 (52). By using HEK293 cells, D'Amino et al. (70) reported a dose-dependent inhibition of COX by exogenous CO, which was enhanced under hypoxic conditions. They also showed that endogenously produced CO inhibited COX. CO derived from HO-1 inhibited mitochondrial respiration by 12% under 20% O2, but by 70% under 1% O2. The K i of CO for COX was 1.44 μM at 20%, whereas it was only 0.35 μM at 1% O2. Similar results were reported previously (113, 239). Considering that tissue CO concentrations are in the micromolar range, it appears plausible that CO inhibits COX in vivo.
CO is known to produce antiinflammatory and antiapoptotic effects, which are apparently regulated at the level of COX and mediated by MRS (260, 262, 369). By using RAW264.7 cells, Zuckerbraun (373) reported that CO production of ROS was derived from the ETC and caused by the inhibition of COX, thereby eliciting MRS. RAW264.2 cells produced TNF-α in response to LPS stimulation through p38 MAPK activity. The antiinflammatory effect of CO is responsible for the inhibition of this TNF-α production. Zuckerbraun et al. also showed that CO inhibits the activation of p38 MAPK though induction of MRS. Furthermore, CO upregulates superoxide dismutase (SOD)2 expression. Because SOD converts O2 − to the signaling molecule H2O2, this characteristic may also enhance the MRS. Downstream signaling pathways of CO-elicited MRS diverge widely, and readers are referred to a review on this subject by Bilban et al. (25).
NO binds COX both reversibly and irreversibly (34, 61). Unlike CO, NO can bind both the reduced and oxidized forms of COX (67). Reported values of K i of NO for COX are 60 nM at 30 μM and 20 nM at 10 μM O2, respectively (34, 309). A recent report showed a much lower K i of 0.2 nM for NO at the O2-binding site on COX (193). Because tissue levels of NO have been reported in a range between 10 and 450 nM (190, 277), it seems possible that NO can bind to COX in vivo. Because the inhibitory effect of NO on COX is much more efficient under hypoxic conditions, a question remains as to the in vivo physiologic effect. This is because the level of NO under hypoxic conditions is suggested to be lower than that under normoxic conditions (174), because NOS also uses O2 as substrate. It is not known, however, whether NO has enhanced inhibitory effects under hypoxia.
It has been suggested that the effect of NO inhibition of COX is the induction of MRS, which secondarily regulates many cellular responses, including ER (endoplasmic reticulum) stress (358), O2 redistribution (109), and acceleration of glycolytic metabolism (7). NO also has a vasodilatory effect through the sGC/cGMP pathway, which can increase the blood flow to the tissue. Thus, both the classic sGC/cGMP pathway and the MRS pathway seem to cooperate to induce the maximal NO effect in vivo. One important consideration is the possibility that NO reacts with O2 − to produce ONOO−, a highly reactive ROS species that might cause harmful effects to the cell (36). If so, MRS elicited by NO must be more tightly regulated as compared with that by CO.
H2S is a competitive inhibitor of COX that has ability to bind to both the oxidized and turnover intermediate of COX (67). Reported K i values are 0.2 μM by using purified COX (239), 10 μM using isolated mitochondria (365) and 30 μM using whole cells (178). Notwithstanding the lack of a reliable method to measure the H2S concentration in tissue, Doeller et al. (79) reported a range of H2S tissue concentration between 1 to 10 μM (79), suggesting that H2S could inhibit COX at the tissue level.
C. Interactions of CO and NO on COX
No report focuses on the interaction of CO and NO on COX, specifically. However, D'Amico et al. (70) reported an interesting finding. They upregulated both NO and CO production in RAW264.7 cells by LPS stimulation and observed the effect of hypoxia on COX inhibition. Although LPS had no effect on CO level, it decreased the NO level. From these results, the authors concluded that COX inhibition was associated with CO, but not with NO. Although the inhibitory effect of these gases is stronger under hypoxic conditions, hypoxia itself might not be a suitable condition for their activity. First, because both NOS and HO use O2 as their substrate, hypoxia might limit the availability of O2, thereby reducing the activity of both NOS and HO. Second, NO has vasodilatory effects through the sGC/cGMP pathway at a much smaller amount of NO, which may increase the tissue O2 concentration beyond the level that increases the affinity of COX to bind O2. And third, the concentration of CO, that induces MRS, depends on the redox state in the cells (74).
HO-1 induction accelerates heme degradation and thereby affects the activity of heme-containing enzymes. HO-1/CO suppresses the activity of mitochondrial NOS (mtNOS) because mtNOS requires heme for its catalytic activity leading to reduced NO production and NO-induced MRS (66). Conversely, HO-1 could reduce O2 − production outside mitochondria by downregulating GP91phox, a component of NADPH oxidase (NOX2) (305). Furthermore, ROS from mitochondria has been shown to increase NOX1 (75). All these findings suggest that ROS induced by HO-1–generated CO could be the net result of ROS production in the cell and not merely from COX inhibition.
D. O2 sensing and hypoxic response: effects of CO, NO, and H2S
Although a detailed description of O2-sensing mechanisms is beyond the scope of this review, we briefly discuss O2 sensing to understand better the role of small gases on O2 sensing. Many excellent reviews are available on tissue O2-sensing mechanisms (172, 272, 343). It is worth stating that, in most cases, MRS is also used as a mechanism for O2 sensing. Previously, mechanisms for O2 sensing were assumed to be different between acute and chronic hypoxic responses. However, a growing body of evidence suggests that they are principally the same because O2 sensing is based on sensing the decrease in O2 concentration through mitochondrial OXPHOS or ETC. Of course, some tissues use apparently different mechanisms, involving NADPH oxidase–related ROS detection.
Two distinct hypoxic responses exist, acute and chronic. The chronic hypoxic response requires transcription of new genes for its effect. Genes newly transcribed are those necessary for adaptation to anaerobic metabolism, such as erythropoiesis, angiogenesis, and glycolysis, most of which are regulated by the HIF pathway. Under hypoxia, stabilization and activation of HIFs depends on the canonic MRS, which results in PHD inhibition, thereby rescuing HIF-1α subunits from proteasome degradation.
Conversely, the acute hypoxic response is executed by specialized tissue such as the carotid body, pulmonary artery smooth muscle cells, and adrenomedulla cells (347). These tissues sense reduced O2 concentration and immediately respond by excreting hormones or contracting smooth muscle cells. The principal purpose of acute O2 sensing is to increase ventilation and circulation to ensure adequate O2 delivery to meet O2 demand. The final step of the acute O2-sensing mechanism is widely accepted to be an increase in intracellular Ca2+ through the opening of the L-type Ca2+ channel (357). Although this opening of the Ca2+ channel is the result of closure of several type K+ channels, mechanisms that close these K+ channels have not been fully investigated. In this part of this review, we discuss the role of these small gases on the acute O2-sensing mechanism.
Williams et al. (353) reported the possibility that HO-2 is an O2 sensor in the carotid body. In the carotid body, the activity of voltage-dependent, Ca2+-sensitive, large-conductance K+ channel (BKCa) is inhibited by CO. Williams et al. showed that HO-2 is physically associated with the channel component, and CO derived from HO-2 is associated with the activity of this channel. It was hypothesized that the activity of the channel needed CO derived from HO-2, of which production is reduced under hypoxic conditions, thereby working as an O2 sensor. However, this possibility has been questioned by subsequent studies using HO-2–knockout mice that showed no compromised response to hypoxia (227). However, HO-2–knockout mice show a lower O2 concentration in the blood (hypoxemia) together with impaired ventilation response to hypoxia (3), indicating the existence of a ventilation–perfusion mismatch in these mice. Accordingly, these results suggest that HO-2 could be an O2 sensor in hypoxic pulmonary vasoconstriction (HPV) or the neuroepithelial body in the lung or both. However, it is not clear whether CO-elicited MRS plays a role in acute O2 sensing.
NO is an important regulatory factor of blood vessel tone, and reports showed that NO inhibits hypoxic pulmonary vasoconstriction (HPV) (104, 174, 348). Actually, NO production has been shown to decrease in hypoxic lung (174). NO has vasodilatory effects through the sGC/cGMP pathway (334). This in turn has given rise to the idea that HPV might result from an impaired vasodilatory effect due to decreased NO production under hypoxia (104, 174, 348). However, Bernarl et al. (24) recently proposed an interesting hypothesis about the role of NO on acute O2 sensing (24). They reported that NO production was increased in pulmonary artery endothelium and that this increase in NO accelerated the release of zinc from metallothionein (MT) by nitrosylation. This Zn activated protein kinase C, which eventually caused HPV (NP/MT/zinc pathway). This hypothesis describes a new role of endothelium-derived NO in HPV. However, one consideration is that knocking out all types of NOS had no effect on HPV (104, 174, 348). Accordingly, further investigation is needed to clarify whether NO is the principal effecter or only a modulator of HPV.
Almost all the investigations on the role of H2S in acute O2 sensing have been made in HPV. HPV is the contraction of middle-size pulmonary arteries in response to acute hypoxia (347). During HPV, blood flow within poorly ventilated lung is redirected to areas with higher pO2, thereby improving ventilation–perfusion. Olson et al. (224) recently reported that H2S is produced in the vessel wall. They also showed that treatment with cysteine, a precursor of H2S, enhanced HPV, and conversely, inhibition of H2S production impaired HPV. They also found that hypoxia and H2S share the same downstream pathways. From these results, they proposed a novel possibility of H2S as an O2 sensor in HPV.
H2S is ubiquitously produced in cytoplasm and oxidized in mitochondria. Olson et al. (223) hypothesized that nonoxidized H2S has biologic activity associated with HPV. Because oxidation depends on O2 availability, O2 concentration is the critical factor that determines the total amount of biologically active H2S. According to this theory, high O2 concentration correlates with lower H2S activity and a dilated pulmonary artery. Conversely, low O2 concentration correlates with higher H2S activity and artery contraction (223). Actually, H2S has been reported to have both vasodilatory and vasoconstrictive effects, depending on its concentration (6, 345). Furthermore, O2 concentration has another effect on H2S-mediated O2 sensing. O2 has been shown to act negatively on the activities of both CBS and CSE (15, 288), which may have an additive effect on the H2S-mediated vascular response. Reduced production of H2S under hypoxia has also been reported (352). Olson et al. (224) reported the possibility of H2S as a universal O2 sensor by showing H2S acted as O2 sensor in trout and zebrafish gills.
In summary, although NO, CO, and H2S seem to be involved in acute O2 sensing in their own way, no direct poof indicates that they use MRS for O2 sensing. This might be due in part to the extremely high sensitivity of O2-sensing tissue of OXPHOS for O2 (80). This might cause decreases of ATP at much higher O2 concentrations compared with other non–O2-sensing cells. One possibility is that eliciting activation of AMP-dependent protein kinase (AMPK) inhibits the K+ channel in these specialized tissues (357). Also, we cannot rule out the possibility of MRS as an O2-sensing mechanism, because ROS production has been shown even in acute O2-sensing tissue under acute hypoxia.
E. Effects of CO and NO on mitochondrial biogenesis
What is mitochondrial biogenesis, and why does it occur? The principal purpose of O2 sensing is to ensure that O2 delivery matches O2 demands at the cellular level. In this context, O2 sensing functions to maintain or increase the energy supply in the tissue. Considering that cell functions depend mostly on mitochondrial OXPHOS, it is reasonable for cells to increase their number of mitochondria to meet their cellular energy demands. Because small gases are involved in O2 sensing, as discussed previously, we next consider the roles of these gases on the mitochondrial biogenesis.
Mitochondrial biogenesis is a complex process involving the coordinated expression of mitochondrial and nuclear genes. Because discussion of the mechanism of mitochondrial biogenesis is beyond the scope of this review, readers are referred to an excellent review in this field (76). The key molecules in mitochondrial biogenesis are transcriptional activator peroxisome proliferators–activated receptor γ-coactivator-1α (PGC-1α) and transcriptional factor nuclear respiratory factor-1 and 2 (NRF-1 and NRF-2). NRFs are the transcriptional factors that control the majority of proteins involved in OXPHOS in mitochondria. Additionally, PGC-1α and NRFs are responsible for the transcription of Tfam, a central molecule controlling mitochondrial DNA replication and transcription of mitochondria-encoded genes. Many physiologic signals control the biogenesis of mitochondria, including exercise (121) and hypoxia (107). Because no reports are available in the current literature concerning the role of H2S, we briefly review the involvement of NO and CO in mitochondrial biogenesis.
Nisoli et al. (217) have reported that in mouse brown adipocyte precursor cells, mitochondrial biogenesis is dependent on NO, and this NO effect is mediated by sGC. The same group has shown, by using different cell systems from different species, that NO can induce mitochondrial biogenesis (218). Because mitochondrial number was reduced in eNOS-knockout mice regardless of the tissue, NO derived from eNOS has been assumed to be responsible for NO-induced mitochondrial biogenesis (217). Recently, however, involvement of nNOS in mitochondrial biogenesis also was reported in rat ischemic brain (107). One characteristic of NO-induced mitochondria biogenesis is that newly synthesized mitochondria can fully produce ATP through OXPHOS (218). Furthermore, ATP synthesis in NO-induced mitochondria was not accompanied by a reduction of ATP produced by glycolysis, which resulted in an increase in total ATP. This may be a characteristic of energy production through NO.
Compared with NO, research into CO-induced mitochondrial biogenesis started only recently. However, contrary to NO, evidence suggests the involvement of MRS in CO-induced mitochondrial biogenesis. Recently, endogenously produced CO, induced by transfection of the HO-1 gene, has been reported to induce mitochondrial biogenesis in rat myocardium (301). HO-1/CO-induced mitochondrial biogenesis required both H2O2 and sGC activity. The production of H2O2 was derived from mitochondria and required the activity of the AKT/PKB pathway for activation of PGC-1α, NRFs, and Tfam. Interestingly, sGC activity was not involved in this AKT activation.
Lancel et al. (173) also reported myocardial mitochondrial biogenesis in septic mice injected with the CO-releasing donor, CORM-3. Additionally, with human muscle biopsy samples, Rhodes et al. (252) reported that inhalation of CO together with exercise induced upregulation of mRNA levels of PGC-1α, NRF-1, Tfam, and DNA-polymerase gamma. Although this CO inhalation also increased COX subunit I and citrate synthase at protein levels, mtDNA was not increased significantly.
Both ischemia and exercise are conditions that increase cellular energy demands. However, whereas the former requires NO, the latter requires CO-induced mitochondrial biogenesis. CO-induced mitochondrial biogenesis accompanies the upregulation of HO-1 and SOD2 (252), which suggests the involvement of mitochondrial oxidative stress. Although brain ischemia seems to be associated also with mitochondrial oxidative stress, involvement of CO was not reported. Conversely, NO-induced mitochondrial biogenesis is associated with the sGC/cGMP pathway. Although NO can elicit a strong MRS, it is not clear why this pathway is not the main pathway for mitochondrial biogenesis. This may suggest the existence of unknown regulatory mechanisms downstream of MRS elicited by different stimuli.
F. Cytoprotective effects of CO, NO, and H2S
CO, NO, and H2S were considered poisonous gases previously. A growing body of evidence now suggests that at physiologic concentrations, these gases have cytoprotective roles against many pathologic conditions. However, the mechanisms whereby these gases exert cytoprotective and pharmacologic effects are not simple and are still largely unknown. In this part, we discuss the protective role of these gases, focusing on the mitochondria. Principally, MRS is derived from the ETC of mitochondria under compromised energy-supply conditions, such as hypoxia. Accordingly, it is probable that most of the signals elicited by MRS are necessary for the protection and survival of the cell.
In this context, stabilization and activation of the HIF system is no doubt the most important downstream pathway under hypoxia. However, MRS elicited by small gases does not necessarily accelerate the HIF pathway. We discuss the characteristic downstream of MRS that is elicited by these gases. Activation of AMPK is the most characteristic downstream of NO-induced MRS. Quintero et al. (248) reported the interesting characteristic of NO-induced AMPK activation in HUVECs. AMPK is usually activated by a decreased ATP level (263). However, in HUVECs, NO-induced AMPK activation was associated with neither the ATP level nor the sGC activity, suggesting that NO-induced mitochondrial redox signaling activates AMPK. This activation was elicited by mitochondrial ROS, although observed independence from the ATP level may be due to the difference of AMPK subunits. AMPK comprises three subunits, and the activity depends on the catalytic α-subunit. Most cells use the α2-subunit, which is sensitive to the ATP level, whereas HUVECs used the α1-subunit. AMPK accelerates glycolysis by upregulating GLUT4, hexokinase II, and 6-phosphofructo-2 kinase. Glycolysis is usually upregulated by HIFs. However, interestingly, NO-induced MRS causes O2 redistribution, thereby increasing O2 concentration in the cell, which in turn inhibits the stabilization of HIF-1α-subunits (109).
Why does NO-induced MRS inhibit HIF-induced glycolysis while enhancing AMPK-induced glycolysis? One possibility is the rapidity of the AMPK pathway to activate the glycolytic system. AMPK is fully activated 15 to 30 min after the hypoxic exposure (171), whereas activation of HIF-1 takes much longer. NO-induced AMPK activation might be an energy stress. One interesting and important finding is the early reduction of the ETC by NO. NO can bind to both reduced and oxidized COX (68). By doing so, NO can control the redox state of the ETC. Interestingly Clementi et al. (62) reported that NO controls COX activity at a basal level (62). Thus, NO may be able to control COX activity to elicit MRS. Considering that mild to moderate inhibition of COX activity does not compromise OXPHOS capacity, NO may use ETC to generate a signal through the regulation of COX. Other downstream targets are translocation of redox-sensitive transcriptional factors and Ca2+ efflux from cells. The former is related to induction of genes necessary for survival. Efflux of Ca2+ may enhance the activity of AMPK and may also inhibit MPT by reducing the sensitivity to oxidative stress (111). The current understanding of the cytotoxic versus cytoprotective effects of NO in the mammalian central nervous system, emphasizing multiple properties of NO, has been well reviewed by others (45).
The role of CO-induced MRS has not been extensively studied, as compared with NO (25, 240). One characteristic is involvement in anti-inflammatory reactions. This may be due in part to the preferential use of macrophage-derived cell lines. Because CO binds only to reduced COX, is it not known whether CO also uses ETC to generate signals that are specific for CO, and not for other gases. CO can stabilize the HIF-1α-subunit, thereby modulating the HIF pathway. However, by CO alone, this activation does not seem to be practical because a large amount of H2O2 is necessary to maintain activation of the HIF pathway.
Currently, no report exists of H2S using classic MRS. Like NO, H2S can bind to both reduced and oxidized COX. It might be possible that H2S can also control COX at a physiologic level. However, despite the similarity to NO at COX binding, no proof is known that H2S elicits MRS.
G. The roles of H2S, NO, and CO in “suspended animation”
Suspended animation is a conserved physiologic response to anoxia in which all life processes reversibly arrest, including movement and development (91, 231, 232). Surprisingly, this characteristic phenomenon completely returns to normal without any damage when animals are released from stress. This characteristic phenomenon is an adaptation to reduced energy demand and is similar to conditions like hibernation and daily torpor. From studies using Caenorhabditis elegans, suspended animation appeared not to be a mere passive response to a compromised state of energy supply, but rather a highly controlled active phenomenon (201, 220). Although suspended animation was originally reported in severe hypoxia, conditions other than hypoxia also can induce this phenomenon. Of particular interest is that gases including H2S can induce this phenomenon.
Blackstone et al. (26) reported that inhalation of a nontoxic level (80 ppm) of H2S in awake self-breathing mice could reversibly reduce the metabolic rate to as low as 10% of the normal state and the body core temperature down to ∼2°C above (15°C) ambient temperature. A subsequent study showed that decreased metabolism and cardiac function were not associated with the reduction of core body temperature (329). The same group further reported that this suspended animation–like condition protects the animals from lethal hypoxia (5 to 3% O2) (27). Similar results were reported in large animals by injecting sulfide into anesthetized pigs (282).
Suspended animation induced by CO was reported in the embryo of Caenorhabditis elegans (221). Caenorhabditis elegans entered suspended animation under anoxia below 0.01 kPa. At modest hypoxia (0.5 kPa), an embryo can survive and continue development by an adaptive response through HIF-1–pathway induction. However, over an O2 range between 0.1 to 0.01 kPa, although the embryo showed development, it eventually died. CO could induce suspended animation in this O2 environment and rescue the embryo from otherwise lethal hypoxia.
Suspended animation induced by NO was reported by using the Drosophila embryo (315). The embryo entered suspended animation under hypoxia. An NO donor also induced suspended animation in the embryo, even under normoxia. This NO-induced suspended animation was inhibited by a scavenger. Interestingly, blocking of respiration by cyanide also induced suspended animation. However, changes in RNA and protein regulation that are observed under hypoxia and NO treatment were not observed in cyanide-induced suspended animation. Currently, no study is using large animals.
In summary, suspended animation including hibernation is a highly controlled adaptive response to hypoxia and related conditions. Recent studies have revealed molecular mechanisms of this interesting and important phenomenon (8). It is not known what signals are involved between O2 sensing at the COX and rapid metabolic downregulation. Although these three gases may have different pathways to induce suspended animation, COX inhibition is the common characteristic. Accordingly, it is highly probable that suspended animation is located downstream of COX inhibition. If so, several questions remain. Is inhibition of COX enough to induce suspended animation? Is MRS necessary for this induction? Do all these gases have their own signals to induce suspended animation? Studies with gases seem to give an important hint, particularly in the Caenorhabditis elegans experiment. Death of an embryo at the O2 concentration of between 0.1 to 0.01 kPa seems to result from energy exhaustion, because the embryos did not stop their embryogenesis and development, although they require a high energy supply. The embryo seems to escape lethal hypoxia by reducing energy expenditure with CO treatment. These situations resemble the septic condition, in which animals are in an exhausted energy condition (179). Moreover, gases with an inhibiting effect on COX improve the survival rate of septic animals (19). Inhibition of respiration is no doubt associated with a reduced energy supply. However, mere blockade of the COX activity does not seem to induce suspended animation. Inhibition of respiration through COX inhibition can be a trigger for suspended animation. However, some additional signals seem to be necessary for complete induction of this interesting condition.
To summarize, NO, CO, and H2S can inhibit COX. Through this inhibition, all three gases elicit MRS, which is necessary for an adaptive response to hypoxia. In addition, these gases display protective roles on mitochondria by inhibiting MPT. Finally, these gases have therapeutic potentials by inducing suspended animation. In this section, we focused on MRS as a gas-mediated signaling mechanism. However, these gases may also regulate cellular signaling without affecting ETC or OXPHOS capacity. COX activity seems to exceed the ETC capacity as seen in muscle tissue (168). Hence, mild COX-inhibition does not necessarily imply a limitation of the ETC. This may allow cellular pathways to be regulated through COX activity without affecting mitochondrial respiration. This may explain how these toxic gases can be fundamental components of complex cellular signaling. Further studies are needed to understand the complex mechanism of COX regulation by these gases.
VIII. Altered Gas Balance in CNS Diseases
What might happen if the gas balance were significantly altered in disorders such as strokes? When and how might we manipulate gas levels to treat disease? Although the majority of studies have suggested both cytoprotective and cytotoxic roles for CO, NO, and H2S gases under pathologic conditions, these suggestions were often made based on either exogenous application of a gas, expression levels of a producing enzyme, or the phenotypes obtained by using knockout mice, but not on actual gas amounts. What is important to study is the direct cause-and-effect relation between gases and their clinical manifestation. Here we consider some of the governing factors controlling local gas amounts, including (a) substrate availability, (b) enzyme control resulting from allosteric control and covalent modification, and (c) genetic control of enzyme expression. With these in mind, we describe the relevance of gas biology in neurodegenerative diseases of the central nervous system (CNS).
A. CO in CNS diseases
CO has been implicated in pathologic processes of the brain. Two isoforms of HO have been identified, the inducible HO-1 and the constitutively expressed HO-2. For HO-2, several mechanisms have been proposed for regulation of its activity. One is through phosphorylation by casein kinase 2 during neuronal stimulation (28). More recently, Yi et al. (362, 363) showed that the C-terminal heme regulatory motifs (HRMs; Cys-Pro signature) act as a thiol/disulfide redox switch controlling the affinity of the HO-2 for heme. Here, HRMs known to control processes related to iron and oxidative metabolism can integrate heme homeostasis with CO signaling. Such regulatory mechanisms are under active investigation.
Substrate availability is an important determinant of CO production. Unlike NO production, which uses readily available
Neural injury can be either ameliorated or exacerbated (336) by the HO-CO system. However, many studies using stroke models suggested cytoprotective effects of HO-1 and HO-2. Overexpression of HO-1 in the mouse brain caused a reduction in infarct volumes induced by middle cerebral artery occlusion. Cell viability of primary neuronal cells of HO-1–knockout mice was decreased when challenged with acute excitotoxicity compared with that of the wild type (4). Moreover, HO-2 appeared to protect against lipid peroxidation–mediated cell loss and impaired motor recovery after traumatic brain injury (54, 55), whereas its deletion exacerbated intracerebral hemorrhage–induced brain edema (337).
HO-1 is induced in response to various prooxidants and stressors. Intense HO-1 immunostaining in the parkinsonian brain has been demonstrated, indicating that HO-1 may be involved in the pathogenesis of Parkinson disease (267). Overexpression of HO-1 in the rat substantia nigra significantly increased the survival rate of dopaminergic neurons against 1-methyl-4-phenylpyridinium–induced neurotoxicity. Furthermore, inhibition of HO activity exacerbated rotatory behavior of the rat, suggesting a role of endogenous CO (125).
Like Parkinson's disease, oxidative stress appears to be involved in the pathogenesis of Alzheimer disease (268). HO-1 immunoreactivity is greatly enhanced in the neurons and astrocytes of the hippocampus and cerebral cortex of Alzheimer disease patients and coexpressed in senile plaques and neurofibrillary tangles (268). Amyloid precursor protein (APP) generates the β-amyloid peptide that has been postulated to participate in the neurotoxicity of Alzheimer disease. APP has been found to bind HO and inhibit its activity (307). However the mechanisms whereby HO-1 or CO or both control these age-related neurodegenerative disorders are not clear at present.
The involvement of the HO-CO system has received considerable attention as a target for the development of effective therapeutic interventions against degenerative and inflammatory diseases in the CNS [see review by Schipper et al. (269)]. It has been proposed that the cytoprotective roles of the HO-CO system are initiated by a series of molecular reactions or interactions or both in response to changes in redox states of the cell, which in turn elicit “homeostatic” responses, as opposed to “chaotic” ones (25). HO-1 has been described as a therapeutic funnel (13, 25, 162, 229) because of the multiple effects mediated by this molecule. Calabrese et al. (43, 46) view the adaptive mechanisms linked to the HO pathways as being crucial for both survival and the physical quality of life. They termed this complex network of adaptive processes, composed of several protective genes, the “vitagene system”. “Vitagenes” encode for heat-shock proteins (Hsps), Hsp32, Hsp70, thioredoxin, and the sirtuin protein systems. Because the system may provide a point at which CO, O2, NO, and H2S interact with one another, the neuroprotective potential of the “vitagene system” as a target for new approaches to neural antidegeneration deserve further investigation.
B. NO in CNS diseases
Unlike the situation for CO and H2S, it has been proven that control of NO synthesis is coordinated with neuronal activity. During neurotransmission, neurons transiently synthesize NO in response to stimulation of the glutamate-mediated NMDA receptor, in which the activity of nNOS is tightly regulated by the intracellular Ca2+ level (32, 159).
NO has been suggested to mediate neurotoxicity in response to the extracellular accumulation of glutamate. With pathologic stimulation, NO derived from iNOS in microglia and astrocytes is associated with neuronal cell death (116). In addition, NO can trigger the activation of transcription factors through its binding to COX (235), leading to gene regulation. NO and other reactive nitrogen species can act as pathologic agents in processes such as neuroinflammation and neurodegeneration (90, 120). These deleterious effects are thought to be attributable to targeted modifications of critical cysteine residues in proteins, including S-nitrosylation and S-oxidation, as well as by lipid nitration (44, 45, 205, 215). Studies support this scenario for neurologic disorders such as Alzheimer disease (71), amyotrophic lateral sclerosis, Parkinson disease (59, 311), and ischemic brain damage (30, 205).
C. H2S in CNS diseases
One possible function of H2S in the brain is to act as a neuromodulator. Currently, however, no definitive mechanism has been reported to link the control of H2S synthesis with neuronal activity. In the brain, CBS is the predominant enzyme responsible for H2S generation. Besides CO, which can inhibit CBS activity, both S-adenosyl-methionine (SAM) (89) and peroxynitrite (ONOO−) (50) can modulate its activity. The human full-length CBS is a homotetramer of 63-kDa subunits, the activity of which is increased by SAM (89). A truncated CBS dimer with a 45-kDa subunit, which is more active, but unresponsive to SAM (153), has been found in HepG2 cells exposed to tumor necrosis factor (372). Unlike NOS, regulation of CBS by Ca2+/calmodulin is most unlikely, as Chen et al. (57) failed to report any stimulation of CBS activity in the presence of Ca2+/calmodulin. Interestingly, Celano et al. (50) showed inactivation of the Fe3+ CBS dimer with ONOO− (50), which could provide a control mechanism for H2S synthesis under pathologic conditions.
One of the suggested targets for H2S is the NMDA receptor in the neuron. Although H2S alone does not induce long-term potentiation, it does so by potentiating glutamatergic transmission through the function of NMDA receptors in neurons (214). Qu et al. (247) raised the interesting possibility that this potentiation by H2S is redox dependent. Given that the neuronal NMDA-receptor possesses thiol groups in the extracellular domain (302), these authors speculate that H2S modulates NMDA activity by its ability to reduce the Cys744 and Cys798 (247). GAPDH is another target for H2S. Mustafa et al. (212) reported the interesting finding that sulfhydration, the formation of SSH in cysteines, of GAPDH augments the enzyme activity by sevenfold (212), implicating a novel H2S-dependent control of cellular energy metabolism in the CNS.
CBS is encoded by a gene on chromosome 21 (21q22-3). One of the manifestations of CBS deficiency is mental retardation. Qu et al. (246) explained this by the inability of H2S to induce long-term potentiation via the potentiation of the NMDA receptor in the hippocampus, leading to the compromised memory function. They also speculate that the memory deficit seen in Alzheimer disease may be related to reduced H2S. By contrast, CBS activity in fibroblasts from Down syndrome patients is ∼150% higher than that in those from normal individuals (51), and H2S is overproduced, based on the amount of excreted thiosulfate in the urine (145). However, the role of H2S overproduction in mental retardation is not well understood.
In stroke, H2S appears to exacerbate ischemic injuries, and thus, its inhibition has been suggested as a potential treatment in stroke therapy (304). Evidence is accumulating to demonstrate that inhibitors of H2S production or therapeutic H2S donor compounds exert significant effects in various animal models of inflammation, reperfusion injury, and circulatory shock (304). However, H2S can also induce a reversible state of hypothermia and a suspended animation–like state in rodents, as discussed in Section VII.G.
IX. Challenges and Perspectives
Although recent advances in gaseous signaling have unraveled uncertainties about their actions in our body, we are still left to understand the biologic potentials of newly emerging gases, such as sulfur dioxide (SO2) (182) and hydrogen cyanide (HCN) (106) and to elucidate mechanisms whereby well-known gases, such as ammonia (NH3) and carbon dioxide (CO2), exert biologic effects. In the last section, we described challenges to make breakthrough in the field of gas biology. We provide some detailed mechanisms of gaseous transductions and their interactions. However, many unknown features are lacking. O2 is by far most well-studied gas as to its reception systems and functions. Looking back at the history of the O2-sensing research, Otto Warburg received the Nobel Prize in Physiology and Medicine in 1931 for “his discovery of nature and mode of action of the respiratory enzymes.” It took more than two and a half centuries to prove the vital mechanism of O2 use in intracellular combustion of energy substances after John Mayow conducted the experiment suggesting the existence of “O2” in the air. Since then, technical advances to measure O2 continuously, accurately, and directly within living tissue (60, 325) were made. These tools that stimulated the field of O2-sensing research and brought about important discoveries.
Many technical advances are needed to detect physiologic levels of O2, CO, NO, and H2S in vivo in a real-time manner with reasonable spatial information. However, a relative lack of technical breakthroughs for detecting gas behavior in vivo appears to be a limit for transforming good speculative ideas into natural philosophical thoughts. Therefore, one of the awaiting challenges to move gas biology forward is development and implementation of reliable methods for continuous, noninvasive, and sensitive measurements of gaseous mediators.
Among CO, NO, and H2S, the field of NO is better established than the others. Since NO was first identified as the endothelium-derived relaxing factor in the late 1980s (95, 129), many approaches have attempted to provide an adequate means for measuring physiologic levels of NO. Several techniques, including the electrochemical sensors (42, 276) and fluorescence-visualization methods (163), have been successful in achieving this aim. With these advanced technologies, the field of NO research has made substantial advances. Conversely, equivalent tools are yet to be made available for CO and H2S. Gas-releasing molecules, however, such as NOC (278) and NOR for NO and CORM (210) for CO, have added great insight into the mechanisms whereby NO and CO exerts their biologic effects.
Although we are still waiting for new tools for visualizing and measuring gaseous molecules in situ, the field of gas biology has added several cutting-edge technologies. We believe that a series of these advanced technologies can help us to make breakthroughs and to unravel mechanisms of gas generation and gas reception. One such technology is metabolome analysis using advanced mass spectrometry to explore systematically gas-responsive regulator enzymes in vivo. We have applied capillary electrophoresis mass-based metabolome analysis to profile small molecular metabolites in pathogenic bacteria or in mouse livers. A comparison of transcriptional expression profiles with metabolome data led us to hypothesize the existence of novel metabolic pathways and their regulatory mechanisms, and eventually to succeed in the discovery of variant TCA cycles in Mycobacterium tuberculosis (317). Furthermore, we found novel pathways to synthesize “pseudo-GSH metabolites” in mammals, which could account for compensatory mechanisms against oxidative stress in the liver (285). This technology has proven to be powerful because simple application of a gas to cultured cells and data collection through differential metabolomic display between disease and control conditions allowed us to hypothesize gas-responsive rate-limiting steps for metabolic pathways of interest.
When combined with metabolome analysis, large-scale computational biosimulation of metabolism is a useful strategy to develop hypotheses on regulatory mechanisms for metabolic systems, as demonstrated by our recent study to predict novel roles of hemoglobin to trigger hypoxia-induced glycolytic activation through multiple enzymes (157). Infrared laser Raman spectrophotometry and related bioimaging devices (IR-RS imaging) are technologies that allow us to examine gas-sensitive structural changes of metal-containing prosthetic groups of enzymes in living cells in culture. Furthermore, two-dimensional mass spectrometry analysis of biologic tissues by means of mass spectrometry imaging (MSI) will make it possible to detect, with high spatial resolution, the abundance of hundreds of molecules that control cell behavior (114, 290, 306). It is our hope that, with the help of these cutting-edge technologies, we will be able to gain new insights into the complexities of gas interactions and to translate experimental work into new therapies to treat human diseases.
Footnotes
Acknowledgments
We thank Mr. Takuya Iwabuchi for preparation of figures and Dr. Yoshinori Yukutake for useful discussion. This work was supported by a Grant-in-Aid for Scientific Research 21500353 (to M. K.) from the Japan Society for the Promotion of Science (JSPS), by a Grant-in-Aid for Creative Scientific Research 17GS0419 (to M.S.) from JSPS, and by Global COE Program for Metabolomics Systems Biology from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to M.S.). M.S. is the leader of ERATO (Exploratory Research for Advanced Technology) Gas Biology Project from the Japan Science and Technology Agency. R.F. is an Assistant Professor supported by Research and Development of the Next-Generation Integrated Simulation of Living Matter, a part of the Development and Use of the Next-Generation Supercomputer Project of MEXT (to M.S.). R.M. B. is an Assistant Professor supported by Global COE Program for Metabolomics Systems Biology from MEXT.
Abbreviations Used
Reviewing Editors: Nader Abraham, Vittorio Calabrese, Enrico Calzia, Carlos Gutierrez-Merino, Hideo Kimura, Giovanni Li Volti, Stefan Ryter, Gregg Semenza, Rui Wang, George Webb, and Matthew Whiteman