
Abstract
The molecular mechanisms underlying thiol-based redox control are poorly defined. Disulfide bonds between Cys residues are commonly thought to confer extra rigidity and stability to their resident protein, forming a type of proteinaceous spot weld. Redox biologists have been redefining the role of disulfides over the last 30–40 years. Disulfides are now known to form in the cytosol under conditions of oxidative stress. Isomerization of extracellular disulfides is also emerging as an important regulator of protein function. The current paradigm is that the disulfide proteome consists of two subproteomes: a structural group and a redox-sensitive group. The redox-sensitive group is less stable and often associated with regions of stress in protein structures. Some characterized redox-active disulfides are the helical CXXC motif, often associated with thioredoxin-fold proteins; and forbidden disulfides, a group of metastable disulfides that disobey elucidated rules of protein stereochemistry. Here we discuss the role of redox-active disulfides as switches in proteins. Antioxid. Redox Signal. 12, 53–91.
I. Introduction
In contrast, the role of Cys in pH-dependent conformational changes has been little studied. Cys is also susceptible to a variety of modifications by reactive oxygen and nitrogen oxide species (ROS/RNS). Cys can be nitrosated, glutathionylated, and can form covalent bonds with other Cys. RNS such as nitric oxide (·NO) can mediate S-nitrosation to yield an S-nitrosothiol (RSNO). Other RNS, such as peroxynitrite (ONOO
Modifications of Cys by ROS were first discovered in diseases of oxidative stress and are also associated with the aging process (90). These modifications were at first thought to be irreversible. Diseases associated with oxidative stress include type II diabetes, cancer, neurodegenerative diseases, and cardiovascular disease. ROS/RNS are generated as a byproduct of metabolic processes, including aerobic respiration in mitochondria. They are also produced by NADPH oxidases and disgorged by macrophages during immune defense (88).
Reductive pathways that reverse the effects of ROS on Cys were discovered by serendipity in plants. A disulfide-mediated thiol-based redox mechanism was shown to differentially regulate the activity of chloroplast enzymes in response to light (31). Light generates electrons which reduce disulfides through the ferrodoxin pathway, but disulfide reduction affects the activity of different groups of enzymes in opposite ways. Biosynthetic enzymes, such as regulated proteins in the Calvin cycle, are activated by light. In contrast, other enzymes such as those linked to carbohydrate degradation behave in the opposite manner: they are inhibited by light. It was later shown that these light-regulated pathways also respond to ROS: a role they fulfil in bacteria, yeast, and higher organisms.
It is now understood that a continuum of redox set points within proteins control cellular processes in response to a range of ROS fluxes. Low fluxes mediate homeostatic control of housekeeping redox processes while higher fluxes mount stress and adaptive responses before a threshold is reached, triggering apoptosis, or programmed cell death. Partial loss of control of these processes is now believed to underlie aging (113). Even higher levels of ROS can cause cellular necrosis.
In addition to its roles in housekeeping and the stress response, thiol-based redox regulation is also important in developmental processes in plants and animals. In plants, thiol-based redox regulation is involved in seed germination (31). Inadvertent activation of proteins involved in these processes by modern food processing techniques may contribute to the allergenicity of seeds and nuts. In animals, the early embryo forms under reducing conditions (93) but its environment becomes more oxidizing as the embryo grows (93, 116). Redox-regulated processes govern the formation of substructures during embryo development by selective apoptosis.
Knowledge of the relevance of these systems to health and disease is growing exponentially, but much remains to be discovered. Although the bulk of research on thiol-based redox regulation has focused on oxidative stress, redox imbalance is emerging as a more general disease category. A mutation in the human αB-crystallin gene that causes a multisystem protein aggregation disease, including cardiomyopathy, was recently linked to reductive stress (218). Thus, both oxidative and reductive stress can cause disease and a more general category of disturbances of redox homeostasis or redox imbalance exists as a disease etiology.
Here we look at redox processes involving disulfide formation with an emphasis on molecular mechanisms.
II. Types of Disulfide Switches
The molecular mechanisms underlying thiol-based redox control are poorly defined. Students of undergraduate biochemistry are still taught that formation of disulfide bonds between Cys residues confers extra rigidity and stability to their resident protein, forming a type of proteinaceous spot weld, and that disulfide bonds occur in proteins in the extracellular environment, but are not found in the more reducing environment of the cytosol. It is not uncommon for structural biologists to routinely ascribe a role in stabilization to any disulfide encountered, without any supporting evidence.
Redox biologists have been redefining the role of disulfides over the last 30–40 years, but this information has still not percolated down to undergraduate textbooks, or out of the field of redox biology into mainstream biological research. Disulfides are now known to form in the cytosol under conditions of oxidative stress. Careful measurements of the stability of Cys mutants versus wild type show that disulfides are not always stabilizing in their resident protein. Some disulfides contribute little to stabilization and others may actually destabilize their resident protein (151, 242). The emerging paradigm is that the disulfide proteome consists of two subproteomes: a structural group and a redox-sensitive group (300).
How can we objectively determine whether a disulfide is structural or redox active? The ultimate arbiter is the disulfide redox potential. Disulfide redox potentials measured in thiol–disulfide oxidoreductases range from −95 to −330 mV (111, 145, 166, 296). For disulfides serving structural purposes, the redox potential can be as low as −470 mV (76).
We have been studying disulfides with the purpose of distinguishing redox-active from structural disulfides. In principle it should be possible to differentiate between the two groups of disulfides by analysis of protein structures, and ultimately, protein sequences.
A. Discovery of the Cross-Strand Disulfide
Our interest in the area of disulfide redox activity arose when we first encountered a subset of unusual disulfides during a structural data mining survey of β-sheets (290). These disulfides, which link adjacent antiparallel strands, attracted our notice because they occur in a secondary structure that is already noncovalently linked and thus, at best, seem redundant. Furthermore, they exhibit unusual protein stereochemistry which disobeys rules expounded in classic structural studies of disulfides by Richardson and Thornton (224, 259).
Using an approach similar to that adopted by Ramachandran to explore conformations available to the backbone of the polypeptide chain (219), Richardson and Thornton defined backbone protein contexts that could not form disulfides between resident Cys residues without introduction of strain into the protein structure. They deduced four rules for forbidden disulfide contexts. With respect to secondary structure, disulfide bonds should not be found between Cys pairs: on (a) adjacent β-strands (224, 259); (b) in a single helix or strand (259); or (c) on nonadjacent strands of the same β-sheet (259). In primary structures, disulfide bonds should not occur (d) between Cys pairs adjacent in the sequence (259). An accompanying data mining study by Thornton confirmed that in 1981 no disulfides existed in protein structures in the Protein Data Bank (PDB) (19) that disobeyed these rules (259).
Fifteen years later, when undertaking a data mining study to find residues that prefer each other's company as cross-strand partners in β-sheet, we expected ion pair-formers such as Glu-Arg and Glu-Lys to be most favored. Indeed these pairs are greatly enriched in β-sheets with enrichments of 3.4 compared to the frequency that would be expected based on their abundances in β-sheet. Surprisingly however, the most enriched pairs were Cys pairs, with enrichments in excess of 9.0. More astonishingly, some of the Cys residues were covalently linked to each other in direct conflict with Richardson and Thornton's rules. Further examination of these disulfides showed they are extremely unusual: the disulfides and the β-sheets around them are significantly distorted (Fig. 1).
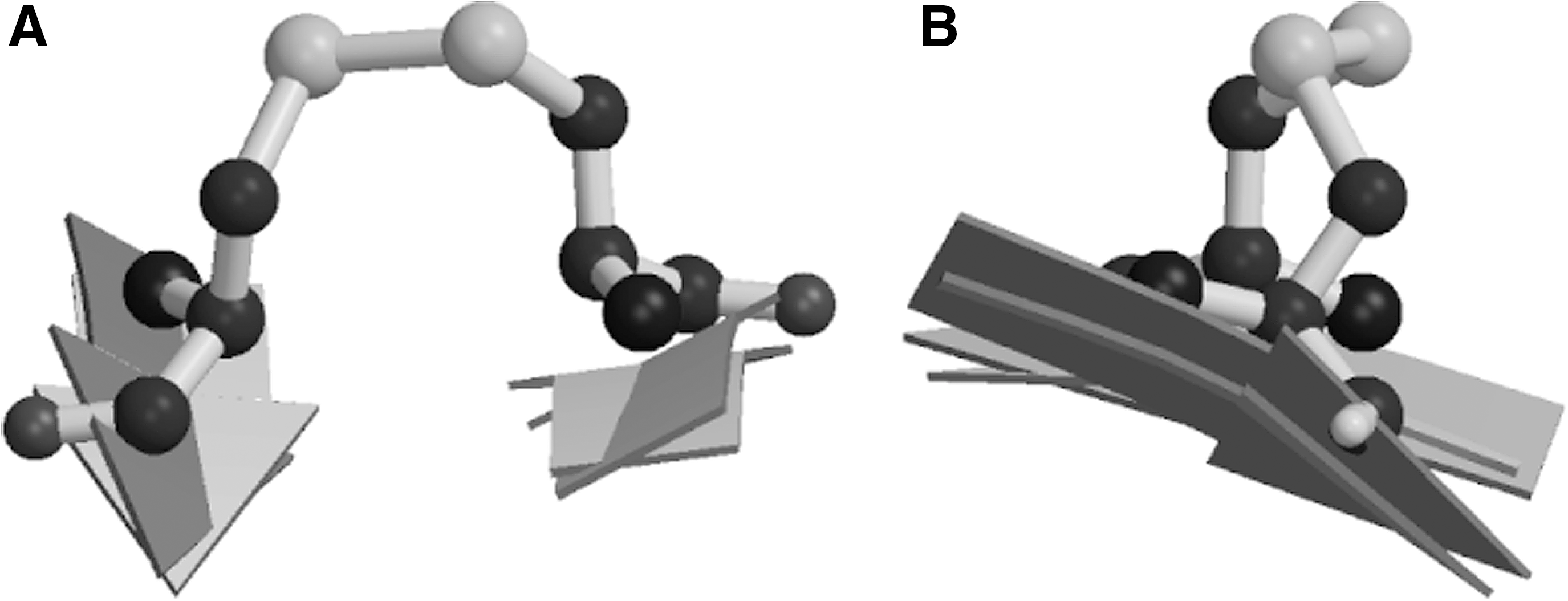
Thus, these covalently-linked Cys pairs do not seem to fit the paradigm of disulfides as structural stabilizers, and in recognition of their special status we gave them a special name: Cross-Strand Disulfides (CSDs). An independent study of disulfide conformations performed around the same time identified a novel disulfide conformation, dubbed the staple conformation, not previously seen in Richardson and Thorntons' studies (92). All disulfides identified in the staple conformation were in fact CSDs, and we later showed that most disulfides adopting the staple conformation are embedded in a CSD structural motif (94). Harrison and Sternberg also noted the prevalence of disulfides adopting the staple conformation on β-hairpins (92). If these forbidden disulfides had a functional rather than structural role, it seemed likely that role would be redox-related, but there was little data to support such a view in 1995.
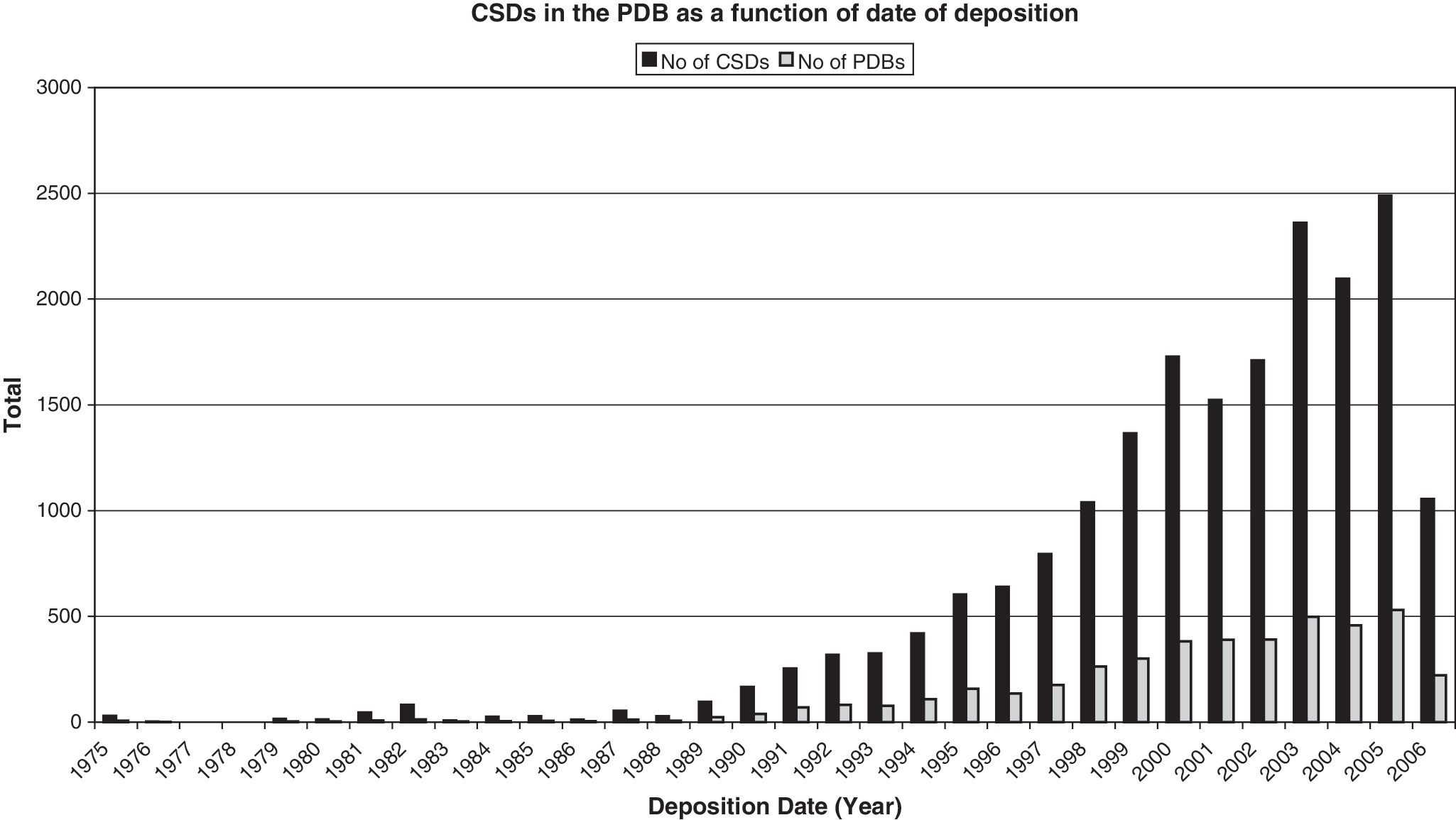
Over the last 10 years, as more structures were deposited in the PDB, more examples of forbidden disulfides came to light (Fig. 2). Data also accumulated linking CSDs to a redox role. In most thiol reductases, the catalytic disulfide is formed between two Cys in a Cys-X-X-Cys motif embedded in a thioredoxin (Trx) fold. However, solution of the structure of the novel thiol disulfide reductase DsbD showed that it contains a novel enzymatically-active CSD embedded in an immunoglobulin (Ig) fold (79). DsbD is a transmembrane electron transporter that activates oxidized periplasmic DsbC by reducing the active site Cys using electrons transported from the cytoplasm. In the final step of the electron transport process, the CSD between Cys 103 and Cys 109 in the N-terminal domain of DsbD is reduced by the Trx-like C-terminal domain of DsbD. The reduced form of the CSD then specifically interacts with the oxidized form of DsbC to form an intermolecular disulfide bond between Cys 103 of DsbD and Cys 98 of DsbC (86). Several other CSDs form catalytic disulfides in other enzymes, including ribonucleotide reductase and several peroxiredoxins (Prxs) (193). All of these proteins have different folds, suggesting the CSD evolved independently many times.
Redox activity of CSDs can also have a regulatory role in proteins, as has been demonstrated for extracellular proteins of the immune system. CD4 mediates cell–cell interactions in the immune system and is the primary receptor for HIV-1. A CSD in the second Ig domain of CD4 is reduced on the cell surface during HIV/cell membrane fusion (181). More recently, reduction of a CSD in Tissue Factor was shown to switch the protein between its coagulation and signaling functions (2). Examples of CSDs found in solved structures in both redox states support the notion that this disulfide motif is redox active (64). These include examples in the Arabidopsis immunophilin FKBP13.
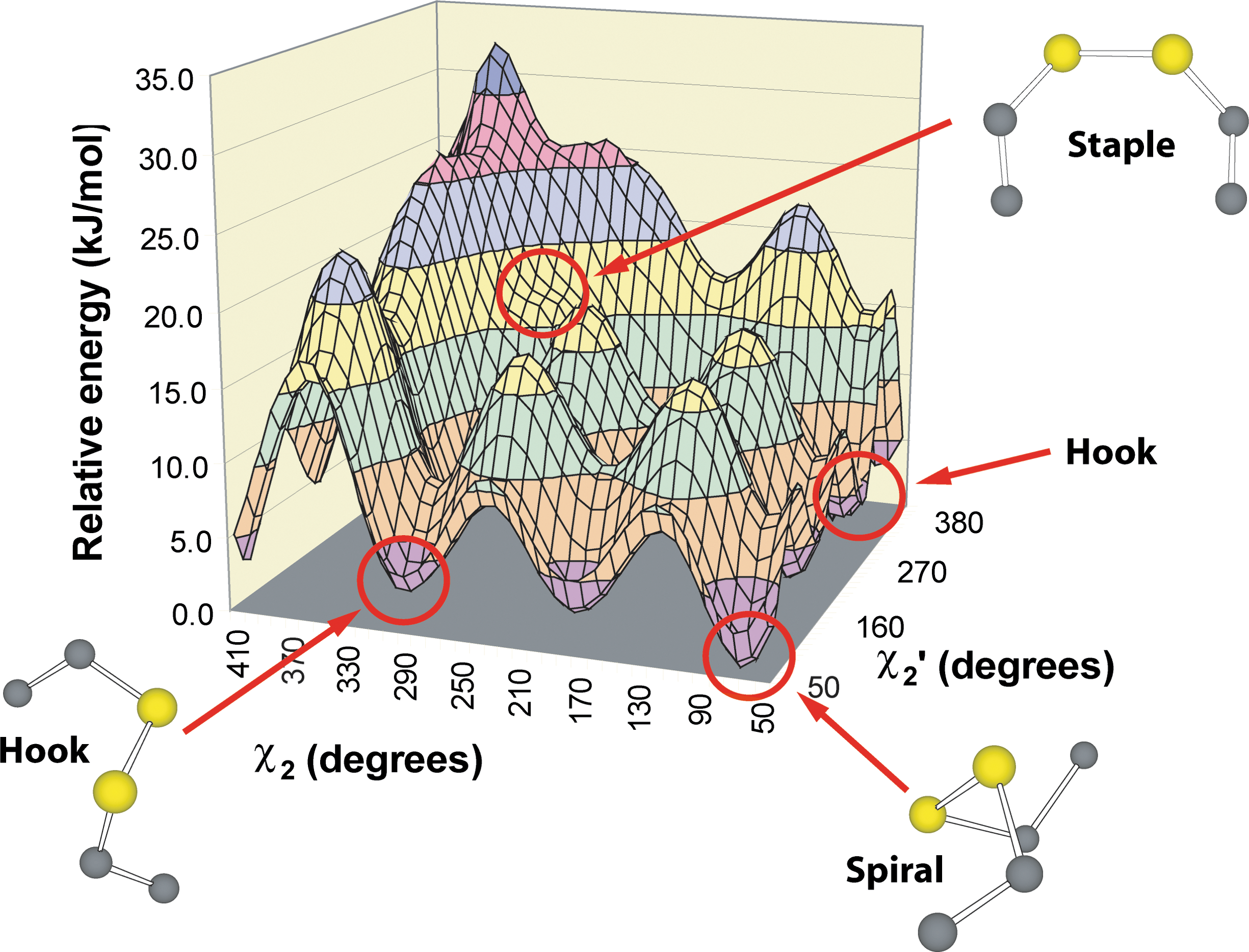
Previous work on physical variables enhancing redox activity suggested disulfides with high torsional energies have an enhanced likelihood of reduction (36, 131, 279, 280, 283). We found that CSDs are indeed associated with high torsional energies and represent 8% of disulfide bonds with torsional energies >12.5 kJ·mol−1 in an unnormalized dataset. Torsional energies in these early studies were estimated using an empirical torsional energy function from the molecular dynamics program AMBER (213). The use of this function is not entirely satisfactory as there was no literature assessing its accuracy. More detailed quantum chemical calculations on the model compound diethyl disulfide subsequently showed that it is a reasonable approximation but has several limitations (95). First, it tends to underestimate most disulfide energies. More worryingly, it is unable to estimate the relative stability of disulfide conformations accurately. Relative stability is important when assessing reaction paths in disulfide relays and cascades. Some examples will be given in Section IV-C. Most importantly, the AMBER torsional energy function completely fails to predict some interesting high energy disulfide conformations. A particularly interesting feature of the more accurate disulfide potential energy surface, not predicted by the AMBER equation, is a high energy feature produced by the eclipsed disulfide staple conformation (95). CSDs sit on a medium energy ledge on this feature (Fig. 3). In contrast, low energy hook and spiral conformations, which are more abundant in the PDB, sit in the lowest energy wells. These low energy disulfides are likely to be structural.
We also began to investigate the types of conformational changes in proteins mediated by CSD redox activity. Many CSDs join short regions of the polypeptide chain and are commonly found bridging β-hairpins (92, 292). The location of many CSDs on β-hairpins suggests the redox status of the disulfide controls presentation and sequestration of epitopes in the hairpin loop. Experimental data supporting this notion is available for Newcastle disease virus. Antibodies against the β-hairpin loop straddled by the CSD neutralize the fusion protein, and two loop mutants escape neutralization (264, 301). It seems likely reduction of the CSD may facilitate opening of the head region and exposure of the fusion peptide sequestered within the head (214). More recently, thiol/disulfide exchange has been demonstrated to be required for membrane fusion (121).
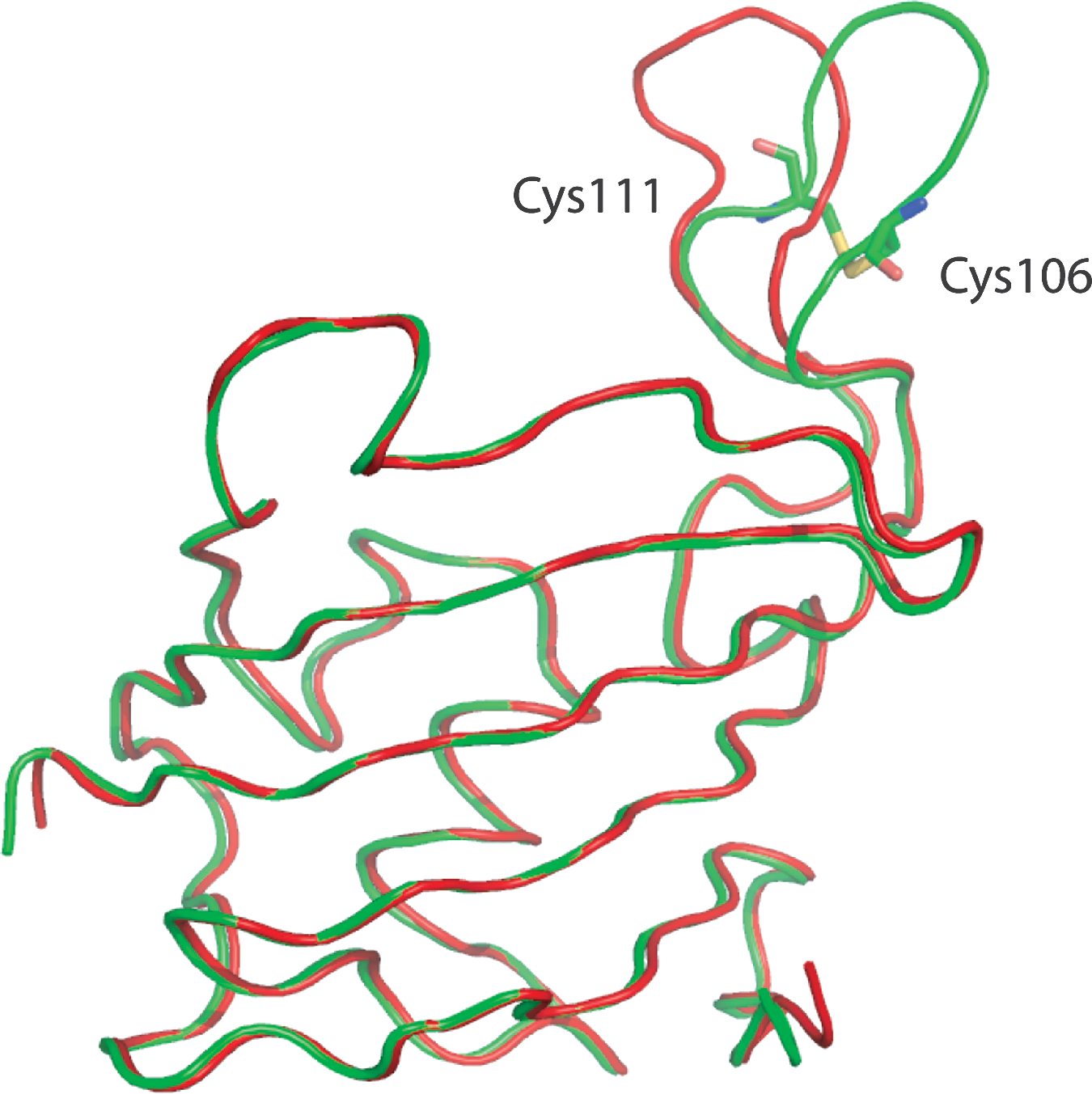
A beautiful structural example supporting the concept of redox-controlled epitope presentation was recently provided by the Arabidopsis thaliana FKBP13, a protein involved in the immune response in plants and animals. Solved in both the reduced and oxidized states, the hairpin loop changes its conformation between the two redox states while the rest of the protein remains largely static (Fig. 4) (78).
In summary, the CSD is a metastable redox-active disulfide which occupies a medium energy ledge on a high energy feature of the disulfide torsional potential energy surface. It functions as a catalytic disulfide in some proteins; may mediate conformational changes important in protein-protein interactions in response to redox signals, such as is likely in FKBP; and also mediates some other functions that will be discussed below.
B. Energetic analysis of allowed disulfide contexts
Disulfides linking Cys in adjacent β-strands, originally postulated to be forbidden by Richardson and Thornton clearly do exist, but they do not appear to be associated with the original role assigned to disulfides as structural stabilizers. Instead they appear to have a functional redox role. The studies of Richardson and Thornton can be re-evaluated in the light of new concepts in protein structure/function relationships.
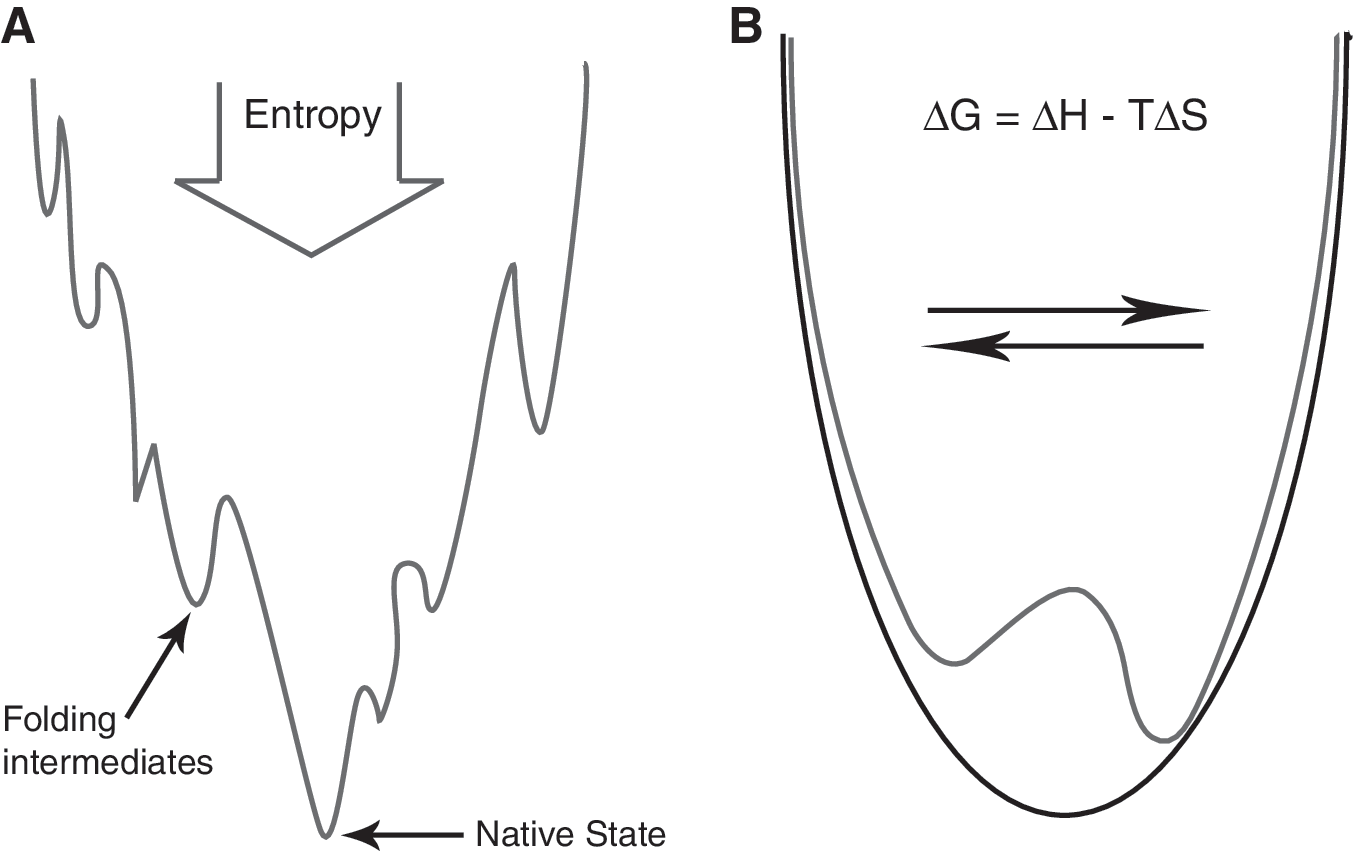
Before the early 1990s, researchers believed that proteins collapsed naturally into a minimally-frustrated fold during the folding process. This concept was based on the work of Anfinsen who showed that a protein's amino acid sequence determines its three-dimensional fold (5). The “thermodynamic hypothesis” states that the three-dimensional structure of a native protein in its normal physiological milieu is the one in which the Gibb's free energy of the system is the lowest (5). While the general basis of this belief still holds, some new results, such as the discovery of molecular chaperones, were difficult to reconcile with this concept. New computational tools, such as profiles-3D (171), originally designed as a check on the structure solution process, showed that natural stresses exist in proteins in functionally important regions. In addition, structural data mining studies provided evidence that locally tolerated stresses are important for function (84, 98, 206). Although researchers were previously aware of stresses in protein structures such as β-bulges, it was posited these “glitches” were temporary structural adaptations to accommodate insertion and deletions in the protein chain (223), suggesting they would be refined in a later evolutionary iteration and “edited out”. Studies on stresses in proteins suggested the alternative view that stresses are central to the protein's function and conserved throughout evolution. Stress studies also suggested that proteins do not adopt a minimally frustrated fold. Many proteins in the PDB adopt folds that are globally close to a minimally-frustrated fold, but contain local tolerated stresses in functionally important regions. The result is a protein folding funnel that does not descend to a point corresponding to the global minimum, but instead is arrested above this minimum energy, with a complex energy landscape at the bottom of the well. Many philosophers have pondered on the physicochemical basis of life (236). From the protein's point of view, being “alive” is not being at the pointy end of a folding funnel. Any protein that occupied the true global minimum of its fold would be functionally dead.
As most proteins in the PDB do adopt folds close to the global minimum, techniques such as threading can successfully predict three-dimensional folds. However, some proteins adopt folds that are significantly different to the minimally frustrated fold for that sequence. Proteins that undergo large irreversible conformational changes, such as influenza hemagglutinin, belong to this group. Redox transitions, some of which are likely to be irreversible, seem also to be fundamental to these large conformational changes (292, 293). In conclusion, it is also interesting to note that Anfinsen observed that “proteins devoid of restrictive disulfide bonds … make more convenient models for the study of thermodynamic and kinetic aspects of the nucleation and subsequent pathways of polypeptide chain folding”.
C. Between-strand disulfides
At their simplest, forbidden disulfides essentially form a bistable switch. A bistable switch requires two stable states, both of which must contain sufficient stored potential energy to help drive the switch into the opposite state in response to the appropriate signal. Regions of strain in a protein structure are candidate potential energy sources. Forbidden disulfides may operate as bistable switches by trading off different terms in the Gibb's free energy (Fig. 5). Forming the disulfide bond decreases the entropy of the chain. However, for forbidden disulfides there is a significant enthalpic cost of forming the disulfide because of the abnormal stereochemistry adopted by the protein chain.
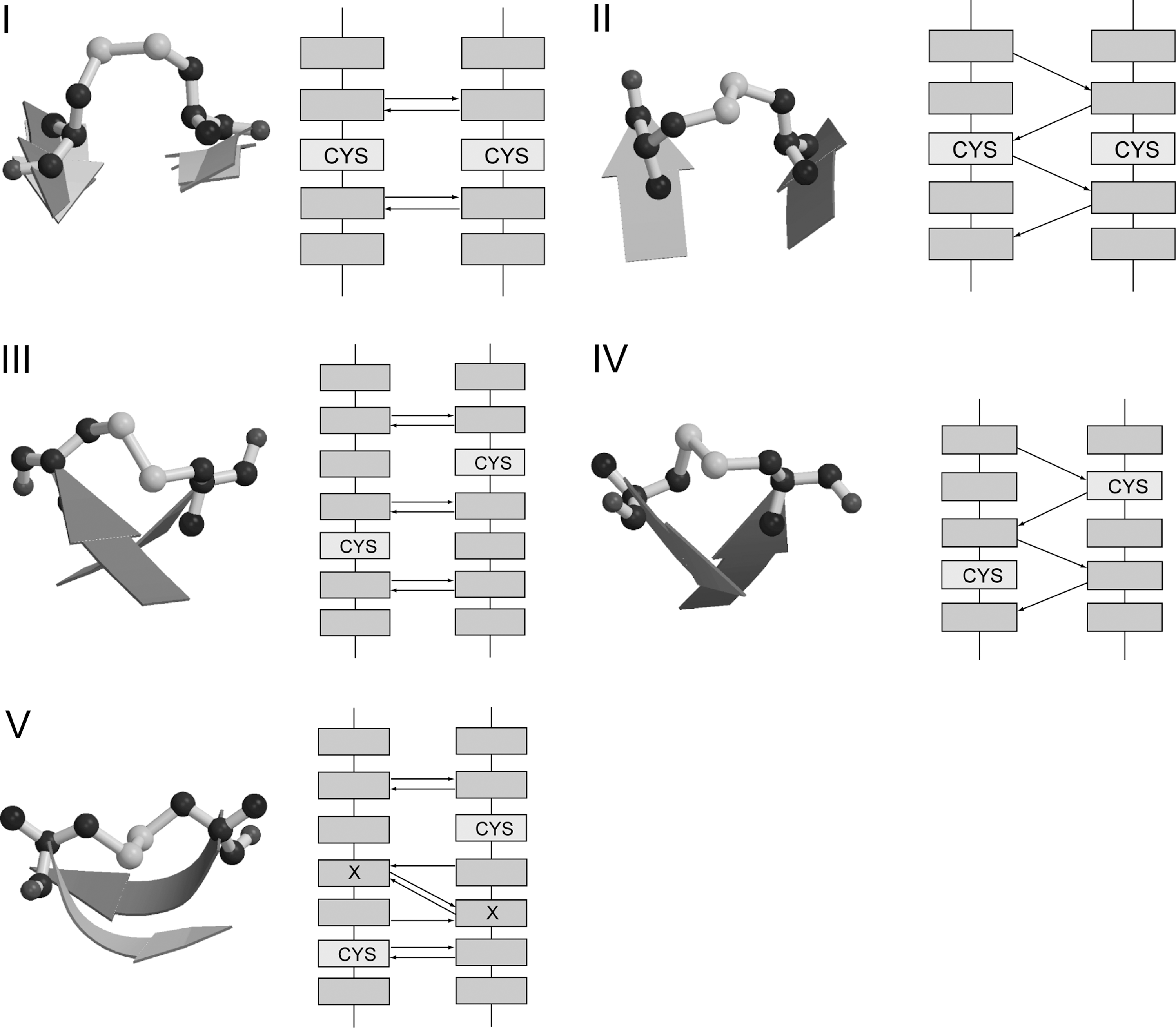
Richardson and Thorntons' original work remains valuable because their energetic analysis provides a basis for understanding disulfide switches and yields a valuable tool for finding redox-active disulfides in protein structures. To find other potentially redox-active disulfides, we searched for other disulfides that, like the CSD, violate Richardson and Thornton rules A-D (Fig. 6).
CSDs come in two flavors: antiparallel (aCSDs), which straddle antiparallel β-strands; and more rarely, parallel (pCSDs), which bridge parallel β-strands. aCSDs are by far the most common type of forbidden disulfide in solved protein structures. Parallel β-sheets are less prevalent in proteins than antiparallel β-sheets, but the rarity of pCSDs compared to aCSDs is likely to be dictated by the more rigid strut-like hydrogen-bonding in parallel sheets which restricts the amount of distortion that can be accommodated in the sheet (Fig. 7). As a result, pCSDs generally have higher torsional energies than aCSDs with modal torsional energies of 20 and 15 kJ·mol−1, respectively (293).
An example of a pCSD is found in Cdc25B, a phosphatase involved in the cell cycle. In Cdc25B, the forbidden disulfide switch acts directly to inactivate the enzyme. Oxidation of the catalytic Cys in Cdc25B is initiated by ROS to generate a sulfenic acid intermediate and proceeds to the oxidized disulfide through the glutathione buffering system (33). In the case of Cdc25B, the reactive Cys of the redox pair, Cys 473, is also the catalytic Cys in the phosphatase reaction, so cysteinyl modification by ROS directly inactivates the catalytic apparatus. The low pKa of the active site Cys, which enhances its role as the nucleophile in the catalytic reaction, also renders it liable to attack by ROS. This suggests a general mechanism of reversible inactivation of Cys-based enzymes (see Section VIII-C).
In addition to CSDs, several other types of forbidden disulfide straddle adjacent β-strands and thus violate Richardson and Thornton's first rule (RT rule A) (293). We refer to all forbidden disulfides that violate rule A as “Between-Strand Disulfides” (BSDs). Five previously identified types of BSD are shown in Fig. 7.
The β-Diagonal Disulfide (BDD) is linked between the ith residue in a strand and the j±2 residue on the adjacent strand, where j is the residue directly opposite i. In order to make the covalent linkage between the two Cys residues, the adjacent strands must adopt a high twist with respect to each other. The BDD is somewhat rarer than the CSD. Like the CSD, it comes in both antiparallel (aBDD) and parallel (pBDD) varieties, with the antiparallel type being more prevalent.
aBDDs have previously been described in several cytokine receptors such as the erythropoietin and interleukin-10 receptors (293), as well as in botrecin, a protein from the venom of the Brazilian viper Bothrops jararaca that mimics the binding of von Willebrand factor to the platelet receptor, glycoprotein Ib-IX-V, initiating thrombus formation (237, 293).
pBDDs are found in several Apo(a) and plasminogen kringle IV domains (293). Stathakis et al. showed the homologous disulfide, Cys 461–Cys 540 in kringle V of plasminogen, is reduced in the process of angiostatin production (246). Reduction of the homologous disulfide is also implicated in autocleavage of ovotransferrin, an iron binding protein abundant in egg albumen that likely has an important role in development (116).
A fifth type of disulfide that violates rule A is the β-flip disulfide. Cys pairs in a β-flip are disulfide-linked between the i residue in a strand and the j±3 residue on the adjacent strand, where j is the residue directly opposite i. In canonical β-sheet, residue side-chains sequential in the polypeptide chain decorate opposite faces of the sheet, alternating along the strand and forming rows perpendicular to the strand direction. Thus, residue i would normally only be on the same side of the sheet as j±2n residues on the adjacent strand, where n is a counting number. In a β-flip disulfide motif, the i and j±3 residues are positioned on the same surface of the β-sheet by the presence of two back-to-back classic bulges in the motif (223). Thus, a β-flip contains two types of canonical stress motifs: the forbidden disulfide and the β-bulge. Previously characterized examples of β-flips include one in Dengue virus fusion protein and another in retinol binding protein (293). Retinol binding protein is a carrier protein of the lipocalin fold that transports vitamin A, in complex with transthyretin, within the blood stream to target cells. Redl et al. suggested that reduction of a disulfide bond in a homologous protein, human tear lipocalin, is of functional relevance because the reduced protein shows a ninefold increase in ligand affinity (220).
D. Other canonical forbidden disulfides
Other previously characterized canonical forbidden disulfides that violate RT rule B-D are shown in Fig. 8. Forbidden disulfides that violate RT rule B, that disulfides should not be found joining Cys residues within a strand or a helix, include Inter-Helical Disulfides (IHDs) and Along-Strand Disulfides (293). Because canonical α-helices have 3.7 residues per turn (211), Cys residues that are three or four residues apart are nearby in the three-dimensional structure. A previously described example of an IHD is found between Cys 268 and Cys 272 in the R2 subunit of ribonucleotide reductase (293). No redox function has as yet been ascribed to this disulfide but the protein itself is well known for its redox activity.
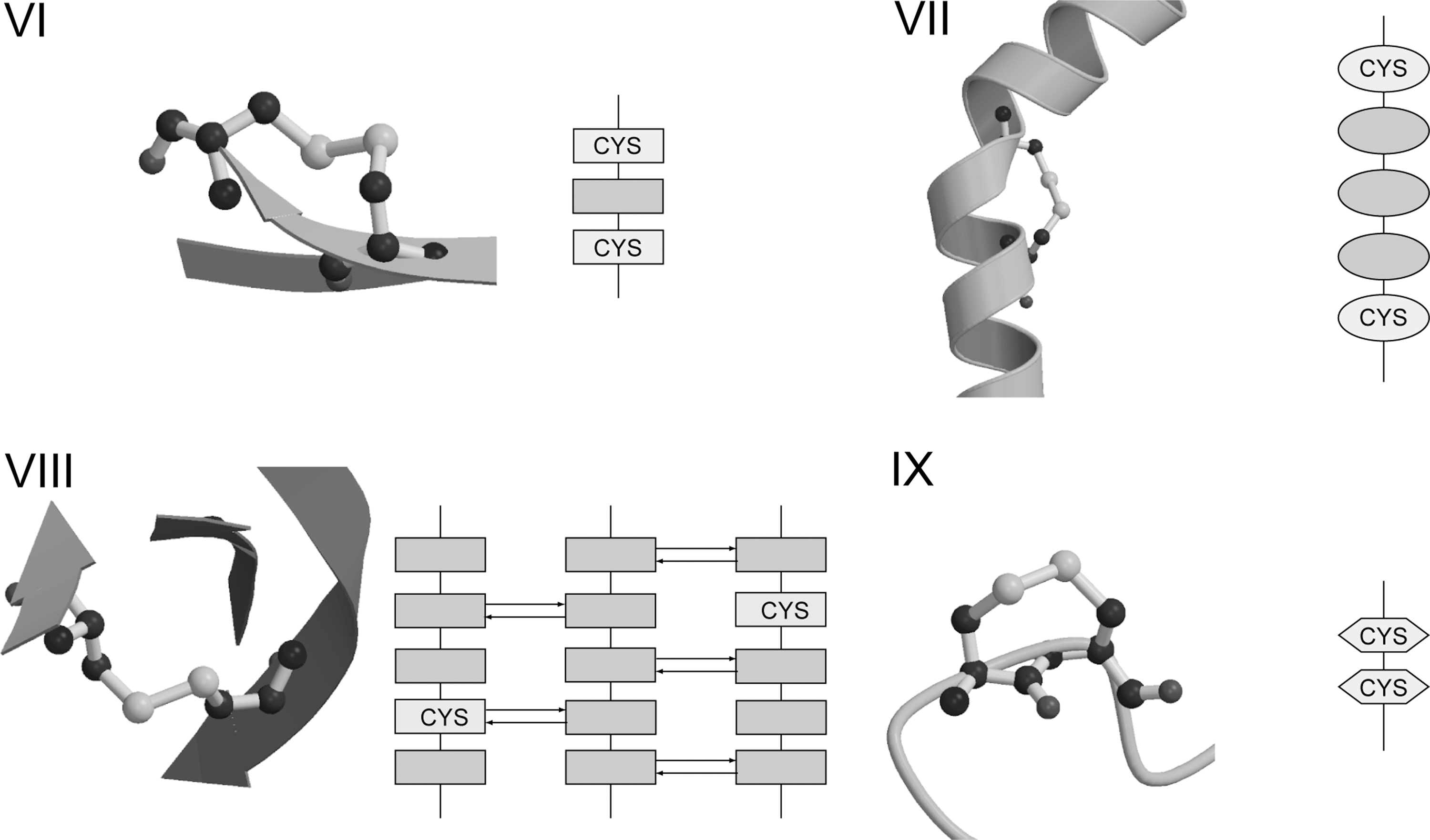
Other well known redox-active disulfides that resemble IHDs, but fail the strict criteria that both Cys, and all residues in-between, must exhibit α-helical Φ, ψ conformations are the catalytic disulfides in well-known thiol oxidoreductases. These include the CXXC motif of enzymes of the Trx family (179), and the i,i+5 motif of the disulfide oxidoreductase (DSOR) family (205). CXXC enzymes include Trx, glutaredoxin (Grx), glutathione peroxidase-like enzymes, as well as DsbA and C (179). In these enzymes the CXXC motif is located at the N-terminus of an α-helix. All residues of the motif are in α-helical conformation except the N-terminal Cys, which adopts an N-cap position [i.e., it is hydrogen-bonded in α-helical fashion but does not adopt helical backbone angles (91, 225)]. The Cys in the catalytic disulfide of the DSOR family are part of an i,i+5 motif (CXXXXC) rather than the i,i+3 motif seen in Trx-like enzymes. The redox-active disulfide is close to the C-terminus of the resident helix. Both Cys, and three of the four residues in-between, adopt helical conformations. The residue preceding the C-terminal Cys generally adopts a β-conformation giving the helix an appearance of being slightly unraveled. Disulfides in both of these motifs generally have high relative torsional energies, in excess of 10 kJ·mol−1 above the most stable conformation. The average relative torsional energy for the CXXC enzymes is 16.1 kJ·mol−1 for a set of 51 structures with oxidized disulfides (293), and the average for the DSOR enzymes is 22.4 kJ·mol−1 (14 structures) (293). Thermodynamic measurements show the disulfide bond of DsbA is so unstable in the folded state that its stability increased by 19 kJ·mol−1 when the protein unfolded (303). Given the resemblance to IHDs, it is possible these enzymes adopt higher energy disulfide conformations as part of their catalytic cycle.
Most Along-Strand Disulfides (ASDs) are found in i,i+2 motifs. The transferrin receptor contains a previously described example (18, 293). Transferrin is the major iron-transport protein in the blood. Cells requiring iron express the transferrin receptor. Iron-laden transferrin binds to its receptor at the cell surface and is internalized through clathrin-coated pits into endosomes. After unloading iron, apo-transferrin and the receptor are recycled to the cell surface. Using FRET imaging, Yang et al. recently showed reduction of disulfide bonds in the folate receptor occurs in an endosomal compartment (299). They were also able to demonstrate co-localization of the transferrin receptor in this compartment, and postulated similar redox activity occurs during trafficking of the transferrin receptor. Another example of an ASD is found in human monoamine oxidase A, an outer mitochondrial membrane-bound flavoenzyme that catalyzes the oxidation of the neurotransmitters serotonin, dopamine, and norepinephrine, generating peroxide as a byproduct. The ASD formed between residues Cys-321 and Cys-323 is located near the entry to the catalytic site (55). Although mutations of these residues did not influence catalytic activity (294), monoamine oxidase A is known to be quite sensitive to thiol oxidation and to thiol reagents (284), suggesting the disulfide may have a regulatory role.
The Jump-Strand Disulfide (JSD) violates RT rule C, that disulfides should not be found joining Cys residues within the same β-sheet. JSDs typically link Cys in the i and k±2 positions where i and k indicate strands hydrogen-bonded to opposite sides of an adjacent strand j. Linkages between i and k, k±1 or k±3 are also seen. Examples include the immune system protein defensin (293), a Cys-rich antibacterial peptide that has cytolytic properties. Bjorkhem–Bergman et al. showed mammalian Trx reductase (TrxR) alters the cytolytic activity of the defensin-related protein granulysin, enhancing its role in the killing of small cell lung cancer cells (23). Another interesting example is found in Scytovirin, a potent antiviral lectin isolated from the cyanobacterium Scytonema varium. Scytovirin has been reported to exhibit significant activity against human immunodeficiency virus (198). Two JSDs are found in the crystal structure: one between Cys 20 and Cys 32, and the other between Cys 68 and Cys 80. Each JSD resides within one half of an internally duplicated region. Interestingly, a disulfide connectivity different from the crystal structure is shown by nuclear magnetic resonance and mass spectrometry data (198), where Cys 32 & 38 and Cys 80 & 86 are paired as aCSDs, suggesting the disulfides isomerize between the CSD and JSD linkages. Further work needs to establish whether this is so. Finally, we recently uncovered a link between JSDs and Zn2+ binding which will be described in Section VI-A-2.
Vicinal disulfides, which violate RT rule D, are formed between two adjacent Cys residues in the primary structure. Formation of the disulfide reverses the direction of the protein chain and distorts the peptide bond between the two Cys residues. Carugo et al. recently reported the properties of vicinal disulfides and reviewed data supporting redox activity for this type of forbidden disulfide (39). A vicinal disulfide is formed as part of the reaction cycle in Drosophila melanogaster TrxR (130). In mammalian TrxRs (155, 305), one of the Cys residues is replaced by a more reactive selenocysteine that forms a vicinal selenenylsulfide during the reaction cycle.
In summary, forbidden disulfides are metastable disulfides likely to be redox active. Like CSDs, other forbidden disulfides can adopt different redox roles within proteins. They can be: catalytic as in the Prx Bacterial Comigratory Protein (BCP); or regulatory, controlling off-pathway events such as in Cdc25B (33), up and down regulating protein function, reversing reactions, or mediating protein function through allostery. Fourteen canonical subtypes have now been identified that disobey the Richardson–Thornton rules of disulfide stereochemistry. A summary is shown in Fig. 6. These classified subtypes likely represent >15% of high torsional energy disulfides. In addition to the previously well-characterized CXXC motif, forbidden disulfides represent a canonical set of redox-active disulfides.
E. Other forbidden disulfides
In addition to the 14 canonical types of forbidden disulfides reported to date, there are other disulfides that disobey the RT rules, but these novel types are currently fairly rare in solved protein structures (M. Wouters, unpublished data). Future growth of the PDB may see this situation change.
In addition, there are likely to be forbidden disulfides that introduce stresses in protein structures outside the Richardson and Thornton canon of disulfide stereochemistry. A possible example is found in the structure of the small Tim protein Tim10. The small Tim family specifically mediates the import of presequence-devoid polytopic proteins into mitochondria. All small Tim proteins contain a consensus twin CX3C Zn-finger motif (141). The motif forms a hairpin-like structure and in the oxidized form two disulfide bonds are nested in the sequence between Cys 40–Cys 65 and Cys 44–Cys 61 (3). Thus, the oxidized structure looks like a pair of tongs with two disulfides clipping it together in a closed position. When Tim 10 is translated in the cytosol, it is loaded with Zn2+. The role of Zn2+-binding is to keep the protein in its reduced, unfolded, and import-competent state in the cytosol, before it is imported into mitochondria. In a low resolution Tim9:10 complex (278), helical backbone angles in the vicinity of the inner disulfide of the pair are distorted in both Tim9 and 10, while all instances of the outer disulfide have torsional energies exceeding 10 kJ·mol−1 (M. Wouters, unpublished data). Thus, there is evidence of stress in the structure around both disulfides which may contribute to their redox activity if Zn2+ binding is reversible.
F. Folds associated with forbidden disulfides
Forbidden disulfides seem to favor some common folds. The Trx fold is well known for its CXXC motif, which can be considered a type of forbidden disulfide that violates RT rule B, see Section II-D (293). But the CXXC motif is not the only forbidden disulfide harbored by the Trx fold. Other forbidden disulfides include CSDs in two distinct 2-Cys Prxs. Prxs have been classified based upon the nature of the catalytic residues. During the reaction cycle, the peroxidatic Cys attacks the substrate, forming a mixed disulfide with it. In 2-Cys Prxs, the substrate is released by a second Cys known as the resolving Cys, whereas in 1-Cys Prxs the substrate is released by other means. In typical 2-Cys Prxs, the peroxidatic and resolving Cys are located in different monomers, whereas in atypical 2-Cys Prxs the two Cys that form the disulfide are within the same monomer. In the atypical 2-Cys Prx BCP, the catalytic disulfide formed between the peroxidatic and resolving Cys is an aCSD (133), whereas in the typical 2-Cys Prx from Aeropyrum pernix, an aCSD formed between the resolving Cys and a 3rd Cys within the same monomer is proposed to be an off-pathway event (193).
Previously, Martin showed that Trx, Grx, glutathione-S-transferase, DsbA and glutathione peroxidase all share the Trx fold (179). Three of these oxidoreductases: Trx, Grx, and DsbA, contain the CXXC motif. In glutathione peroxidase, the reactive N-terminal Cys of the motif is substituted by a more reactive selenocysteine and the C-terminal resolving Cys is replaced by threonine. In glutathione-S-transferase both Cys are absent, and the reaction is catalyzed by a tyrosine residue located nearby in the aligned sequences and positioned in three-dimensional space such that the substrate Cys residue docks in the same relative location of the Trx fold. Despite the differences, all these enzymes of the Trx fold interact with substrates that possess a thiol or disulfide group and have a common active site suggesting a common evolutionary origin (179). Sinning et al. proposed this common ancestor was glutathione binding (244). Copley et al. showed that Prxs also evolved from a Trx-like ancestor (50). They proposed an alternative classification of Prxs based on phylogenetic relatedness rather than reaction mechanism. Four subgroups of Prxs (Prx1–4) were identified using eight blocks of sequence identity, some of which are shared by several subgroups and others that are unique to particular subgroups. Five of these conserved blocks are also shared with cytochrome maturation proteins like Escherichia coli CcmG, thus providing the missing link between Prxs and Trxs (50). In Prx1s, the C-terminal resolving Cys of the CXXC motif is conserved but the N-terminal Cys is replaced by Ser or Thr. A subset of Prx1s has an additional novel Cys located five residues C-terminal to the remaining Cys of the CXXC motif. In this subgroup, which includes BCP, the two Cys are embedded in strands and form an aCSD. In Prx2s the additional Cys is located in the central insertion.
Another common fold harboring forbidden disulfides is the immunglobulin (Ig) fold, including the related fibronectin III domains. CSDs are found in Ig folds in the N-terminal domain of the E. coli folding protein DsbD (79), numerous cytokine receptors (293), immune molecules like CD4 (181), and proteins that mediate cell adhesion like the E. coli pilus-forming protein PapD (120). Evidence of redox activity is given in the associated references. The CSDs do not occupy homologous positions in these molecules, suggesting they evolved independently many times.
Given the repeated independent evolution of these elements, the question arises, “Why are the same folds regularly involved?” Possibly, these folds are reused because they are associated with promoter elements that are governed by redox-regulated transcription factors and are thus amenable to positive selection of new redox-sensitive switches. The introduction of potential disulfide-forming thiol pairs may be facilitated by the fact that both Cys do not need to be introduced in the protein chain simultaneously. Incorporation of a single Cys may make the protein immediately responsive to a range of oxidative modifications, including nitrosation and glutathionylation. Introduction of a second Cys at a later stage may then enable disulfide formation. An example where one protein is glutathionylated and a homolog forms disulfides is the Grx–Trx pair. Trx forms a disulfide between Cys residues of the CXXC motif as part of its reaction cycle. Grx contains only the reactive N-terminal Cys that is glutathionylated as part of the reaction cycle. Another example is the 1-Cys and 2-Cys Prxs. When the resolving Cys is present, Prxs are reduced by Trx. When it is not, they are reduced by Grx. In this way, protein regulation can switch from the Grx pathway to the Trx pathway.
G. Latency in bistable switches
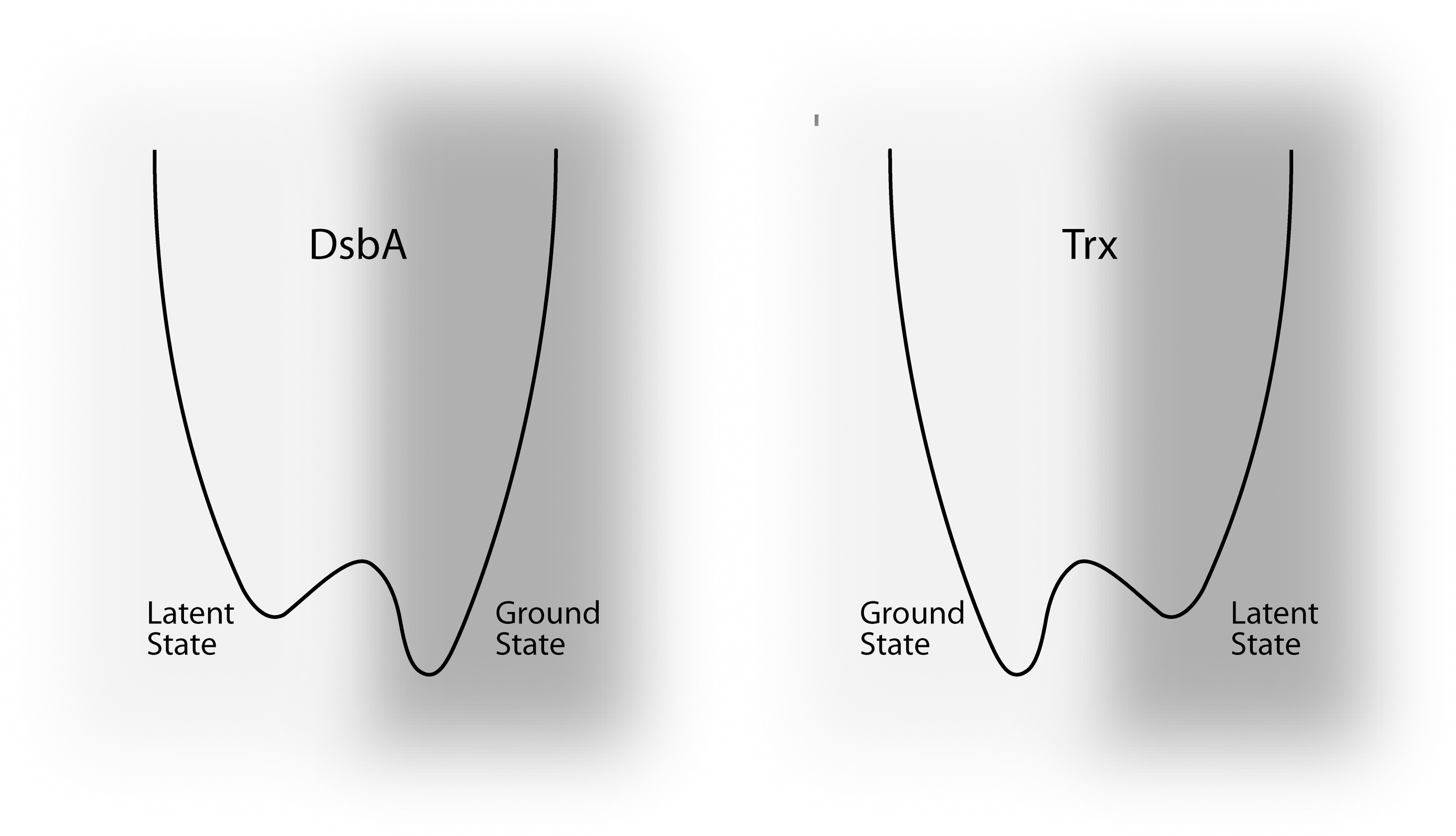
The latent redox state is an important consideration in the operation of a disulfide switch. This is the state in which the thiol pair spends most of its time under normal physiological conditions. As a result, it is normally the most populated state, and this is dependent on the normal redox conditions of the cellular compartment. Comparison of the redox potentials of thiol-pairs in proteins relative to the cellular compartment they occupy is a means of predicting whether a disulfide switch is latently oxidized, and would be reduced during the switching process; or latently reduced, and oxidized during switching. The two principal systems that maintain the cellular thiol/disulfide redox state are the glutathione (GSH) and Trx systems (194). These are buffered at different values in different compartments and are dependent on cell type and state. In the cytoplasm, the GSH/GSSG ratio is held between −260 and −200 mV, whereas the Trx1/TrxR1 system is buffered around the lower redox potential of −300 mV. In mitochondria, the two systems are buffered at still lower redox potentials, around −280 and −330 mV, respectively (77). To understand whether a particular thiol-pair is likely to be reduced, oxidized or left unaffected upon perturbation of the local redox state, its redox potential needs to be compared to that of the relevant buffers in the cellular compartment in question. For example, the cytosolic reductase Trx1 has a lower redox potential (−270 mV) than the cytoplasm (−220 to −240 mV) and hence acts as a reductant in this compartment. The redox potential of the strong oxidant DsbA is −124 mV (8, 80, 252), well above the periplasmic redox potential of −165mV (187). With regard to latent state, the cellular reductant Trx1 exists predominantly in the reduced form (277), whereas the oxidant DsbA is mostly oxidized (140).
However the latent state of these proteins should not be confused with the ground state (Fig. 9). The folded conformation adopted by the oxidized form of Trx is more stable than the dithiol form (166), that is, the protein is poised to return to its oxidized ground state, and like a mouse-trap, must continually be reset into a higher energy dithiol state to perform its function as a reductant. The opposite is true for DsbA which prefers to be reduced (296, 303), and must continually be pumped into its oxidized state to function as an oxidant.
As part of their normal function, redox-active disulfide switches undergo transient switching away from their latent states. However, the latent redox state of switches can be perturbed through modification of the buffered redox potential or disruption of the redox homeostasis system. Thiol oxidoreductases are not true catalysts. A true catalyst is unchanged by its reaction, whereas thiol oxidoreductases need an additional helper molecule to return to their latent state. Thus, the entire system acts as a catalyst rather than an individual protein. This is not the case for other oxidoreductases that transfer an electron from a cofactor to a substrate. Problems are caused in these systems by removal of components of the electron chain. For example, an E. coli strain lacking the enzyme TrxR is no longer able to regenerate Trx1 and 2, and the cells accumulate the oxidized forms of both (247). Similarly, in an E. coli strain lacking DsbD, DsbC is found in the fully oxidized state (226).
1. Latently reduced switches
Latently reduced switches (dithiols) occupy reducing compartments such as the cytoplasm. Disulfides can form in the cytoplasm under conditions of oxidative stress, which leads to a more oxidizing (positive) cell redox potential; or in response to localized generation of ROS. The latter process is likely to be important for normal tissue function both during development and in the adult organism. Different levels of ROS induce specific cellular processes. In general, low levels of ROS modulate homeostatic switches in proteins. Moderate levels are mitogenic or promote differentiation. Higher amounts of ROS favor growth arrest, while even higher concentrations activate apoptosis. Thus, the thresholds within proteins that effect these pathways must be set at appropriate levels so that the correct pathways are activated once a particular level of ROS is achieved. Excessive amounts of ROS cause necrosis.
Diurnal regulation of plant enzymes in response to varying light levels is the archetypal example of a homeostatic response (31). Adaptive responses include the activation of transcription factors. In bacteria such as E. coli, the transcription factor OxyR mediates transcription in response to oxidative stress by binding to ∼30 target genes. The yeast protein Yap1 performs an analogous role in eukaryotes. The formation of disulfides is transient and may be reversed by proteinaceous reductants such as Trx or by glutathione.
Oxidative stress responses are likely to be maladaptive when continually stimulated in diseases such as motor neuron disease, Alzheimer's disease, diabetes, heart disease, aging, and possibly graying of hair (7), where degeneration of the oxidative control processes is implicated. Finally, very high levels of ROS can induce apoptosis. This may be maladaptive but can also be an important programmed event during development. Selective apoptosis mediated by hydrogen peroxide governs the formation of substructures during embryo development (97). Expression and regulation of antioxidant enzymes in the developing limb suggest ROS mediates interdigital cell death (235). Motoneuronal death during spinal cord development is mediated by oxidative stress (233).
2. Latently oxidized switches
Latently oxidized switches are generally found in oxidizing compartments such as the endoplasmic reticulum (ER) and extracellular compartments. Reduction, or possibly isomerization, of disulfide bonds is important for transient switching in these compartments. Processes such as blood coagulation and development are likely to be dependent on transient switching of latently oxidized disulfide switches. In the case of membrane-resident proteins such as receptors and ion channels, redox regulation of disulfides on the extracellular surface may mediate signal transduction from the outside to the inside of the cell. A redox-active disulfide in cell surface Tissue Factor can switch the protein's function between coagulation and cell signaling (2). Research in this area is still in its infancy.
Factors influencing the reduction of latently oxidized switches include mechanical force such as twisting and stretching, as well as exposure to reducing agents. The ratio of [GSSG] to [GSH] is higher in oxidizing compartments: in the ER ∼1:1–3 compared to 1:30–100 in the cytosol, resulting in a redox potential of −230 mV. GSH reduces protein disulfide isomerase (PDI) and secretory proteins, allowing isomerization of disulfide bonds. Reduced thiol groups in PDI interact with ERO1p to be reoxidized. The ER is highly enriched with PDIs but it is generally assumed their action in the ER is confined to protein folding. This view is being modified by work such as that by Park et al. (208) which recently showed that PDI-mediated redox activity of the α2 disulfide of the MHC I complex optimizes peptide-binding in the groove by ejecting nonoptimal peptides. PDI's involvement in peptide loading is probably more accurately described as protein assembly than protein folding. The ER contains a plethora of PDI-like proteins whose exact functions have not yet been determined.
In the extracellular environment, Trx and PDIs have been shown to act as reductants. Trx is generally perceived as a cytosolic protein, but is also actively secreted by a variety of normal cells, including fibroblasts, airway epithelial cells, activated B and T lymphocytes, as well as transformed cells (57, 229). In addition, PDIs, like Trx, are found outside the ER and have been shown to modulate cytokine receptor signaling (2). Formation of a complex with the substrate via a mixed disulfide seems to occur.
The population of latently oxidized switches is perturbed under conditions of reductive stress. Reductive stress in the ER prevents formation of disulfide bonds and leads to an unfolded protein response (14). Hypoxia and the forced production of thiol-rich secretory proteins are postulated as principal causes of ER reductive stress. Reductive stress can also be maladaptive. The autosomal dominant R120G mutation in the human αB-crystallin gene causes a multisystem, protein aggregation disease including cardiomyopathy. Transgenic mice overexpressing cardiac-specific hR120GCryAB had a cardiomyopathy phenotype and were found to be suffering reductive stress. The redox potential of the glutathione buffering system is altered in the myopathic hearts. Increased recycling of glutathione from its oxidized form to its reduced state by augmented expression and enzymatic activities of glucose-6-phosphate dehydrogenase, glutathione reductase, and glutathione peroxidase was responsible (218).
III. Determinants of the Redox Potential
A. Role of primary structure in determining the redox potential
Experiments to determine how the redox potential of a dithiol pair is encoded in a resident protein have most extensively studied the CXXC motif embedded in the Trx fold. Evolutionary adjustment of the redox potential of the CXXC motif is quite remarkable: measured redox potentials for the CXXC motif in E. coli range from −287 mV for EcTrx to−95 mV for DsbL, a dynamic range of 182mV (Table 1). The CXXC motif was originally characterized as a latently-reduced switch. However, its frequent appearance in isomerases and its later characterization as an oxidant demonstrates its versatility and it has been described, in an evolutionary sense, as a molecular rheostat (43). The molecular basis of this variability has been extensively studied. The location of the CXXC motif at the N-terminus of a helix was proposed to contribute to the extremely low pKa of the reactive Cys via the helix dipole effect (144). The internal XX sequence is an important determinant of the disulfide redox potential (111). The effects of changing the XX residues in Trx and DsbA are opposite (195, 196). Trx becomes a less potent reductant, but DsbA becomes a less potent oxidant, suggesting the two oxidoreductases represent the extremes to which the redox potential motif can be tweaked by manipulation of the internal sequence. Within the same context, the redox potential is a function of the pKa of the catalytic Cys (80). However, the redox potential is not solely determined by the pKa and is also a function of pH-independent factors (43). These include the dihedral strain energy and other mechanical aspects of the polypeptide chain such as entropic factors.
Protein column: ¶CSD status based on homology to BCP. Disulfide column: *interchain disulfide; Redox Potential (RP) column: Method equilibrated against †DTT, §GSSG/GSH, ‡cysteine/cystine, °NAD/NADH, ^NADP/NADPH. Numerical superscripts equilibrated against substrate, Key: 1-Grx1; 2-Trx, 3-TrxPDI, 4-DsbDγ. Redox potentials of compartments are indicated as superscripts, Key: aperiplasm −165 mV (187); bchloroplast −330 mV<x<−280 mV (209); cIMS −255 mV (109); dER −180 mV (115); ecytoplasm −232 mV (115); fMM mitochondrial matrix −280 mV (134). Abbreviations: species: Bj, Bradyrhizobium japonicum; Bs, Bacillus subtilis; Ce, Caenorhabditis elegans; Ec, E. coli; Hs, Homo sapiens; Hv, Hordeum vulgare; Mm, Mus musculus; Nm, Neisseria meningitides; Pa, Pseudomonas aeruginosa; Pf, Plasmodium falciparum; Pt, Populus tremula; Sc, Saccharomyces cerevisiae; So, Spinacia oleracea; St, Salmonella typhurium; cyt c, cytochrome c; other abbreviations: IMS, intermembrane space, MM, mitochondrial matrix.
Although the structures of many thiol oxidoreductases have been solved in both oxidized and reduced forms, the dynamics of their reactions are hidden from us. The reaction mechanics are likely important determinants of the redox potential (286). Two steps in the reaction need to be considered: formation of the mixed disulfide accompanied by the first electron transfer; and resolution of the mixed disulfide accompanied by the second electron transfer. We need to dissect how mechanical and electrostatic effects contribute to each of these steps. In a protein that is latently oxidized, stress in the disulfide, which is increased by twisting or stretching upon encountering the substrate, should facilitate formation of the mixed disulfide. For example, the first residue of the XX sequence in both Grx and DsbA is proline, a residue conformationally-restricted by its ring structure to a greater extent than any of the other 18 amino acids with sidechains. The residue preceding proline is also sterically restricted in any polypeptide chain and this residue is the catalytic Cys in both these oxidoreductases. The most reducing DsbA mutant with a Pro–Pro sequence (80), has an extremely restricted conformational space. Entropic factors may also be important. For a latently reduced protein, conformational freedom of the active site thiols may favor its interaction with the more conformationally-restricted thiol in its substrate. The XX residues in E. coli Trx also include a proline, but in the second position, in which it only marginally modifies the large conformational space available to the sidechain-less glycine in the first position. In addition to these purely mechanical considerations, the electrostatic environment and its dynamics are important. Many of the CXXC oxidoreductases of the Trx fold have a histidine residue in the first or second position that likely stabilizes the nearby Cys residue as a thiolate (75). However, the internal residues are not the only important determinants of the redox potential. The internal residues of Grx1 and 3 are the same (PY) but the redox potentials differ by 35mV, giving some measure of the influence of other variables.
Similar internal residue preferences are apparent in ferrodoxin-thioredoxin reductase (FTR), the disulfide-reducing protein of chloroplasts. Several of the Cys ligands of the redox-active iron center are embedded in CXC motifs which feature internal Pro and His residues. The C55PC57 and C85HC87 sequences each donate their N-terminal Cys as ligands to the 4Fe–4S center, with the C74PC76 sequence donating the other two ligands. In the latent state, a disulfide is formed between the two free Cys, 57 and 87. In the C55PC57 motif, the peptide bond between the conformationally-restricted Pro residue and the mixed disulfide-forming C-terminal thiol adopts a higher energy cis conformation. After activation by receiving electrons from ferrodoxin, the disulfide is reduced and Cys 87 penta-coordinates the [4Fe–4S] cluster. In the first step of the reaction, Cys 57 forms a mixed disulfide with Trx to form the one-electron reaction intermediate. In the second half of the reaction, His 86 of the CHC motif moves towards the Fe–S cluster in response to the additional positive charge on the cluster, detaching the fifth Cys ligand.
The redox potentials of other forbidden disulfides in which the involved thiols are near each other in primary structure are also likely to be greatly influenced by the internal and flanking residues. These forbidden disulfides include IHDs such as CXXC motifs and the i,i+5 motifs in DSOR thiol oxidoreductases, as well as ASDs and vicinal disulfides.
B. Role of secondary structure in determining redox potential
For other forbidden disulfides, where the involved Cys are further separated in the sequence, secondary structure seems to play a major role in determining the latent state of the redox switches. In crystal structures, antiparallel CSDs are commonly oxidized while parallel CSDs are more often found reduced, suggesting the two types of CSD may favor different latent states. The two types of CSD also tend to occupy different cellular compartments. Many proteins containing pCSDs are cytosolic and thus likely to spend most of their time in the reduced state. Three pCSDs identified to date function in the cell cycle: a process known to be redox-regulated (33, 49). These include the eukaryotic cell cycle regulator Cdc25 phosphatase (33), and two enzymes involved in peptidoglycan synthesis in E. coli: glmU and murD (20, 30, 94). Thus, there seems to be a loose relationship between the forbidden disulfide motif, the cellular compartment, and the latent state of the switch (94).
Redox potentials of CSDs measured to date range from −229 to 325 mV (Table 1), but the data are very limited. Thus, there is overlap between known redox potentials of the CXXC motifs in the Trx fold and CSDs. It has previously been observed that the flow of electrons in redox pathways alternate between proteins of the Trx fold and proteins of other folds, or from a disulfide perspective, that CXXC motifs alternate with disulfides arranged otherwise. The CSD is an example of a disulfide arranged otherwise. One example is the DsbD pathway. The disulfide formed between Cys residues in the CXXC motif of the Trx-like DsbC is a substrate of thiols which form the aCSD in the Ig-like N-terminal domain of DsbD, and this in turn is reduced by the CXXC motif in Trx-like C-terminal domain of DsbD. Another example is the CSD in PrxQ, which is a known substrate of Trx y (47).
C. Influence of conformational changes and measurement technique
Disulfide torsional energies and hence redox potentials are dependent on the conformation adopted by the cystine. Redox-potentials may change upon docking a ligand. An example is the OOC-5 protein from the Torsin-family AAA+which contains a critical disulfide adjacent to sensor-II that couples redox state to nucleotide binding. The equilibrium redox potential of OOC-5 is −210 mV in the absence of nucleotide. However, when ADP was present, the redox potential decreased from −210 mV to less than −240 mV (306). In the absence of nucleotide the redox potential of OOC-5 is very close to that of the lumen of the ER under normal conditions, enabling it to respond to the changes in ER redox state. It has been proposed that the Torsin-family AAA+ OOC-5 functions during oocyte or early embryo development as an integrator of redox and nucleotide concentrations: two key parameters reflecting the metabolic status of the cell.
For proteins where conformational changes affect disulfide redox potential, the measured redox potential may be dependent on the measurement technique. Two methods commonly used to determine redox potentials are titration against small molecular weight compounds such as DTT and GSH/GSSG, and equilibration between proteins (8). For proteins where large conformational changes occur in the reaction mechanism, the protein equilibration method should give more accurate results than titration against small molecular weight compounds. However, this may only be true if the wild-type redox couple is used. This might explain large discrepancies in redox potentials measured for proteins like DsbB (Table 1).
Measurement of disulfide redox potentials is complicated further if disulfide cascades are involved, as is the case for DsbB and Ero1. These additional disulfides may act as step-down transformers between the redox potential of the inorganic molecule and the substrate. A similar principle has recently been demonstrated for the [4Fe–4S]2+/3+ redox couple of FTR. The redox potentials of both the [4Fe–4S]2+/1+/[4Fe–4S]2+/3+ couples of the [4Fe–4S] cluster differ substantially from those of the electron donor ferrodoxin and the electron acceptor Trx. An additional Cys residue (Cys 87), which transiently forms a penta-coordinated intermediate, lowers the redox potential of the [4Fe–4S]2+/3+ redox couple from +420 to −210 mV, close to the redox potential of the Trx active-site disulfide (53). It is likely a similar mechanism operates for the electron transfer between ferrodoxin and the [4Fe–4S]2+/1+ redox couple of FTR. Alternatively, intraprotein disulfide isomerization has been proposed to act as a kinetic driver of thermodynamically unfavored reactions (127). The redox potential of DsbA(−124 mV) is higher than the redox potential of the 104–130 disulfide of DsbB (−186 mV). After formation of the mixed disulfide between Cys 30 of DsbA and Cys 104 of DsbB, intraprotein electron transfer by reduction of the 41–44 bond of DsbB has been proposed to occur faster than the interprotein reverse reaction of reduction of the DsbA disulfide. Finally, it is possible that the redox potentials of DsbB have still not been accurately measured. If the redox potentials of the disulfides being measured are dependent on activated states of the other disulfides in the system, measurement of the redox potentials of single disulfide mutants (81, 221) may not give accurate results.
IV. Switching of Redox-Active Disulfides
The modes by which redox-active disulfides are reduced are slowly being established. Stretching and twisting of the bond can enhance reduction. For example, DTT-mediated reduction of disulfides can be accelerated by applying an external stretching force to the bond (285). However, applying a similar external force to Trx lessened its ability to reduce disulfide bonds (286). Within the context of a thiol-oxidoreductase the force must be applied discriminately so that it aids and does not hinder the mechanics of the reaction. Different mechanisms will be in competition for a nucleophilic addition on the disulfide. Depending on the angle of attack, an SN2 reaction may be favored over disulfide-thiolate exchange (11, 12).
Little is known about formation of forbidden disulfides during folding or function. Given that forbidden disulfides store energy in their protein chains, it seems unlikely they would form spontaneously in a fully-folded protein. We speculate they are regenerated by an oxidase as part of the dithiol/disulfide cycle. The scant evidence available points to a role for Trx-like proteins in oxidation. The aCSD in the oxidoreductase DsbD is oxidized by its substrates DsbC, DsbG, and CcmG (132). The aCSD-containing molecular chaperone PapD in E. coli mediates pilus assembly, but is unable to do so in a DsbA negative background (120).
Within a protein, autoreduction of a disulfide is possible if a conformational change brings a more reactive Cys or disulfide within proximity. Autocatalysis is also possible if a basic residue such as Lys or Arg is brought into the vicinity of a redox-active disulfide by a conformational change. This might be the case for integrin which has been demonstrated to have endogenous disulfide isomerase activity.
A. Cellular transport as a switching mechanism
Because different cellular compartments are buffered at different redox potentials, transfer of a protein from one compartment to another may be sufficient to reduce or oxidize disulfides. Cellular transport has already been demonstrated to be important in thiol-based redox switching for proteins entering endosomes. Using FRET imaging, Yang et al. recently showed that reduction of disulfide bonds in the folate receptor occurs in an endosomal compartment (299). They were also able to demonstrate co-localization of the transferrin receptor in this compartment and postulated that similar redox activity occurs during trafficking of the transferrin receptor. A number of proteins containing forbidden disulfides including the transferrin receptor, retinol-binding protein, and numerous viral and bacterial cell proteins are trafficked through endosomes. Disulfides in viral and bacterial cell entry proteins likely co-opt the change in redox environment that occurs upon transit between cellular compartments as part of their cell entry strategy (292, 299).
In eukaryotes, nucleocytoplasmic transport is also implicated in disulfide switching. Early work suggested alternate mechanisms exist in bacteria and eukaryotes for control of transcription factors mediating the oxidative stress response. In E. coli, formation of disulfides in OxyR controls the oligomeric state of the protein, and thus its DNA-binding activity. In yeast, DNA binding by the analogous transcription factor Yap1 is not affected by oxidation (148). Instead Yap1 regulates the oxidative stress response via a completely different mode involving nucleocytoplasmic transport. Upon cytoplasmic oxidative stress, an intramolecular disulfide is formed masking a nuclear export signal. Upon the basis of these experiments, it was postulated that bacterial redox mechanisms work directly to regulate proteins, whereas in eukaryotes nucleocytoplasmic transport is used to control transcription factors instead. Given that other eukaryotic proteins have been shown to be directly redox regulated (123), it seems unlikely that eukaryotic transcription factors should be exempt from this direct mode of redox control. Both mechanisms were later shown to control the eukaryotic transcription factor Hmgb1 demonstrating both mechanisms are utilized in eukaryotes (106), a far more likely scenario from an evolutionary point of view. Thus, nucleocytoplasmic transport is an additional mode of redox control that has been added in eukaryotes.
B. Redox gradients at the organismal level
At the organismal level, temporal transitions of redox conditions which occur during mammalian embryonic development are related to the gradient of redox conditions that exist along the oviduct. Pre-implantation, the fertilized egg encounters increasingly more reducing conditions as it travels down the oviduct (93). Embryonic substructures are formed by a ROS-mediated program of apoptosis. Cell death activation during cavitation of embryoid bodies is mediated by hydrogen peroxide. Expression and regulation of antioxidant enzymes in the developing limb support a function of ROS in interdigital cell death (235). Motoneuronal death during spinal cord development is also mediated by oxidative stress (233).
C. Disulfide relays and cascades
Disulfide switches may have different set points allowing electron transfer through disulfide cascades. These cascades may occur either between proteins, as occurs in the Dsb folding pathways, or within a single protein, as occurs in S. aureus ArsC. In all cases, the redox potential of the substrate must be higher than the electron donor so that a downhill flow of electrons occur. However, because redox potentials are dependent on the conformation of the disulfide, conformational changes of the protein can enable these cascades within a single protein. In addition, apparent “uphill” transfer of electrons may occur if a disulfide redox potential is ligand-dependent. However these proteins are not perpetual motion machines and the protein needs to be reset for the next cycle by the addition of energy through an exogenous factor. Although the term “cascade” is used for both intra- and interprotein electron transfers to indicate the downhill flow of electrons, we reserve the term in the following discussion for intraprotein transfers and use “relay” for interprotein electron transfers.
1. Disulfide relays
Because thiol oxidoreductases are not true catalysts, their reactions must proceed through disulfide relays. The Trx buffering system is well known. In bacteria and animals, Trx is reduced by TrxR with electrons donated by NADPH/NADH, while in chloroplasts of plants, Trx is reduced by FTR with electrons donated by ferrodoxin. Another well-known example of an interprotein cascade occurs in the Dsb folding pathways in E. coli. As previously noted, alternation occurs between CXXC motifs and disulfides arranged otherwise. In the DsbD pathway this alternation proceeds from a CXXC motif (DsbDγ) to an aCSD (DsbDα) back to a CXXC motif (DsbC/G).
2. Disulfide cascades
Well known examples of intraprotein disulfide cascades occur during the reactions of ribonucleotide reductase and thioredoxin reductase. In the class I ribonucleotide reductase of E. coli, a disulfide is first formed between Cys 225 and Cys 462 in the active site. Thiol–disulfide exchange transfers electrons to a disulfide on the surface of the protein between Cys 754 and Cys 759 which is ultimately reduced by Trx or Grx.
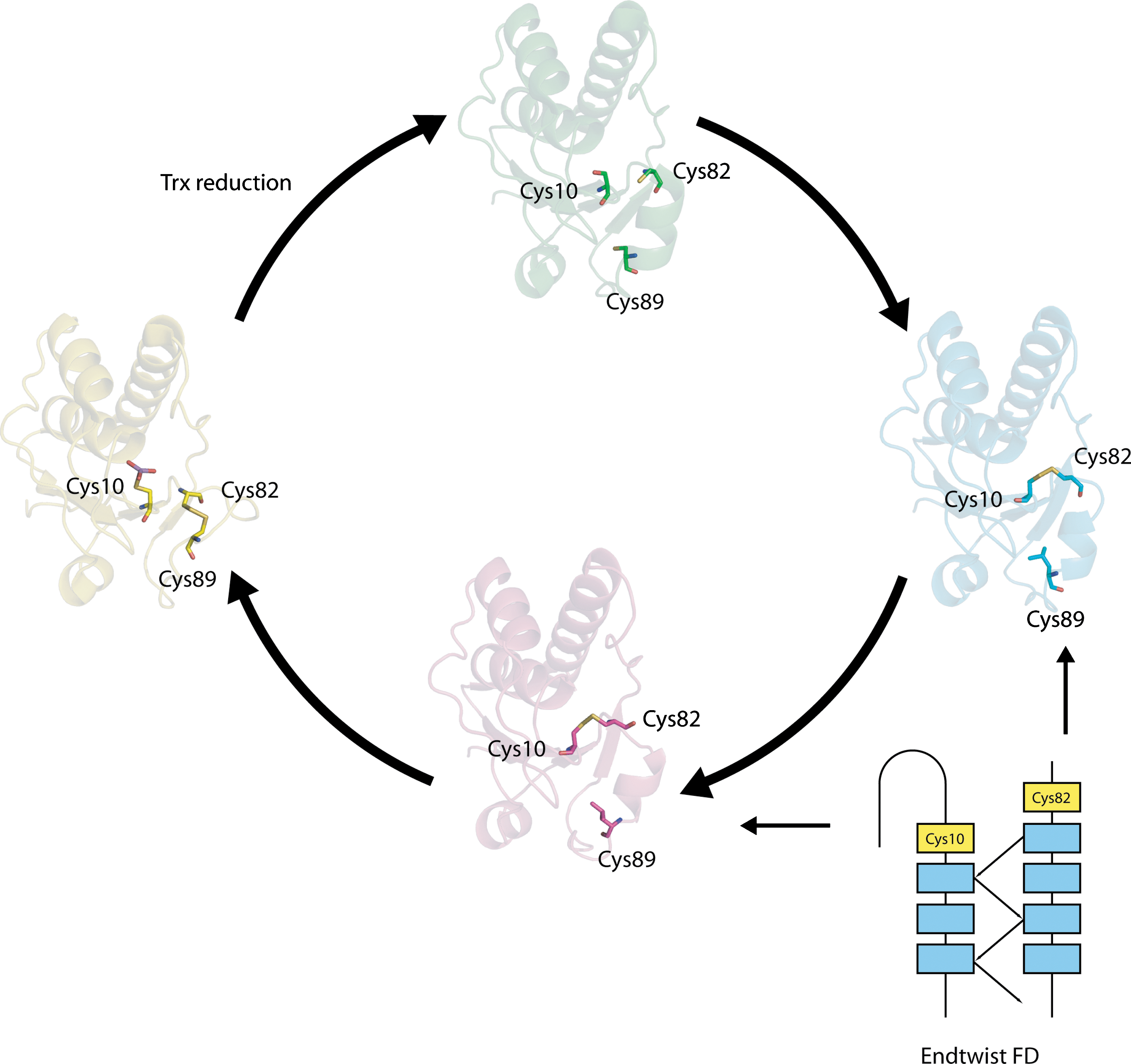
Another example of an intraprotein cascade occurs in Staphylococcus aureus arsenate reductase, in which stepwise formation of the Cys 10–Cys 82 and Cys 82–Cys 89 disulfides form part of the reaction cycle to detoxify arsenic (186). Upon activation, a forbidden BSD forms between Cys 10 and Cys 82 that conforms to an End-Twist motif (N. Haworth, unpublished results). It initially likely adopts the lower energy conformation present in chain A of the structure (Fig. 10) with a relative torsional energy of 14.9 kJ·mol−1 (95). Recondensation of additional helical structure at the N-terminus of the helix comprising residues 82 to 97 likely twists the carboxylate of Cys 82 into the higher energy disulfide conformation found in chain B with a relative torsional energy of 24.0 kJ·mol−1, rendering it susceptible to attack by the nearby Cys 89 thiolate. The subsequently formed Cys 82–Cys 89 disulfide adopts a conformation with a lower energy of 12.2 kJ·mol−1, and is sufficiently stable that it must be reduced by Trx in order to regenerate arsenate reductase for the next reaction cycle.
An example of a protein containing multiple disulfide switches which may communicate by allostery is the sarcoplasmic reticulum calcium pump SERCA. SERCA1a pumps Ca2+ from the cytosol of skeletal muscle cells into the sarcoplasmic reticulum store: terminating the process of muscle contraction. Four key states in the reaction cycle have been structurally characterized (Fig. 11). In the Ca2+-binding E1 state both disulfides are reduced. In the E2P state in which Ca2+ is released into the lumen, a low torsional energy disulfide (∼8 kJ·mol−1) is formed on the lumenal side of membrane between Cys 876 and Cys 888 (Fig. 11). In the E2P* state, a second disulfide is formed between Cys 636 and Cys 675 in the cytosol. In this state, the torsional energy of the Cys 876–Cys 888 disulfide increases. Both E2P* disulfides have extremely high torsional energies of ∼40 kJ·mol−1. Disulfides with such high energies may be reduced mechanically without the aid of an exogenous reducing agent or reductase. On transiting to the E2 state, the Cys 636–Cys 675 disulfide is reduced and the torsional energy of the Cys 876–Cys 888 disulfide decreases to ∼25 kJ·mol−1.
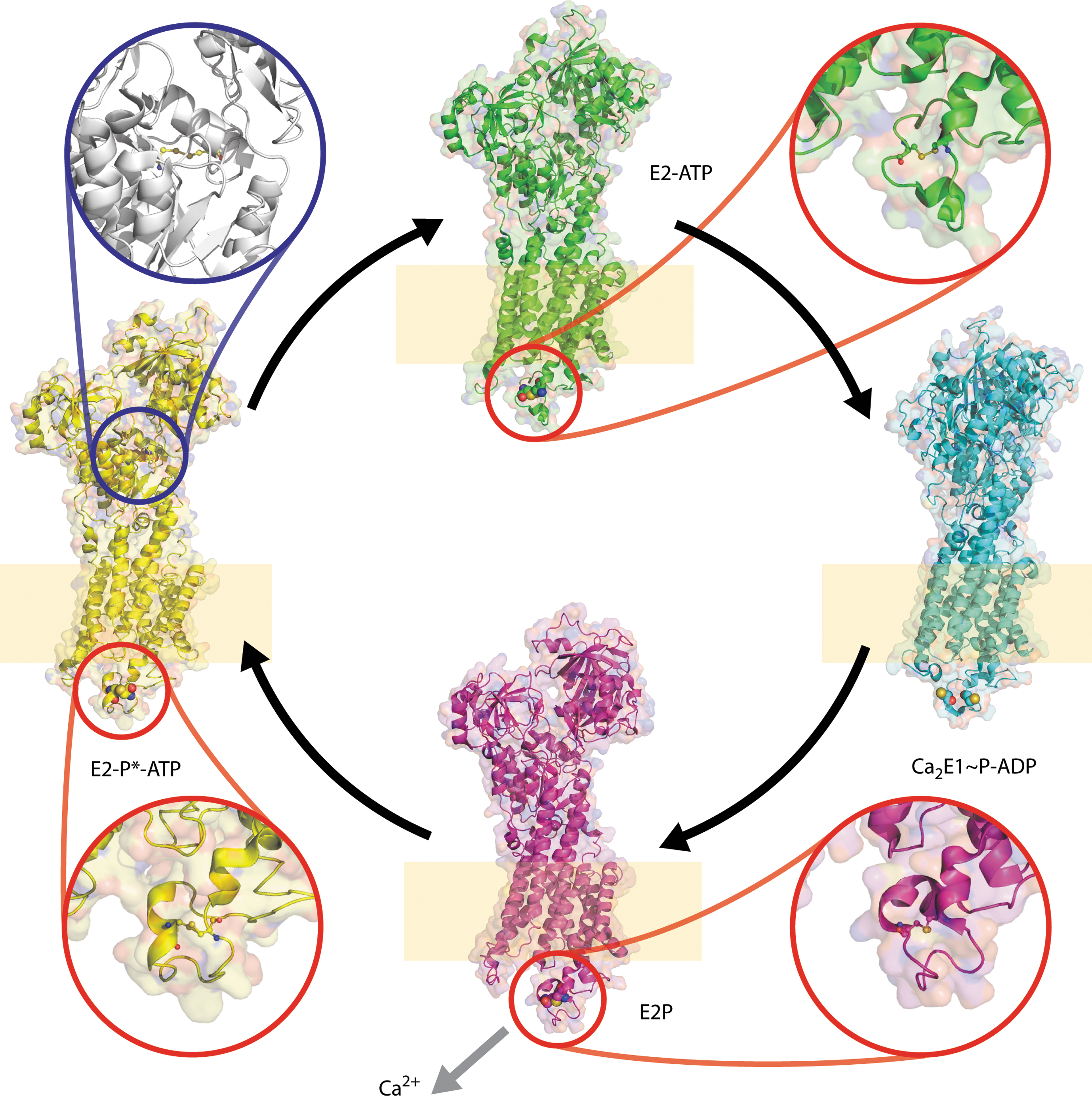
It is unclear whether these disulfides are physiologic and if so, part of the reaction cycle or off-pathway events. Redox regulation has been demonstrated for the homologous protein SERCA 2b which is expressed in all tissues at low levels. Previous work showed ERp57 modulates the redox state of ER-facing thiols in SERCA 2b in a Ca2+-dependent manner, providing dynamic control of ER Ca2+ homeostasis (164). In addition, Cys 674, homologous to Cys 675 in SERCA1a, can be glutathionylated following modification by NO, and Cys 674 and Cys 875, are sulfonated in atherosclerosis.
3. Ramping and alternate pathways
It has been proposed that some thiol-based switches may be multistate rather than simple binary (ON/OFF) switches. Disulfide formation may only be one option of an alternate pathway, or may be the ultimate step in discrete ramping of oxidation (204). For the redox-sensitive transcription factor, OxyR, formation of a disulfide between Cys 199 and Cys 208 is only one potential modification. Cys 199 can be differentially oxidized depending on the extent of redox imbalance and the nature of the oxidizing molecules. Kim et al. showed S-hydroxylation, S-nitrosation, or S-glutathionylation of Cys 199 could produce discrete outcomes of gene expression in vivo. Alternate modifications of the Cys residue differentially affect DNA binding which is dependent on cooperative effects between OxyR subunits (136).
D. Irreversible redox switches
In addition to the latency of the switch, another important variable is the frequency of use. Switches can be reversible or single-use switches. Conformational constraints imposed by primary and secondary structure influence the reversibility of the redox switch. For some forbidden disulfide types, the primary structure of the polypeptide chain provides a basic constraint on the conformational space that must be searched in order to reoxidize the reduced thiol pair. These include CXXC motifs, CXC motifs such as those found in ASDs, and vicinal disulfides (CC motif ). Forbidden disulfide motifs associated with ordered secondary structures may provide additional constraint on the polypeptide chain. For example, the CXXC motif in a helical context is additionally constrained by the hydrogen bonds of the helix. The majority of CSDs which straddle β-hairpins covalently link Cys residues which are nearby in sequence. On the other hand, when disulfides are formed between Cys residues distal in the primary structure, the polypeptide chain provides little constraint. Long-range forbidden disulfides rely more on their context secondary structures for stability. If these constraints are of low energy, for example, constituting only a few hydrogen bonds, their disruption may cause an irreversible conformational change in the protein. By this we mean that without the help of folding chaperones, it is improbable the protein could attain its former conformational state.
Common traits associated with proteins that undergo irreversible redox-controlled conformation changes are the long range nature of the covalent linkages involved and cleavage of the polypeptide chain. Nicking of the chain may increase the redox potential by increasing the entropy of the chain (292). An example is the activation mechanism of matrix metalloproteases. Matrix metalloproteases are synthesized as zymogens. The pro-peptide contains a Cys ligand which contributes to tetrahedral coordination of Zn2+. Upon cleavage of the pro-peptide, the resident Cys ligand releases its hold on the Zn2+ and is replaced by water, activating the enzyme.
Viral entry proteins contain both long-range forbidden disulfides, likely to be irreversible, associated with cleavage sites; and short-range forbidden disulfides, such as those on β-hairpins, which are likely to be reversible. What drives these large irreversible conformational changes? Work on influenza hemagglutinin provides some illumination. Influenza A hemagglutinin (HA) is expressed as a single chain. During transit from the ER, the polypeptide chain is cleaved in two. The cleavage site is straddled by a forbidden disulfide between Cys A4 and Cys A466 (B137 after HA cleavage). The two halves of the polypeptide chain are linked only by the single covalent bond of the forbidden disulfide after cleavage. The two-chain HA is in a metastable state until it is exposed to low pH, whereupon the smaller C-terminal chain spontaneously adopts a new conformation (34). It appears, therefore, that during folding, the larger N-terminal chain kinetically traps the C-terminal chain in a non-native fold. Exposure to low pH tips the balance in favor of the native fold which is adopted spontaneously. This scenario is supported by the observation that the smaller chain adopts the low pH conformation when it is expressed in the absence of the larger chain. Although the low pH structure is often thought of as the ground state, further conformational changes are implicated during fusion of the viral and host membranes. The protein appears to transit through a series of metastable states before reaching the ground state. Another way to view this is that the forbidden disulfide forms a lock on regions of protein structure of higher potential energy. Reduction of the forbidden disulfide releases this stored energy, which is used for conformational changes that trigger the action of the protein. Again this notion seems to contravene the thermodynamic hypothesis of Anfinsen (5) that proteins spontaneously adopt a minimally frustrated fold. Anfinsen's discussion of insulin demonstrates he realized the importance of cleavage of the protein chain to irreversibility in folding.
Analysis done on disulfide formation during folding of HA is also illuminating. Disulfides are formed in HA in the ER during the folding and assembly process. In HA, six disulfides are formed: four between Cys that are nearest neighbors in the sequence; and the other two between Cys that are distal in sequence. Both disulfides formed between the distal Cys pairs are forbidden. N-glycosylation plays a role in selecting these two particular disulfide bonds for special treatment (54). A glycosylation site proximal in sequence to the N-terminal Cys of each thiol pair recruits calnexin or calreticulin, and the disulfide linkage is formed by the PDI-like protein ERp57. Calnexin is specifically involved in the formation of the aCSD between Cys A4 and CysA466 (B137 after HA cleavage), whereas calreticulin targets the pCSD formed between Cys A47 and Cys A274. It is not clear whether ERp57-calnexin/calreticulin system targets these two disulfides because they are forbidden, or merely because they are the only two nonlocal disulfide linkages in the chain. Several other substrates of the ERp57/calnexin/calreticulin system contain forbidden disulfides, but the association has not been investigated at the level of specific disulfides. These include HIV gp120, Semliki forest virus E1, integrins, and possibly RNase B. For all these proteins, glycans influence choice of disulfide partners and the disulfide isomerase ERp57 is recruited by calnexin/calreticulin (54).
E. Occult sites
Solvent accessibility is required to enable access by the reductant whether it is ROS, glutathione, or an exogenous protein molecule. In some cases such as the engineered redox sensitive GFP, the disulfide always remains solvent accessible and such cases are straightforward to study. However, in other cases the disulfide may be protected from solvent and a conformational change is required to expose the disulfide for reduction. Examples include: the CSD in influenza HA, which is exposed during a pH-dependent conformational change (292); the CSD in CD4, which has been proposed to be solvent exposed by the process of domain swapping; and the CSD in DsbD, where a cap structure normally protects the disulfide (79). Systematic approaches for recognizing these occult sites are necessary.
V. Conformational Changes in Redox Transitions
Distinct, and sometimes spectacular, conformational changes can be associated with disulfide formation/reduction during protein function. Study in this area is difficult because X-ray structure solution only gives a snapshot of a protein structure. Conformational changes must be inferred from multiple independently-solved structures in different redox states. Unfortunately, circumstances have conspired to thwart the solution of multiple redox states. Granting agencies and journals discourage crystallographers from solving structures twice unless there is an overwhelming biological reason to do so. In addition, crystallographers often remove “troublesome” Cys residues from proteins of interest by mutation to improve their handling during purification and crystallization. The wayward behavior of these Cys residues is likely to be linked to their redox-activity. Nonetheless, some themes have begun to emerge from individual studies, but more work is required in this area.
A. Redox-associated morphing transitions
For some proteins, disulfide formation and reduction is associated with large-scale plastic deformations of the polypeptide backbone (63). These transitions, which have been referred to as morphing transitions, typically involve unravelling of portions of secondary structure (200). The well-known redox-sensitive protein OxyR belongs to this group. In OxyR, a protein of 305 residues, significant refolding of residues 196–221 occurs between redox states (44). In the reduced structure, residues 200–204 form a short 310 helix flanked by regions of coil (Fig. 12). Upon oxidation, residues 196–221 adopt a different fold, with residues 189–193 and 213–218 forming two parallel β-strands joined by a loop with a disulfide linkage between Cys 199 and Cys 208. In the reduced OxyR structure, the two Cys that form the disulfide are separated by almost 13Å. Cys 199 in OxyR is known to undergo alternate modifications such as oxidation, nitrosation, and glutathionylation. Because of the spectacular nature of the transition between the reduced and oxidized structures, and the existence of alternate modes of redox regulation, the physiological relevance of the disulfide-linked structure has been called into question (136). From a theoretical standpoint, it seems unlikely that a nonphysiological state could be uniformly adopted by every monomer in the crystal as required for the structure-solution process. Nonetheless, it is easy to understand why people are uncomfortable with this notion as this also challenges cherished notions of a one-to-one mapping of protein sequence to fold put forward by Anfinsen.
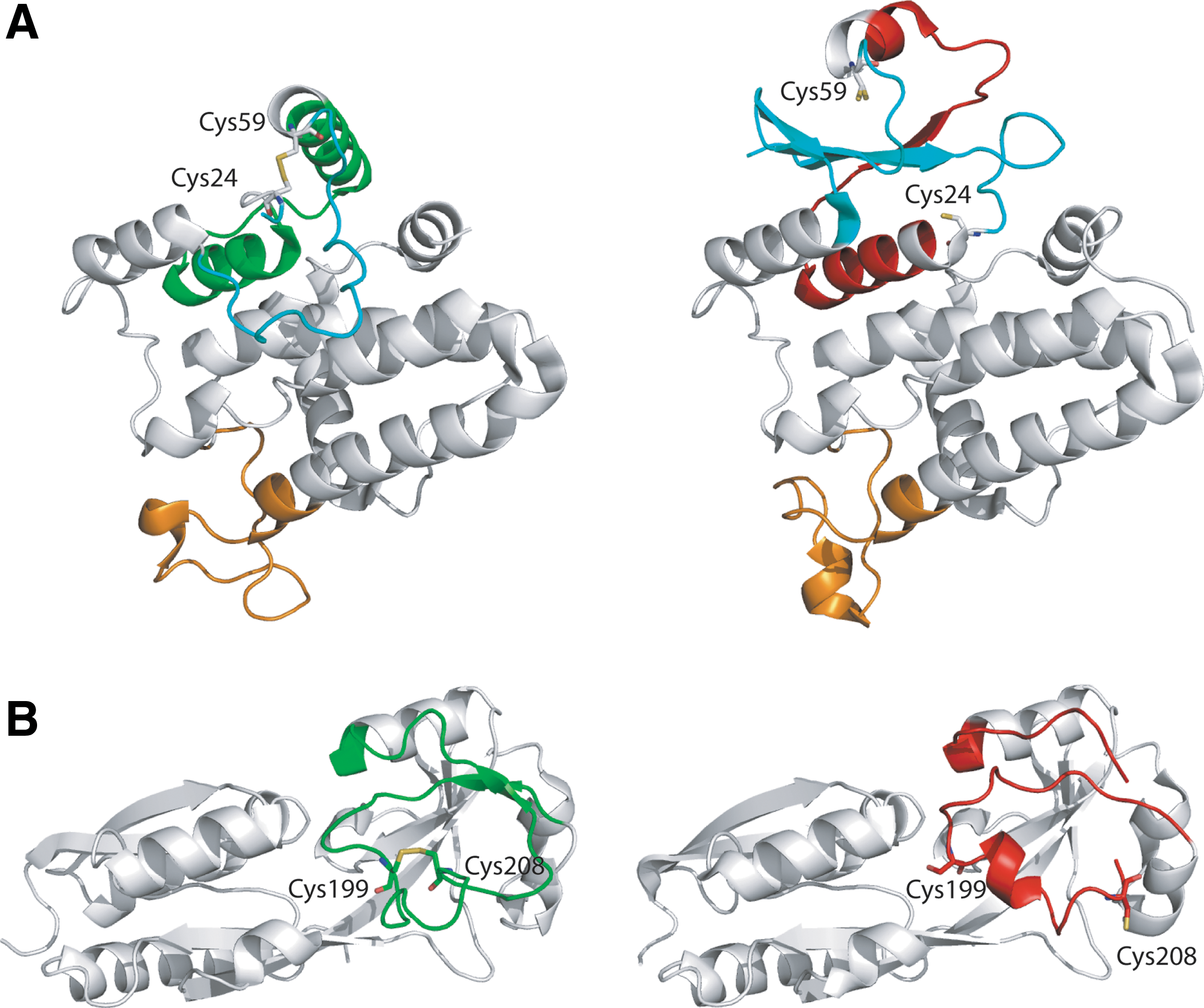
Another protein with an even more spectacular redox-associated conformational change is the chloride channel CLIC1: a protein that forms ion channels by inserting itself into membranes in response to oxidation (168). CLIC1 is homologous to soluble proteins of the glutathione-S-transferase superfamily. Unlike conventional membrane-resident ion channels, CLIC1 is synthesized without a leader sequence, and appears to reversibly insert itself into cell membranes. The latent form is the soluble reduced form which is present in the cytosol. Upon oxidation, a strand peels from a three-stranded β-sheet and adopts a helical conformation, destabilizing the remaining β-hairpin and forming a disulfide between Cys 24 and Cys 59. The sulfur atoms of these two residues are 13.5 Å apart in the reduced state. Reorganization of the CLIC1 subdomain promotes its incorporation into membranes. Like OxyR, the physiological relevance of the CLIC1 conformational change associated with disulfide formation is controversial.
Finally, as previously discussed, the detoxification enzyme arsenate reductase undergoes smaller multiple plastic conformational changes associated with a disulfide relay as part of its reaction cycle (95, 186) (see Fig. 10). The Cys residues of the second disulfide intermediate (Cys 82–Cys 89) are 10.7 Å apart in the reduced structure.
Large physical separation of the reduced Cys pair seems to be a common feature of many of the reduced states of morphing proteins and does not preclude disulfide formation. Possibly attack by a helper molecule, such as glutathione, may be required as an intermediate in disulfide formation to bridge the intervening distance. In the case of OxyR, glutathione is involved in returning the protein to its original state. Large physical separation of partner Cys residues may represent an example of negative design against inadvertent disulfide formation under mild oxidizing conditions.
B. Redox regulation of the quaternary state
Redox regulation of the quaternary state has previously been associated with cooperative behavior in proteins. Oligomerization may mediate binding with large repetitive molecules like DNA, or may switch recognition of an endogenous ligand on or off.
The previously discussed conformational change in the OxyR monomer modulates its quaternary structure state. OxyR binds to RNA polymerase when oxidized, inducing gene transcription. Oxidized active OxyR has the same DNA binding affinity as reduced inactive OxyR. But whereas reduced OxyR binds only to two adjacent major DNA grooves, oxidized OxyR binds to four grooves and also binds RNA polymerase (149, 276). A similar mechanism is utilized by HoxB5, a protein involved in embryonic development. HoxB5 binds DNA in vitro when either oxidized or reduced, but formation of an intramolecular disulfide between HoxB5 monomers is necessary for cooperative binding (74).
An example of a different type of redox-regulated quaternary structure change is cytoglobin, a member of the vertebrate hemoglobin family. Cytoglobin, which resides in the nucleus and cytosol, is upregulated under conditions of hypoxia and certain types of stress. Although cytoglobin is a dimer in both the oxidized and reduced structures, the dimer interface changes between redox states. Modulation of the quaternary structure by intermolecular disulfide bonds may regulate the ligand-binding properties of cytoglobin (248). The homologous lamphrey hemoglobin is in an equilibrium state between a low O2 affinity oligomer and a high-affinity monomer which contributes to its cooperativity. Cytoglobin has recently been implicated as a tumor suppressor (241).
C. Redox switches, domain swapping, and amyloid formation
Redox processes may be involved in large conformational changes in proteins associated with protein aggregation diseases. Several common complex diseases such as Alzheimer's disease and type II diabetes are associated with deposition of amyloid fibrils but have not been proven to be caused by such deposits (215). Both of these diseases are associated with oxidative stress. In addition, familial forms of amyloidosis are associated with mutations in genes whose protein products form amyloid. Around 17 or 18 different proteins have been identified as being amyloidogenic (255) and redox-active disulfides are found in many of these.
Amyloid diseases are characterized by deposition of protein fibrils. Amyloid formation results from a conformation change in a normal protein because of mutation, aging, a change in the environment, or proteolysis. Although amyloid is formed by proteins of different folds, the proteins are believed to adopt a common cross-beta ultrastructure demonstrated by the ability to bind the histological dye Congo Red (24). Redox processes have been implicated in formation of amyloid. Reduction of disulfide bonds in the amyloidogenic Bence–Jones protein, α-lactalbumin, and lysozyme leads to formation of amyloid-like fibrils in vitro (138, 253). A post-translational cysteinyl modification has also identified in an amyloidogenic kappa 1 light chain protein (165).
Previous work noted a correlation between proteins that undergo domain swapping and amyloid (234). In domain swapping, two or more identical proteins exchange homologous parts of their structure to generate an oligomeric unit. Domain swapping may facilitate a number of biological functions. First, it allows proteins, which tend to be globular, to cooperatively bind large repetitive biological structures such as DNA and oligosaccharides. The intertwining of the polypeptide chains during polymerization probably serves to improve the structural integrity of the polymerized unit. This was recently established for human and chicken cystatins and appears to be a feature of the cystatin superfamily. Domain swapping may also regulate protein–protein recognition. Cystatin inhibits two families of cysteine proteases as a monomer. Upon formation of domain-swapped dimers, cystatin loses its inhibitory effect on C1 cysteine proteases because the inhibitory L1 loop is co-opted by the cross-over β-ribbon (1). The domain-swapped dimers are still inhibitory towards C13 cysteine proteases. Thus, the specificity of the inhibitor is modulated by the domain swapping process. Ligand binding can also mediate domain swapping. Diphtheria toxin domain swaps upon binding to its receptor (169).
Forbidden disulfides are common in proteins that domain swap, such as diphtheria toxin and cystatin, and proteins that form amyloid, including cystatin, gelsolin, the amyloid precursor protein, and serum amyloid component P (Table 2). Thus there seems to be an anecdotal three-way relationship between redox-active disulfides, proteins that form amyloid, and proteins that domain swap (Fig. 13). Not all proteins that undergo domain swapping contain forbidden disulfides but an appreciable number of them do, suggesting that redox switching of protein conformation may be one of several mechanisms used as regulators of domain swapping. Single reactive Cys residues may also be involved in redox regulation. Glutathione binding to Cys 60 regulates domain swapping in glyoxalase I (231).
APP, amyloid precursor protein; IL5, interleukin 5; KH97, the common beta subunit of granulocyte macrophage-colony stimulating factor, interleukin-3 and interleukin-5; Mup, major urinary protein; PrP, prion protein; SOD, superoxide dismutase; TrK, neurotrophin receptor; u, unclassified.
Domain-swapped dimer involves interchain disulfide.
An example of a domain-swapping protein that contains a forbidden disulfide and also forms amyloid is cystatin C, an endogenous cysteine protease inhibitor. Cystatin C is accumulated within amyloid-beta (Abeta) amyloid deposits in Alzheimer's disease brain as well as the hereditary disease Icelandic hereditary cerebral amyloid angiopathy; and is proposed to play a role in Alzheimer's disease pathogenesis. In both cystatin and another domain-swapping protein, the neurotrophin receptor TrK, the forbidden disulfide is on strands immediately adjacent to a β-strand that constitutes part of the swapped subdomain. Formation of the swapped dimer requires temporary disruption of the hydrogen bonds between one of the forbidden disulfide strands and the adjacent swapped strand. Redox regulation of the forbidden disulfide may be involved in the forward or reverse process. Biochemical evidence supporting this notion is available for cystatin. In vitro, the metastable monomer transits to the dimer with the help of pre-denaturing elevated temperatures or levels of denaturant. In vivo, reduction of the CSD has been implicated in the dimerization process. Formation of domain-swapped dimers in TrK occurs spontaneously on binding of its ligand. The BDD disulfide in TrK becomes more solvent exposed in the domain-swapped dimer, possibly allowing PDI or Trx to return it to its monomeric form (Fig. 14).
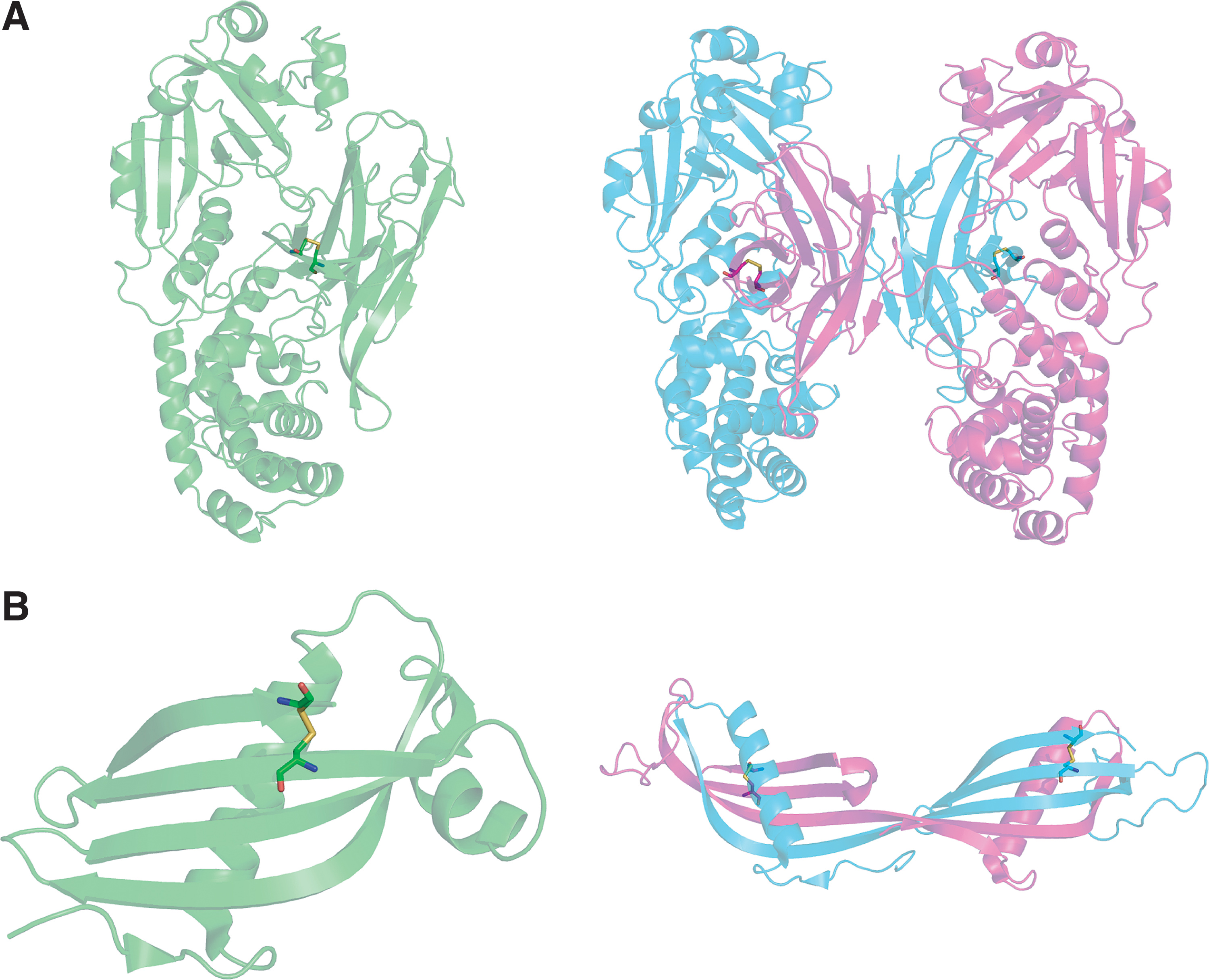
In Finnish hereditary amyloidosis, the extracellular form of the actin-interacting protein gelsolin forms deposits. The structure of gelsolin consists of six homologous repeats of the same fold but only the second domain forms amyloid (37), and it is this domain that harbours the CSD between Cys 188 and Cys 201. The CSD is conserved in mammals such as mouse, human, and pig, but is not found in lobster, Drosophila, or frog (281). It is close to a high affinity calcium binding site and calcium is required for oxidation of the disulfide to occur (281). The protein exhibits different susceptibility to proteases with and without the disulfide linkage (281). For proteins that form amyloid generally, redox-activity of the disulfide may expose a cryptic protease cleavage site which contributes to amyloid formation.
Redox processes have been implicated in conformational changes in the amyloidogenic prion protein (PrP). Recombinant hamster PrP is converted to a second form, redox PrP, by a redox process in vitro and this redox PrP form seeds the conversion of other PrP molecules to the redox PrP form (157). The converted form shows properties of oligomerization and seeded conversion that are characteristic of the scrapie form of PrP, the form of the protein found in amyloid deposits. Oligomerization can be reversed in vitro. X-ray fiber diffraction suggests an amyloid-like structure for oligomerized PrP. In crystal structures of domain-swapped dimers of PrP and major urinary protein, the disulfide straddles the hinge loop, the linker that joins the two swapping subdomains. Disulfide isomerization during the domain-swapping process is required to switch between the intrachain disulfide bond in the closed monomer and the interchain disulfide bond in the domain-swapped dimer.
In complex diseases, increased oxidative or reductive stress may overwhelm the redox homeostasis system and affect the ratio of domain-swapped dimers to monomers. In genetic disease, mutations in the vicinity of a critical disulfide may adversely change its redox potential, and if it is involved in domain swapping, impact the domain-swapped population. This seems to be the case for superoxide dismutase (SOD1) mutants associated with familial lateral sclerosis (FALs), an age-dependent degenerative disorder of motor neurons in the spinal cord, brain stem, and brain. SOD1 is largely localized to the cytosol but is also found in the intermembrane space of mitochondria. SOD1 is activated by formation of the disulfide and insertion of Cu by the copper carrier CCS. The conserved disulfide in SOD1 is essential for activity and unusual because it remains oxidized in the cytosol. Although the disulfide is intrachain, it is thought to play a critical role in stabilizing the SOD1 homodimer. Formation of the disulfide is part of the SOD1 maturation process which is regulated by physiological oxidative stress. 14 FALs mutants have the common property of being more susceptible to disulfide reduction than wild-type protein (261). Toxicity associated with disulfide-linked multimerization of SOD1 mutants has recently been implicated in the disease mechanism (73).
Not all proteins that undergo domain swapping contain redox-active disulfides. For example, barnase, calbindin, and cyclin-dependent kinase domain swap, but do not contain Cys residues. However, an appreciable number of domain-swapping proteins do contain redox-active disulfides, suggesting that redox switching of protein conformation may be one of several mechanisms used as regulators of domain swapping.
VI. Systems Biology Approaches
A. Phosphorylation, calcium, and peroxide
It has become evident that reduction and oxidation of disulfides is another post-translational modification akin to phosphorylation. Indeed, the two paths are linked. ROS inhibits a superfamily of protein tyrosine phosphatases (262, 263). Reversible oxidation of the catalytic Cys residue likely extends the timeframe of the kinase signaling response by delaying removal of the phosphate group (222). Thus, multiple signals are integrated by these phosphatases. In classical Tyr-specific phosphatases, ROS modification converts the catalytic Cys to a cyclic sulfenamide by reaction with the main chain nitrogen of the adjacent Ser residue: inhibiting the enzyme, preventing its further oxidation, and likely facilitating its conversion back to the active form (232, 271). Dual specificity phosphatases dephosphorylate Thr and Ser residues in addition to Tyr, as well as inositol phospholipids. Oxidation inactivates dual-specificity phosphatases by formation of a disulfide between the catalytic Cys and another nearby Cys. Examples include PTEN (156), Cdc25 (33), and PRL-1 (250) (Fig. 15). In all of these phosphatases, a forbidden disulfide is formed, but the type is not conserved across the entire family. The disulfide formed in Cdc25 is a forbidden disulfide of the pCSD variety (33, 293). The forbidden disulfide in PRL-1 and PTEN is a novel type of BSD known as an End-Twist (N. Haworth, unpublished data). A similar disulfide forms in Staphylococcus aureus ArsC as part of the reaction cycle.
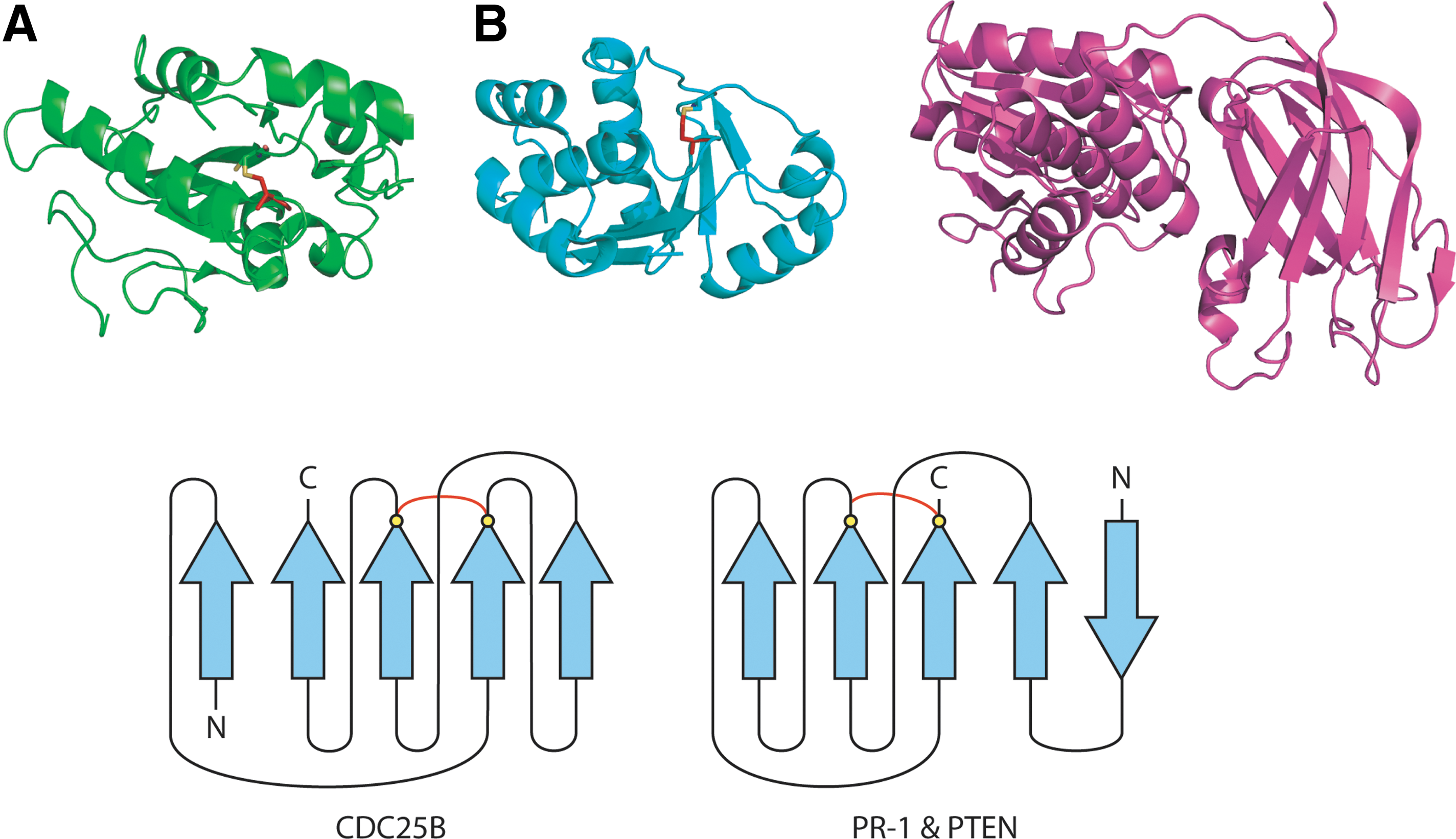
Ca2+ handling pathways are also linked to disulfide redox status (99, 105). For example, stimulation of the B cell antigen receptor induces oligomerization, phosphorylation, and activation of downstream signaling components such as phospholipase Cγ, which in turn generates inositol triphosphate (IP3) and mobilizes Ca2+ (243). The ryanodine receptor, a Ca2+ release channel located on the sarcoplasmic reticulum–ER, is also a redox sensor (249, 297, 302). The oxidized channel correlates with the open state, whereas the reduced form correlates with channel closure. The IP3 Receptor (IP3R) may also sense the redox potential because it contains Cys in the ER lumenal facing loop that lines the channel pore. Consequently, modulation of the redox potential of reactive thiols in the ER might be a general mechanism by which SERCA 2b, IP3R, and the ryanodine receptor control ER Ca2+.
Homeostasis of some physiologically important metals is also regulated by disulfides. The PDI homolog ERp57 has been proposed to modulate Ca2+ release and uptake in coordinate and opposite directions. In particular, oxidization appears to favor Ca2+ release by opening Ca2+ channels and inhibiting Ca2+ pumping. On the other hand, reduction seems to favor Ca2+ uptake by channel closure and increasing Ca2+ pumping. The combined effect of these two actions helps to minimize the loss of ER Ca2+ and maximize refilling of Ca2+ stores, thereby protecting physiological functions of the ER such as glycoprotein folding. Finally, Zn2+ homeostasis is also linked to disulfide redox status. This will be discussed in Section VI-B.
The redox state outside the cell is linked to the redox state in the cell by signaling pathways. Redox regulation of disulfides on the extracellular surface of membrane-resident proteins such as receptors and ion channels may mediate signal transduction from the outside to the inside of the cell. For example, forbidden disulfides are abundant in cytokine receptors (293). An example of this is cell surface Tissue Factor that can switch the protein's function between coagulation and cell signaling (2). Intracellular H2O2 is transiently generated upon activation of receptors for peptide growth factors and cytokines as well as integrin-mediated cell adhesion (222). In addition, ROS generated outside the cell can also directly affect intracellular responses. Phagocytic cells, such as macrophages, contain a NADPH oxidase complex that produces a burst of ROS as part of the innate immune response to infection. Peroxide-producing NADPH oxidases also produce ROS in other cells at lower levels (153). Peroxide can freely diffuse through cellular membranes, but it was recently discovered that extracellular H2O2 is actively transported into the cell by some types of aquaporin, a type of membrane-resident channel (22).
B. The role of Zn2+ in thiol-based redox activity
1. Zn2+ sites in proteins
Unlike transition metals such as copper and iron, Zn2+ does not participate directly in redox reactions (i.e., it is not involved in reactions that alter its valence). However, it is involved in redox regulation via Cys ligands in proteins (178). Zn2+ binding changes the properties of the thiol group, lowering its pKa (178). Just as there seems to be a bipartite distribution of redox-active and structural disulfides in proteins (300), Zn2+ sites can also be either redox-active or structural (178).
Protein X-ray crystallographers typically differentiate two types of Zn2+ sites: catalytic Zn2+ sites, which in their simplest mononuclear form have three amino acid sidechains and one water molecule as ligands; and structural Zn2+ sites, which have four amino acid ligands (270). From a redox point of view, a second variable, lability, is important. Some Zn2+ sites expel Zn2+ upon variation of physiological redox conditions, whereas others bind Zn2+ tightly, and would not be expected to release Zn2+ under physiological conditions. We refer to Zn2+ sites in this first redox-regulated structural group as “labile” Zn2+ sites, and those in the second, purely structural group, as “inert” (Fig. 16).
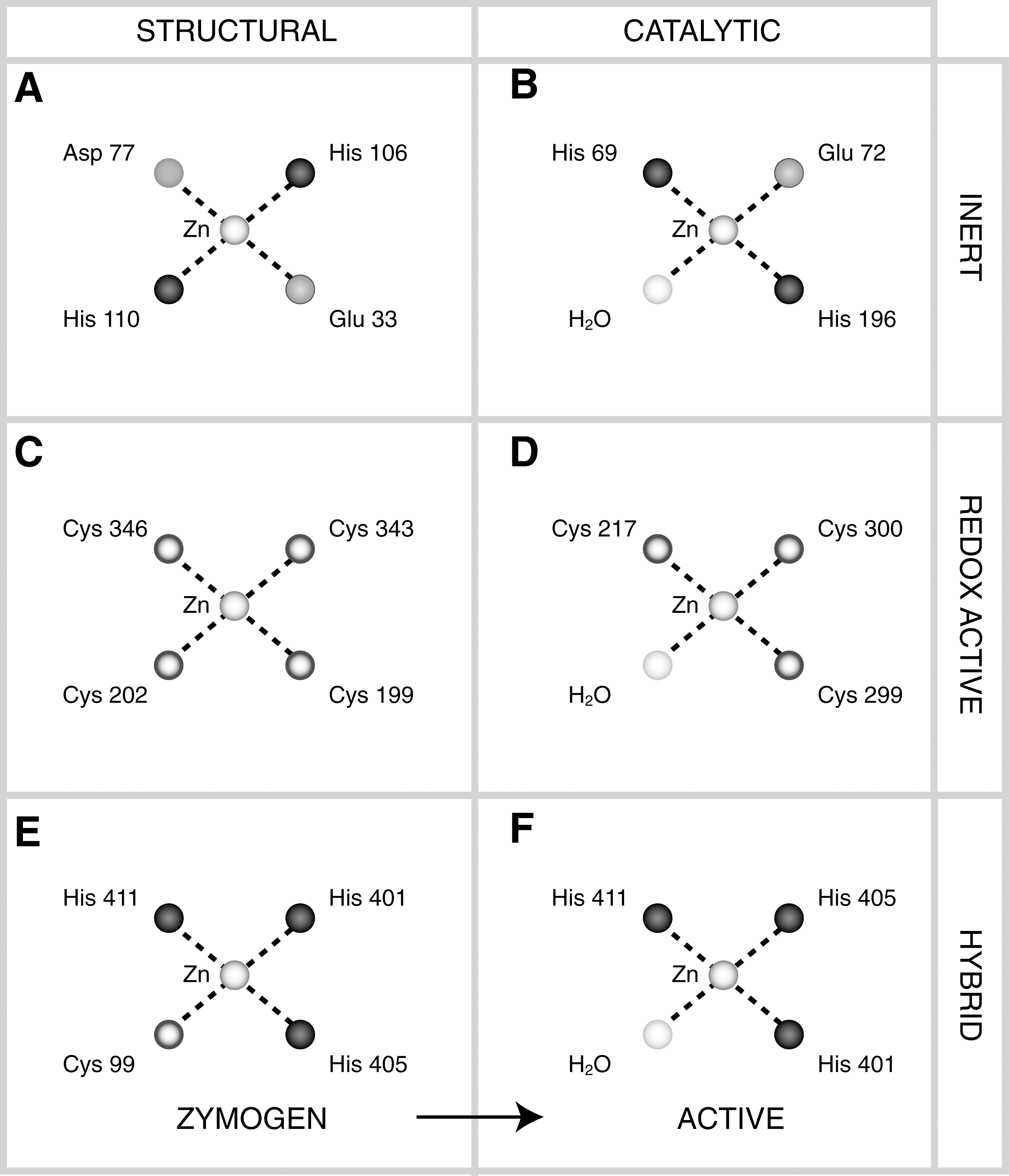
Zn2+ ligand identity is a key indictor of Zn2+ site function. For redox-inert catalytic Zn2+ sites, residues with carboxylates (Asp, Glu) predominate in binuclear Zn2+ sites, whereas His is more common in mononuclear catalytic sites (269, 291). In labile catalytic sites, Cys residues predominate as ligands. Redox-active catalytic Zn2+ sites have previously been characterized in betaine-homocysteine S-methyltransferase (BHMT) and formaldehyde-activating enzyme. These catalytic Zn2+ sites are highly unusual because of their utilization of Cys residues as ligands instead of the more usual His, Glu, or Asp. BHMT and MetH are enzymes of sulfur metabolism, which are redox-regulated in plants (32). In labile structural Zn2+ sites, Cys residues predominate, sometimes in combination with His. In sites with four Cys ligands, Zn2+ is bound by four sulfur (S) ligands, each donated by a Cys residue (S4 site). S4 sites are the most reactive type of structural Zn2+ site and are almost as reactive as a free thiol (184). Substitution of a single His residue for Cys in a redox-active Zn2+ site lowers the reactivity of the site. His sidechains ligate Zn2+ via the nitrogen (N). Hence, the order of reactivity of labile structural sites is largely determined by the Zn2+ ligands with S4 > S3N >S2N2. However, other factors modify the redox potential of Zn2+ sites including the electronic environment and entropic factors.
2. Proteins harboring Zn2+ sites and their conformational changes
Previously described proteins with redox-active structural Zn2+ sites include transcriptional regulators such as SMAD, anti-TRAP, and SIRT2; and the molecular chaperone Hsp33 (117). These proteins reversibly expel Zn2+ to form disulfides under conditions of oxidative stress. However, Zn2+ regulation is not confined to the oxidative stress response. It is also important for other cellular functions such as intracellular transport. In small Tim proteins, Cys residues required for disulfide formation are kept in a transport-competent and functionally inactive state by Zn2+ ligation until delivered to the mitochondrion where disulfides form in the intermembrane space in more oxidizing conditions compared to the cytosol (170).
Structural changes that occur upon Zn2+ loss and subsequent oxidation of these proteins are varied. In enzymes, such as BHMT, very small conformational changes can be sufficient to have a major effect on activity (58). However, large conformational changes also occur. In T. thermophilus Leu-tRNA synthetase, a large order/disorder transition is associated with reversible Zn2+ binding. In the pre-transfer complex, a disordered region of polypeptide chain is partially stabilized by a disulfide formed between Cys 439 and Cys 484 (167). In the more ordered post-transfer complex, the polypeptide chain forms a small β-sheet that tetrahedrally coordinates a Zn2+ atom (266). The Zn2+ site is conserved in eukaryotic mitochondrial Leu-tRNA synthetases but not in the cytosolic form of the enzyme. When four Cys residues are bound to Zn2+, the conformational space of the resident polypeptide chain is more constrained than if the Cys residues form one, or even two, disulfide bonds. Expulsion of Zn2+ and subsequent oxidation increases the entropy of the chain. Reversible Zn2+ binding is also associated with changes in quaternary structure. Most commonly, Zn2+ binding can induce oligomerization. An example occurs in iNOS where a redox-active ZnS4 site is formed at the dimer interface. Oxidation of the Cys ligands results in dissociation of the dimer to two monomers, uncoupling nitric oxide synthesis (307). However, the opposite behavior is also known: the Zn finger motif in the monomer of cellulose synthase (CesA) is proposed to participate in oxidative dimerization of the enzyme. Zn2+ maintains the protein in its monomeric state and a covalent dimer is formed upon oxidation (150).
Upon Zn2+ expulsion from a redox-active site, disulfides do not form randomly between free thiols. In an S4 site, six different disulfides are possible but one of these tends to be selected. This preferred disulfide is known as the trigger disulfide (304). Some known variables that influence selection of a preferred disulfide are the presence of His residues in the site (deselection), and electrostatic effects of nearby residues.
Our work suggests the structural context of Cys Zn2+ ligands may also be important in function. Two forbidden disulfides motifs, the JSD and BDD, are associated with likely redox-active Zn-binding sites in proteins (64). An example of a Zn2+-associated JSD is found in nicotinate phosphoribosyltransferase (NAPRTase) from Thermoplasma acidophilum, an archaeal thermophile. NAPRTase catalyzes the first, rate-limiting reaction in NAD salvage synthesis. The C-terminal Zn knuckle domain contains the JSD (239). This domain is unique to T. acidophilum and is not found in other NAPRTases, but has structure and sequence similarities to other Zn knuckle domains such as one found in the DNA replication initiator from Methanobacterium thermoautotrophicum, which mediates intersubunit interactions (66). In both cases, Zn2+ is bound by a CXXC(14 –24)XCXC motif. In NAPRTase, the JSD forms between the first and third Cys of the motif. Another example is in the previously mentioned bacterial Leu-tRNA synthetase that is conserved with the mitochondrial homolog in eukaryotes. The post-transfer complex (266) contains a reduced JSD motif that binds Zn2+ in a CXXC41XCXXC motif. In the pre-transfer complex, the β-sheet is disrupted and a disulfide formed between the first and third Cys of the motif (167). These two examples and others shown in Table 3, suggest there may be a relationship between the JSD motif and reversible Zn2+ binding. Another characterized JSD-containing protein is defensin. While there is no direct evidence that defensins bind Zn2+, they mediate Zn2+ tolerance in plants (190). Human defensins contain two potential Zn2+ binding sites.
Shaded rows contain composite sites. Site column-catalytic sites are in italics; sequence motif column-Zn2+ ligands are
The other forbidden disulfide motif commonly associated with Zn2+ binding is the antiparallel BDD. In most instances, the two Cys residues are in the hydrogen-bonded site (h) of adjacent strands (aBDDh) on a β-hairpin (290). Most examples are embedded in a CXXC sequence motif but C3XC and CXC sequence motifs are also represented. The shaded examples in Table 3 are structures that contain both a JSD and an aBDDh within a single Zn2+ site. The significance of these composite motifs is unclear. Differential modification of composite sites may modulate different responses to ROS/RNS, or alternatively, disulfide isomerization may occur during the Zn2+ redox cycle.
The co-occurrence of forbidden disulfide motifs of the aBDDh type with labile Zn2+ binding sites is particularly interesting because of the recent characterization of the oxidative process leading to activation of the bacterial chaperone Hsp33 (117). In Hsp33, oxidative modifications of the N-terminal Cys residues of the Zn2+ site in the linker region are proposed to release Zn2+ from the structure. The aBDDh disulfide is formed between the two unmodified C-terminal Cys of the Zn2+ site (117). The function of disulfide formation between Cys 269 and Cys 272 in the Zn2+ binding site has not been fully delineated. Potential benefits are protection from other oxidative modifications and restriction of the conformational freedom of the protein chain, allowing easier refolding upon return of the protein's milieu to normal conditions. However, these benefits apply to any disulfide and the repeated appearance of the aBDDh in labile Zn2+ sites suggests a more specific function, possibly recognition by a foldase or metallochaperone. We hypothesize the forbidden disulfide would become stressed upon condensation of the β-sheet in its vicinity (293), possibly allowing easier re-insertion of Zn2+ (Fig. 17).
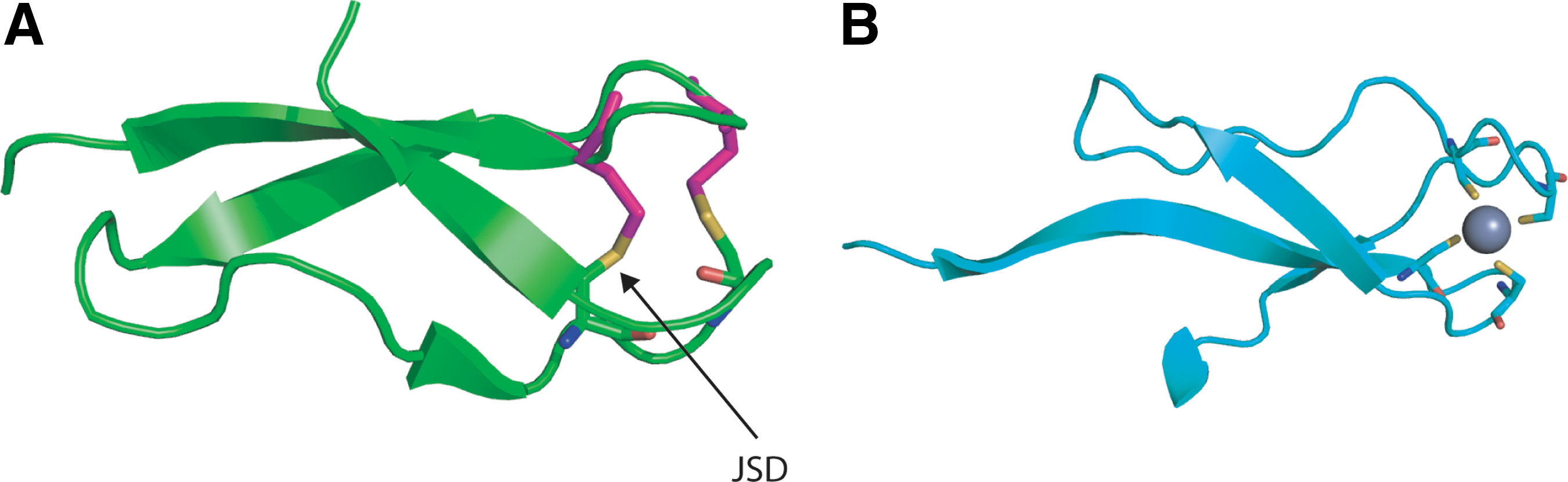
A common fold associated with reversible Zn2+ binding is the nucleic acid-binding Zn finger. Whereas other DNA-binding proteins generally make use of the twofold symmetry of the double helical structure of DNA for binding, Zn fingers do not. As such, Zn fingers can be arranged linearly in tandem to recognize DNA sequences of different lengths (139). This special property also makes Zn fingers equally at home binding RNA as well as DNA (139). Many RNA-interacting proteins contain likely redox-active Zn2+ sites including amino acid tRNA synthetases, RNA/DNA polymerases, ribonucleotide reductase, and the ribosome. Zn fingers have been classified into eight different structural types (146). The aBDDh motif seems to be a common feature of many of these structural types. In C2H2-like Zn fingers and GAG knuckles, the N-terminal pair of Cys ligands can be found as a reduced aBDDh motif. In C2H2 Zn fingers, the two C-terminal Zn2+ ligands are His residues. If the function of C2H2 Zn fingers is analogous to activation of Hsp33, pH changes rather than oxidative stress may initiate Zn2+ expulsion from these folds. A third major structural group is the treble clef Zn finger which consists of two distinct subgroups: one of these ligates a single Zn2+ atom; while a second group, which ligates two Zn2+ atoms, are known as cross-braced Zn fingers (180). Among treble clef Zn fingers, aBDDh motifs are mostly associated with cross-braced treble clefs that bind phospholipids including FYVE, PhD, and DAG-binding Zn fingers. aBDDh motifs are generally not found in the other major group of cross-braced Zn fingers, RING domains, and are only occasionally found in non-cross-braced treble clefs, mostly in LIM domains where the N-terminal pair of Cys ligands of the first of two Zn2+ sites is found in an aBDDh motif. A fourth structural group are the Zn ribbons. In some Zn ribbons, such as rubredoxin-like domains, the N-terminal thiols are often present in a reduced aBDDh motif, while in others, such as members of the general transcription factor IIB and IIS families, the C-terminal Zn2+ ligands form a reduced aBDDh motif. In summary, in ∼50% of Zn fingers with defined secondary structures, two of the Cys Zn2+ ligands are present in a reduced aBDDh motif. While only a handful of Zn fingers with aBDDh motifs have been crystallized in the oxidized state, the behavior of Hsp33 may represent the general behavior of the aBDDh motif in Zn fingers: where nonmotif Zn2+ ligands release Zn2+, triggering formation of the aBDDh forbidden disulfide in the oxidized state. Further work needs to be done to determine whether this is the case.
Zn2+ binding is not associated exclusively with these specialized domains. Zn2+ sites may alternatively be added to other existing non-Zn2+ binding proteins both by point mutation such as in BHMT, and by domain insertion such as in TaNAPRTase.
3. Zn2+ as a second messenger and its physiologic significance
Upon release from a protein under oxidative stress, Zn2+ can act as a second messenger. Extracellular and intracellular free Zn2+ can have important signaling functions, such as inhibition of gamma-aminobutyric acid (GABA)ergic neurotransmission (107) or modulation of protein kinase C signaling pathways (143).
Zn2+ is buffered in the cell by the metallothionein/thionein pair (35, 176, 207). Thioneins are small proteins, typically ∼6–7 kDa, with a high proportion of Cys residues which can coordinate multiple Zn2+ atoms. In the most common types, up to seven Zn2+ atoms can be bound by 20 Cys residues (173). Zn2+ binds with high affinity to the Cys within metallothionein lowering intracellular free Zn2+ to the pico- to nanomolar range. Oxidation or nitrosation of metallothionein Cys residues can release Zn2+ (174, 175, 203) that can subsequently bind to other proteins. Physiologic changes of the glutathione redox potential affect the redox state of metallothionein and the amount of free Zn2+ available for binding by proteins. The redox potential of metallothionein is less than −366 mV. Recently, thionein was identified as a reducing agent for methionine sulfoxide reductase (230).
Low intracellular free Zn2+ concentrations are maintained under normal homeostatic conditions. Under basal physiological conditions, Zn2+ buffering systems keep the concentration of free Zn2+ in the subnanomolar range (48, 178). Like ROS, the amount of free Zn2+ correlates with different cell states. The amount of free Zn2+ is 614 pM in resting cells, 1.25 nM in undifferentiated cells and >2.5 nM in apoptotic cells. Cellular free Zn2+ is also controlled by membrane importers of the Zip family and exporters of the ZnT family (56), as well as the metal response element transcriptional factor (270).
Zn signaling seems to have a special role in the brain. In neurons of the mammalian cerebral cortex, Zn2+ is selectively stored with the neurotransmitter glutamate in synaptic vesicles, released into the synapse upon excitation, and enters postsynaptic neurons through Zn2+-permeable gated channels (6, 40, 71, 108, 258, 273). Physiologically relevant concentrations of Zn2+ alter the activity of GABA and glutamate (NMDA receptor subtype) gated channels and other synaptic proteins. Zn2+ is thought to be important in synaptic plasticity (29, 163), required for memory and learning. The importance of Zn2+ in brain function is also reflected in the brain's Zn2+ content. The amount of Zn2+ in the mammalian brain averages ∼10 μg/g (wet weight) (70). Zn2+ concentrations in the serum and extracellular fluid are estimated to be 0.15 μM (254). The concentration of Zn2+ inside synaptic vesicles is micro- to millimolar (69). Thus Zn2+ may play a central role in the effect of oxidative stress on the brain, of importance to neurodegenerative diseases such as Alzheimer's disease. Zn2+ is also essential to proper development of the nervous system (217).
In summary, for structural Zn2+ sites an analogous situation may exist to the bipartite distribution of redox-active and structural disulfides in proteins, with labile Zn2+ sites being redox-active and inert Zn2+ sites corresponding to the purely structural group. Zn2+ can inhibit enzymes such as caspase-3, fructose 1,6-diphosphatase, glyceraldehyde 3-phosphate dehydrogenase, aldehyde dehydrogenase, tyrosine phosphatases and yeast enolase. Addition of thionein can restore activity by acting as a Zn2+-chelating agent (174). On the other hand, release of Zn2+ can inactivate other proteins such as DnaJ which must be reactivated by Trx reduction. The reader is referred to more extensive reviews on the role of Zn2+ regulation and its importance for neurochemistry (147, 178, 270).
VII. Global Approaches to Identifying Redox-Active Cys
A. Proteomic approaches to identification of redox-active Cys
Developed over the last 10–15 years, experimental approaches to detection of reactive Cys residues by labeling are sufficiently refined to be applied at the proteome level. H2O2 and iodoacetamide selectively and competitively react with Cys of low pK a. In early work in 2000, a decrease in labeling of cell lysate proteins with a biotin-conjugated iodeacematinide reagent was observed following exposure of cells to H2O2, or to an agent that induces H2O2 production (135). Labeling was monitored by streptavidin blot analysis. Modified proteins were identified by immunoprecipitation, a technique which requires preparation of specific antibodies against each protein of interest. A novel modified peptide within creatine kinase was detected by trypsin-digestion, followed by separation of the fragments by high performance liquid chromatography (HPLC). Further analysis by partial Edman degradation and mass spectrometry identified the peptide sequence containing a single modifiable Cys residue. In an advance that increased the scope for discovery of novel proteins, iodoacetamide reagents of different mass were conjugated to reactive thiols in separate reactions. Pooling of the results, followed by trypsinization and purification by HPLC potentially allowed novel peptides to be identified in fractions by mass spectrometry through iodoacetamide-conjugate doublets (238). Both these techniques, which were performed on fully folded proteins, are limited by access of the reagent to the reactive Cys (135), particularly when large reagents such as biotin are involved.
Differential thiol trapping techniques attempt to solve the problem of reagent access by denaturing proteins before conjugation. In differential thiol-trapping, the folded proteome is exposed to oxidative or nitrosative stress, resulting in a spectrum of modifications to susceptible thiols. The proteome is then denatured and unmodified thiols are labeled irreversibly with an iodacetamide reagent. Reversible thiol modifications (i.e., disulfides or sulfenic acid, but not sulfinic and sulfonic acid) are then reversed using a strong reductant and labeled with a visualizable form of the same iodacetamide reagent. For example, the second iodacetamide reagent used to label modifiable thiols may be radioactive (158) or fluorescent (102). Modified proteins are commonly visualized using two-dimensional gel electrophoresis. Proteins spots can potentially be excised, digested using trypsin, and modified peptides identified by mass spectrometry (158).
A recent improvement on the differential thiol-trapping technique is the OxICAT method which adds the advantages of detection by iodoacetamide-conjugate doublets (160). In OxICAT, unmodified thiols are labeled irreversibly using a “light” 12C-based form of an iodacetamide reagent called ICAT. Reversible thiol modifications are then reversed using the strong reductant Tris(2-carboxyethyl)phosphine and labeled with the “heavy” 13C-based form of the same ICAT reagent. After trypsin digestion, peptides with single reversible thiol modifications are 9 Da heavier than their unmodified form, allowing modifiable proteins to be identified, and the relative abundances of the modified and unmodified forms to be quantified using mass spectrometry. Tandem mass spectrometry can be used to identify modified peptides and hence potentially oxidized Cys.
Proteomic approaches are currently limited by the quantities of proteins that can be obtained. Many proteins identified to date are components of redox homeostasis systems, or highly abundant target proteins such as ribosomal proteins in easily cultivated organisms (160). The resolution of these systems is constantly improving. Only proteomic approaches with the potential to identify individual redox-reactive Cys have been discussed briefly here. For additional methods, the reader is referred to more detailed recent reviews on the subject (159, 191).
B. Bioinformatic approaches to prediction of redox-active Cys
Early studies that attempted genome-wide identification of redox-regulated proteins using computational approaches looked for the presence of Zn2+ or selenocysteine in sequences. Conour et al. (49) used sequence-based Zn2+ signatures from InterPro (114) to search for redox-regulated proteins involved in regulation of the cell cycle. InterPro is a metadatabase of motif and domain-based sequence signatures integrated from multiple sources. While several likely redox-regulated proteins were found, this study was not exhaustive. Significantly, many Zn2+ sites are not found by these signatures (177). Furthermore, no attempt was made to distinguish structurally stable Zn2+ sites from labile Zn2+ sites which could be involved in redox activity (177, 184). Further studies are necessary to identify the most reactive thiols in these sites.
A more recent study by Fomenko et al. (68) used a sequence-based technique which predicts redox-active Cys based on homology to selenocysteine (Sec) in other organisms. Sec is an isostere of Cys where the sulfur is replaced by selenium. Sec has a significantly lower pKa than Cys and thus is more reactive in the same context. It is known as the 21st amino acid acid in ribosome-mediated protein synthesis and its specific incorporation is directed by the UGA codon. Because UGA is also used as a stop codon, recognition elements within the mRNAs, known as SECIS elements, are essential for translation of UGA as Sec. Unique tRNAs that have complementary UCA anticodons are aminoacylated with serine. The seryl-tRNA is converted to selenocysteyl-tRNA that binds specifically to a special elongation factor and is delivered to the ribosome.
The study by Fomenko et al. represents the first time inherent redox properties of a biological molecule have been used to identify redox activity in a resident protein, however the technique has several limitations. One limitation of the Cys–Sec pair method is that it is restricted to proteins found in reducing compartments. Work to date on redox regulation in bacteria and eukaryotes has concentrated on redox regulation in the cytosol. Only a few cell surface proteins such as CD4, integrins, and Tissue Factor have been demonstrated to contain redox-active disulfides (2, 104), but it is likely these studies represent the tip of the iceberg. In addition, only those Cys that carry out catalytic functions (e.g., attacking nucleophiles) are identified by the Cys–Sec pair method, whereas Cys that serve supporting functions (e.g., resolving Cys) are not detected (68).
Physicochemical approaches to discriminate between redox-active and structural disulfides in protein structures have also been attempted. The calculation of individual redox potentials within protein structures is currently not computationally feasible. Disulfide redox potential is a function of electrostatic screening, entropic effects, and physical stress applied to the disulfide bond both through stretching and twisting. The redox potential contains both pH-dependent and pH-independent parts (43). Thus pKa, which is often used to estimate redox potential, is not entirely predictive. Nor is the calculation of pKa possible in a high-throughput manner. Estimation of redox activity via some of these variables represents a more accessible approach.
Whitesides and co-workers used molecular mechanics calculations to investigate factors that affect the redox potential of two thiols in molecules with similar structure and thiol pKa (36). The most important parameters were the three central dihedral angles of the disulfide bond (χ2, χ3, χ2′). Experimental work has confirmed that torsional energy is an important variable influencing disulfide redox activity (131, 280, 283). The AMBER forcefield (213) was previously used to estimate disulfide torsional energies in a high-throughput manner using solved protein structures (292). More recently, interpolation on a potential energy surface calculated using more detailed quantum chemical calculations of the model compound diethyl disulfide has allowed more accurate estimation of disulfide torsional energies in protein structures (95). Disulfides with very high torsional energies are likely to be redox-active and may be reduced by purely mechanical means such as stretching and twisting. For example, a short-lived disulfide intermediate formed between Cys 10 and Cys 82 as part of a disulfide cascade during the reaction cycle of S. aureus arsenate reductase is estimated to have a torsional energy of 24.0 kJ·mol−1 (95).
However, as discussed earlier, these very unstable high torsional energy disulfides are not the only ones likely to be redox-active. Most forbidden disulfides have torsional energies in the intermediate energy range. Forbidden disulfides account for only ∼10% of disulfides with torsional energies >10 kJ·mol−1 and constitute an even smaller proportion of very high energy disulfides. Their metastability is likely an important feature of their biologic function. While they are unlikely to undergo spontaneous reduction, they are likely to be far more susceptible to redox attack than low energy, structural disulfides. Although forbidden disulfides were originally identified by physiochemical analysis of stresses in crystal structures, once canonical motifs have been identified they can easily be mined from crystal structure coordinates.
Structural data-mining methods based on torsional energy analysis and forbidden disulfide motif identification only consider static mechanical energy contributions to increasing the redox potential. Other variables likely to influence the redox potential include the electrostatic environment; and dynamic stress applied to the disulfide through directed stretching and twisting, as well as by Brownian motion. To identify disulfides that rely on these other means for reduction, an alternative method that adopts an entirely empirical approach has recently been developed. In this technique, redox-active disulfides are identified empirically by mining redox pairs of structures from the PDB (64). In protein redox pairs, Cys are disulfide bonded in one crystal structure but reduced in another structure of the same, or a highly similar, protein. Redox-active disulfides are more likely to be in alternate redox states in a crystal structure than structural disulfides whose disruption is likely to greatly impair the protein's stability. For example, redox-active disulfides are likely to be selectively reduced by X-ray radiation during the structure solution process and their oxidation state is more susceptible to changes in pH. Potential redox-active disulfides identified by this purely empirical technique include high torsional energy disulfides and forbidden disulfides. A limitation of the Redox Pair technique is that it requires the protein structure to be solved independently at least twice under different redox conditions.
The three structure-based techniques using torsional energy analysis, recognition of forbidden disulfides motifs, and detection of Redox Pairs have recently been applied to identification of proteins harboring redox-active Cys in the yeast genome (63). The results showed reasonable agreement with reactive Cys deduced experimentally as well as predicting many novel proteins. Further experimental investigation of these results will hopefully allow the accuracy of the methods to be better delineated.
The structure-based methods described are not restricted to the cytosol and can be used to identify redox-active Cys in other redox-regulated compartments such as plasma and mitochondria. For instance, several vacuolar proteins were implicated in the yeast genome study (63). Because disulfides are used as the starting point, predictions are not limited to catalytic Cys. For instance, resolving thiols can be predicted. Furthermore, the methods are not restricted to redox-active Cys in catalytic sites and can predict redox-active Cys other than catalytic disulfides, such as regulatory redox-active Cys. Structure-based prediction methods can be extended to the sequence level by homology-based sequence searching.
Theoretical studies to identify redox-regulated Cys (49, 68) are a valuable alternative approach to high throughput experimental methods which will hopefully help define the area of thiol-based redox regulation and act as a catalyst for further experimental studies. Computational methods are complementary to high-throughput approaches such as microarrays and two-dimensional gels, which can identify particular proteins that are under redox control but are unable to identify particular thiols through which redox control is exerted. Specific residues critical to redox activity can be identified and subsequently tested by mutagenesis. While not limited by protein abundance like proteomic methods, computational methods have their own drawbacks. For example, while a specific Cys may be identified as redox-active, the method may not identify under what conditions it is redox active (i.e., by which assay redox-activity would be uncovered). Inspection of individual predictions may be helpful here, as well as considerable experimental insight. Combining computational predictions with experimental results would be useful, and can also provide an additional level of quality control with high throughput approaches such as yeast deletant screening.
VIII. Evolution of Thiol-Based Redox Regulation
A. Evolution of protein cysteine content
Early studies on protein sequences indicated Cys is more abundant in more complex organisms (61, 192). This has been confirmed by more recent studies on whole genomes that have shown the abundance of Cys in proteins has been increasing since divergence from the Last Universal Common Ancestor (27, 288), and this increase is a continuing pan-species phenomenon which is evident within the last 10 million years (126).
This has important implications for protein structure. Structural domains in proteins are independent folding units (282). From a domain viewpoint, Cys is more likely to be present within similar domains if they are present in more complex organisms. Given that disulfide-forming Cys can have either a redox role or a role in structural stabilization, two hypotheses present themselves: first, that any given domain is increasingly unstable in more complex organisms, and needs additional covalent cross-links; or alternatively, thiol-based redox signaling is more prevalent in organisms of increasing complexity.
Although the prevalence of Cys in proteins is increasing, the abundance has not yet reached the neutral frequency (i.e., the frequency that would be expected based on the number of Cys codons in the genetic code) (27, 137). Based on the absence of Cys and some other amino acids from the Murchison meteorite, and the inability to produce these same amino acids abiotically in spark experiments (189), it seems likely that Cys belongs to a group of amino acids that were added late to the genetic code after development of biotic pathways to enable their synthesis (289). Being a latecomer, it has been suggested that insufficient time has passed to allow Cys to reach its equilibrium frequency (28, 126). However, Phe was also likely recruited late to the genetic code, and postdates the introduction of Cys (265). In contrast, the abundance of Phe in complex organisms is higher than would be expected based on neutral frequencies (27).
Although prebiotic amino acids do seem in general to be decreasing in abundance while late-coming amino acids are increasing, other factors may affect the rate of acquisition. Although there is still some disagreement over which amino acids are increasing in frequency, it is agreed that Cys and Phe are increasing (27, 126). One study suggests Tyr might also be increasing (27), while another suggests Met, His, and Ser are becoming more abundant (126). These data are based on a small number of completely sequenced genomes and the methods examine different epochs in evolutionary time. They may yet be reconciled by specific trends within subgroups or epochs. A common property of these increasingly more prevalent residues is their role in signaling either through post-translational modifications by ROS (Met, Cys), phosphorylation (Ser, Tyr); or other signaling modes such as pH responsive elements (His, Cys), and π–π signaling (Tyr, Phe). An alternative explanation for the failure of Cys to reach its equilibrium frequency is that incorporation of additional Cys into proteins may come at a cost. In the case of Cys, increased signaling and control may be gained at the cost of deleterious effects of over-oxidation. Our preliminary data suggests redox-active Cys has been increasingly incorporated into genomes all the way up to mammals, which represent the most extensive users of thiol-based redox signaling on the planet.
Thus, increased usage of Cys correlates with increased organismal complexity. Before multiple genome sequences were complete, it was generally believed increased complexity correlated with gene number. The small differences in gene number between simple unicellular eukaryotes and mammals forced revision on how complexity is encoded. For example, the yeast genome contains 5,000 genes, yet the human genome contains only five times more: 25,000. Some geneticists now argue that additional complexity at the organismal level must be encoded at the molecular level by noncoding DNA, as this is considerably different between these organisms (182).
While additional control elements in genomes almost certainly play an important role in encoding additional complexity, the studies on changing amino acid content suggest increased complexity may also be encoded at the protein level. It was previously recognized that concatenation of existing domains through gene fusion, also known as protein domain mosaicism, encodes new functions in more complex organisms (210). We will call this the “Lego” hypothesis. However, studies on the changing amino acid content of proteins show that domains too are not static structures. Nature is continually tinkering with these independent folding units.
This is a somewhat surprising realization. For many years structural biologists believed the planetary proteome consists of a limited set of 1,000–5,000 folds that form a universal construction set (45). The “divide and conquer” approach and automation of structure solution posit that solution of the universal construction set would give us complete knowledge of the molecular structure of the planetary proteome. But here is a caveat: a domain from archaea may not have the same sophisticated set of switches as the homologous domain from a mammalian protein.
Additional complexity added to protein domains in the form of redox and other switches likely increase the signaling capabilities of individual domains. It also suggests the energy landscape at the bottom of the folding funnel is likely to be more complex in a domain from a higher organism than a homologous domain from a lower organism. Pursuit of a complete knowledge of fold space is worthy, but it does not give us a complete knowledge of proteins.
B. Origins of thiol-based redox signaling
The origin of thiol-based redox signaling is very ancient and rather murky. Evolutionary studies comparing the protein complement of genomes have determined Trx is found in every lineage in the Clusters of Orthologous Groups (COGs) database (256, 257). Thus the Last Universal Common Ancestor is proposed to contain a Trx. Most other proteins for which this is true are involved in transcription and translation.
The earliest evidence of life on earth comes from analysis of 3.5 billion year-old cherts that suggest anaerobic photosynthetic prokaryotes inhabited marine environments around this time (260). Based on these data, the Last Universal Common Ancestor is often dated at 3.5 billion years (15). However, given the increase in the oxygen content of the atmosphere only began ∼2.5 billion years ago (212), before which the earth is believed to have had a reducing atmosphere, it seems likely that Trx was only incorporated into a genome after this change in atmospheric content. This suggests that the Last Universal Common Ancestor occurred <2.5 billion years ago and whatever life existed on the planet during the first billion years was supplanted by organisms capable of dealing with the new oxygen-based atmosphere.
The enzyme capable of reducing Trx, however, is not universal. In bacteria and animals, reducing power is provided by the NADP+-dependent TrxR. In plants, this enzyme operates in the cytosol but an additional enzyme, FTR is used in chloroplasts. Plastids themselves are thought to have originally been free-living organisms that were incorporated into plants in a symbiotic event. The FTR system was previously assumed to be present in all oxygenic photosynthetic cells, but was recently determined to be absent from some cyanobacterial species likely to be of very early origin (67). In these slow-growing species, NADP+-dependent TrxR meets the limited needs of these organisms for photosynthesis. Thus, FTR appears to have been added in plants as an auxiliary system to deal with high electron fluxes.
Other evidence that redox control was acquired very early in the evolution of life on Earth come from archaea. Evidence has emerged of specialized use of Cys within some archaeal subgroups, particularly in the cytosol. Disulfide bond mapping suggests >40% of Cys in cytosolic proteins of crenarchaeal genomes participate in disulfide bonds, and >30% in the Pyrococcus branch of euryarchaea (172), far more than predicted for cytosolic proteins in bacteria and eukaryotes. The propensity of these organisms for even numbers of thiols in the cytosol was also noted. Additional support for enrichment of cytosolic disulfides was provided by discovery of protein disulfide isomerase-like enzymes encoded in a subset of archaeal genomes predicted to have disulfide-enriched cytosols (152). It was hypothesized that the increased number of intracellular disulfides in these hyperthermophilic subgroups was a mechanism for achieving thermostability. However not all hyperthermophiles have disulfide-enriched cytosols (16). Alternatively, the increased number of disulfides could be involved in the process of redox regulation. Although redox regulation was briefly considered as an alternate hypothesis in the original work, it has not been systematically investigated. It is interesting to note that of the two disulfide-enriched subgroups, the crenarchaeal group contains aerobic organisms (133, 275), whereas hyperthermophiles of the Pyrococcus group are anaerobic. It is possible that distinct disulfide-enriched subgroups have adopted disulfide-enriched cytosols for different reasons: one for redox control, and the other for thermostability. It should also be noted that preference for redox regulation by disulfides over free thiols may be driven by the presence or identity of other redox homeostasis systems. Among prokaryotes, the well-studied glutathione system of eukaryotic redox regulation operates only in purple bacteria and cyanobacteria (62, 172). A redox homeostasis system based on coenzyme A may be utilized in disulfide-enriched archaea (112).
Finally, it is also interesting to note that two of three internal cellular structures thought to have been acquired by symbiosis: plastids and mitochondria, have important redox functions. This suggests acquisition of redox control by the Last Universal Common Ancestor was a major determinant of success of living organisms in evolution of life on earth.
C. Redox-regulation of cysteine-based enzymes
Hartley originally classified proteases into four classes based on their ability to be inhibited by specific classes of inhibitors. These four classes are cysteinyl proteases, serine proteases, aspartic proteases, and metalloenzymes.
Interestingly, the catalytic apparatus of cysteinyl and serine proteases is astonishingly similar. Cys and Ser, which act as the nucleophile in reactions in these proteases, are isosteres; and two other common members of the catalytic triad, His and Asp, are identical and similarly arranged in three-dimensional space in both classes of proteases. Furthermore, Brenner noticed that Ser is specified by two types of codons which differ by more than one base in multiple groups of homologous serine proteases of different folds (26). For example, in most trypsin-like serine proteases the catalytic Ser in encoded by AGN, where N is any of the four bases. However, in blood trypsin-like serine proteases such as the clotting factors, Ser is encoded by TCY, where Y is a pyrimidine. In order for this differential codon usage to have arisen, these enzymes must have transited through an evolutionary step that involved Cys or Thr to remain viable proteases. Indeed, there have since been many cysteinyl proteinases found with identical folds to serine proteases.
It seems likely this evolutionary step has not occurred multiple times by coincidence, but is instead a result of selection. Cys may be chosen over Ser in these enzymes when it is advantageous for the enzyme to be under redox control. Thus, differential codon usage may have arisen by transit through such a Cys evolutionary step. Redox control of Cys-based enzymes by modification of the reactive Cys has been demonstrated for several examples. One example is the previously described regulation of activity of the protein tyrosine phosphatase (PTP) family (Section VI-A) which encompasses 100 members in humans alone (263). The mechanism of inhibition of PTP family members suggests a general mechanism of redox-based inhibition of Cys-based enzymes. Another example of redox regulation of Cys-based proteases in Arabidopsis thaliana has recently been demonstrated. PDI5 is expressed in endothelial cells about to undergo programmed cell death in developing seeds. Three different Cys proteases interacting with PDI5 have been identified in yeast two-hybrid screens. PDI5 regulates proper seed development and the timing of programmed cell death by chaperoning and inhibiting Cys proteases during their trafficking to vacuoles (4).
In addition to cysteinyl proteases, Brenner identified multiple other Cys-based enzymes with differential codon usage. Many other enzymes have Cys-based reaction mechanisms including transglutaminases, cathepsins (96, 216), calpain (85), and caspases (89). These enzymes are also likely to be under redox control.
IX. Conclusions
Categorizing disulfide switches allows us to recognize them in novel contexts and should eventually enable their recognition in protein sequences as well as structures. Understanding the physicochemical basis of redox-active disulfide switches should enable extrapolation of their behaviors from well-characterized examples to novel types. More work needs to be done to map particular motifs to specific functions. The association of two forbidden disulfide motifs, the JSD and the aBDDh, with reversible Zn2+ binding is a step in this direction. Insight into the molecular mechanisms involved in the etiology of known redox-related diseases such as Alzheimer's disease, diabetes, and cancer will undoubtedly lead to prevention and cures.
Footnotes
Acknowledgments
The authors gratefully acknowledge funding from the Australian Partnership for Advanced Computing (APAC) National Facility and the APAC Australian Centre for Advanced Computing and Communications (ac3). Thank you to Drs. Lisa Martin (Monash) and Kieran Scott (UNSW) for enlightening conversations, Jason Liu and Sara Ballouz for help with figures, and Siiri Iismaa (VCCRI) for critical comments of the manuscript.
Abbreviations Used
Reviewing Editors: Miriam Cortese-Krott, Jay Hanas, Klaus Kroencke, Jason Larabee, Hui Lu, F. Wayne Outten, and Peep Palumaa