
Abstract
The infiltration and persistence of hematopoietic immune cells within the rheumatoid arthritis (RA) joint results in elevated levels of pro-inflammatory cytokines, increased reactive oxygen (ROS) and -nitrogen (RNS) species generation, that feeds a continuous self-perpetuating cycle of inflammation and destruction. Meanwhile, the controlled production of ROS is required for signaling within the normal physiological reaction to perceived “foreign matter” and for effective apoptosis.
This review focuses on the signaling pathways responsible for the induction of the normal immune response and the contribution of ROS to this process. Evidence for defects in the ability of immune cells in RA to regulate the generation of ROS and the consequence for their immune function and for RA progression is considered.
As the hypercellularity of the rheumatoid joint and the associated persistence of hematopoietic cells within the rheumatoid joint are symptomatic of unresponsiveness to apoptotic stimuli, the role of apoptotic signaling proteins (specifically Bcl-2 family members and the tumor suppressor p53) as regulators of ROS generation and apoptosis are considered, evaluating evidence for their aberrant expression and function in RA. We postulate that ROS generation is required for effective therapeutic intervention. Antioxid. Redox Signal. 12, 743–785.
I. Introduction to Rheumatoid Arthritis
A. Histopathology of the rheumatoid joint
The histological features of the inflamed rheumatoid joint shed some light on the key cells and mediators that contribute to localized joint swelling and pain; the most notable features are the proliferation of cells within the membranous capsule that lines any articular joint (25). The synovial membrane is comprised of two or three intimal layers of fibroblast-like and macrophage-like synoviocytes embedded in a dense extracellular matrix. During the progression of RA, the synovial membrane expands and chemokine secretion by resident synoviocytes triggers the recruitment of many immune and inflammatory cells (19, 25, 69). Although the mechanism mediating the extravasation of these cells is far from understood, their accumulation, activation, differentiation, and persistence within the rheumatoid joint are considered to drive the autoimmune process. Recruited inflammatory cells (e.g., neutrophils and monocytes, which differentiate into macrophages, dendritic cells or bone resorbing osteoclasts according to the local cytokine environment) contribute to degradation of cartilage and bone through release of proteolytic enzymes, reactive oxygen species (ROS), and reactive nitrogen species (RNS; 80, 175). Despite the synovial tissue being highly vascularized, the rheumatoid joint is recognized as a site with typical biochemical features of hypoxia-induced oxidative stress; these characteristics are postulated to arise because of increased pressure in the synovial cavity due to inflammatory swelling, reduced capillary density relative to mesenchymal cell proliferation, and an increased metabolic rate (275). Correspondingly, changes in gene expression profile are reported that vary according to the degree of hypoxia (169, 212). There is conjecture over whether ROS are generated from the mitochondria during hypoxia, or whether this remains an artefact of the fluorescent probe detection systems. However, it is clear that on reperfusion ROS are key players in the degradative changes seen in many tissues. Therefore, the repetitive cycles of ischemia and reperfusion (169, 212) are also important contributors to an increased flux of ROS and RNS that are implicated in the pathophysiology of RA.
B. Genetic involvement in disease etiology
The etiology of RA remains unknown, although there is strong evidence for genetic association; despite only 15% concordance rates in monozygotic twins (∼24% of cases), the majority of RA patients carry one of three variant amino acid sequences between residues 70–74 of the beta chain of human leukocyte antigen-DRB1, which is expressed on cells of the immune system, the so-called “shared epitope” (220, 221). The importance of this molecule in discriminating between self and non-self, and therefore in determining whether an immune response will be developed against an autoantigen supports its role in the aetiology of RA. However, this genetic association is not absolute. Individuals with the shared epitope do not always develop RA and this genotype appears to be more important in predicting severity rather than the development of disease (175, 221). It is therefore likely that a combination of genetic and environmental factors is important in the etiological process (221).
C. Environmental influences on disease severity
RA susceptibility may be determined genetically, however, it is most likely that disease onset is dependent on stochastic nongenetic or epigenetic events. Indeed, increased incidence of stochastic events such as modifications to protein and lipid (e.g., by ROS/RNS, accelerated telomere shortening, DNA damage, and somatic mutations) have been observed in RA patients (94, 305). It has been postulated that the presence of a specific genetic variant, the so-called “shared epitope” within the beta chain of human leukocyte antigen-DRB1 (219) increases susceptibility to RA by favoring a pro-oxidative environment, thereby increasing the risk of deleterious stochastic events. Consistent with this model, a gene–environment interaction between the “shared epitope” and smoking (known to enhance oxidative stress) has been reported. The relative risk ratios for development of RA is 7.5 in “shared epitope”-positive smokers; 2.4 in “shared epitope”-negative smokers; and 2.8 in “shared epitope”–positive, never-smokers. The study also identified a gene–dose effect, with a relative risk of 15.7 in smokers carrying two copies of the “shared epitope” (221).
Other examples of environmental agents that may interact with genetic risk factors to predispose to RA include exposure to an infectious event without adequate resolution or a steroid hormone imbalance (154).
D. RA as an inflammatory autoimmune disease
The autoimmune component of RA is typified by the presence of an antigen-driven immune response where autologous antigens may be generated from damaged joint tissues and apoptotic cells in response to joint injury (61, 94, 214). It is not clear whether such damage occurs prior to, or is a consequence of disease. The first identified high-affinity autoantibody, known as rheumatoid factor was discovered by Rose and Waaler in the 1930s and recognizes the Fc portion of immunoglobulin G (IgG) (72, 270).
More recently, anticitrulline autoantibodies, which are generated by post-translational deimination of arginine residues on proteins by peptidyl arginine deiminase (114), have been adopted as diagnostic markers of disease; the most commonly tested antibody is the anti-cyclic citrullinated peptide antibody (anti-CCP). In either case, the antigens are autologous, highly abundant, and therefore able to support increased antigen–antibody complex formation within the synovium (301). This in turn enhances activation of the complement cascade, thus exacerbating inflammation by release of chemotactic stimuli and recruiting further inflammatory cells to the site of immune complex deposition.
The accumulation of oxidized DNA, proteins, and lipids within the inflamed rheumatoid joint provides evidence for the damaging effects of radicals in this pathology. Oxidized biomolecules may arise for any one of at least three reasons including [a] increased rates of oxidized damage; [b] defective degradation of oxidized molecules; or [c] ineffective repair. Oxidized proteins and DNA express neoantigenic determinants that can promote autoantibody production, as reviewed elsewhere, and drive the autoimmune response (61, 94). The normal control of B cell maturation to antibody producing cells should include the elimination of self-reactive autoantibodies and this is considered further in Section IIID.
II. Amino Acid Oxidation Chemistry and Its Relevance to Autoimmune Inflammatory Disease
Oxidation of amino acids within the protein backbone may be either reversible in the case of methionine oxidation to methionine sulfoxide or cysteine oxidation (see Section IIB); or irreversible, for example, in tryptophan oxidation to N-formylkynurenenine or proline oxidation to 4-aminobutyric acid, when ring breakage occurs (93). Oxidative changes to proteins can be induced by a range of reactive metabolites produced during inflammation by phagocytic cells, including hypohalous acids and peroxynitrite; the latter can also induce specific modifications such as amino acid nitration, bromination, or chlorination. Detailed description of the chemistry of amino acid modification by ROS and RNS are reported elsewhere (93). Such damage can affect protein conformation and has been shown to affect the function of many proteins, contributing to loss and gain of function changes, including gain of antigenicity (94).
A. ROS/RNS mediated generation of neoantigens as primers of the immune response in RA
Several autoantibody isotypes (IgM, IgA, and IgG) can be detected against many antigens in the sera and synovial fluids of patients with RA (301). Whereas some of the autoantibodies can bind efficiently to native proteins, their binding affinity is enhanced if the protein is modified or denatured in some way (93). The classicial serological test for RA is based on presence of the autoantibody rheumatoid factor (RF) against self-IgG; it is of interest to note that the assay for RF is based on binding to aggregated IgG. In addition, there is evidence for IgG aggregates in the plasma and synovial fluid (SF) from RA patients, although it is not clear whether these aggregates are immune complexes (72, 247).
Protein oxidation is characterized by protein aggregation, either due to irreversible crosslinking (e.g., between tyrosine residues or between newly formed protein carbonyls and existing amine groups); or reversible crosslinks may be formed through inter-chain disulfide bridge formation (93). While the disposal of oxidized, intracellular protein aggregates is mediated via the proteasome that displays reduced activity with age, the extracellular aggregates are most likely removed by cell-mediated clearance mechanisms (134, 238, 285, see also Section IIIE). Levels of oxidized proteins are elevated in RA, probably arising from increased inflammatory and respiratory burst activity, raising the possibility that protein oxidation may perpetuate if not trigger autoantibody production. In support of this, oxidized IgG has been shown to be a better antigen for RF than native IgG. These concepts have been reviewed extensively elsewhere and therefore are beyond the scope of this article (61, 94). Table 1 summarizes the evidence for oxidized antigens in the etiology of RA. RA is characterized by the presence of circulating autoantibodies against IgG, but there are several other abnormal immune responses observed in the disease. Autoantibody titers against oxidized LDL are elevated in early RA patients compared to healthy controls (61). We have also observed increased levels of autoantibodies towards nitrated LDL in RA patients with cardiovascular complications, where nitrated LDL was more avidly scavenged by macrophages than native or oxidized LDL (95). These data represent an association between protein modification and change of function but do not imply causality; indeed recent studies have shown that myeloperoxidase-catalyzed LDL-phospholipid oxidation can elicit a proinflammatory phenotype on aortic endothelial cells (53). The clearance of oxidized or nitrated LDL via scavenger receptors, such as CD36 present on macrophages, may contribute to the incidence of cardiovascular complications frequently observed in RA patients and reduction of CD36 expression (e.g., by anti-inflammatory drugs) is expected to exert an antiatherogenic effect; it has been shown that CD36 levels are reduced on erythroid-lineage cells by anti-TNF drugs, although whether this is true for leukocytes remains unknown (222). This effect seems at first glance to be counterintuitive, however, extensive studies of atherosclerosis induced in mice that do not express the critical apoliproprotein E on LDL, have shown that vascular disease is prevented in CD36 knockout mice (160).
B. Controlled oxidation/nitration of the amino acid residue, cysteine, can mediate redox signaling
Cysteine is a nonessential amino acid derived from amino acids methionine and serine and is the key amino acid in most redox-mediated cellular reactions (39, 133, 156). The functional importance of cysteine lies in its sulfur-containing functional group thiol, which is also known as sulfhydryl or mercapto (-SH) group. The sulfur atom has an outer valence shell electron configuration of 3s2 3p4, which allows oxidation states ranging between +6 to −2. The sulfur moiety in the cysteine thiol group is fully reduced and under catalysis or in an oxidative environment can undergo a range of oxidative reactions acquiring different oxidation states (Table 2).
G, glutathione.
The pKa value of thiol groups on cysteine is 8.7 (i.e., close to neutral) and this allows it to act as nucleophile, which can be easily oxidized to the thiolate anion. The structural rationale for low pKa and high reactivity of cysteine thiol is clearly an important aspect of the structure/function relationship of any protein (133). The pKa value of the cysteine and the redox potential also determine the distinct reactivities of the CXXC proteins (i.e., sequences of two cysteine residues interrupted by any two other amino acids). Not all thiols are involved in redox-mediated reactions, as most proteins do not react with oxidants under the normally reduced cellular environment. However, the local pKa value of active site thiols is determined by its environment and the proximity to positive charged amino acids such as arginine (133), rendering specific cysteine moieties more or less susceptible to oxidation. Therefore, strong nucleophilicity of thiols is promoted by its local environment, thus providing the specificity for thiol-mediated redox reactions. For example, in all known members of the thioredoxin family, the pKa value of cysteine residues are significantly lower than the pKa of free cysteine; this is true for thioredoxin (Trx) reductase, glutaredoxin (Grx) (thioltransferase), and protein disulfide isomerase. In some Trx-like proteins, where positively- charged residues are not found in the immediate vicinity, the resulting thiolate anions are stabilized by surrounding reducing environment such as the partial positive charge of the helix dipole interactions (156). However, the local pKa value primarily does not exclusively define the strength of thiol reactivity, where other factors such as three-dimensional structure and accessibility of oxidants are also important.
Apart from its role as a constituent amino acid in proteins, cysteine availability is the substrate limiting precursor for glutathione synthesis; glutathione is a tripeptide (γ-glutamylcysteinylglycine) which is the major low molecular weight thiol present in cells (300). Reduced glutathione (GSH) has long been recognized to act as a co-factor in the reduction of ROS and lipid hydroperoxides by glutathione peroxidases and glutathione-S-transferases (GSTs). The oxidized form of GSH (GSSG) is then recycled back to GSH by glutathione reductase and requires the essential co-factor, nicotinamide adenine dinucleotide phosphate (NADPH). Usually, the cellular GSH/GSSG ratio is carefully controlled to maintain a high value (only 1%–5% of cellular GSH is present in the disulfide form) indicative of a reducing environment in the cytoplasm and any change in the redox potential can be used an index of the oxidative stress level in the cell (300). Therefore, the GSH/GSSH couple serves as a redox buffer in cells/plasma that maintains the redox state.
Metabolically active cells or aerobic pathogenic organisms produce reactive oxygen species (ROS) as intermediates or as by-products resulting in an oxidative environment. ROS are also important effectors of innate immune cell functions for bacterial killing (59, 80, 296). Low levels of ROS are quickly detoxified by various antioxidant enzymes and low molecular weight scavengers. The cysteine thiol (-SH) group is particularly sensitive to oxidation reactions. These moieties also can interact with a variety of oxidants, to form in many cases a reversible covalent modification (39, 133, 156). The reversibility of oxidative modifications, which include disulfide bond formation and cysteine sulfenic acid formation, is important for maintaining the reduced cellular environment (300).
Since thiolates are stronger nucleophiles than their protonated counterparts, they easily oxidize to sulfenic acid (Cys-SOH). Once it has been formed, sulfenic acid can either be further oxidized to a higher oxidation state [e.g., sulfinic (Cys-SO2H) or sulfonic (Cys-SO3H) acid] or can be stabilized within the protein environment. Biochemically, formation of the lower oxidation state cysteine acids (sulfenic) can be reversed in the presence of reducing enzymes/agents such as Trx, Grx, or GSH, and the sulfinic acid may be reduced to the sulfenic acid oxidation state under the action of the glutaredoxin system (77, 139, 260, 284).
Equation 1: Reversible oxidation of cysteine thiol.
The cysteine sulfenic acids were initially identified as intermediates of biochemical reactions that are readily reduced to their original thiol but because of their high reactivity, their detection is difficult. In some proteins the formation of higher oxidation states (e.g., sulfonic acids) causes irreversible changes where the protein may lose functionality due to structural alterations (38, 237).
As reviewed in Ref. 237, it is evident that protein sulfenic acids have more biochemical importance than to act as simple intermediates. The nonflavin redox center in NADPH peroxidase contains only one single cysteine residue per subunit and unusually stabilizes the cys-sulfenic acid during catalysis in contrast to NADPH reductase, which forms disulfide bonds (38). This redox-active element, when appropriately stabilized by the respective protein environment, appears to play key roles in both the catalytic and regulatory aspects of oxidative stress.
Thiol groups are capable of forming covalent bonds known as disulfide bonds between nonadjacent cysteine residues (Equation 1). Formation of intraprotein disulfide bonds within the endoplasmic reticulum (ER) is an essential step in protein folding and in defining the tertiary structure. Similarly, many disulfide bonds are important in the quaternary structure of proteins, by the formation of homo- or hetero-multimers (e.g., for antibodies). Therefore, reduction or the disruption of disulfide bonds could dramatically affect proteins by changing their three-dimensional conformation.
Equation 2: Disulfide bond formation.
Within an oxidative environment, the thiol groups of cysteine act as strong nucleophiles, which react with adjacent cysteinyl thiols forming protein disulfide bonds. Disulfides can be formed as a result of thiol disulfide exchange reactions, reaction of thiyl radicals, or by reversible oxidation followed by reduction involving proximal thiol. Protein cysteines can form disulfide bonds within the protein (intraprotein), between proteins or protein moieties (interprotein), between protein and glutathione (mixed disulfides). This is illustrated in Fig. 1.
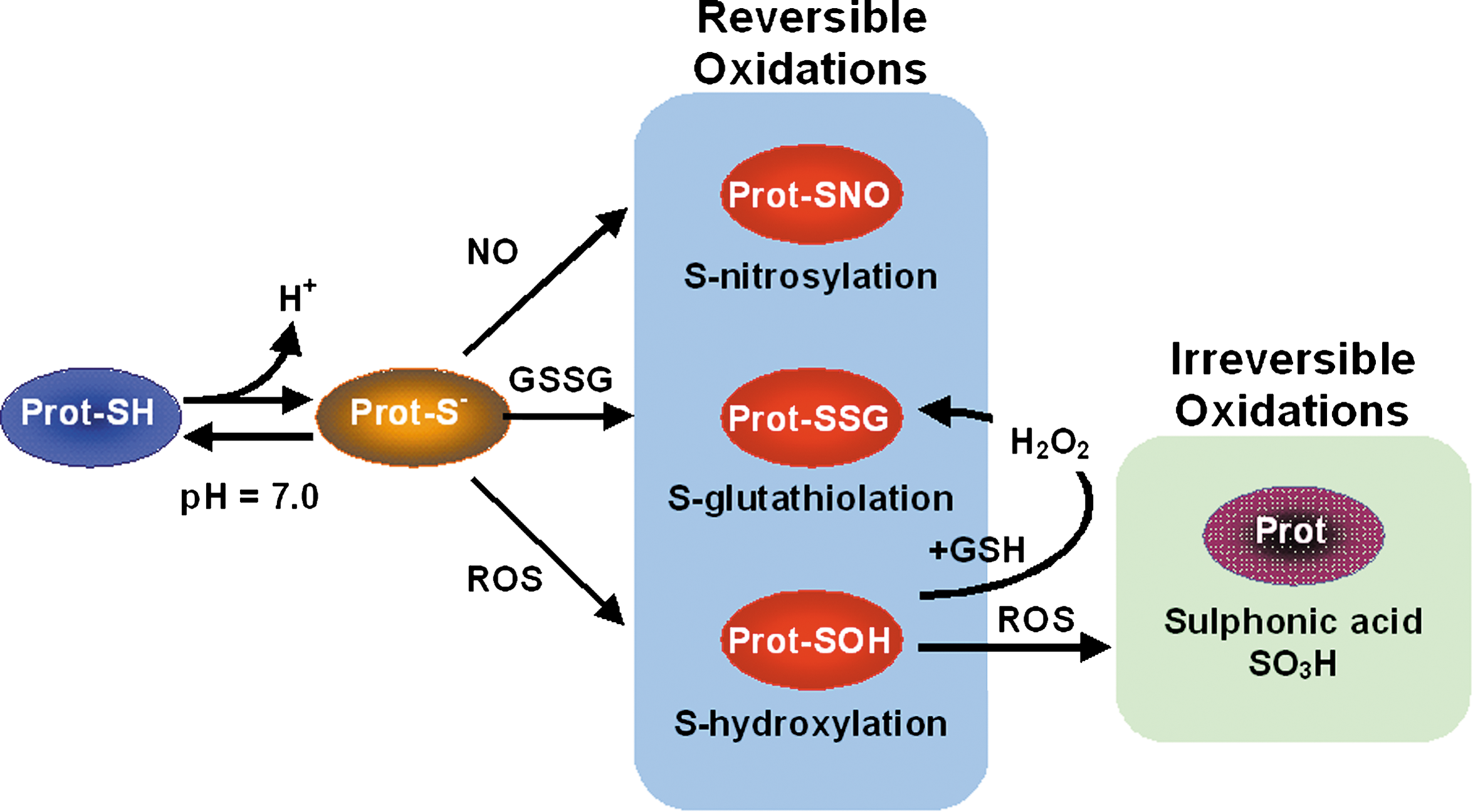
C. Molecular redox sensors
The toxicity of ROS on cellular systems has been investigated for many decades. It is now evident that passing the information of cellular redox state onto biomolecular reactions can switch molecular activity and in turn can influence gene transcription (46, 260). The reaction between oxidants and biomolecules is the foundation for a redox sensing molecular switch. Within this context, oxidation of cysteines acts as the principle mechanism by which signal transducing regulators sense cellular oxidative stress.
In response to many physiological signals (e.g., growth factors, tumor necrosis factor, p53), cells generate intracellular hydrogen peroxide (H2O2), which acts as a second messenger (52, 132, 148). H2O2 generates an oxidative environment, where most protein thiols are either oxidized to form disulfide bonds or cys-sulfenic acids (38). Most of the oxidized forms of mixed disulfides and sulfenic acids are recovered by one of several thiol/disulfide systems; GSH/GSSH, thioredoxin/thioredoxin reductase or Trx1 (–SH2/–SS–), and cysteine/cystine (300). Cys-sulfenic acids are transferred onto thioredoxin molecules which then condense with the proximal cysteine to form a disulfide; this is in turn reduced by the reducing enzyme, thioredoxin reductase (77, 156)
Dynamic rearrangement of thiol groups and disulfide bonds is responsible for receptor signaling and cellular activation across a range of cellular activities, including maturation, proliferation, differentiation, survival, and cell death by apoptosis. Evidence (264) suggests that the cellular redox state is involved in regulating protein tyrosine phosphatase (PTP) activity through the reversible oxidation of the catalytic cysteine to sulfenic acid via oxidized glutathione. This implies that receptor activation can inhibit PTP. H2O2 also induces activation of protein kinases in vitro, although the mechanism for some kinases appears to be similar to that of phosphatase inactivation (i.e., cysteine oxidation) and whether such reactions can occur at physiologically significant rates remains to be determined. Instead, the oxidation of lipids to produce longer-lived oxidizing species (93) may provide a mechanism for signaling via oxidation of specific thiol moieties on kinases.
D. Redox signaling to transcription factor activation: Evidence in RA
The earliest redox sensitive transcription factors that were identified over 15 years ago were activator protein 1 (AP1) and nuclear factor kappa B, NF-κB. These transcription factors have been shown to play important roles in the expression of many genes under pro-oxidant conditions both within cells of the immune system [e.g., following cytokine activation (NF-κB)] and in a variety of cell types under stress [e.g., UV light treatment (AP1)]. Despite apparent similarities in mechanisms of activation, both transcription factors have been shown to be subject to modulation by thioredoxin in opposing ways; while AP1 is activated by thioredoxin resulting in increased expression of its component genes fos and jun, NF-κB is inhibited by thioredoxin (260).
Considering the activation of the NF-kB and the associated ROS-dependent signaling events, generally it is understood that oxidants can activate the p38 mitogen activated protein kinase (MAPK) cascade. In a nonactivated state, NF-κB is maintained in the cytoplasm by conjugation to its inhibitor molecule, inhibitor kappa B (I-κB), and activation is controlled by redox-modulated phosphorylation. One oxidant which can elicit the MAPK cascade in vitro is H2O2, although it is likely that this effect is indirect through changes to intracellular GSH levels in favor of oxidized glutathione or lipid peroxides. The activated MAPK cascade catalyzes the phosphorylation and therefore activity of one of two serine inhibitor kappa kinases (IKK), IKK1 or IKK2 (17, 85, 132, 286). In turn, IKK1 and/or IKK2 trigger the phosphorylation of one of seven members of the IkB family. Phosphorylated IkB then dissociates from NF-κB and is targeted by the ubiquitin-dependent proteasomal system for degradation. The nuclear localization signal, previously masked by IkB, promotes the migration of released NF-κB (a heterodimer of p50 and p65 in the canonical NF-κB pathway) to the nucleus where it effects gene expression (see Fig. 9). This process specifically requires a reducing environment and the reduction of oxidized cysteine residues in the nucleus is mediated by thioredoxin.
In an attempt to mimic the chronic oxidative, inflammatory scenario that is prevalent in the rheumatoid joint, repeated exposure of fibroblasts in culture to low concentrations of H2O2 has been reported to promote cell survival in a p38 MAPK- and NF-κB-dependent manner (31). The central role of NF-κB in inflammatory arthritis has been illustrated in recent experiments in which NF-κB inhibitors were targeted to dendritic cells to prevent their differentiation. When these cells were subsequently exposed to arthritogenic antigens, the autoimmune response was suppressed in an established mouse (C57BL/6) model of arthritis (190), indicating that the NF-κB inhibition drives a phenotype switch from a specific effector phenotype to a Th1 phenotype (183, 190). Further evidence for the role of NF-κB in the pathogenesis of RA comes from immunohistochemical analysis of biopsied human synovial membrane, where the presence of nuclear-localized NF-κB has been detected at higher levels (7).
There are many genes under the control of NF-κB that are central to RA pathogenesis, including proinflammatory cytokines, chemokines, and cyclooxygenase-2, which promote osteoclast differentiation and resistance to apoptosis; this evidence suggests that NF-κB is an efficient and feasible therapeutic target for RA, either directly or indirectly [e.g., via glucocorticoid receptor competition for nuclear activation of transcription (149, 201)]. The NF-κB response is also negatively regulated by IkB family expression; some IkB proteins are targets of NF-κB itself and will inhibit NF-κB in a negative feedback loop (296). NF-κB rapidly upregulates I-kBα, as it is has 11 NF-κB promotor sites upstream of the gene and therefore signaling is rapidly switched off in IkBα-dependent cells. In contrast, cells that show dependence on IkBβ for inhibition show prolonged NF-κB signaling as expression of IkBβ is not regulated by NF-κB and has slower induction kinetics.
Transcription factors rarely exert their effects in isolation; the peroxisome proliferator-activated receptor-α ligands inhibit IL-1-induced production of IL-6 by negatively interfering with NF-κB transcriptional activity, probably by increasing expression of IkB. Additionally, peroxisome proliferator-activated receptor (PPAR)-γ ligands also inhibit disease progression of inflammatory diseases, including RA (147, 251). Correspondingly, novel PPAR agonists are proving efficacious in reducing the severity of disease in animal arthritis models (279).
Taken together, these findings support the hypothesis that aberrant control of intracellular ROS generation may contribute to cellular dysfunction through activation of the p38 MAPK and NF-κB pathways.
III. ROS Regulate the Function of Hematopoietic Cells: Relevance to RA
In the inflamed rheumatoid joint, recruited immune cells (T and B cells) survive and persist through support from cytokines, chemokines, angiogenic factors, resident synoviocytes, antigen-presenting cells, and the extracellular matrix (19). On presentation of antigen by macrophages or dendritic cells, the naïve CD4 T cell population differentiates under control of the local cytokine network to one of at least four subtypes (316) (Fig. 2). In a healthy individual, the regulatory T cell network (Treg) inhibits the proliferation and cytokine production of other T cells, including autoreactive T cells, thus supporting the tolerance of self. However, there is growing support for the hypothesis that Treg cell-mediated suppression is defective in RA. This supports the expansion of TH1 and TH17 populations that are implicated in RA; these T cell populations support the recruitment and activation of inflammatory cells and the maturation of B cells to produce autoantibodies (Fig. 2) (225). It is increasingly recognized that ROS have a role to play in signaling for survival and proliferation of recruited hematopoietic cells and that the disease itself is not limited to the joint. A significant cause of morbidity and mortality in RA patients (e.g., the onset of cardiovascular disease) arises from the presence of systemic inflammatory vascular disease (152).
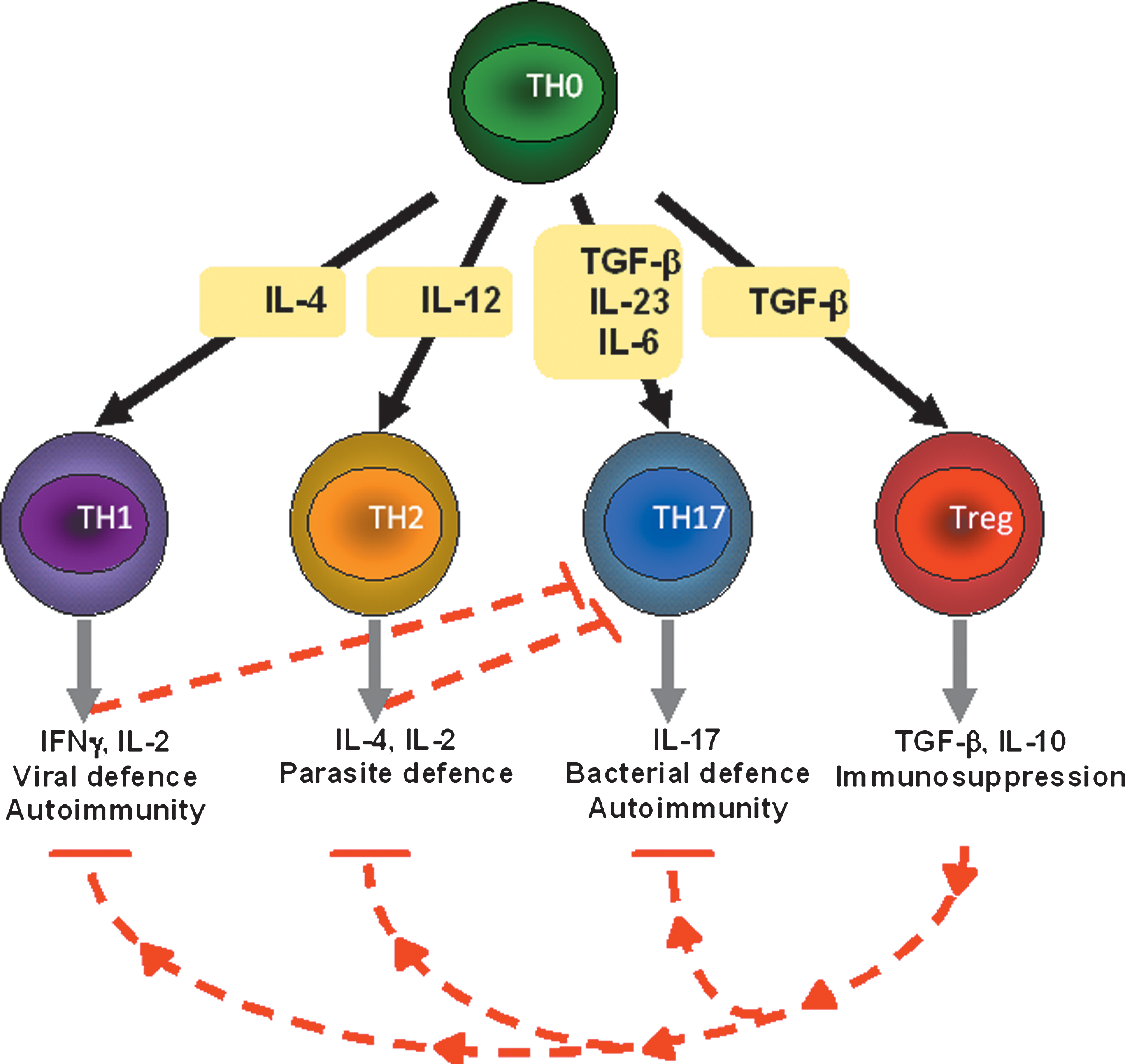
The following sections discuss the evidence for ROS/RNS as modulators of hematopoietic function and the indications for defective redox signaling in RA pathogenesis that may contribute to the development and persistence of disease.
A. Involvement of ROS in antigen processing by antigen-presenting cells
Processing of the 30–100,000 naturally occurring human protein isoforms by proteolytic apparatus within the endosomes of antigen-presenting cells such as macrophages and dendritic cells is estimated to yield in the order of 30 million self-peptides, however, there is some preference over peptide usage. As a consequence, a maximum of 10,000 self peptides may be presented. The effect of oxidative modifications to antigens on their processing and presentation has been investigated only to a limited extent and the emerging data are mixed; some groups report increased presentation of modified or oxidized protein antigens, and others report less favored usage of these epitopes (114, 224, 278). It is likely that the degree of oxidative modification may govern the likelihood of antigen processing and presentation, as reported for the proteolytic degradation by the immunoproteasome (278).
Putative autoantigens may be directly presented to T cells by B cells (outside of the thymus), but many antigens are processed by antigen-presenting cells, principally dendritic cells (Fig. 3). Recent studies that have focused on the potential for ROS to modulate the activity of antigen-presenting cells have reported conflicting data. Tse et al. (282) have reported that modulation of the redox balance with a catalytic antioxidant effectively inhibited antigen-driven T cell responsiveness by diminishing intracellular ROS production in antigen-presenting cells. This resulted in a decrease in interferon gamma (IFN-γ) production by CD4+ T cells, which is required for TH2 suppression, macrophage activation and promotion of leukocyte recruitment to inflamatory sites, and a subsequent impairment of immune effector function. These observations support the earlier work of Matsue et al. (193), who demonstrated that antigen-specific bi-directional communication could be blocked by the antioxidant ebselen. Similarly, Gong et al. showed that the antioxidant molecules, rutin and N-acetyl cysteine, were able to inhibit antigen-presenting cell activity in the absence of any effect of antioxidants on T cell responses (89). Endosomal NADPH oxidase (NOX-2) activity is also critical for effective antigen processing by antigen-presenting cells, where associated acidification of endosomes prevents complete proteasomal degradation of peptides (131). Correspondingly, patients with chronic granulomatous disease have an impaired antigen presentation function, possibly due to excess degradation of antigen in endosomes or to impaired cross-talk between antigen-presenting cells and T cells at the immunological synapse. Conversely, studies utilizing anti-B7 antibodies to block intercellular signaling between T cells and antigen-presenting cells via the major histocompatibility complex (MHC) and accessory molecules showed an enhancement of ROS production, which was associated with the triggering of the innate immune response (149). Additionally, Khan et al. (148) have shown that macrophage-derived ROS production induced by infection with Mycobacterium tuberculosum, prevents HLA expression and the surface expression of processed antigens by antigen-presenting cells, and they suggest that this is a result of inhibiting the nuclear translocation of the transcription factor c-rel. Infection with M. tuberculosum also caused the downregulation of the proinflammatory cytokine, IL-12. Furthermore, these authors showed that catalase pre-treatment prevents the downregulation of the stimulatory cytokine, IL-12, and also enhances antigen processing (148) and so in this latter scenario, ROS production appears to prevent effective antigen processing and immune response. Clearly impeding the cross-talk between T cells and antigen-presenting cells via the MHC and accessory molecules represents a promising avenue for preventing autoimmunity. However, the role that ROS play in this mechanism is currently considered contradictory (888, 89, 148, 281). Further work is required to gain a better understanding of ROS/RNS in the control of antigen processing in RA. An even greater challenge lies in manipulating the immune system to restore the ability to discriminate self from non-self and whether this can be achieved through interfering with nitric oxide signaling that is induced following activation of the rheumatoid shared epitope remains to be established (see Section IIID; 174, 175, 220).
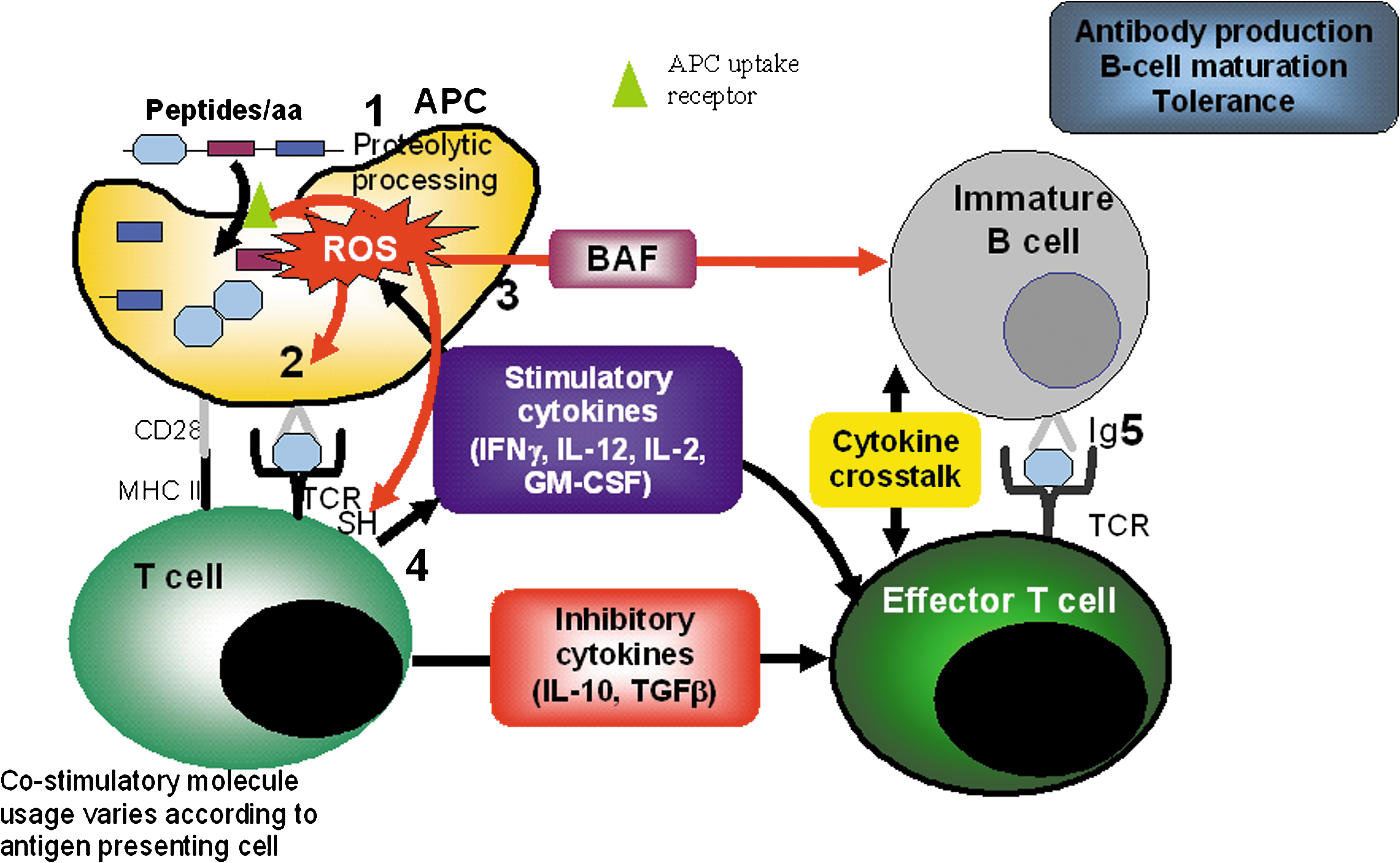
B. T cell activation pathways and ROS signaling
The T cell receptor (TCR) present on the surface of T cells is a positively charged heterodimer (normally comprising alpha and beta chains) that can recruit accessory molecules. Activation of the receptor typically causes crosslinking of adjacent chains. The cytoplasmic tails of T cell alpha and beta chains are considered too short to mediate intracellular signaling directly and instead associate with accessory molecules such as CD3 and zeta chains. The TCR, in association with CD4 or CD8, respectively, are responsible for recognition of antigen presented in association with human leukocyte antigen II or I molecules, via the variable region of the beta chain (296). TCR molecules are suggested to prolong the engagement between presented antigen; formation of the TCR complex, comprising of the crosslinked TCR chains by antigen, triggers the subsequent recruitment of essential tyrosine kinase intracellular components (289). Several intracellular signaling cascades may operate in parallel and cross-talk to each other, ensuring efficient and effective response to antigen including Ca2+, phosphoinositides, kinases, and ROS; thus, the engagement of the TCR and accessory molecules is associated with a rapid induction of ROS production by a T cell NADPH oxidase isoform (21, 273). However, it is important to note that the presence and importance of T cell NADPH oxidase in ROS generation during signaling is far from clear, and mitochondrial ROS production (116) or ROS generated from bystander cells such as macrophages or neutrophils may be more significant modulators in intracellular ROS in T cells (122). In addition, exposure to oxygen during reperfusion of the ischemic joint is proposed to contribute to ROS production during active rheumatoid inflammation, probably mediated via the action of xanthine oxidase which has been localized within the rheumatoid joint (275).
One of the earliest reports linking a redox mechanism to dysregulated T cell activity came from the work of Nakashima et al. (210). These workers exposed murine thymocytes or spleen cells to a thiol reactive agent, HgCl2, which is known to induce autoimmune proliferative disorders. Bivalent mercury was able to stimulate the crosslinking of transmembrane CD4, CD3, and CD45 and glycosylphosphatidylinositol-activated Thy1 leading to intracellular aggregation and activation of intracellular T cell proteins. The magnitude of TCR triggering by mercury was in the order of 10 times greater than with conventional mitogen activation or antibody-induced crosslinking of surface receptors. The assimilated data from the study showed that T cell receptor dysregulation by mercury causes an increase in IL-2 production and prolonged cellular proliferation and survival, which are typical consequences of efficient receptor-mediated intracellular signaling processes involving conventional ligand-induced dimerization. Post-hypoxic re-oxygenation may also contribute to enhanced T cell survival through altered gene expression. Re-oxygenation causes the degradation of Von Hippel Landau protein which normally stabilizes the hypoxia-induced transcription factor, HIF-1, during low oxygen tension. HIF-1α is enhanced in the nucleus of synovial cells (186) where it supports the survival of antigen receptor driven activated T cells during hypoxia.
Studies using mice, which are genetically deficient in NADPH oxidase component p47 phox and therefore respiratory burst function, have confirmed that p47 phox deficient T cells are refractory to proliferation and activation-induced death (123, 217). Moreover, the refractory nature of p47 phox-deficient T cells to stimulation can be reversed by increasing the extent of cell surface protein oxidation. Interestingly, the loss of extracellular thiols was observed in the absence of intracellular changes in redox state (83). An important facet to this study is that changes to T cell behavior in the absence of p47 phox occur in the thymus. Lack of ROS production by macrophages at the immunological synapse during antigen presentation results in fewer thiols being oxidized on the surface of T cells. Transplantation of T cells from the thymus of p47 phox deficient mice was able to affect an increase in susceptibility to RA, indicating that the effect of p47 deficiency was not local to the joint.
In contrast, others have shown that downstream activation of intracellular signaling cascades may be impaired by oxidation (97, 98, 194, 195). In this way, compartmentalization of ROS production either within the cell or in specific tissues and the independent modulation of ROS levels in each compartment is likely to be critical in maintaining normal T cell responsiveness.
A central protein in TCR signaling is the Linker for Activated T cells or LAT protein, which is anchored into the membrane through a glycolipid tail. LAT normally undergoes rapid tyrosine phosphorylation by protein tyrosine kinases, but modulation of intracellular redox state by buthionine sulfoximine with concomitant depletion of GSH alters the cellular localization of LAT. The membrane-displaced protein shows altered conformation and function (83, 99). However, after a cysteine to serine mutation in LAT at positions C9, C26, or C29 (which are located either in, or proximal to, the transmembrane alpha helix) LAT activity is maintained in the presence of oxidative stress, again supporting the hypothesis that oxidation-induced changes in conformation of specific cysteine residues may contribute to loss of activity (97). Other reports have described that oxidation of the C-terminal domain of TCR zeta and the membrane proximal domain of p56 lck may also contribute to loss of TCR function (29). Alterations in T cell redox state and phosphorylation are common observations in the elderly and associate with increased risk of autoimmunity (236).
Using activated neutrophils or diethylmaleimide to induce oxidative stress, Kanner et al. have suggested that oxidation of phospholipase C-γ1 inhibits its activation following TCR engagement and may contribute to T cell hyporesponsiveness (144). Other pathways, whose activity is dependent on proteins with redox-sensitive thiol moieties such as caspases, remain functionally active (144). Specificity of thiol oxidation is likely to reflect the different local redox environments associated with particular protein domains, rendering some susceptible, and others resistant to oxidation in the presence of oxidants (46). Extracellular ROS production may influence the oxidation state of surface proteins, but endogenous sources of oxidants (e.g., mitochondria) or endosomal ROS production may play an important role in the regulation of intracellular signaling. Remans et al. (244 –246) have undertaken studies to determine how intracellular pathways that are associated with ROS production may be regulated. They identified that a failure of RapI to regulate the RaI-mediated increase in ROS production, which lies downstream from Ras activation, may contribute to a hyporesponsive T cell phenotype; using a Jurkat T cell line transformed to overexpress ras constructs, this group determined that following anti-CD3 driven activation of T cells, intracellular ROS levels increased, as demonstrated by dichlorofluorescein diacetate (DCF) fluorescence. These cells were not sensitive to diphenylene iodinium (DPI) nor were these effects associated with changes in mitochondrial integrity. In the absence of RapI, which acts as a suppressor of ras activation by sequestration of ras effector targets such as RaI into an inactive complex, chronic intracellular ROS production ensues and diminishes TCR dependent ERK phosphorylation (245). Chronic elevation of intracellular ROS appears to favor the switch in intracellular signaling from a p38 MAPK mitogenic response to a refractory or potentially apoptotic ERK-dependent response.
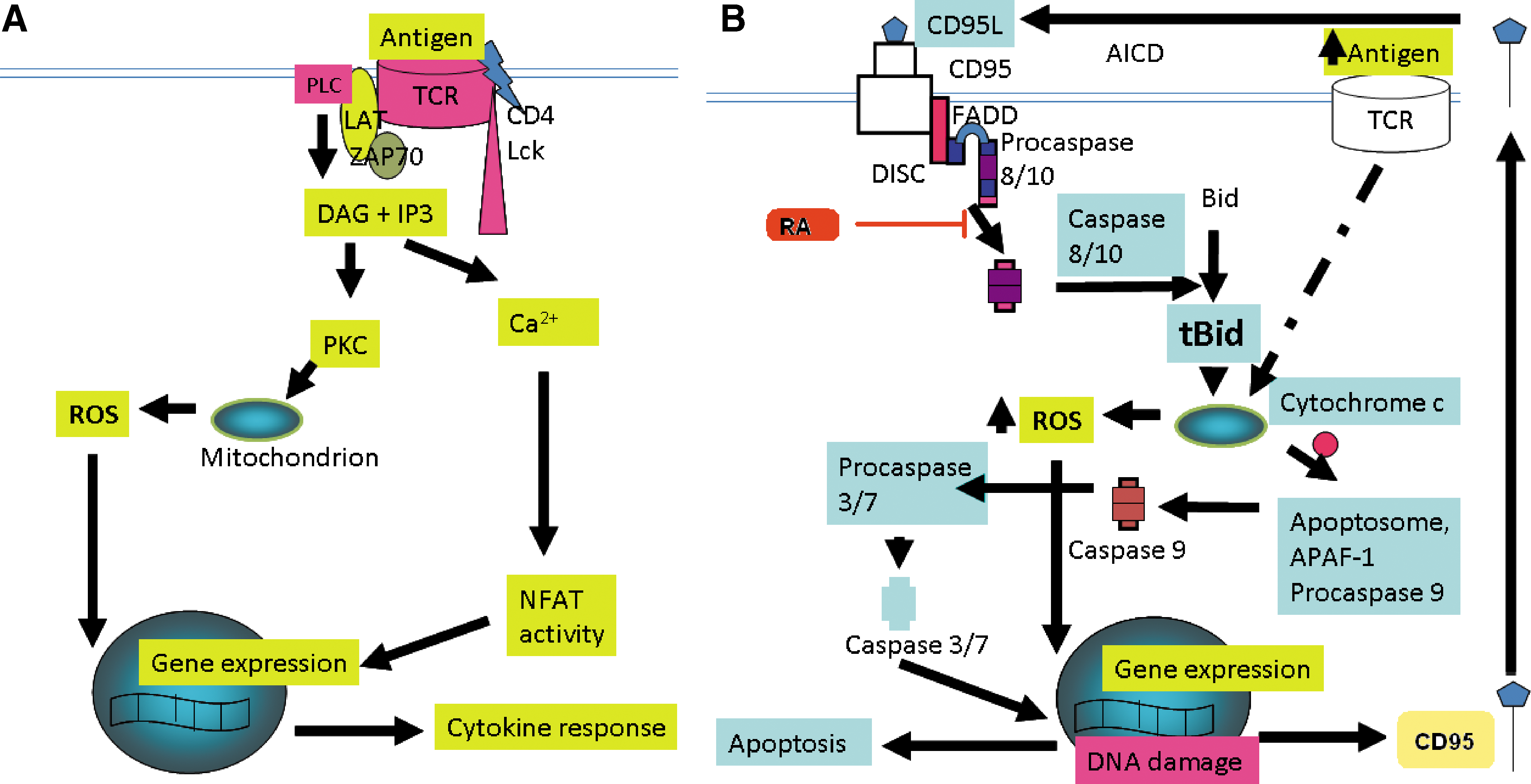
The picture of ROS involvement in hematopoietic cell death is a complex one and is likely to be dependent on ROS dose and influenced by cell maturity. On the one hand, the inhibition of activated/memory T cell death has been reported in the presence of hydrogen peroxide (187), however, recent evidence suggests that the generation of ROS is required for the induction of activation-induced cell death of T cells (4, 88, 112, 113, 129, 143). Activation-induced cell death is one of two processes that are required to “shut-down” the immune response and consequently, protect against the development of autoimmunity (21). Activation-induced cell death describes the induction of apoptosis in pre-activated T cells through the re-stimulation of the TCR during the contraction phase of the immune response and can proceed through a death receptor dependent (intrinsic) or independent (extrinsic) apoptotic pathway. Essential to activation-induced cell death via the intrinsic apoptosis pathway is the upregulated expression of the death receptor ligand CD95, otherwise known as Fas ligand (CD95L or FasL). Following TCR engagement, zeta chain-associated protein kinase 70 (ZAP70) is activated and phosphorylates LAT (32, 70). Phospholipase C (PLC)γ1 is subsequently recruited to LAT and mediates IP3 and DAG generation that respectively increase Ca2+ flux and protein kinase C (PKC) activity. The mitochondrial translocation of a particular isoform of PKC, PCKθ, results in mitochondrial ROS generation in T cells. Therefore, the proximal TCR machinery is essential for mitochondrial ROS generation and cell death subsequently proceeds through the redox dependent upregulation of FasL expression. Importantly, activation-induced ROS generation is diminished in respiratory incompetent (mitochondria deficient) Jurkat T cells and as a result, apoptosis inhibited (143). NADPH oxidase activity may also contribute to the ROS generated in response to TCR stimulation (129), although more recent data suggest that NADPH oxidase-derived ROS is dependent upon ROS generated at complex I of the mitochondrial electron transport chain (138). ROS-driven expression of FasL in activation-induced cell death is indirect, and is dependent upon the activation of redox sensitive transcription factors including NF-κB (4).
TH2 cell apoptosis occurs independently of the extrinsic apoptotic pathway and is dependent upon the serine protease granzyme B (52). Similarly, activated T cell autonomous death is an apoptotic process that also proceeds independently of death receptors and is also referred to as “death by neglect” or “passive death”, occurring through cytokine withdrawal (107). Activated T cell autonomous death requires the pro-apoptotic Bcl-2 family member Bim (107), and may require additional members from this family of apoptosis regulators (64, 135, 270). The role of Bcl-2 family members in RA and ROS generation are discussed later in this review and are important in the context of both activation-induced cell death and activated T cell autonomous death. The interrelationships between ROS and T cell death are illustrated in Fig. 4.
An inability in rheumatoid T cells to upregulate ROS in response to activation as described here or in animal models (83, 232) will therefore have a profound consequence for the efficiency of activation-induced cell death and activated T cell autonomous death. Correspondingly, a failure to eliminate activated T cells not only results in the hypercellularity associated with the rheumatoid joint but also, their persistence will support pro-inflammatory effector functions. This presents an apparent paradox; on the one hand, ROS/RNS appear to promote inflammation and autoimmunity by inducing post-translational and antigenic changes in extracellular proteins that may drive autoimmune responses and by upregulating pro-inflammatory genes, but on the other hand, ROS are required to repress the T cell response possibly by inducing apoptosis.
This paradox can be addressed by considering the organelle, the subcellular compartments, the cell type, and endogenous antioxidant defenses, the kinetics and the extent of ROS/RNS production. Rapid fluxes of intracellular ROS/RNS production appear important for mitogenesis and signaling for proinflammatory events. Any extracellular ROS/RNS leakage may promote protein damage and autoantigen formation. As signaling responses, such fluxes should be rapidly controlled and homeostasis restored by removal of modified molecules.
In contrast, the balance of production of ROS/RNS during antigen presentation may be important for deletion of autoreactive T cell clones and if there is insufficient signal, either in duration or in the extent of redox shift, there may be a failure of apoptosis and therefore self-reactive clones may survive.
C. ROS and redox signaling in RA T cells
The hyporesponsiveness of T cells in the synovial joint, evidenced by a failure to proliferate or to undergo apoptosis in situ, correlates strongly with levels of oxidative stress and is mimicked in vitro by depletion of intracellular glutathione (97, 98). Cellular GSH levels are reduced in synovial T cells and responsiveness may be restored by N-acetyl cysteine. Gringhuis et al. have proposed that a redox imbalance in synovial T cells leads to hyporesponsiveness, where oxidation of C117 in LAT leads to its displacement from the membrane and failure to be phosphorylated by ZAP70 (97). In association with a failure to respond to receptor-mediated activation, T cells from the synovium of rheumatoid patients have been shown to produce constitutively increased levels of ROS compared to peripheral blood T cells, which only exhibit a small transient increase in ROS on stimulation with anti-CD3 (98). Remans et al. further demonstrated that Ras and Rap1 signaling are dysregulated in rheumatoid synovial T cells, with constitutive activation of Ras and inactivation of Rap1 (244). While the inhibition of Ras signaling with a dominant negative Ras peptide blocks ROS production by synovial T cells compared with peripheral T cells, constitutive ras activation is associated with defective TCR mediated ERK activation and little production of cytokines IL-2, IL-4, IFNγ, transforming growth factor (TGFβ) or tumour necrosis factor (TNFα) in vitro or in situ. Whether such T cells drive autoimmune responses in RA is therefore questionable (73).
Attention over the past 10 years has moved towards considering the roles that other resident cells of the autoimmune synovium play in supporting cellular persistence in RA (19); enhanced survival of T cells in the rheumatoid synovium has been linked to the production of survival factors from synoviocytes (69). Many growth factors can exert redox modulatory effects and this may contribute to the pro-survival support mechanisms for T cells in the synovium. Alterations in nitric oxide metabolism may also play a role in T cell survival; nitric oxide is a potent activator of apoptosis which can be attenuated by TNFα secreted from fibroblasts (35). These findings contrast with the work of Migita et al. (199) who showed that nitric oxide protects synovial cells from Fas-induced apoptosis by inhibiting caspase 3 activity. Using SNAP as an NO-donor, NO is able to modify the redox active thiol in caspase 3 via S-nitrosylation (10), resulting in apoptosis inhibition, even in the event of mitochondrial cytochrome c and pro-caspase 8 activation. It is likely that both responses to NO may occur during autoimmune diseases with the outcome being dependent on both the levels of NO produced and the responding cell type.
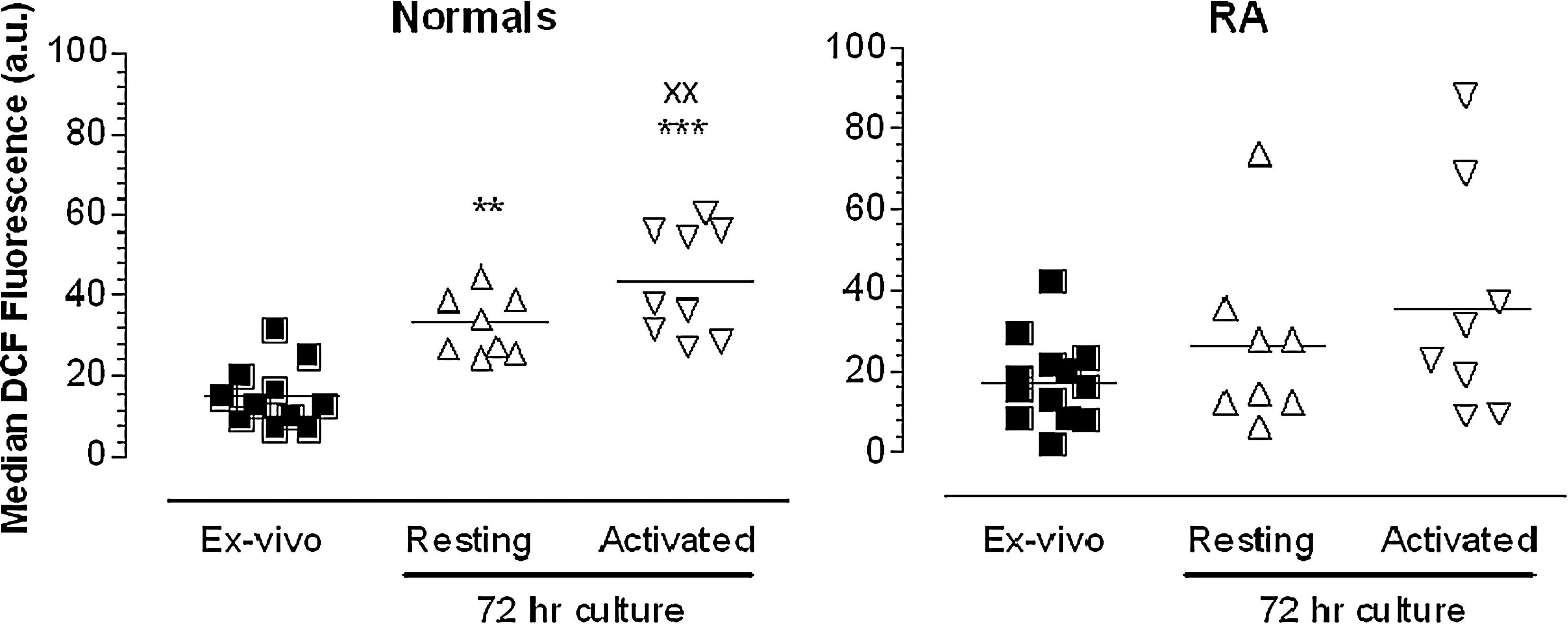
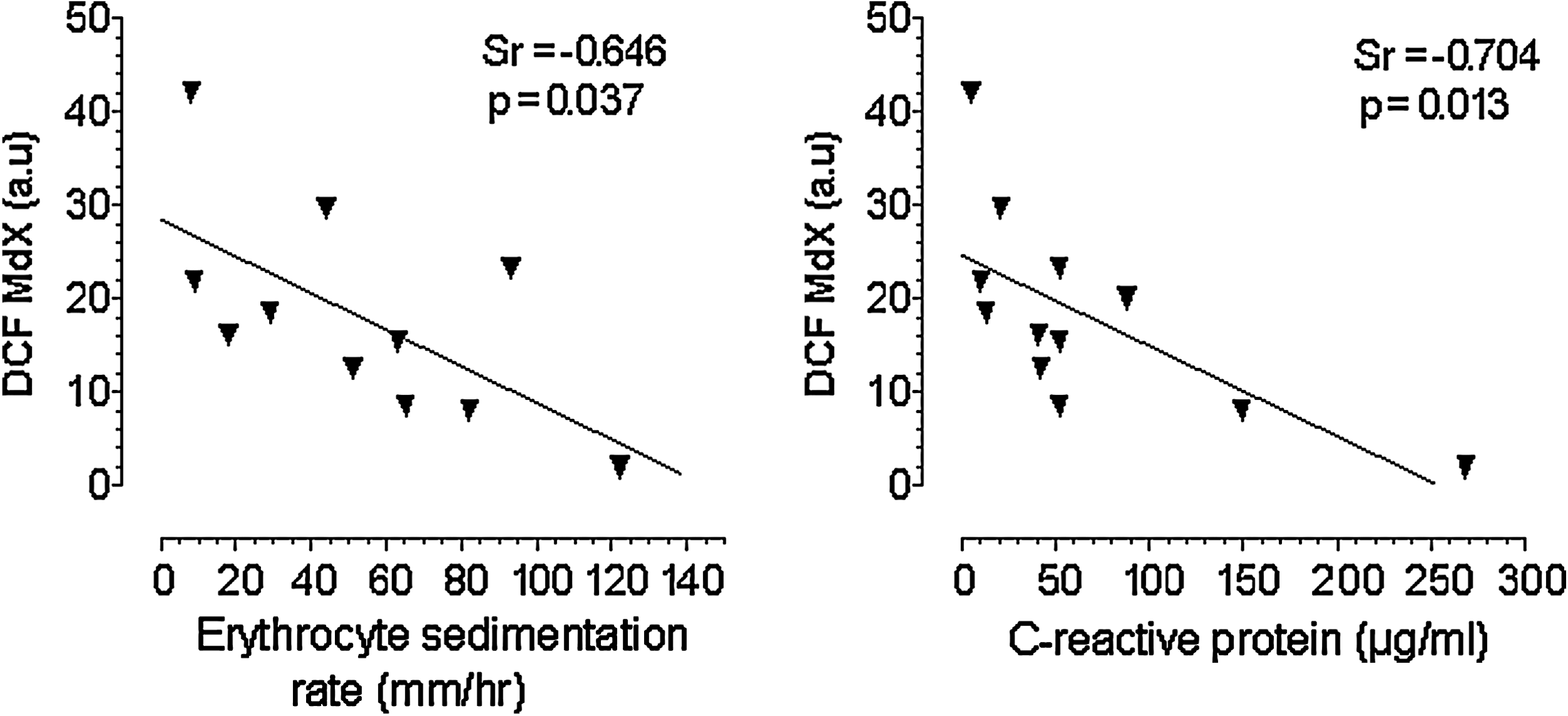
Our studies on peripheral blood T cells isolated by negative depletion have shown, in contrast to T cells from normal subjects, that rheumatoid T cells do not show an elevation in cytosolic ROS when cultured ex vivo in the presence or absence of a mitogenic stimulus (Fig. 5) (223). Moreover, the levels of intracellular ROS were inversely proportional to disease activity; RA peripheral blood T cell DCF fluorescence is inversely proportional to disease activity measured by C-reactive protein (CRP) levels and erythrocyte sedimentation rate (ESR) (Fig. 6). In the light of the capacity for surviving cells to elicit an adaptive response under oxidative stress conditions, we have postulated that peripheral RA T cells show altered redox biochemistry either because they have been preconditioned to ROS during inflammation increasing protein glutathiolation, or by increased RNS which contributes to nitrosation (96); both are critical regulatory processes leading to gain of function (via ras; 39) or loss of function (via p53, AP-1 or NF-κB; 161, 197). As T cell ROS production is important for TCR signaling, these data support the hypothesis that failure to elicit an effective intracellular ROS flux may lead to a refractory T cell phenotype. ROS are important priming molecules within the immune system, particularly for T cells. In this regard, a reduction in intracellular ROS levels is known to impair T cell function; mice and rats with mutant Ncf1 (neutrophil cytosolic factor 1; also known as p47phox) which lack an active p47phox component of the NADPH oxidase complex, show a reduced capacity to generate ROS and this is accompanied by an increased susceptibility to, and severity of, pristane-induced RA (83, 122). Furthermore, with the defective ability to produce ROS, T cells from the Ncf1 rat also possessed enhanced surface levels of reduced thiol groups (-SH). Artificially increasing reduced –SH groups with N-acetyl cysteine or GSH treatment consequently increased T cell activation and proliferation (83) whereas oxidation of surface T cell thiols with GSSG afforded protection. Collectively, these data suggest that loss of intracellular ROS associated with RA, as observed here in human T cells ex vivo or in animal models (83), may promote T cell-mediated autoimmunity.
Investigation of NO production by RA T cells is in its infancy; one recent report used diaminofluorescein (DAF)-2M to determine intracellular NO levels in RA patient's peripheral blood T cells ex vivo. Unstimulated T cells from RA patients were shown to express approximately twofold more NO than healthy donor T cells, derived from e-NOS and/or nNOS but not iNOS and the increased cellular NO levels were associated with increased intracellular calcium concentration (208). It is of interest to note that NO selectively promotes proliferation of the proinflammatory T cell population, TH1 (155). In addition, the treatment of mice with NOS inhibitors reduced the severity of experimentally induced arthritis (155).
Synovial T cells from rheumatoid patients typically exhibit markers of activation on their cell surface (CD45RO++) but possess low proliferative responses that are associated with decreased production of IL-2 and IFNγ. Although the reduced efficiency of TCR/CD3 signaling has been attributed to loss of GSH as considered above, the CD28 response remains unaffected (194). Consequently, T cell-associated CD28 interacts effectively with antigen-presenting cell co-stimulatory molecule, B7, thereby enhancing survival of memory cells in the absence of T cell receptor activation.
Analysis of the transcriptome of rheumatoid synovial cells has revealed the upregulation of thioredoxin reductase 1, the activity of which restores the thioredoxin redox couple (139) and an enzyme which works with the glutaredoxin system to maintain a reduced state for cellular protein thiols. These data suggest that synovial cells are able to adapt and to survive in an oxidizing environment where GSH is lost; the thioredoxin/thioredoxin reductase cycle may be preferentially induced to provide an alternative strategy for restoring oxidized cysteine residues. Further evidence for thioredoxin reductase expression as part of an adaptative response to oxidative stress is provided during models of ischemia/reperfusion where nonadapted tissues express lower levels of thioredoxin reductase than adapted tissues (284). In addition, the thioredoxin system is an important regulator of the apoptotic cascade, where TRX1 acts as a negative regulator of apoptosis signal-regulating kinase 1 (ASK1), thus preventing apoptosis and promoting cell survival (77).
Taken together, T cells survive longer during RA, most likely through refractory apopotic signaling. This leads to the aberrant production of T cell cytokines, which drive B cell maturation and the production of autoantibodies, resulting in increased help for autoantibody production during active RA.
D. Receptor-dependent ROS signaling for B cell survival and function in RA
Over the past few years, interest in a pathogenic role for B lymphocytes in RA has re-emerged (157). A population of refractive B cells has been described within the RA synovium that fails to respond to fibroblasts, activated T cells, or stimulation via the B cell receptor. This abnormal B cell population probably arises due to a blockade at both proximal (e.g., serine phosphorylation, protein translocation between membranes and cytosol) and distal (e.g., protein expression, NF-κB activation, and cell proliferation) events, although the mechanisms underlying this change have not been fully evaluated (247).
B cells are critical in the initiation of peripheral tolerance to antigen and in the humoral immune response by activating T cells, manipulating lymphoid structure, and secreting cytokines. Consequently, chronic autoimmune disorders are also characterized by atypical B cell functions that include impaired receptor editing, defective central tolerance, and abnormal B cell activation and proliferation (105, 312). In RA, B cells accumulate and mature in the inflamed synovium, forming ectopic germinal centres for maturation, antibody production (110), and providing support for the activation of synovial T cells (277). These processes have been studied to a limited extent in the context of redox regulation, and several groups have used N-acetylcysteine to invoke the importance of intracellular ROS without considering effects of potential chemical reduction of extracellular receptors (83, 89, 98, 101, 234, 236).
In common with T cell activation, the activation of B cells involves a number of different surface receptors and intracellular signaling cascades. Antigen-induced signaling via the B cell receptor (BCR) initiates elimination or silencing of self-reactive B cells and additionally activates B cells to recognize foreign antigens (87, 213). Co-stimulatory signals provided by T cells via B cell-expressed CD40 result in B cell activation without the establishment of BCR-induced anergy or apoptosis (9). CD40 and BCR activation both result in the activation or protein kinase B (PKB), ERK, Jun N-terminal kinase (JNK), and p38, ultimately triggering NF-kB activation (3, 8, 87, 239). The collective activation of these signaling cascades are essential for BCR/CD40-driven B cell survival, pro-inflammatory IL-6 secretion, IgG production, B cell activation, proliferation and differentiation (45, 68, 76, 168). BCR and CD40 activation induce ROS generation (66, 167, 168, 266) via NADPH oxidase in B cells (101). The mechanism by which the BCR activates NADPH oxidase to generate ROS is poorly understood. However, using N-acetyl cysteine as a ROS scavenger, it has been suggested to require Rac1, PI3K, and TNF receptor family protein, TRAF-3 (101). Lee et al. have investigated the involvement of ROS in CD40-mediated signaling events in B cells and have shown a requirement for ROS in CD40-mediated proximal and distal events, which were inhibited in the presence of N-acetyl cysteine (168). Following B cell receptor activation, B cell-linker (BLNK; a functional integrated homologue of the T cell LAT and SLP-76) is phosphorylated and recruited to the plasma membrane. However, whilst possessing three cysteine residues in its cytoplasmic tail, BLNK remains remarkably insensitive to oxidation (127). Given the increasing importance ascribed to B cells in peripheral tolerance and autoimmunity, there is a need for closer evaluation of the effects of oxidative stress on B cell signaling.
Induction of B cell apoptosis as a means to prevent the development of autoimmunity requires elements of both the intrinsic and extrinsic cell death pathways. BCR-mediated apoptosis of immature autoreactive B cells, an essential process in the deletion of autoantibody producing clones, is reported to proceed in a Fas-independent but Bcl-2/Bcl-xL-dependent fashion (15, 66). Consequently, Bcl-2/Bcl-xL overexpression diverts autoreactive B cells to undergo receptor editing rather than apoptosis following BCR ligation (66, 164). Additionally, mice genetically deficient in Bim (the pro-apoptotic BH3 only protein) accumulate autoreactive lymphocytes, develop background-specific systemic lupus erythematosus (15), and display a prolonged and more severe inflammatory arthritis when induced by collagen (258).
In contrast to immature B cells, mature B cells are exquisitely sensitive to induction of apoptosis by Fas; CD40 driven B cell activation dramatically upregulates Fas expression and correspondingly, the sensitivity of B cells to Fas-mediated apoptosis (82, 242). Fas-mediated B cell death plays a key role in the maintenance of self tolerance, and failure of this mechanism can lead to autoimmunity (191). Although maintenance of normal Fas signaling is also required for T cell homeostasis, studies in mice where deficiencies in Fas are restricted to the B cell compartment show identical autoantibody production and hence autoimmunity to that of the unrestricted animal (78, 79). However, autoantibody production is lost in MRL-lpr/lpr mice expressing Fas in both the T- and B cell compartments (79), indicating defects in B cell Fas signaling result in the persistence and differentiation of B cells with tolerance against self-antigens. A recent investigation into B cell sensitivity to apoptosis in RA confirmed that RA B cells are resistant to Fas-mediated cell death, and this effect was attributed to overactivity of sphingosine kinase 1 (234).
Investigations into the association of shared epitope-encoding DRB1 alleles, which correlate not only with RA susceptibility in population studies (220) but also with pathogenically diverse autoimmune diseases that do not share any apparent antigen or species specificity, have led to the hypothesis that the shared epitope may have nonantigen-specific effects. In this regard, the shared epitope has been shown to act as an allele-specific signaling ligand that activates an NO-mediated pro-oxidative pathway (174). Furthermore, shared epitope positive B cells produce increased NO compared to shared epitope negative cells irrespective of the presence of RA; shared epitope positive B cells are resistant to cytolytic elimination by T cells and this resistance to death can be overcome by pre-incubating B cells with NOS inhibitors (175).
Collectively these data demonstrate that resistance to B cell activation and consequently apoptosis through inappropriate regulation of ROS/RNS levels may be key contributors to B cell persistence within the RA joint; the maturation of and production of autoantibodies by B cells that subsequently form immune complexes, renders these cells as critical effectors of autoimmunity. Considering the primary role that aberrant B cell function plays in the development of peripheral tolerance and autoimmunity, and the association with oxidative stress, there is a need for closer evaluation of the effects of ROS/RNS on B cell signaling.
E. Receptor-mediated immune complex clearance
RA is not a target-organ restricted autoimmune disease; while much of the damage is localiszed to the joint, it differs from other autoimmune diseases in having systemic manifestations as the target antigens for B cell-derived autoantibodies (e.g., IgG, collagen, LDL) that are not restricted to the joint. Therefore, the “complications” of this systemic autoimmune disease are manifest as a chronic inflammatory disorder which occurs through ineffective clearance and systemic activation of phagocytic cells by autoantigen-autoantibody immune complexes (37) (Fig. 7); this invokes intracellular ROS signaling processes further (238) and contrasts with the phenotype of target organ specific autoimmune diseases, which present as loss of the antigen bearing tissue via apoptosis in a noninflammatory process (37). It is this process which links the adaptive and the innate immune systems; the adaptive immune system has responded to an antigen by supporting specific antibody production; subsequent contact with the antigen enables it to be captured by (a) B cells bearing receptors for antigen which drives further B cell proliferation and further antibody formation; and (b) circulating specific antibodies that are present in interstitial fluid and plasma, resulting in immune complex formation that must be cleared by cells of the innate immune system, the phagocytes.
There are several receptors for human IgG that bind to the Fc region of the gamma chain of IgG (FcγR) that are responsible for triggering the effector functions of inflammatory cells, such as phagocytosis or activation of the respiratory burst via the NADPH oxidase complex (reviewed in Refs. 134 and 230). Within the three families of the FcγR, there are several genetic variants which possess different structures and functional properties. Salmon et al. (255) have demonstrated that crosslinking of FcγR IIIB directly leads to activation of FcγR IIa via the release of ROS and that this amplifies receptor function in neutrophils. Moreover, the magnitude of the influence exerted by FcγR IIIB is allele specific and is probably dependent on chlorinated oxidants. The effect is rapid and is likely to reflect a change in conformation of the antigen binding site rather than a change in expression of receptor mediated via redox sensitive transcription factors such as NF-κB.
In support of this notion, there are several reports that describe RA synovial neutrophil activation by IgG containing immune complexes (57, 58, 248, 249). However, there is a need to re-evaluate these data and to consider whether complement receptors are activated in RA neutrophils. Recent evidence suggests that neutrophils from RA patients have a functional impairment in FcRγIIa- and FcRγIIIb-mediated ROS production compared to age- and sex-matched controls (65). In this latter study, the defect in neutrophils from RA patients to produce ROS upon FcγIIa/FcγIIIb heterologous crosslinking was not a consequence of changes in FcγIIa/FcγIIIb expression, since identical levels of FcγIIa/FcγIIIb were observed in neutrophils from RA patients when compared to controls (65). Collectively these data suggest that neutrophils from RA patients may have impaired amplification of Fcγ receptor function that contribute to neutrophil hyporesponsiveness and further exacerbate the susceptibility and morbidity to infection.
IV. Innate Immune Responses Within the Rheumatoid Joint
The cellular innate immune response is activated locally in the rheumatoid joint. It is mediated principally within the synovium by resident fibroblasts and infiltrating monocytes/macrophages and neutrophils. The chief function of these cells is the removal of pathogens, macromolecular aggregates/particles, and apoptotic cells by phagocytosis. Phagocyte activation elicits the activation of many nonspecific damaging molecules and enzymes and their role in RA is considered below.
A. Respiratory burst and nitric oxide synthase (NOS) activity of phagocytic cells in RA
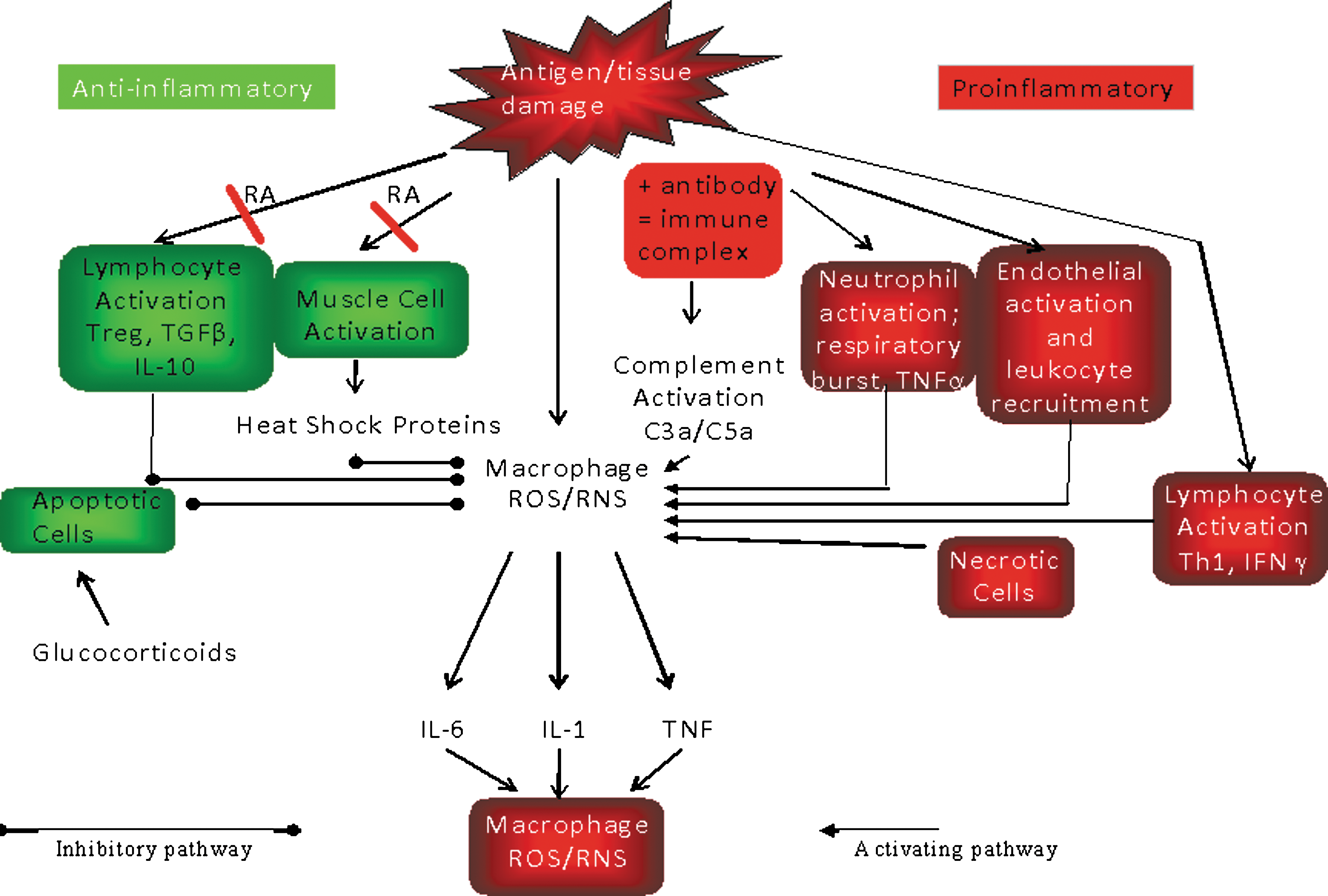
Central to the phagocytic function of macrophages and neutrophils is the generation of the ROS, superoxide, hydrogen peroxide, and hypohalous acids; the RNS, nitric oxide and peroxynitrite; (12) and the action of various proteases that collectively destroy the engulfed material. This process occurs within discrete endocytic vesicles that restrict destructive action of ROS, RNS, hypohalous acids, and proteases from damaging the host cell and associated tissues. Additionally, protease inhibitors and scavenger proteins contained within serum and extracellular matrix act as a second level of protection against tissue damage. However, despite these protective mechanisms, activated phagocytic cells release significant concentrations of ROS/RNS into the extracellular environment, causing further damage and predisposing the RA joint to chronic inflammation and autoantigen production (as reviewed in Refs. 94 and 297).
Within inflammatory joints and the systemic circulation, the expression of mononuclear cell- and endothelial cell-derived cytokines and chemokines, particularly IL-1β, TNFα, IL-6, IL-8, IL-17, IFN-γ, monocyte chemotactic protein 1 and macrophage inflammatory protein 1, are increased (109). Together, these molecules contribute to further leukocyte recruitment into inflammatory sites and perpetuation of inflammatory responses. Further evidence for the likely role of proinflammatory cytokines in the etiology of RA is provided by animal models. For example, the Fas-deficient mouse model of autoimmunity (lpr/lpr) is characterized by a more severe experimental inflammatory arthritis than the wild-type mouse and is associated with elevated levels of IL-1 and CCL2, which supports the increased recruitment of macrophages (22).
Additionally, cells localized to the synovial tissue demonstrate increased expression of cell adhesion molecules, iNOS, anti-apoptotic genes (such as Bcl-2, Bcl-xL and FLIP), and metalloproteinases. The majority of these proinflammatory genes are regulated by the redox sensitive transcription factor, NF-κB and several cytokines, including TNFα and IL-1β, which are initiators of, and regulated by NF-kB in a feed-forward loop (223). The mechanisms of redox control are likely to be cell-type and ligand-dependent, but it is clear that the pathway is dysregulated in RA, either at the stage of activation or through ineffective resolution resulting in a pro-inflammatory/pro-survival phenotype.
B. Requirement for ROS in cytokine processing by the inflammasome
Recently, the requirement for two independent signals in the activation of IL-1β, and its related family member, IL-18, has been recognized; the first drives the NF-kB transcription activation pathway and subsequent translation of the mRNA species yields a precursor form of the protein, pro-IL-1βb. The second signal is required to activate processing of IL-1β by caspase 1 within the NALP-3 containing inflammasome (47). It is unknown what drives these responses in chronic inflammatory conditions although in vitro studies indicate that purinergic receptor-dependent (P2X) pathways are involved and ATP is considered the principal physiological ligand. Recent studies have shown that treatment of macrophages with ATP results in the production of ROS via membrane associated NADPH oxidase. In turn, glutathionylation and inactivation of the tumour suppressor gene PTEN occurs; subsequently, the phosphatidylinositol 3-kinase (PI3K) pathway is activated (47) and in turn downstream activation of protein kinase B (PKB) ensues. The first report on the role of ROS in inflammasome activation and IL-1 processing indicates that the ROS-dependent PI3K pathway is essential for processing. ATP treatment also leads to a rise in intracellular calcium and this likely supports membrane re-modelling via phospholipase C and A2 activation. Indeed, in vitro studies have shown that activation of a calcium-independent PLA2 is observed in macrophages and that this stimulates the co-localization of caspase-1 and pro-IL-1 in secretory lysosomes; the latter supports a model for lysosomal activation of caspase-1, pro-IL-1 cleavage, and its subsequent release (47).
C. Neutrophil survival in RA
Neutrophils are the predominant infiltrating cell observed in the synovial fluid contributing up to 90% of the cells present during the active phase of RA. Neutrophils survive for 12–24 h in tissues, and turnover is in excess of 109 cells per day in a 30 ml joint effusion (59). In vivo depletion of neutrophils in mouse RA models impairs the recruitment of mononuclear cells (monocytes and lymphocytes) to inflammatory sites, probably as a result of lower levels of tissue damage, which in turn reduces the levels of autoantibody in affected joints, thus preventing progression of the disease (262, 302).
Constitutive apoptosis is essential to maintain the equilibrium between neutrophil production and their contribution to the resolution phase of the immune response. However, in synovial neutrophils from RA patients, this process is defective; in tissue neutrophils, NF-kB is activated on isolation without any stimulation in vitro, and is not further induced by addition of TNF-α. In addition, treatment of neutrophils with an NF-kB inhibitor does not produce any morphological apoptotic changes which are typically induced by TNF-α in tissue neutrophils (118). These findings have been further investigated by Wang et al. (292) who showed that type I IFNs, present at high concentrations in the rheumatoid joint, inhibit neutrophil apoptosis in a PI3K-dependent manner. This process requires the activation of protein kinase C-delta and induction of NF-kB-regulated genes. Enhanced survival is also supported in the RA synovium by excessive priming by growth factors (G-CSF) and pro-inflammatory cytokines such as IL-1β, TNF-α, and IFN-γ (142, 298). Data obtained from G-CSF −/− mice provides further supportive evidence for G-CSF and neutrophils as primary instigators of arthritis development; G-CSF −/− mice are resistant to collagen-induced arthritis; in addition, the administration of antibodies against G-CSF to wild-type mice prevented the onset of collagen-induced arthritis (166). G-CSF is an essential regulator of neutrophil production and survival, partly by stabilizing Mcl-1 expression and preventing truncation of Bid and Bax activation (54, 173, 185). In addition, neutrophils stimulated with G-CSF produce and release higher concentrations of the B cell activating factor, BAFF, a key cytokine responsible for B-cell proliferation and maturation (257). Together these observations suggest that increased activation of the redox sensitive transcription factor, NF-κB from cytokine and growth factor signaling pathways, drives neutrophil survival with the likelihood of increased ROS production via NADPH oxidase activation.
In health, activated neutrophils initiate superoxide anion-dependent apoptotic processes via triggering cathepsin D relocalization to activate pro-caspase 8 (41). The apoptotic neutrophils are recognized and consequently phagocytosed by macrophages, thus preventing the release of the cytotoxic contents of neutrophils and promoting resolution of the inflammatory event by the upregulation of the anti-inflammatory cytokine TGF-β. Failure to clear apoptotic neutrophils by phagocytosis results in secondary necrosis of the dying neutrophil, release of cytotoxic molecules, and consequently amplification of inflammation. Indeed, the potentiation of neutrophil apoptosis in vivo by genetic deletion of Foxo3a in a passive mouse model of RA (K/BxN arthritis) inhibits the joint swelling and tissue destruction that is observed in the WT mouse (137). Conversely, the reconstitution of the Foxo3A −/− mouse with wild-type neutrophils restored susceptibility to arthritis in the passive RA K/BxN model, further demonstrating the key role that neutrophil apoptosis plays in development of RA. Pro-inflammatory cytokines and hypoxia both prolong neutrophil survival (18, 51, 104), at least in part through regulating the balance of anti-apoptotic and pro-apoptotic Bcl-2 family members (174, 204, 205). Indeed, the synovial fluids from RA joints with significant synovial membrane hypertrophy are hypoxic and this environment lends itself to enhanced neutrophil survival (169). More recent data from Wong et al. describes how neutrophil survival in the RA joint of patients with established disease may be mediated by increased concentrations of the iron binding protein, lactoferrin (304). The effects of lactoferrin on intracellular redox state remain unknown.
Neutrophils from RA synovial fluids produce elevated levels of ROS when compared to those from the periphery (27, 226) and may persist as a result of the excessive concentrations of pro-inflammatory cytokines and growth factors that override apoptotic-signaling cascades (18, 40, 51, 104, 174, 204). Under normal physiological conditions, such activated neutrophils would initiate apoptotic mechanisms that mediate their clearance through phagocytosis by macrophages. Collectively, these data paint a complex picture of the proinflammatory joint, where hypoxia, cytokines, and growth factors can drive neutrophil survival, and this is associated with the increased production of ROS and the promotion of oxidative damage in the joint, since ROS-dependent apoptosis is overriden. An accurate understanding is required for the role and control of neutrophil-ROS generation in the activation of neutrophil cell death pathways in health and in RA.
D. Nitric oxide synthases and their activity in RA
In contrast to our understanding of the enzymatic production of ROS by the NADPH oxidase in neutrophils, we have only relatively recently understood the role of nitric oxide synthases in the physiological synthesis of nitric oxide. Nitric oxide synthases catalyse the conversion of L-arginine into nitric oxide (NO) and citrulline. Brain nitric oxide synthase was first described in 1991, and in 1992, the first report of increased NO synthesis in synovial fluid was described by Farrell et al. (67). Using the indirect measure of nitrite by the Griess assay, these authors described an increase in serum nitrite concentrations in RA and osteoarthritis when compared to age-matched healthy controls. Moreover, nitrite levels in RA patient sera were always higher than OA sera. Closer examination of the rheumatoid synovial compartment revealed that synovial fluid nitrite levels were significantly higher than serum levels, leading the authors to suggest that the principal site of NO production was in the joint itself. Whilst initial interest was focused on iNOS expression in macrophages as the source for elevated nitrite in synovial fluid, histopathological analysis of RA synovial membranes by McInnes et al. (196) identified that 90% of synovial cells expressing iNOS did not stain for the macrophage marker CD68, with only a minor 10% of iNOS expression being detected in macrophages. Instead, the major cell type expressing NOS in the RA synovium was the fibroblast, again highlighting the importance of stromal cells in the pathology of RA. A follow-up study by Grabowski et al. (90) again confirmed significant increases of iNOS expression in the RA synovium, however, the authors suggested that macrophages were expressing more iNOS than fibroblasts. The reasons for this inconsistency are unclear although they may relate to antibody specificity.
Tissue expression of iNOS is driven by proinflammatory cytokines, particularly IFN-γ, itself under the regulatory control of NF-κB. IFN-γ is produced by TH1 cells which promote macrophage iNOS expression, whereas the TH2 cell population affords inhibitory control of iNOS expression through secretion of IL-4. In addition, IL-18 and its receptor are also expressed at higher levels in RA tissues which are able to elicit IFN-γ and nitric oxide production in synovial tissue (91). In turn, IL-18 secretion has been found to be independently regulated by TNF-α and IL-1β. The importance of IL-18 in the pathogenesis of RA is further supported by administration of the cytokine to a collagen-adjuvant mouse model of RA, where it facilitated the development of an erosive inflammatory arthritis (91), however, this study did not evaluate whether the IL-18 pro-arthritogenic effect was due to enhanced NO production.
The irreversible process of bone erosion is a key feature of RA. Severe erosive damage to joints can only be repaired by joint replacement and this is more effective for larger joints than for the smaller metacarpal and phalangeal joints of the hand. Improved understanding of the mechanisms of joint erosion has come from studies examining mechanistic control of osteoclast activity. These macrophage-like cells normally work in partnership with synthetic osteoblast cells, ensuring continued and balanced bone-turnover and resynthesis. However, in RA, osteoclast activity is enhanced and bone erosion predominates. The major cytokine associated with bone erosion is IL-1β which is also a potent inducer of iNOS in bone cells. To investigate whether iNOS is an important effector of bone erosion, van't Hof et al. (287) examined the effects of iNOS deficiency on bone resorption. These authors describe that iNOS-deficient mice were profoundly defective in osteoclast bone resorptive activity in response to IL-1. However, osteoclasts from these animals showed a normal response to calciotropic hormones, indicating that the loss of iNOS activity only affected the resorptive response to IL-1β, probably via modulating NF-κB activation.
The attribution of causality of disease to NO production has been limited by the poor specificity of available NO inhibitors. To address this issue, Ohtsuka et al. (215) describe the synthesis of a novel imidazole derivative which inhibited NO production by macrophage-like RAW264.7 cells and prevented iNOS dimerisation. When the therapeutic potential of the novel imadazole to inhibit two models of arthritis was examined, the development of erosive diseases in both collagen-induced and adjuvant arthritis were suppressed. This effect was paralleled by a decrease in plasma nitrite levels, implying that strategies to inhibit NO production could be valuable in the treatment of RA. Further investigation of the role of NO production in the joints from animals with inflammatory arthritis has been undertaken using an ESR/spin trapping method (310). NO activity peaked at day 10, and this effect could be inhibited by administering s-2-aminomethylisothiourea, a selective iNOS inhibitor which concomitantly decreased paw swelling in the animals. Histological analysis revealed significantly increased levels of tyrosine nitration in chondrocytes at day 10, an effect that was prevented by iNOS inhibition.
NO synthesis is controlled not only by the level of the iNOS enzyme and its activity but also by the availabaility of its substrate, L-arginine. In addition to being the substrate for NOS isoforms, L-arginine is also used by arginase, a ubiquitous enzyme which is also expressed in synovial membranes. Moreover, its expression can be enhanced by the pro-inflammatory prostaglandin PGE2. Enhanced expression of arginase can prevent detectable NO production by synovial cells even in the presence of the TH1 cytokine, IFNγ (42). In contrast, induction of NO by IFN inhibits arginase activity. This complicated pathway of autoregulation paints an interesting picture with early inflammation being characterized by the induction NO production and tissue damage, and subsequently arginase activity increasing as NO levels reduce, thereby providing a source of ornithine for subsequent tissue repair.
The evidence for involvement of the innate immune system in arthritis, via generation of ROS and RNS, has been extended into the study of juvenile idiopathic arthritis (JIA; 231); this complex, often multisystem disease, poses a real challenge in approach to treatment for pediatric rheumatologists, and further insight into the disease mechanism are needed to support clinical interventions. Nitrite and nitrate production have been shown to be greatly enhanced in the joints of JIA patients when compared with serum from non-JIA control patients. These levels were positively correlated with the number of infiltrating lymphomononuclear cells. Similarly, the levels of 3-nitrotyrosine in synovial fluid showed a high correlation with degree of bone erosion. Together these lines of evidence suggest a high level of in situ production of NO with possible ROS/RNS involvement in the observed joint destruction (231).
V. Mitochondrial-Derived ROS and the Consequence for Cell Death and Survival
Apoptosis is a cellular process defined by the activation of numerous evolutionarily conserved cell-signaling cascades that permits self-contained cellular deletion without the initiation of inflammation. Inappropriate execution causes loss of tissue homeostasis, eventually and is associated with proliferative (e.g., cancer or autoimmunity) or degenerative conditions (e.g., Alzheimer's disease). In the context of chronic inflammatory conditions and specifically RA, failure of cells to apoptose results in their persistence and exacerbation of autoimmunity. This is illustrated in several knockout mouse models (e.g., Bim −/− where apoptosis is ineffective). Bim −/− mouse models are characterized by a more aggressive inflammatory and proliferative pathology in collagen-induced arthritis (15, 16, 64, 258).
ROS are generated within several discrete and restricted sites within the cell. The role of NADPH oxidase generated ROS in RA has been extensively reviewed by Hultqvist and Holmdahl in 2005 (122), and we refer the reader to this excellent review for more information. In brief, these authors provide evidence that NADPH oxidase-like activity in macrophages/antigen presentating cells is essential to cause oxidation of T cell surface thiols and sensitivity to apoptotic factors; the presence of T cell surface oxidized thiols, but not intracellular thiols, can reduce the severity of induced RA in an animal model. Indeed, Pedersen–Lane et al. (228) have characterized lymphocytes from RA patients and have observed fewer surface thiol groups but also lower intracellular GSH concentrations compared to control lymphocytes. Whether there are differences in the mechanisms by which intracellular and surface thiols are oxidized, for example, due to lack of expression of functional NADPH oxidase-like activity in macrophages from the animal models, remains to be determined.
Although originally considered as representing a major source of ROS production in the oxidative burst of phagocytic cells such as neutrophils or myeloid cells, the contribution of NADPH oxidase activity to cellular ROS production by NADPH oxidase-like (NOX) isoforms in other cell types has triggered significant interest (5). Although of less importance for nonphagocytic cells in terms of ROS generating activity, NOX isoforms are considered important in generating ROS within different cellular compartments (286), intracellular signaling cascades (e.g., p38 MAPK and JNK) and in signaling to transcription factor activation (e.g., NF-κB, AP-1, and p53), thereby controlling expression of multiple downstream genes associated with inflammation and the control of cell survival/death. The major source of ROS production within nonphagocytic cell types is the mitochondria. Mitochondria are primarily responsible for the aerobic respiration and ATP production by oxidative phosphorylation, acting as the cell's key organelle for supplying the appropriate energy to drive metabolism. Consequently, mitochondria are ideally placed to dictate cellular outcome (i.e., the decision to live or die). It is this cellular property that highlights a secondary function of mitochondria, as a point of convergence for apoptosis mediated by both the intrinsic (mitochondrial poisoning pathway) and extrinsic [receptor dependent (e.g., Fas or TNF-α)] signaling pathways. In this way, ROS derived from a mitochondrial origin play an important regulatory role as a signaling molecule for apoptosis and autophagy.
Approximately 90% of cellular oxygen is consumed by the mitochondria for oxidative phosphorylation and ATP production. The mitochondrial membrane potential (ΔΨm) is required for the maintenance of the proton motive force that drives ATP production, and additionally for mitochondrial protein import and metabolite transport. This bioenergetic process takes place within the inner mitochondrial membrane, utilizing the electron transport chain to generate an electrochemical gradient that not only produces ATP but also, as an unavoidable by-product, the ROS superoxide (O2 ·−) from complexes I and III. O2 ·− is released into the intermembrane space and to the matrix where mitochondrial superoxide dismutase converts the O2 ·− to hydrogen peroxide that can diffuse through the membrane. Under normal conditions, ROS production is “managed” by SOD. However, disruption of ΔΨm leads to disruption of oxidative phosphorylation, loss of ATP synthesis, Ca2+ and GSH release, oxidation of NAD(P)H and GSH, that collectively result in the decreased generation of ROS. The production of O2 ·− by complex I is attenuated by uncoupling agents, and using JC-1 it has been shown that in lymphocytes from RA patients, the mitochondrial membrane potential is lost. This effect has been attributed to mitochondrial DNA/lipid oxidation and results in loss of energy production/utilization and is inversely associated with disease activity (202). As such, several classes of signaling proteins that are associated with cell survival and death also control mitochondrial function. The appropriate and timely resolution of an inflammatory response, which is necessary to evade pathogens, is critical, as failure to resolve the inflammation can contribute to chronic inflammatory disease if left unchecked. Therefore the balance between cell survival and cell death must be tightly regulated.
In the following section, the role of two such protein family pathways in the context of ROS production and RA will be discussed: Bcl-2 family members and p53. Further, we will evaluate how the aberrant expression and function of these proteins may contribute to the pathology of RA and the efficacy of agents utilized in the treatment of this disease.
A. Regulation of apoptosis by Bcl-2 family members
Defects in the ability of cells to generate or detoxify ROS in RA have a major impact on the cellular response to physiological signals that regulate tissue homeostasis. Cells that are ineffective in ROS generation in response to stimulation of the extrinsic pathway (e.g., Fas, TRAIL) or following administration of therapeutic agents for the treatment of RA which require activation of the intrinsic apoptotic pathway are likely to be persistent and refractory. The Bcl-2 family contributes an important role in apoptosis resistance and regulation of the cellular redox state, but is also associated with the etiology, persistence, and exacerbation of chronic inflammation in RA. In vitro, Bcl-2 or Bcl-xL hyperexpression prevents methotrexate (MTX)-induced apoptosis and dissipation of ΔΨm (120). However, it is not known whether Bcl-2 overexpression in RA can contribute to resistance to MTX through suppression of ROS toxicity in vivo.
The relationship between ROS production and survival is a complex one, with cell survival also being an outcome of cellular ROS generation dependent on the cell type under study and the context of the signals that it receives. One potential explanation for this controversy is that transient, low level ROS production triggers a corresponding level of MAPK activation that is associated with NF-kB activation, gene expression, proliferation, and differentiation, whereas prolonged ROS generation triggers extended MAPK activation that ultimately promotes cell death, by a mechanism that may be mediated by JNK regulated gene expression but also by post-translational activation of precursor enzymes.
Critical regulators of cell death at the level of the mitochondria are members of the Bcl-2 family of proteins. These can be broadly divided into two classes; (a) the anti-apoptotic members (Bcl-2, Bcl-xL, Mcl-1, Bcl-w, A1/Blf-1) which contain up to four Bcl-2 homology domains (BH1-4), and (b) the pro-apoptotic members that can be further subdivided into the multidomain BH1-3 proteins (Bax, Bak, and Bok) and BH3 only proteins (Bim, Bik, Bid, Bad, BMF, bNIP3, HRK,
Mitochondrial outer membrane permeabilization (MOMP) is thought to initiate the selective release of mitochondrial apoptogenic factors localized within the inner mitochondrial space through the formation of pores or channels involving Bcl-2 family members (49, 254). Alternatively, some have suggested that Bcl-2 family members form supramolecular weight channels (162), or, interact with components of the mitochondrial permeability transition pore (PTP) resulting in loss of ΔΨm. In the latter scenario, subsequent influx of fluid into the mitochondrial matrix results in mitochondrial swelling, the eventual rupture of the mitochondrial outer membrane and the nonselective release of mitochondrial factors (20, 189). Once released into the cytosol, mitochondrial-related apoptotic factors such as cytochrome c, for example, initiate caspase-9 activation by binding to and activating apoptosis protease activating factor-1 (APAF-1) in a multidomain protein complex called the apoptosome. In turn, caspase-9 may subsequently activate other effector caspases (caspase-3, -6, -7) and caspase-dependent nucleases that induce the apoptotic phenotype (268). Cytosolic Smac/Diablo assists cytochrome c in activating post-mitochondrial caspases by sequestering X-linked inhibitors of apoptosis (IAPs) that ordinarily inhibit caspase-3 or caspase-9 (268). ROS are intimately linked to mitochondrial-mediated apoptosis, although the relationship between ROS and mitochondrial function (e.g., cytochrome c release and Δψm reduction) are not clear; ROS levels may be enhanced as a consequence of and as a mediators of ΔΨm dissipation (128, 177, 184). NO is also a potent regulator of apoptosis; NO-dependent apoptosis has been shown to associate with a decrease in the concentration of mitochondrial membrane cardiolipin, decreased activity of the mitochondrial electron transport chain, and release of mitochondrial cytochrome c into cytosol (23).
B. Bcl-2 family members, ROS generation, and RA
The last 15 years has seen an intensive period of investigation into the functionality of Bcl-2 family members, some of which mediate pro-survival functions and others contribute to pro-apoptotic functions; collectively these proteins exert their effects in regulating apoptosis primarily at the mitochondria. Despite this, investigations studying the integration of Bcl-2 family members with normal mitochondrial respiration, their effects on normal mitochondrial ROS production, and the consequences for Bcl-2 family member function upon excessive ROS production have been lacking. This is somewhat surprising given that this family of proteins primarily reside at the mitochondria—the principal intracellular source of ROS in nonphagoctytic cells. Indeed, Bcl-2 itself was initially considered to function in an antioxidant-like capacity. The notion that Bcl-2 possessed antioxidant-like activity was first proposed by Hockenbury et al. (116), and subsequently, Bcl-2 was shown to protect against apoptosis induced directly by oxidants (H2O2 and menadione), or by treatments that induce ROS (TNF-α, glutathione depletion, and cyanide/aglycemia) (116, 198). Bcl-2 has also been shown to prevent lipid peroxidation, probably induced as a consequence of apoptosis (116). Similarly, shifting the redox state of the cell to that of a pro-oxidant environment by depleting cellular GSH is prevented by Bcl-2 overexpression (198), suggesting that Bcl-2 can protect against oxidants by increasing the reducing capacity of the cell. Correspondingly, the phenotype of Bcl-2−/− mice is typified by chronic oxidative stress and includes elevated levels of oxidized brain proteins, resulting in fewer cerebellar neurons (115). However, the antioxidant-like behavior of Bcl-2 appears to be indirect. Bcl-2 expression does not prevent ROS generation (116), but elevates the capacity of cells to detoxify their effects or essentially allows the cell more time to cope with the ROS generation (158). Initial data suggested that there were no disparities between the ability of Bcl-2 overexpressing cells to generate ROS under resting conditions compared to the parental line (116), although subsequent research has challenged this view. Recently Chen and Pervaiz (36) have provided evidence that cells overexpressing Bcl-2 actually generate more ROS than the parental cells. This latter study describes higher levels of cytochrome C oxidase (complex IV of the electron transport chain), oxygen consumption, and mitochondrial respiration in CEM leukemia cells overexpressing Bcl-2 (36). One potential explanation for this apparent paradox lies in the finding that Bcl-2 has a GSH binding domain in the BH3 groove. Bcl-2 found within mitochondrial or nuclear membranes appears to sequester GSH and regulate its level. If Bcl-2 is overexpressed within these organelles, the GSH levels are also elevated. Consequently, organellar proteins are less oxidized (317). This observation may explain the increased activity of cytochrome oxidase described by Chen and Pervaiz (36), which is known to be very sensitive to oxidation, despite the increase in flux of electrons through the electron transport chain and increased likelihood of leakage to form ROS. Overall, these observations are consistent with reports of elevated antioxidant levels that are associated with Bcl-2 overexpressing cells (158), presumably to detoxify the higher rate of mitochondrial ROS production, prevent mitochondrial permeability transition (MPT), and subsequently provide resistance to apoptosis. An alternative hypothesis is that Bcl-2 can negatively regulate ROS generation by modulating cytochrome oxidase activity in response to elevated ROS production by the mitochondrial electron transport chain (36). These data appear contradictory whereby Bcl-2 at first glance exerts an antioxidant-like effect, yet permits further ROS generation by the respiratory chain. This inconsistency may allow the cells, such as those found in RA or in other diseases characterized by unchecked proliferation, to survive otherwise detrimental cellular insults that are mediated by excessive ROS. A summary of the Bcl-2 family members that modulate cellular ROS and their effect on the phenotype of haematopoietic cells is tabulated in Table 3.
The importance of trophic cytokine production by synovial cells for the survival of infiltrating immune cells within the joint has been described earlier (19, 25, 69). In the event that trophic factor support is withdrawn, it has been shown that T cells accumulate ROS either through increased production or ineffective scavenging. Furthermore, increased ROS is associated with the induction of the pro-apoptotic BH3 only proteins Bim (253) and Puma (180) and for caspase-dependent apoptosis (143). However, pretreatment of T cells with ROS scavengers prevents Bim accumulation and caspase activation, suggesting a causal relationship between ROS production, Bim expression, and apoptosis (253). This work also provides further evidence for the hypothesis of Geldermann and colleagues that ROS production is essential to enable T cells to apoptose and failure to generate signaling ROS from NADPH oxidase-like complexes can result in more severe RA. Although Bcl-xL has been described to share some of Bcl-2's “antioxidant-like” functions (13), the contribution(s) of other members of the Bcl-2 family towards basal mitochondrial ROS production and the control of the cellular redox state are relatively unknown at this time.
Genetic deletion of a variety of the pro-survival Bcl-2 proteins reveals their requirement for normal tissue development in a variety of different systems, with particular emphasis upon the hematopoietic system. Bcl-2 is essential for survival of mature naïve lymphocytes (211); Mcl-1 and Bcl-xL are important for survival of immature lymphocytes (203), the former being essential for survival of of hematopoietic stem cells (203, 219). Correspondingly, mice transgenic for Mcl-1 demonstrate enhanced viability in a wide number of hematopoietic subtypes (315). In contrast, activation of death signaling pathways, principally effected by the JNK cascade, leads to inhibition of anti-apoptotic and upregulation of pro-apoptotic Bcl-2 family members and ultimately mitochondria-dependent cell death. Inactivation of Bcl-2 and Bcl-xL can be mediated via JNK-catalyzed multi-site phosphorylation of these proteins while proapoptotic proteins Bim and BMF are activated by phosphorylation (171). It appears likely that RA cells, which have been activated by proinflammatory cytokines and which demonstrate strong activation of NF-κB, may show increased expression of antioxidant enzymes such as SOD. In this way, the cells may control levels of ROS such that the JNK cascade is not activated resulting in cell death resistance (209). In support of this hypothesis, the induced expression of the pro-survival Bcl-2 protein, Bfl-1, has been illustrated in Jurkat T cells after priming with ROS in an NF-kB-dependent manner and promotes survival in the presence of subsequent exposure to an apoptotic Fas stimulus (150).
Considering the aggressive involvement of the hematopoioetic system in RA pathogenesis, a limited number of animal data and associative data obtained from RA patients suggest the involvement of the Bcl-2 family in the development and perpetuation of this disease. Transgenic mice harboring the pro-survival human Bcl-2 cDNA under the control of an immunoglobulin heavy chain enhancer (Eμ) possess an excessive number of B lymphocytes in the absence of modifications in T cell number or function. Further, these Bcl-2 transgenic mice develop autoimmune disease-like symptoms that are synonymous with that of the RA-like disease, systemic lupus erythematosus in humans (270). It is interesting to note that the pro-survival protein Mcl-1 is highly expressed in the rheumatoid joint, particularly in synovial fibroblasts and in the synovial lining which is replete with macrophages (178, 179). In addition, it has been shown that macrophages isolated from the synovial fluid of RA patients possess elevated expression of Mcl-1 when compared to those from normal subjects (178). Further, the silencing of Mcl-1 expression in RA synovial fibroblasts or RA macrophages in vitro results in cell death by apoptosis, indicating that expression of this protein is required for survival (178, 179). Elevated levels of the pro-survival protein Bcl-xL are also observed in the RA synovium (26) and in atherogenic B cells (71). The enhanced expression of Bcl-xL in RA autoreactive B cells not only predicates a resistance to apoptosis but also alters their ability to produce cytokines and modulate the inflammatory response (71). Similarly, primary murine B cells that are genetically predisposed to overexpress Bcl-xL show elevated production of IFN-γ and mediate an enhanced TH1 response. Bcl-xL transgenic mice also develop more severe and sustained collagen-induced arthritis than wild-type mice (314). Finally, an increased number of cells are positive for Bcl-2 in the RA synovium when compared to that of the osteoarthritic synovium where the frequency of Bcl-2 positive cells correlated with the extent of synovial hyperplasia and inflammation (229). These data indicate that the enhanced expression of anti-apoptotic Bcl-2 family members contribute to the hyperplasia of the RA synovium through inhibition of apoptosis, permitting cell survival and persistence in the presence of high levels of extracellular ROS that may otherwise induce cellular dysfunction or apoptotic cell death. Additionally, overexpression of Bcl-2 may prevent the enhancement of ROS levels that are required in a cellular signaling capacity, such as that necessary for efficient T cell activation (129) and may reduce the efficacy of drugs used in the treatment of RA which are dependent on ROS production (see Section VI) to exert their cytotoxic effects. Further examination of the likely mechanisms for upregulation of the pro-survival protein Bcl-2 have shown that exogenous application of MnTBAP to T cells in culture, an antioxidant with SOD-like activity, increases T cell expression of Bcl-2 (112). Moreover, activation of T cells, which renders them more susceptible to cell death, elicited an increase in ROS and a decrease in the expression of Bcl-2 by a mechanism that is independent of proapoptotic Bim expression (112). Taken together, these data suggest that ROS sensitize activated T cells to death by decreasing expression of Bcl-2.
Analogous to the observations that overexpression of anti-apoptotic Bcl-2 family members prevent appropriate apoptosis, with consequential exacerbation of autoimmunity, a failure to express pro-apoptotic Bcl-2 family members can also result in similar catastrophic phenotype. On a mixed C57BL/6x129Sv background, mice genetically deficient in the pro-apoptotic protein Bim, develop a fatal systemic lupus erythematosus-like autoimmune disease (16) that is consistent with that described in mice that have been engineered to overexpress Bcl-2 (270). However, genetic ablation of the other pro-apoptotic Bcl-2 family members (Puma−/−, Bid−/−, Noxa−/−, Bax−/−, Bak−/−, Bik−/−, HRK−/−, Bad−/− ) in mice does not confer phenotypes that are consistent with autoimmune-like diseases (44, 135, 153, 241, 290, 309), although hyperplasia or hypertrophy is commonly observed. (15, 153). Consistent with this, cells derived from pro-apoptotic protein knockout mice show defects in their abilities to undergo apoptosis in response to genotoxic or physiological stress in vitro and in vivo. However, it is important to note that the degree of resistance is both cell type- and agent-specific (43, 81, 135, 153, 241, 290, 295, 307, 309).
Apoptotic stimuli are capable of activating several BH-3 only proteins and therefore functional overlap is likely. Similar to the phenotype of the Bim−/− (15) or the Bcl-2 overexpressing mouse, inducible deletion of both Bax and Bak in adult mice results in the development of severe glomeluronephritis and arthritis (275a). Loss of function or expression of a single pro-apoptotic Bcl-2 family member may therefore not be sufficient to induce autoimmunity in its own right. That said, the SLE-like disease associated with the Bim−/− mouse is not exacerbated by the additional deletion of Puma, and showed an enhanced resistance to cell death (64). Interestingly, the SLE-like phenotype associated with Bim deficiency in mice on a mixed C57BL/6x129SV background is not recapitulated by Bim deletion in C57BL/6 mice, despite mice of either background possessing identical defects in lymphocyte apoptosis, negative selection, and immune response (15, 16, 271). This suggests that these immune functions are related to the development of SLE in a more complex manner, possibly due the presence of synergistic factors present in one model but not the other. Importantly, these data indicate that genetic background contributes to the observed phenotype and further, suggest that additional loci to that of Bcl-2 family members are required for the development of spontaneous arthritis.
The pro-apoptotic protein, Bid, is essential for maintaining myeloid homeostasis in mice and its truncation to tBid following activation of the death receptor pathway of apoptosis integrates the extrinsic apoptotic pathway with the mitochondria, ultimately resulting in dissipation of ΔΨm (128, 268, 313). Monocytes and macrophages contribute directly to synovial inflammation in RA, hyperplasia of the synovial lining, destruction of bone and cartilage, and additionally serve as the main source of pro-inflammatory TNF-α and IL-1β (206). Using a serum transfer model of RA, Fas-deficient mice (lpr/lpr) on a C57BL/6 background, show enhanced systemic inflammatory arthritis that is associated with elevated numbers of inflammatory monocytes, increased levels of tissue macrophages, and an upregulation in IL-1β and CCL2 (22). To our knowledge, mutations or genetic deletions in Fas have not been observed in RA; however, downstream inhibition of Fas-induced apoptosis is likely to contribute to RA pathogenesis. In this regard, Bid−/− mice display increased inflammation, bone destruction, pannus formation, and a delay in the resolution phase of arthritis when compared to wild-type mice in models of K/BxN serum transfer-induced arthritis (259). These pathologies are commonly associated with increased leukocyte number within the synovium (259) and fewer apoptotic cells were observed in the synovium of the pro-apoptotic Bid−/− knockout mouse model compared to the wild-type mice (259). Local levels of pro-inflammatory cytokines and chemokines were similar in Bid−/− mice to that of wild-type mice, which is in contrast to that seen in Bim−/− identically conditioned to develop arthritis; in this case, Bim−/− mice possess elevated levels of pro-inflammatory cytokines (IL-1β, IL-6, TNFα) and chemokines (murine CXC) and decreased joint and serum production of the anti-inflammatory cytokines (TGFβ1, IL-10). This phenotype again points to a defect in regulatory T cell activity (see Fig. 2) and the failure to delete autoreactive T cells. Coupled with a reduction in the incidence of apoptotic cells when compared to wild-type mice, Bim−/− mice correspondingly display increased severity and prolongation of arthritis (258). ROS have been shown to regulate Bim expression levels in cellular systems, where activated integrins that are required for cell survival, stimulate intracellular ROS generation in a rac-dependent process (86). Rac-dependent ROS trigger the redox-dependent phosphorylation of EGFR, activate extracellular signal-regulated protein kinase (ERK) and Akt signaling pathways, culminating in degradation of the pro-apoptotic protein, Bim, and promoting cell survival (86). In contrast, Bax−/− and Bak−/− mice showed no difference in the severity of experimentally-induced arthritis when compared to wild-type mice, indicating functional redundancy between the two (258). Evidence for Bcl-2 involvement in autoimmune disease is summarized in Table 4.
Tg, transgenic.
A further level of activity control is mediated by post-translational phosphorylation of Bim; it has been shown that JNK1 activation triggers phosphorylation of Bim in a mitochondrial-ROS dependent manner, increasing its pro-apoptotic activity and thereby promoting cell death (171). Thus, there is evidence for both a stimulatory and an inhibitory effect of ROS on Bim-dependent apoptosis, suggesting that the cellular context, including kinetics and subcellular localization of ROS production may be important to governing the outcomes. A summary table describing known cellular effects of ROS on Bcl-2 family member expression is illustrated in Table 5.
The aberrant expression of p53 in RA synovial tissue (30, 75) has major consequences for gene expression and modulation of the redox state (see Section VE for in-depth discussion within this review). In addition, the p53 target gene and pro-apoptotic Bcl-2 family member, Puma, is poorly expressed within the synovial intimal lining; Puma is required for apoptosis mediated by numerous cytotoxic signals (135, 290) and is a redox modulatory gene since Puma disturbs mitochondrial respiration and results in enhanced ROS production (180). However, Puma is not induced following infection of RA fibroblast-like synoviocytes with adenoviral-p53 (30). These data indicate an inability of RA synovial fibroblasts to upregulate Puma expression in response to p53 induction, thereby contributing to apoptotic resistance in RA (30), although the authors did not investigate why p53 failed to upregulate Puma in RA synovial cells. Several questions remain to be addressed in order to better understand the importance of this pro-apoptotic Bcl-2 family member in RA: does p53 bind to the promoter of Puma in RA synovial fibroblasts? If so, why is Puma not upregulated? Although Puma−/− mice do not possess any overt defects in hematopoietic tissues or cell number (135, 290), it is likely that their resistance to apoptotic insult within the RA joint will contribute to the complex etiology.
Recent studies in B cells obtained from mice genetically predisposed to develop Burkitts lymphoma have indicated that Puma expression is silenced through DNA methylation resulting in an apoptotic resistant phenotype (81). The involvement of DNA methylation as a mechanism to regulate expression of genes which exert a pro-apoptotic effect in RA has not been extensively studied; other potentially important epigenetic pathways for controlling gene expression include histone acetylation and small ubiquitin-like modifications. The activity of histone deacetylase (HDAC) is depressed in rheumatoid arthritis patient synovial tissue, suggesting that strategies aimed at restoring HDAC function may be therapeutic (138). Paradoxically, HDAC inhibition has proved beneficial in RA models (138) and has been shown to increase synovial cell sensitivity to apoptosis. A failure to induce pro-apoptotic gene expression is likely to diminish cell clearance in RA and the previous evidence indicates that one important pro-apoptotic mechanism that may be inhibited is mitochondrial ROS generation.
Collectively, these data suggest that inherent or acquired mutations that affect the functionality of Bcl-2 family members, or their expression through, for example, epigenetic silencing, will contribute to the accumulation of unresponsive cells that is systematic of the pathology of RA. Such abnormalities will have profound effects on the capacities of rheumatoid cells to either generate ROS in response to apoptotic signals or buffer their death inducing capacities. Defining the functional contributions for aberrant Bcl-2 family member expression and activity in particular cell types that infiltrate the rheumatoid joint will enhance our understanding of the causes of RA. In addition, improved knowledge of genetic defects will also contribute to a better understanding of the failure of treatment regimens that utilize the induction of apoptosis in RA. Thus, therapeutic manipulation of Bcl-2 functionality and expression represents a particularly attractive treatment strategy in the clinical management of RA and is discussed later within this review (see Section VI).
The pathophysiological development of RA in humans is widely believed to combine gene–environment interactions. As such, animal models to investigate the contribution of Bcl-2 family members to RA etiology should be also undertaken in models which are dependent on the induction of experimental RA (e.g., collagen-induced arthritis).
C. Involvement of p53 in mediating and mitigating effects of ROS in apoptosis
Hyperplasia is a striking pathological characteristic of the rheumatoid synovium where hyperproliferating fibroblast-like synoviocytes (FLS) resemble a transformed phenotype that with additional recruitment of macrophages, ultimately forms the tumor-like panus (163). These proliferating FLS express activated proto-oncogenes such as early growth response gene-1 (egr-1), c-myc, Bcl-2, c-fos, c-jun (55), and ras; for a recent review on the dual role of factors associated with chronic inflammation and cancer, see Hold and Omar (117). Complementing this cancer-like phenotype of the RA synovium is the aberrant expression and function of the transcription factor and tumor-suppressor p53 (74, 75). p53's tumor suppressor function is underscored by its ability to modulate cell growth by dictating cell cycle progression, DNA repair, genomic stability, senescence, and apoptosis (291). Central to these cellular responses mediated by p53 is its transcriptional activity, regulating the expression of a series of p53-induced genes. Such p53-induced genes include p21cip1/waf1 , GADD45, and 14-3-3, their transactivation resulting in growth arrest, a quiescent-like state that allows for the repair of damaged DNA prior to replication in mitosis. However, in the scenario of excessive cellular damage or disruption of the arrest response, transcription of pro-apoptotic p53-induced genes is upregulated and this ultimately results in programmed cell death. Pro-apoptotic p53-induced genes can be subdivided into two classes. The first group belongs to the “extrinsic” pathway or death receptor pathway that includes the receptors CD95 (Fas) and DR4/DR5, and their respective ligands CD95L (FasL) and TRAIL. The second group, termed the “intrinsic” pathway primarily consists of numerous pro-apoptotic members of the Bcl-2 family including Bax, Noxa, and Puma (218).
Initiation of cell death via both the intrinsic and extrinsic pathways are influenced by the redox state of the cell and have each been reported to induce ROS production at their point of intracellular convergence in apoptosis signaling, the mitochondria. For example, the p53 target genes Bax and Puma disrupt the efficiency of the electron transport chain that results in ROS generation (252). Further, through the transcriptional regulation of other redox-active genes, p53 modulates the expression of enzymes responsible for the production and detoxification of ROS (6, 24, 311). An added level of complexity exists whereby ROS can act as upstream initiators of the p53 pathway through oxidative DNA damage. The cellular redox state can be modulated by p53 expression and function in numerous ways and it is therefore important to consider the regulation of the redox state by p53 in the context of either its normal physiological expression, its overexpression, and its mutation. To understand the role of p53 and redox state it is first necessary to describe the regulation of p53 function, although for more in-depth discussions see reviews (218, 294).
Under normal physiological conditions, the cellular expression of p53 is maintained at low levels through its sequestration with murine double minute 2 (MDM2; the human homologue is known as hDM2), an E3 ligase ubiquitin protein that promotes nuclear export of p53 to the cytosol for proteasomal degradation (201). As a result, p53 has an extremely short half-life. In response to a variety of cellular stresses including DNA damage, genomic instability or hypoxia, p53 is stabilized and activated via post-transcriptional modifications which direct the ensuing cellular responses primarily through its transcriptional activity (218, 294). Cellular DNA is continuously exposed to oxidative damage as a direct consequence of cellular metabolic activity or by external oxidants and pro-oxidants of environmental, endogenous, or xenobiotic source. The resulting appearance of apurinic and apyrimidinic sites, single or double strand breaks (SSB or DSB, respectively), or base modifications such as oxidation of guanine at C-8 that results in the formation of 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodG), initiates activation of the DNA damage response pathway that in turn activates p53 to dictate cell cycle progression, DNA repair or apoptosis (108). Further, the collective sum of these processes is responsible for maintaining the genomic integrity of organisms. In addition, p53 has a direct role in the recognition of DNA damage induced by ROS; physical interaction of p53 with DNA damage repair proteins has been demonstrated (1) and is functional in most cellular DNA repair systems including mismatch repair (MMR), nonhomologous end-joining (NHEJ), homologous recombination, nucleotide excision repair (NER) and base excision repair (BER) (for review, see Jänicke et al; 130).
In addition to the essential role that p53 plays in determining cell death or survival, more recent studies have suggested that this tumor suppressor is an indirect regulator of ROS levels through modulation of antioxidant and ROS synthetic enzyme expression. Several genes that regulate ROS production are transcriptionally activated by p53 (24, 136, 177, 252, 311) and ROS can directly activate p53 in a positive feedback loop (34). Thus, the control of the cellular redox state by p53 is directly related to its “tumor suppressor function,” exercising differential effects that are attributed to its level of expression, activation status, and transcriptional activity. At physiological levels, the roles of p53 in the regulation of cellular redox state are subtle, maintaining normal expression of antioxidant p53-induced genes (SESN1, SESN2, and GSH-Px1). Suppression of p53 function consequently reduces the transcription of the aforementioned antioxidant genes without affecting the expression of pro-oxidant genes Bax, Puma, and NQO1 (252). This subsequently results in the increased generation of ROS, oxidative DNA damage, and the activation of the DNA damage response pathway (as reviewed in Ref. 108) through p53. Increased transcription of antioxidant enzymes [
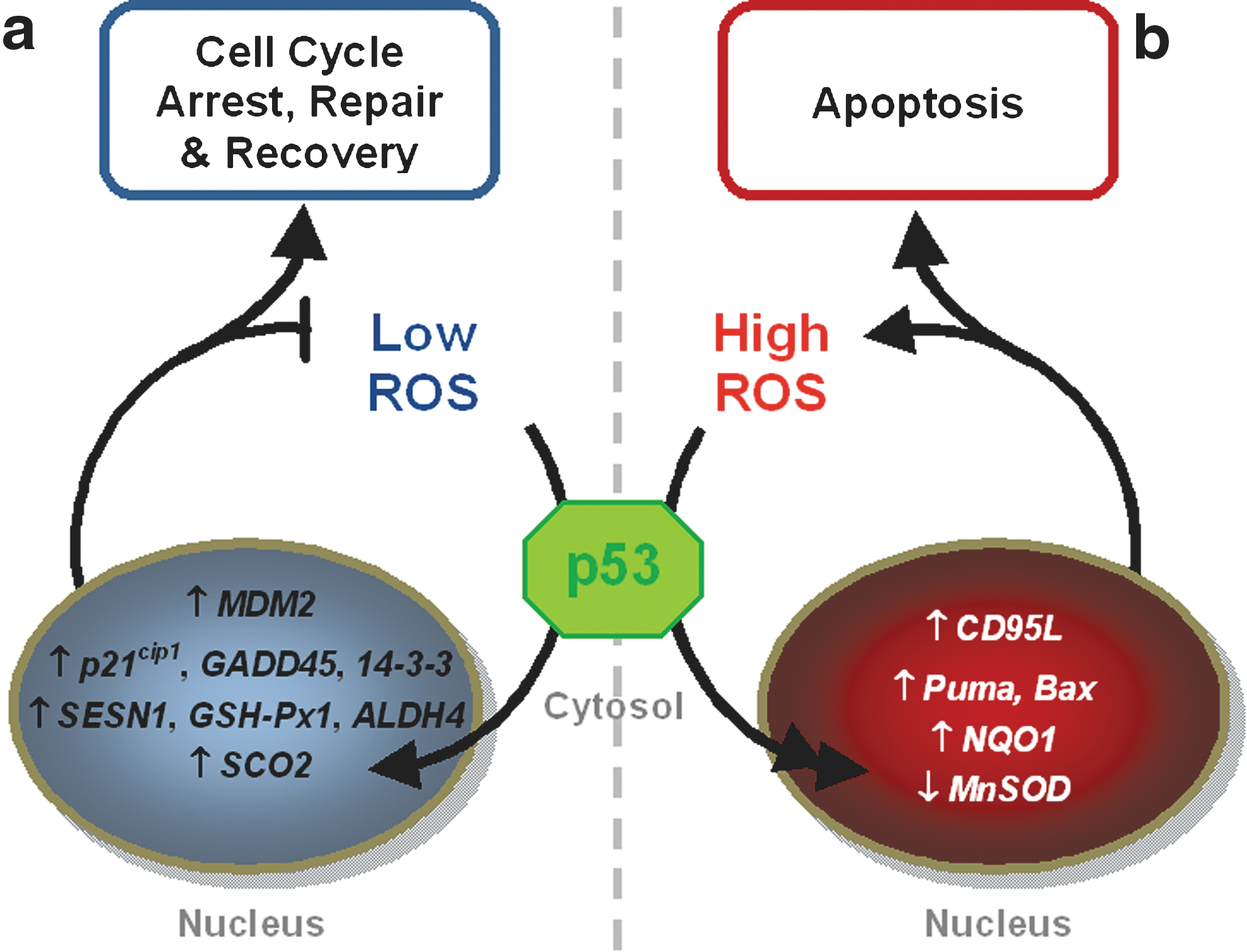
Hyperexpression of p53, however, results in differential modulation of the redox status. In the scenario of excessive ROS production and oxidative DNA damage, p53 activity surpasses the threshold for repair and control of ROS production, to initiate cell deletion. Hyperexpressed p53 promotes a pro-oxidant redox environment through the direct transcriptional regulation of ROS generating enzymes NAD(P)H: quinone oxidoreductase 1 (NQO1; 235), and proline oxidase (POX; 247a), upregulation of redox active pro-apoptotic genes (Bax and Puma; 252), and suppressed expression of the antioxidant enzyme MnSOD (56). The net result is the elevated production of ROS and apoptosis (235) (Fig. 8). Paradoxically, overexpression of p53 can upregulate specific antioxidant enzymes such as microsomal glutathione transferase (235), ALDH4 (311), MnSOD, and GSH-PX, but not catalase (125). However, in this latter study, oxidative stress was still evident, and was hypothesized to result from the increased production of hydrogen peroxide via elevated MnSOD activity in the absence of a corresponding effect on catalase, indicating an imbalance of the antioxidant defense mechanism (125). TIGAR however, is capable of decreasing cellular sensitivity to p53-induced apoptosis and other ROS-dependent apoptotic signals (6). The expression of these antioxidants and p53 target antioxidant genes have yet to be evaluated in RA, but it will be intriguing to determine their expression in the different cell types that contribute to RA pathology in the context of p53 status.
D. p53, ROS, and RA
The p53 gene is mutated in 50% of all tumors and the mutations in p53 which are commonly associated with human tumors have also been found within the RA synovium (74, 103). Many mutations found in RA are transition base changes and are consistent with those mediated by localized oxidative and nitrative stress (74, 299). Such mutations will have substantial effects on gene expression, control of cell cycle, and repair of DNA damage, promoting the existence of an anti-apoptotic and proliferative phenotype that collectively have dramatic pro-inflammatory effects. For example, Han et al. (103) demonstrated that the p53 mutants that are associated with RA, downregulate expression of the pro-apoptotic Bcl-2 family member Bax, and enhance production of the pro-inflammatory cytokine IL-6 (103), whereas expression of the p53-281G mutation in fibroblast-like synoviocytes results in a failure of wild-type p53 to repress the expression of the metalloproteinase MMP13 that is responsible for the destruction of articular cartilage (272). p53 missense mutations have only been detected within the intimal lining of the synovial tissue and not the sublining (305, 306). The authors suggest that these gene mutations are present in type B fibroblast-like synoviocytes and potentially type A macrophage-like synoviocytes. However, sublining T cells do not possess any p53 abnormalities (306). Specific p53 mutations impede wild-type p53 mediated repression of IL-6 expression (103) and are associated with the sites of p53 mutations within the intimal lining of the RA synovium (306). In other words, mutation of p53, as seen in RA, supports IL-6 overexpression (256) that is a key pro-inflammatory cytokine present within the RA joint (119) and contributes to hematopoietic cell expansion. Therefore, the prevalence and functions of, for example, T cells, that do not harbor p53 mutations (306), can be affected by other cell types such as the fibroblast-like synoviocytes within the RA joint that possess such mutations in p53. Clearly, mutations in p53 have paracrine and autocrine consequences in addition to those of the host cell.
Evidence from animal models of RA provides further support for a role of defective p53 signaling in the aetiology of RA. Using p53−/− mice, Yamanishi et al. (305) showed that the onset of RA induced by immunization with type-II collagen occurred earlier than wild-type mice. Further, delivery of p53 into the joints of rabbits predisposed to develop RA by collagen II injection, promoted apoptosis, and these animals also presented with lower rates of leukocyte infiltration, indicating a reduction in inflammation (307). An intriguing question is whether p53 delivery has any effects on the redox state in such animal models of RA. Introduction of the negative regulator of p53 function, MDM2, or mutant p53, results in enhanced ROS production and increased DNA mutation rates as a result of a loss in p53 function, although whether this is due to enhanced oxidative DNA damage or lack of effective cell cycle arrest alone is unknown. This response is also observed upon gene silencing or deletion of p53 (252). Recent studies have described that the human homologue of MDM2 is overexpressed in RA (276), thereby supporting a hypoapoptotic phenotype in RA synovial tissue and rapid proliferative capacities of RA fibroblast-like synoviocytes through sequestration of wild-type p53 activity. These observations may partly explain the low rate of apoptosis within the RA synovium. Further, a single nucleotide polymorphism (SNP) has also been identified in MDM2 (SNP309) that results in higher levels of MDM2 expression (14) and may be of relevance in RA. In this regard, indirect activation of p53 through suppression of MDM2 function (e.g., via nutlin), may have therapeutic potential in the management of RA. Another SNP has been reported at proline 72 in p53 and this mutation has been associated with severity of erosive disease but not the presence of RA in a population of Italian rheumatoid arthritis patients (182). Collectively these data suggest that activation of wild-type p53 is not only required for normal prevention of RA progression, but defects in the expression and function of p53 are likely to have profound effects on the generation of ROS, maintenance of the normal redox state, and the responses of cells to excessive ROS production. Increased rates of DNA oxidation and mutagenesis are associated with deletion of p53 and can be prevented by overexpression of sestrins in p53−/− cells in vitro or by maintaining p53−/− mice on a diet supplemented with N-acetyl cysteine (252). Thus, the manifestation of mutations associated with oxidative DNA-damage may contribute to the hypercellularity associated with the RA joint that culminates in its neoplastic-like histology. In addition, the failure of RA T cells to generate ROS in response to activation and to undergo apoptosis may be an indirect consequence of dysfunctional expression and activity of p53.
Correspondingly, aberrant expression and function of p53 are likely to have detrimental effects upon the efficacy of several agents used in the management of RA. However, the importance of p53 and its functional status in the mechanism of drugs utilized in RA treatment remains unknown. In addition, it is important to bear in mind that many of the studies that implicate NF-κB, Bcl-2, and p53 in pathogenesis of RA have been undertaken in hematopoietic cells from mouse models, and there are fundamental differences between the mouse and human immune systems. The availability of animal models has facilitated the manipulation of specific genes in primary cells and this has advanced our understanding of the RA process. Similarly, human cell-based models that have been manipulated to under- or overexpress NF-κB, Bcl-2, and p53 have been important in improving our understanding of their roles in inflammatory and apoptotic processes. Importantly, they have highlighted many pathways and closely related proteins that can be induced by agonist activation and which differ according to inflammatory stimulus, duration of stimulus, or immune cell type. They also provide useful resources for investigating novel drugs for efficacy and mechanisms. However, it is important to note that the capacity for cell lines to divide continually compared with macrophages that are resident in RA joints and which are nonreplicative is also likely to influence the relevance of observations made on the control of apoptosis in cell lines compared to primary human cells. Recently developed molecular techniques may be applied to primary cells isolated from RA patients to specifically target the knockdown of genes or to introduce gene amplifications. These approaches will be important to better understand any role for aberrant control of ROS/RNS and apoptosis in disease. This may facilitate the development of novel approaches for the resolution of RA that are effective in the clinic.
VI. Effects of Therapeutic Approaches on ROS/RNS
A. Management of RA
A key objective in the therapeutic approach of confirmed RA is to arrest disease progression. In the past, symptomatic therapy was eventually escalated to the use of modifying antirheumatic drugs (DMARDs), corticosteroids, and immunosuppressives. Over the last 20 years, this treatment pyramid has been upturned such that any patient with confirmed RA begins early aggressive DMARD therapy. There is evidence that this approach reduces and/or delays the severity of joint damage, as well as systemic complications such as vascular disease. Although the overall increased mortality does not appear be affected, despite the fact that the general population has enjoyed significant survival benefits over the same period; 50% of RA patients still die prematurely within 20 years of diagnosis (152). The class of DMARDs used to manage RA includes methotrexate, leflunomide, sulfasalazine, and hydroxychloroquine, while D-pencillamine and gold are rarely used nowadays. Invariably these drugs are slow to take effect, with the normal time window of 3–6 months before a decision can be made on whether a treatment is affording any significant benefit. Of these drugs, methotrexate is the most effective and best tolerated, and is currently the most widely used DMARD in the routine clinical setting. According to the National Institute of Clinical Excellence UK guidelines, failure to respond to DMARDs, one of which must have been methotrexate, is a prerequisite for intervention with the newer biological DMARDs, such as the anti-TNFα agents infliximab, adalimumab, or etanercept, or later additions such as anti-B cell therapy (Rituximab), Abatacept (cytotoxic T-lymphocyte antigen-4 (CTLA-4)-immunoglobulin (Ig) (CTLA-4-Ig); an inhibitor of CD28-mediated T-cell costimulation) or anti-IL-6 receptor therapy (Tocilizumab) are amongst an ever expanding list. Anti-TNFα drugs are now being used successfully in the treatment of refractory RA (126).
Prior to confirmed diagnosis of RA, the physician has little armory in defense against progression to chronic inflammatory disease. Conventional approaches include the use of NSAIDs to control pain, but despite the significance advances described above, glucorticoids continue to hold an important place in the treatment of RA since they induce a rapid anti-inflammatory/immunosuppressive effect. Exogenously applied corticosteroids increase the expression of lipocortin 1, an autologous protein that inhibits prostaglandin production and has also been shown to inhibit iNOS activity, measured as NO production, within cultured synovial macrophages (308). The addition of antilipocortin antibodies to synovial explants or cells that have been preincubated with corticosteroid, neutralized the effect of lipocortin on NO production but not iNOS expression, suggesting that corticosteroids may reduce synovial inflammation through inhibiting enzymatic activity rather than enzyme expression.
Corticosteroids bind to nuclear glucocorticoid receptors and can exert a direct influence on gene expression (e.g., promoting lipocortin 1 expression). However, GRs also exert indirect effects on gene expression by competing with other transcription factors (e.g., the pro-inflammatory NF-κB), for C/EBP binding or for interaction with the basal transcription initiation complex. The net effect of activated GRs is therefore to reduce the transcription of NF-κB-dependent genes such as adhesion molecules, proinflammatory cytokines (IL-1β, IL-6, TNF-α), and iNOS, thereby suppressing inflammation.
The evidence presented above has highlighted a role in RA for aberrant ROS production by inflammatory cells and cell signaling sequelae which promote inflammation and reduce the sensitivity of inflammatory cells to activation-induced cell death. If these pathways are important in the pathogenesis of disease, it is anticipated that therapeutic approaches which are effective in management of autoimmune disease may also modulate ROS production and signaling, and this is illustrated in Fig. 9. In addition, effective modulation of the disease process is achieved by promoting the activation-induced cell death of inflammatory cells, for example, by some of the disease modifying antirheumatic drugs. Steroids such as dexamethasone have successfully been used to induce cell death in leukemia cells. However, although dexamethasone-induced cell death proceeds in a p53-independent fashion, efficient induction of apoptosis requires expression of pro-apoptotic Bcl-2 family members (64, 135, 290) and is inhibited by Bcl-2/Bcl-xL overexpression (33, 263). Therefore the aberrant expression of the Bcl-2 family of proteins in RA is likely to impact on the anti-inflammatory efficacy of dexamethasone. Using synovial biopsy material from RA patients receiving intra-articular glucocorticoids confirmed that synovial T cells but not macrophage numbers were reduced. However, synovial tissue apoptosis levels were unchanged following treatment, virtually absent from lymphoid aggregates, and minimal in CD3+ cells both before and after treatment, implying that the mechanism underlying the reduced T cell numbers was more likely related to recruitment and homing mechanisms, rather than apoptosis. Indeed, RA synovial fluid T cells were shown to be susceptible to apoptosis in isolation but resistant to glucocorticoid-induced apoptosis when cultured in the presence of monocytes due to the secretion of survival factors by monocytes (19). In contrast, to the resistance to dexamethasone-induced apoptosis of RA macrophages and of co-cultures of macrophages and T cells, some of the complications observed in patients receiving long-term steroids as anti-inflammatory agents do appear to involve the induction of apoptosis; osteoporosis and arthropies are common place as a consequence of accelerated apoptosis of osteoblasts and chondrocytes, respectively. Taking the range of positive and negative benefits of corticosteroids together supports the use of monocyte-targeted steroid therapies to induce apoptosis and to improve the benefit:risk ratio.
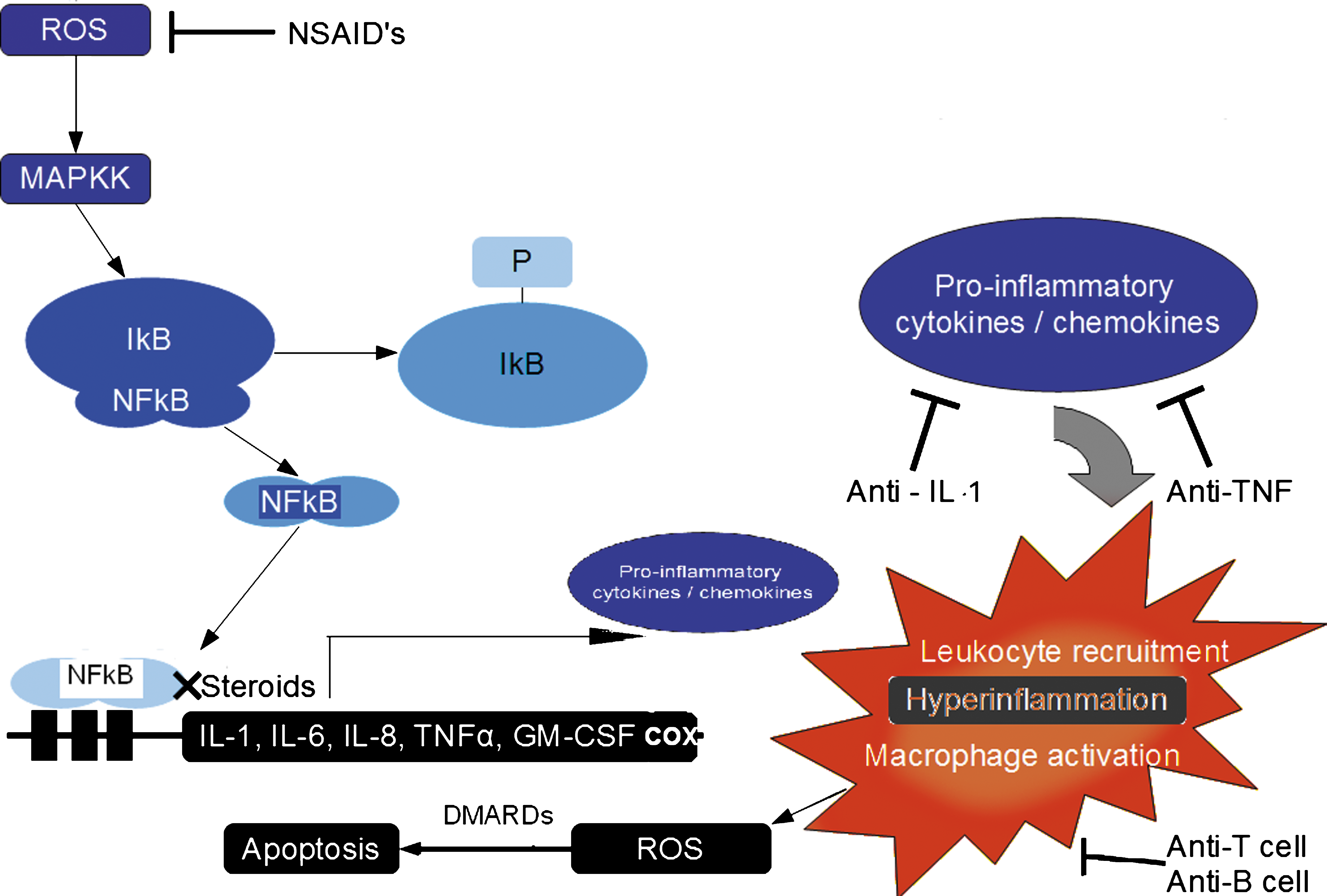
B. Dietary antioxidants, ROS damage, and RA
The pro-oxidant environment of the RA synovium has prompted several clinical investigations into the use of antioxidant-based interventions for the effective management of RA. Despite a distinct lack of therapeutic benefit observed in cardiovascular disease and cancer, antioxidant intervention studies have had some success, albeit in small sample sizes, in reducing disease activity in RA. In a recent report, van Vugt et al. (288) have reported that the disease activity score improved in RA patients receiving a combined supplement of lycopene, α-carotene, tocopherol, lutein, and ascorbic acid over 10 days while being maintained on existing therapies. Plasma oxidative stress markers were not affected. Despite the observation that disease activity worsened within 4 days of supplement withdrawal, the study lacked a placebo control arm and therefore it is not possible to make any strong conclusions about the value of antioxidant supplementation in this instance. The lack of impact of antioxidant vitamin supplements on oxidative stress parameters in RA patients supported findings from an earlier study using a Mediterranean diet intervention in 25 RA patients compared with 26 RA patients maintaining normal diets; the levels of retinol, vitamin C, and uric acid were negatively correlated to disease activity variables, however, antioxidant levels in plasma were unaffected by diet (102).
Additional studies have confirmed that clinical parameters such as pain, grip strength, morning stiffness (60) are improved in patients receiving antioxidant-based interventions, but overall the findings of these studies are not dramatic; there is a lack information concerning the consequences of drug modality, antioxidant dose, and the small number of patients enrolled into the studies. Importantly, whether these antioxidant-based treatment approaches have detrimental consequences for the efficacies of other DMARDs that require ROS generation, such as MTX (see Sections VIC and VID; 233), needs to be determined.
C. DMARDS and ROS
Several disease modifying antirheumatic drugs DMARDS, such as gold, sulfasalazine, cyclosporine, and the recently developed agent leflunomide, have also been shown to inhibit NF-κB activation and downstream expression of pro-inflammatory cytokines, possibly via inhibition or scavenging of ROS required for NF-κB activation in the cytoplasm in vitro. A search of current literature for articles that reported the effects of anti-RA drugs on ROS or ROS-induced damage in vivo yielded few results. The limited studies that were retrieved are summarized in Tables 6 –9. Lemarcechal et al. (172), Table 6, examined the effect of infliximab therapy in combination with methotrexate on protein oxidation in 71 RA patients. The objective of this study was to examine protein oxidation in RA and evaluate the effect of therapy on protein oxidation. Of the 71 RA patients included in the study, 30 were treated with infliximab (3 mg/kg) infusions at weeks 0, 2, 6, 14, 22, and 30 in combination with methotrexate 7.5–20 mg weekly. A non-RA inflammatory control group also received six infusions of infliximab at the same dosage. Blood samples were obtained at week 0, 6, and 30. At baseline, significantly lower levels of protein thiols and higher levels of protein carbonyls were reported in RA patients compared to control subjects. Moreover, these workers observed an increase in protein thiols and reduction in protein carbonyls in patient plasma after 30 weeks of treatment with infliximab which is indicative of a reduction in oxidative stress, and these changes correlated with an improvement in disease activity.
Extracted data from reference 172 expressed as mean ± SD).
P < 0.0001 compared to Control; † p < 0.002; ‡ p < 0.001 compared to Refractory RA week 0.
Data from reference 269.
No significant effect of DMARD treatment or stimulus was observed on neutrophil superoxide production.
Data from ref. 50.
p < 0.04 RA vs. controls; † p < 0.05 RA + Adalimumab versus controls.
Data from ref. 27.
p < 0.05 RA + treatment vs. control.
Several other independent clinical studies have also described anti-TNF-based therapies to mediate systemic antioxidant-like effects. A more recent study by Kageyama et al. (140) examined the effects of infliximab in RA patients on markers of oxidative damage to lipids and DNA, and also measured glycoxidation of protein over 30 and 54 weeks. Despite a smaller cohort of 23 patients at the start of the study, and with only 16 subjects completing the 54-week study, the authors reported a significant reduction in urinary oxidation products from lipid (15-isoprostane F2a) and DNA (8oxodG) at both 30 and 54 weeks. Moreover, DNA oxidation correlated with disease activity and C-reactive protein levels. The presence of urinary pentosidine, a marker of glycoxidation, declined after 54 weeks but did not associate with disease activity (140). Similarly, the same group have reported that Etanercept reduces numerous markers of serum and urinary oxidative stress (pentosidine, N-hexanoyl lysine, and 8-hydroxy-deoxy guanosine) within 6 months of treatment and these were associated with a reduction in the number of swollen joints/tender joints and disease activity score (141).
A smaller study by Tunez et al. (283) examined 13 subjects that were designed to examine the specificity of the infliximab effect in reducing oxidative stress in RA patients, compared to the response of treatment in patents with ankylosing spondylitis and psoriatic arthritis. A range of oxidative stress outcome measures were used, including protein carbonyl formation, glutathione levels, lipid peroxidation products, and antioxidant enzymes. Again, infliximab was shown to be effective in reducing markers of oxidative damage, even within the small cohort, and the effect was more marked for patients with active inflammatory disease rather than presence of specific disease (e.g., RA). Finally, Nagy et al. (208) studied 5 patients for the effects of infliximab (3 mg/kg) on T cell NO levels and have shown that after 6 weeks of treatment, NO levels were significantly decreased compared to pretreatment. Taken together, these data confirm that inhibition of TNFα signaling reduces systemic markers of oxidative stress and nitric oxide production which associates with an improvement in the clinical condition. However, the clinical benefit of anti-TNF therapy is likely to be multi-factoral. For example, reducing macrophage numbers within the rheumatoid joint is associated with enhanced apoptosis and clinical response in patients treated with etanercept and infliximab (28). In addition, inhibition of TNF-α and IL-1 activity suppresses synovial inflammation and joint destruction in RA, probably due to decreased monocyte/macrophage recruitment to the affected joints (267).
Leflunomide is a pyrimidine biosynthesis inhibitor that has been approved for a decade in the treatment of RA. It specifically inhibits the proliferation of T and B cells that are dependent on the de novo pyrimidine synthesis pathway in order to expand the pyrimidine pool eightfold, as required for proliferation. Other effects have also been ascribed to leflunomide, including inhibition of NF-κB activation, downstream pro-inflammatory gene expression, and osteoclast suppression. Leflunomide suppresses TNFα-induced ROS generation and associated lipid peroxidation in Jurkat T cells (188). Further, leflunomide reduces plasma levels of malondialdehyde and carbonyls (145), inhibits the production of NO in human synovial cells ex vivo (63), and reduces NO production in RA patients (243).
Other studies designed to investigate whether effective therapies for RA exert any effect on ROS have focused on ROS production by inflammatory cells following therapeutic intervention in vivo; one of the first studies of this kind was performed by Laurindo et al. (165) who examined the superoxide production of neutrophils from RA patients who had received methotrexate and found the respiratory burst in treated patients to be decreased when compared to untreated controls or non-RA neutrophils. Furthermore, these authors reported that the suppressive effect of methotrexate could be transferred to neutrophils from normal subjects by co-incubating normal neutrophils with methotrexate-treated RA patient sera. More recently, a larger study undertaken by Storgaard et al. (269) examined the effects of clinical intervention in RA patients with methotrexate (n = 27), sulfasalazine (n = 16), and sodium aurothiomalate (n = 15) on superoxide production from either PMA- or fMLP-stimulated neutrophils compared to controls: there was no difference in superoxide production between neutrophils from normal or RA subjects, nor was any effect of treatment observed in RA neutrophils (Table 7). In another study by Den Broeder et al. (50), therapy with adalimumab was investigated for effects on the production of superoxide and ROS by neutrophils from patients with RA (n = 21) or controls (n = 25). Spontaneous ROS production and the ROS production in response to stimulations with opsonised zymosan or PMA were recorded. Assessments were performed at baseline and 2 weeks after the first administration of adalimumab. Den Broeder et al. (50) reported elevated ROS production by PMA-activated RA neutrophils compared to controls but did not observe a reduction in ROS production after 2 weeks of intervention with adalimumab (Table 8). Whether patients showed any clinical benefit from the drugs over this limited period is unclear. A lack of effect of adalimumab on neutrophil function was also reported by Capsoni et al. (27) in 10 RA patients treated for up to 12 weeks (Table 9). This study evaluated the effect of therapy with adalimumab, 40 mg administered subcutaneously every other week, on phenotypic and functional aspects of neutrophils. Ten patients with RA and 20 healthy control individuals were studied. All patients were receiving a stable regimen of hydroxychloroquine, methotrexate, and prednisolone for 3 months before and during the trial. Peripheral blood neutrophils were obtained at baseline and during adalimumab therapy at weeks 2, 6, and 12. Neutrophil chemotaxis, phagocytic and reactive oxygen production were measured but no significant changes were observed. It is difficult to draw conclusions from so few reports that may be limited by small sample sizes and lack of power to determine an effect. However, apart from the initial report by Laurindo et al. (165), the more recent studies do not show any difference in neutrophil ROS production from RA patients compared with normal subjects, and more importantly, these studies could not confirm a reduction in ROS production after treatment despite clear evidence of successful therapeutic intervention. On the other hand, elevated oxidative stress markers in RA can be restored to levels seen in normal subjects during the course of effective therapeutic intervention (172), bringing about the intriguing possibility that extracellular neutrophil ROS production may play a significant secondary role in the pathogenesis of RA and that targeting the intracellular redox shift may provide the primary clinical benefit. In addition, the accumulation of oxidized proteins may not represent an increased level of oxidant production but rather a decreased rate of oxidation product removal. The latter should be investigated by determining the rates of oxidized protein clearance in autoimmune disease and by considering the rate of activation-induced apoptosis pre- and post-treatment.
In vitro studies focused on the modulatory effects of anti-RA drugs upon ROS production by activated neutrophils have shown that infliximab (226) and methotrexate (269) can independently suppress the neutrophil respiratory burst after induction by phorbol myristate acetate or GM-CSF, respectively. Moreover, the ROS-inhibitory effects of methotrexate were also evident in unstimulated primary neutrophils (216, 269). This observation contrasts with the lack of effect of methotrexate treatment in vivo on ROS production by RA neutrophils and may be due to reduced bioavailability of methotrexate in target tissues in vivo after drug absorption, metabolism, and distribution. Our own studies investigating the mechanisms of methotrexate-induced apoptosis have shown that intracellular ROS production is essential for the cytotoxic effects of methotrexate on Jurkat T cells or U937 monocytes. ROS-dependent cytotoxicity could be reversed by the presence of thiol donors such as N-acetyl cysteine (233). Importantly, our studies differ from those described above (e.g., 269) in the subcellular localization of ROS measurement. Whilst others have described the extracellular production of ROS by neutrophils using chemiluminescent probes that detect singlet oxygen and superoxide anion radicals (216), we have determined intracellular oxidant levels in T cells and monocytes using the peroxide-sensitive dye DCF (233). Another explanation for the contrasting observations on ROS production after methotrexate treatment may relate to the drug dose and endpoint under study; the induction of apoptosis by ROS in immune cell populations such as T cells is an important outcome for anti-RA therapies whilst inhibition of proinflammatory molecule production (ROS release) from neutrophils may limit further inflammatory tissue damage.
D. Requirement for ROS generation in DMARD-dependent apoptosis of hematopoietic cells
At the intracellular level, the generation of intracellular ROS appears to be a prerequisite in the mechanisms of action of several DMARDs and may be important for mediating cell death. However, the extracellular release of ROS that produces a pro-oxidant environment, which typifies the rheumatoid joint, will eventually mediate its destruction. In this regard, several antirheumatic treatment strategies specifically target the pro-oxidant environment of the RA synovium. Gold(I)-containing compounds have been used in the treatment of RA for decades and possess disease modifying activities. While the exact mechanism behind the antirheumatic action of gold(I)-containing compounds is unknown, it has been demonstrated that these agents inhibit ROS production from activated macrophages (48). More recent data describe these agents as capable of activating the transcription factors Nrf2 and small Maf. Upon DNA binding, these transcription factors induce gene transcription of the antioxidant stress genes heme oxygenase-1, gamma-glutamylcysteine synthase, and NADP(H):quinone oxidoreductase (146) that contribute to ROS detoxification and consequently exhibit an anti-inflammatory action.
Similarly, both sulfasalazine (SSZ) and D-penicillamine mediate at least some of their anti-inflammatory action through the generation of ROS; D-penicillamine has been shown to cooperate with copper sulfate to generate H2O2 that subsequently increases expression of Fas antigen on rheumatoid synovial fibroblasts and enhanced apoptosis of these cells (2, 100, 107).
Recent studies have focused on the capacity of MTX to improve the balance of TH1 and TH2 cells via a ROS-dependent mechanism, where a TH1-driven response promotes inflammation and TH2 response exerts an anti-inflammatory effect; in a cohort of 28 patients, Herman et al. (111) reported that MTX induces secretion of the TH2 cytokine, IL-10, and significantly reduces TH1 cytokine profile in peripheral mononuclear cells isolated from active RA patients. In addition, the authors observed that MTX modulates the immune status towards an anti-inflammatory TH2 dominance by decreasing the expression of receptors specifically observed on TH1 cells, namely IL-12 receptor and the CXCR3. MTX was also found to inhibit the production of NO from the patients' peripheral mononuclear cells, an observation that may offer at least part of an explanation towards the MTX effects on cytokine homeostasis. In support of this relationship, a significant correlation was found between the induction of IL-10 by MTX and NO inhibition in active RA patients. One possible explanation for these observations is that the shift in intracellular redox favours the production of IL-10 by TH2 cells and in turn promotes apoptosis of pro-inflammatory TH1 cells, leading to the homeostatic return of the TH1:TH2 cell ratio. Indeed, patients who demonstrate an effective shift in redox in response to MTX with an increase in circulating IL-10 may prove to be the best clinical response to treatment, although this remains to be investigated.
Additional indirect mechanisms of inducing a pro-oxidant environment may also contribute to the mechanism of action by MTX and ultimately trigger apoptosis. MTX reduces the level of intracellular antioxidants such as GSH (233), spermine, and spermidine (121). Orthine decarboxylase (ODC) is the rate limiting enzyme responsible for the decarboxylation of L-ornithine to putrescine that is subsequently metabolized to the polyamines spermine and spermidine. Overexpression of ODC results in elevated levels of polyamines which associates with a reduction in MTX-mediated ROS production, dissipation of inner mitochondrial membrane potential, and inhibition of apoptosis (121). However, the ability of MTX to induce apoptosis in T cells is restricted to activated T cells and not resting T cells (120, 233). It has been shown that RA T cells and other hematopoietic cells fail to upregulate ROS production in response to activation in RA (124). Whether this refractory phenotype reduces the efficacy of MTX, which is dependent on changes in intracellular redox state to induce apoptosis either directly (via the intrinsic pathway) or via induction of a death ligand (extrinsic pathway), is unknown.
The majority of studies examining the effects of p53 mutations on cytotoxic agents that are used in RA treatment have been examined from the perspective of cancer. It is possible that the impact of p53 status on the sensitivity to these agents may differ between the two diseases. MTX can induce DNA damage and p53 induction (62) is required for MTX-mediated apoptosis (62, 207). MTX upregulates caspase-8 in a p53-dependent manner and caspase 8 activity is required for its' apoptotic mechanism (62), indicating that MTX-mediated apoptosis may also involve death receptor signaling that subsequently converges at the mitochondria. Similarly, the p53–273H gain of function mutation results in resistance to MTX-induced apoptosis due to, at least in part, the downregulation of caspase-8 (303). However, the consequence for MTX-mediated ROS production (233) in the context of p53 status has not been evaluated. One could postulate that MTX-induced ROS generation in the absence of normal p53 function would not only contribute to resistance against the antirheumatic action of MTX, but also increase the frequency of oxidative DNA damage, mutation, and the development of a more aggressive or refractory pathology.
A reduction in macrophage numbers within the rheumatoid joint is associated with enhanced apoptosis and successful clinical outcome in patients treated with etanercept and infliximab (28). However, lymphocyte numbers are not significantly reduced. This presents a conundrum whereby the cells responsible for the non-inflammatory clearance of dying lymphocytes are lost from the synovium, whereas those cells driving the cellular immune response and producing antibodies do not change. This suggests that the therapeutic targeting of TNF-α reduces inflammatory cell numbers; and the loss of these inflammatory macrophages appears to be primarlily responsible for suppressing synovial inflammation and joint destruction in RA (140, 141, 267). Early insight into the mechanisms by which TNFα neutralization by drugs such as infliximab may contribute to loss of inflammatory cells has focused on the lack of autocrine survival signals. However, more recent investigations have suggested that the membrane-bound form of TNFα may be the target for induction of apoptosis rather than the released, soluble form (200). Antibody-mediated cross-linking of transmembrane TNFα by infliximab has been shown to activate intracellular ROS accumulation, activation of JNK and p53 dependent upregulation of Bax and Bak triggering apoptosis (200) offering a direct “outside to inside” approach for cell death signaling.
E. Managing vascular complications in RA
Much of the foregoing discussion has focused on ROS-dependent therapeutic effects of anti-RA drugs which are effective in improving joint inflammation. However, it is increasingly recognized that RA associates with complications from prospective studies, particularly cardiovascular diseases which contribute to the increased mortality of RA patients. Early scepticism that the increased prevalence of vascular disease in RA patients could be attributed to high dose steroid therapy has not been discounted and the involvement of the chronic systemic inflammatory response in causing accelerated atherosclerosis has been implicated. The presence of elevated levels of modified LDL in the plasma of RA patients, a putative biomarker of atherosclerotic risk, further supports the role of ROS/RNS-mediated processes in the vascular complications of RA (94, 95). Previous reports have also shown increased circulating levels of autoantibodies against modified LDL in RA, suggesting an antigenic role for modified LDL in stimulating autoantibody formation (94, 172). Two recent studies have examined whether existing therapeutic regimens for the treatment of RA can reduce circulating oxidiszed LDL autoantibodies; in a cohort of 58 early diagnosed RA patients treated with MTX and prednisolone, Lourida et al. observed that the LDL was more oxidized and autoantibody titers against mildly oxidiszed LDL were higher, and were independently associated with disease in RA patients compared to 63 normal controls (181). Moreover, standard therapeutic intervention for confirmed RA which aims to halt progression and minimize inflammation elicited a decrease of autoantibody titer and a corresponding increase in high density lipoprotein cholesterol. It remains to be confirmed from prospective studies whether the reversal of the treatment pyramid at the turn of the century, to aggressively treat RA disease, will improve long-term survival and reduce cardiovascular morbidity and mortality in RA patients, but results from observational studies appear promising.
The clearance of oxidized LDL by macrophages and its accumulation in plaque is principally mediated by the scavenger receptor CD36, whose expression is increased as monocytes mature and differentiate into macrophages. CD36−/− mice when crossed with ApoE−/− mice do not develop vascular disease in contrast to the CD36+/+/ApoE−/− mice (160). Papadaki et al. investigated whether the expression of several markers on bone marrow-derived cells were different between RA patients and healthy controls (222). Other phenotypic changes in cells isolated from 24 RA patients and controls, demonstrate an increase in the number of apoptosing CD36+ve cells in the periphery as well as a diminished number of CD36-ve cells in the bone marrow, probably due to their increased rate of maturation and release into the circulation to replace apoptosing cells. Additionally, these findings correlated with anemia. Treatment with infliximab restored the compartmental balance of monocytes, reduced anemia, and the number of CD36-ve cells in the bone marrow was increased. The primary objective of the above study was to investigate the benefits of infliximab on anemia and the findings relating to CD36 expression after treatment merit further analysis in the light of their potential benefits for cardiovascular disease.
F. Future perspectives for harnessing redox modulatory processes for management of RA
Insights into the role of ROS in the RA disease process derived from studies of therapeutic drugs in vitro, ex vivo, and in vivo lead to the hypothesis that macrophages and T cells may be refractory due to aberrant control of intracellular ROS production (92). Local cells or mediators produced within the synovium may exert an influence on intracellular ROS and thus the response to anti-RA drugs; while synovial fluid T cells display a high basal rate of intracellular ROS production (measured as DCF fluorescence) compared with peripheral blood T cells, co-incubation of peripheral blood T cells within adherent synovial cells but not synovial fluids, is able to enhance intracellular ROS production by peripheral blood T cells (246) and protein interactions between T cell CD28 and CD80/86 on antigen presenting cells (250). Subsequent human clinical trials with Abatacept have shown some promise in modulating disease activity (84, 160, 250, 261). These latter studies demonstrate the complex inter- and intracellular interactions that may contribute to altered ROS metabolism in RA and presents difficulties in developing a theoretical rationale of how to effectively manage RA. There is striking evidence for widespread tissue-associated oxidative damage that mediates protein, lipid, and DNA modifications, induces production of pro-inflammatory cytokines and secretion of matrix metalloproteases that collectively induce joint destruction, and may mediate secondary complications such as vascular disease. This evidence suggests that therapeutic approaches that prevent excessive ROS production are warranted. However, more recent genetic and pharmacological evidence indicates that at the intracellular level and in a signaling capacity, ROS generation are required for the induction of apoptosis as a physiological means to prevent autoimmunity and for therapeutic efficacy. The requirement to increase or decrease ROS production is dictated by the subcellular compartment and cell or tissue type highlighting the importance of developing targetted drug delivery approaches if ROS and nitric oxide production is modulated therapeutically.
An alternative approach is to develop more specific interventions that affect gene targets downstream of ROS generation. For example, cell lines, animal models, biopsied human material, and transformed primary human cells from RA patients have identified NF-κB as key in creating the proinflammatory and pro-survival environment that typifies RA pathology (see Sections IID and IVC) and therefore renders this protein an important target for inhibition. The pharmaceutical industry is focusing efforts on inhibition of the canonical IKK proteins. However, given the critical role of this pathway in regulating cellular response to pathogens, it is unclear whether the benefit of such pharmacological intervention can be greater than the associated risks. Instead, kinases act downstream of ROS signaling via p38MAPK and JNK pathways are also potential targets by modulating gene transcription in favor of preventing proliferation or activating apoptosis. Therefore, the pro-inflammatory effects of ROS mediated through gene expression can be prevented while allowing the apoptotic events associated with ROS generation to progress without interference, which are required for effective therapeutic intervention.
More recently, in the cancer field, an intensive research effort has focused upon circumventing the pro-survival function of anti-apoptotic Bcl-2 family members as an anti-tumor treatment strategy. The similarities between RA and cancers have been drawn upon in the past and include rapidly dividing cell populations that can be effectively treated with cytotoxic drugs such as methotrexate and cyclophosphamide, which are indicated in both conditions. Small molecule inhibitors of the anti-apoptotic Bcl-2 family members [ABT-263; Obatoclax/GX15-070 (282, 293)] or anti-sense approaches to prevent Bcl-2 expression [Genasense/Oblimersem/G3 (139, 151, 265)] have considerable tumoricidal action both in vitro and in vivo and are currently being evaluated in numerous human clinical trials for the treatment of cancer. HA14-1, a small cell permeable nonpeptide Bcl-2 inhibitor, induces apoptosis through the release of cytochrome-c and dissipation of ΔΨm (106). Considering that the hyperexpression of Bcl-2, Bcl-xL, and Mcl-1 are characteristic of the rheumatoid synovium, the evaluation of novel drugs that target these anti-apoptotic Bcl-2 family members in a cell-specific manner either as single agents or in combination with current DMARDs such MTX presents an exciting future opportunity for disease management.
VII. Conclusions
There is evidence for involvement of aberrant levels of many reactive species including reactive oxygen and nitrogen species, which may be either increased or decreased according to subcellular compartment, cell type, and localization in different autoimmune diseases. Alterations in ROS and RNS are reported to contribute to autoantigen formation, to regulate antigenic processing, to control the extent of the cell-mediated immune response via intracellular signaling, to mediate cross-talk between cells, and to control apoptosis. In other words, ROS/RNS may be involved in the initiation and the resolution of disease in normal individuals. However, in those who are genetically susceptible to RA and in whom there is an additional trigger (e.g., risk of RA is increased with smoking for SE positive individuals), we can envisage that the apoptotic response may be blunted due to adaptive changes to increased ROS levels in T cells and neutrophils which combine to allow cell survival.
Given such complexity, it is not surprising that interventions with antioxidant molecules have failed to show any benefit; indeed, this review has highlighted data which describe a requirement for ROS or RNS production in the arrest of autoimmune disease. This has led to the development of respiratory burst inducing compounds, phytols, which have been shown to be as effective in modulating arthritis in the Ncf1DA rat model as proven treatments such as methotrexate and etanercept (124).
The challenge for the future is to improve our understanding of the kinetics and cellular and subcellular sources of ROS/RNS that are important for regulating the immune system in RA. This will further support the selective targeting of ROS/RNS-modulatory molecules, thereby allowing selective immunosuppression and/or anti-inflammatory effects without inflicting damage to bystander cells and tissues.
Footnotes
Acknowledgments
The authors gratefully acknowledge the extraction of data from References 26, 48, 164, and 259 by Pavandeep Assi. Figures 5 and ![]() comprise part of the PhD thesis of Darren Phillips, who was funded for this work by an Aston University PhD studentship. The authors gratefully acknowledge careful reading of the manuscript by Sean Garrison, St. Jude Children's Hospital (USA).
comprise part of the PhD thesis of Darren Phillips, who was funded for this work by an Aston University PhD studentship. The authors gratefully acknowledge careful reading of the manuscript by Sean Garrison, St. Jude Children's Hospital (USA).
Abbreviations Used
Reviewing Editors: Giovanni Davi, Carolyn L. Geczy, Rikard Holmdahl, Maureen McMahon and Ricardo M. Xavier