
Abstract
Disulfide bond formation is probably involved in the biogenesis of approximately one third of human proteins. A central player in this essential process is protein disulfide isomerase or PDI. PDI was the first protein-folding catalyst reported. However, despite more than four decades of study, we still do not understand much about its physiological mechanisms of action. This review examines the published literature with a critical eye. This review aims to (a) provide background on the chemistry of disulfide bond formation and rearrangement, including the concept of reduction potential, before examining the structure of PDI; (b) detail the thiol-disulfide exchange reactions that are catalyzed by PDI in vitro, including a critical examination of the assays used to determine them; (c) examine oxidation and reduction of PDI in vivo, including not only the role of ERo1 but also an extensive assessment of the role of glutathione, as well as other systems, such as peroxide, dehydroascorbate, and a discussion of vitamin K–based systems; (d) consider the in vivo reactions of PDI and the determination and implications of the redox state of PDI in vivo; and (e) discuss other human and yeast PDI-family members. Antioxid. Redox Signal. 11, 2807–2850.
I. Introduction
In several compartments, catalyzed disulfide bond formation occurs, including the endoplasmic reticulum (ER), mitochondria (118, 172), and the bacterial periplasm (141). Although many similarities appear between disulfide bond formation in these different compartments, many differences are noted, and this review focuses only on disulfide formation in the ER, the cellular compartment in which protein disulfide isomerase (PDI) resides.
PDI was the first folding catalyst ever reported (101, 316). It is an ER-located protein with a classic KDEL-ER retrieval motif (234) and is present in the ER (177) in high concentrations. It is a multifunctional protein, being able to catalyze disulfide bond formation, breakage, and rearrangement in all nonnative protein and peptide substrates reported to date. In addition to its role in disulfide bond formation, PDI is the β-subunit of prolyl-4-hydroxylase (161) and microsomal triglyceride transfer protein (336). PDI is also implicated in peptide loading onto MHC class I (232), and in regulating NAD(P)H oxidase (133) in the ER. It has also been reported to be involved in a wide range of other biological functions in nearly every cellular compartment (305), although it is unclear for many of these reactions how it escapes the ER. Recently, a number of reports indicated that PDI is, in effect, a marker for the release of intracellular contents from damaged cells (for example, activating tissue factor via catalysis of thiol-disulfide exchange and thus initiating the blood-clotting cascade at the site of wound damage) (253). This review focuses only on its role in disulfide bond formation in the ER.
After 30 years of trials, structural studies on full-length PDI-family members are starting to bear fruit with two crystal structures of human PDI-family members, two full-length yeast PDI-family members, as well as the crystal and NMR structures of the
II. Identification and Initial Characterization of PDI
The first identification of PDI-like activity was independently published by two groups in 1963. The group of Brunó Straub (315, 316), later president of Hungary, found that extracts from both pigeon and chicken pancreas were able to stimulate the reoxidation of reduced ribonuclease. In parallel, Anfinsen and co-workers (101), as part of the work on ribonuclease that earned Anfinsen the 1972 Nobel Prize in Chemistry with Moore and Stein, detailed studies showing acceleration of reactivation of ribonuclease by a microsomal system from rat liver. Anfinsen's group subsequently partially purified the enzyme responsible (99, 100, 102) and showed that it was able to catalyze thiol-disulfide exchange reactions. The material they obtained contained several protein components and was “a faint yellow” (99). In addition, a low-molecular-weight component that was essential for activity and was lost during dialysis could be replaced by the addition of FAD (101). These results predate the discovery and characterization of the FAD-cofactor containing enzyme Ero1, which works with PDI, by three decades (see Section VIII). Subsequently, the “sulfhydryl-disulfide exchange enzyme” was purified to near homogeneity from beef liver (57) and was found to contain at least one disulfide bond that was sensitive to reduction by dithiothreitol (92). PDI was given the enzyme classification number EC 5.3.4.1 in 1972, along with its subsequent officially accepted name, a name that was used for the first time in a publication in 1975 (113).
For 20 years, the significance of PDI was not widely recognized, nor were its activities well defined. However, other proteins were identified as substrates of PDI in vitro, including soybean trypsin inhibitor (284), insulin (313, 314), immunoglobulins (58, 217, 256), vasopressin and oxytocin (314), bovine serum albumin (295), cholera toxin (216), bovine pancreatic trypsin inhibitor (BPTI) (43), ricin (17), and procollagen (82). In addition, the subcellular localization of PDI to the ER was confirmed (177). Furthermore, its detailed action in catalyzing a specific folding pathway was defined (44), and confusion between PDI (EC 5.3.4.1) activity and glutathione-insulin transhydrogenase (EC 1.8.4.2) activity was resolved with the finding that PDI is one of the cellular enzymes that has glutathione-insulin-trans-hydrogenase activity (25, 120).
The major breakthroughs in the study of PDI came in the mid-1980s with the publication of the first highly cited review specifically on the enzyme (87), which raised awareness of the enzyme, and the publication of the sequence of rat PDI (70). This sequence predicted two regions with a high degree of homology to thioredoxin, a small cytoplasmic enzyme involved in thiol-dependent redox reactions (see refs. 93, 180, and 182 for recent reviews of different aspects of thioredoxin). In particular, it showed that, as previously postulated (121), the active sites of PDI contain vicinal thiol groups, in particular, the sequence WCGHC, which changes between the dithiol and disulfide states during the catalytic cycle of PDI.
Before discussing the role of the active-site cysteines and the structure of PDI in its mechanisms of action, we must look first at the chemistry of disulfide bond formation.
III. The Chemistry of Disulfide-Bond Formation and Rearrangement
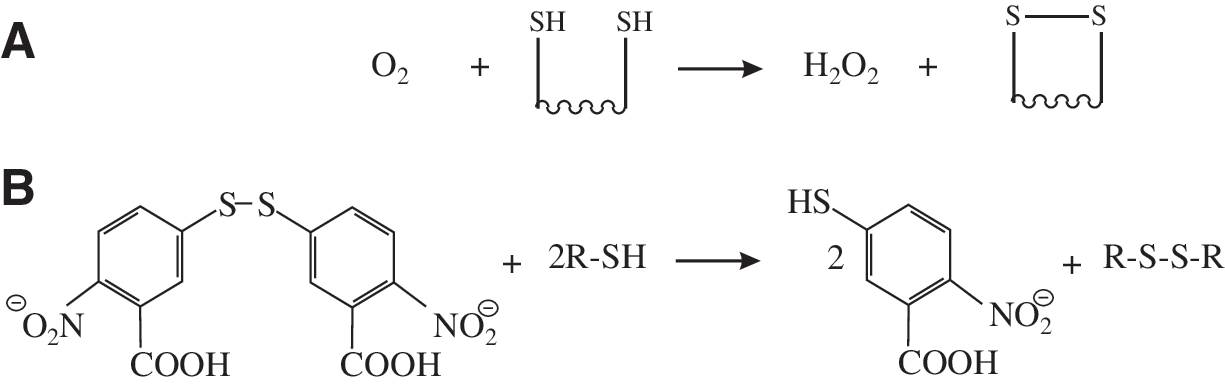
The chemistry of disulfide bond formation is the chemistry of redox reactions and of thiolates. Redox reactions, or reduction–oxidation reactions, are, as the name suggests, reactions in which one chemical species attains a higher oxidation state through the loss of electrons or gain of oxygen, while another is reduced, i.e., attains a lower oxidation state, through the gain of electrons or a loss of oxygen. For disulfide bond formation, the starting species is the thiol group found on the side chain of cysteine residues in proteins. The thiol group represents the −2 oxidation state of the sulfur atom. When two thiol groups are oxidized to form a disulfide, both of the sulfur atoms involved attain the −1 oxidation state, and two electrons and two protons must be transferred to the oxidant. The simplest oxidant that can be used to form a disulfide bond is molecular oxygen, which, in the process of forming the disulfide, is itself reduced to hydrogen peroxide (see Fig. 1A). The formation of disulfide bonds by using molecular oxygen as the oxidant is a thermodynamically favored process, and so it occurs spontaneously, as long as the two thiol groups can be juxtaposed. However, this oxidation process is kinetically slow, but it is used, in combination with metal ion catalysis, in vitro for the refolding of proteins produced as insoluble inclusion bodies (73). This reaction also mirrors what happens, at least in part, in vivo. However, in vivo, the reaction is catalyzed by enzymes known as sulfhydryl oxidases, including the ER-resident protein Ero1 (see Section VIII). Other oxidants can also be used to form disulfides, including reactive oxygen species (ROS) (for example, hydrogen peroxide, and other species such as dehydroascorbate, the oxidized form of vitamin C) (see Section VIII). Another category of oxidants is proteins, for example, PDI, peptides, for example, oxidized glutathione (GSSG), and other molecular species, for example, dithionitrobenzoate, which contain a disulfide bond and transfer this to a folding protein, becoming themselves reduced to the dithiol state in the process (see Fig. 1B). PDI is able to act in such a way as an oxidant of protein dithiols in vivo and in vitro (see Sections VI and VIII). In addition to the disulfide state, other oxidized forms of cysteine thiol groups can and do exist in vitro and in vivo. These include cysteine sulfenic acid (−SOH, oxidation state 0), cysteine sulfinic acid (−SO2H, oxidation state +2), and cysteine sulfonic acid (−SO3H, oxidation state +4). All of these higher oxidation states are reached by the reaction of ROS with cysteine thiols. Cysteine sulfenic acid has a number of biological functions. For example, it is used in intracellular signaling as well as being an intermediate in some enzymatic reactions (38, 240). It is also a potential intermediate in disulfide bond formation in the ER (see Section VIII). In proteins, cysteine sulfinic acid has no reported function. It is thought to be a nonfunctional product caused by the action of ROS on reactive cysteines and requires the action of sulfiredoxin to reduce back it to the free cysteine thiol (24). Cysteine sulfinic acid is, however, an important cellular metabolite lying at the branch point between the formation of sulfate and taurine in cysteine metabolism (285). It has also been reported to be an agonist of various cellular receptors (for example, see ref. 154). In contrast to these oxidation states of cysteine, cysteine sulfonic acid has no known biological functions.
Any two cysteine residues in a protein have the potential to form a disulfide bond. With two cysteines in a protein, only one intramolecular disulfide can be formed; with three cysteines, three different disulfides could be formed; with four cysteines, six different disulfides, etc. Because the protein could have none, some, or all of its cysteines in disulfide bonds, this gives 10 different intramolecular thiol/disulfide redox states for a protein with just four cysteine residues and >13,000 different intramolecular redox states for a protein with 10 cysteine residues. Usually a protein will have only one pattern of disulfide bonds and free thiols in the native state (unless it is involved in redox processes via thiol chemistry, such as PDI, or it has redox-regulated activity).
How does a protein attain its native disulfide state? Some proteins [for example, bovine pancreatic trypsin inhibitor (BPTI)] appear to proceed via a folding pathway where predominantly native disulfide bonds are formed in the folding protein (331). This is presumably linked to the formation of regular secondary structure, which limits the juxtaposition of cysteine residues in the folding intermediates to native-like combinations. However, many proteins presumably form both native and nonnative disulfide bonds during oxidation, and processes are necessary to convert the nonnative disulfide bonds into native ones. This can occur via two distinct pathways: (a) cycles of reduction–oxidation, and (b) direct isomerization. PDI is able to catalyze both of these pathways (see Section VI). Cycles of reduction–oxidation are as described. First, the incorrect nonnative disulfide bonds are reduced back to the dithiol state, and then a second oxidation step occurs, as described earlier. The reduction of nonnative disulfide bonds can be done by a variety of reducing agents, which themselves become oxidized in the redox reaction. For example, in vitro, strong chemical reductants can be used, such as sodium borohydride. In vivo, disulfide bond reductants tend to be based on thiol chemistry [for example, reduced glutathione (GSH), equivalent to the use of β-mercaptoethanol or dithiothreitol (DTT) in vitro (see Fig. 2A)] or linked to NAD(P) or FAD redox cycles [for example, thioredoxin reductase (see Fig. 2B; 218) or glutathione reductase (11)]. In contrast to the two-step reduction–oxidation cycle, intramolecular isomerization of disulfide bonds is a one-step process based on nucleophilic attack by a free thiol group on a disulfide bond to form a new disulfide and releasing a new thiol group (see Fig. 3A). Intermolecular isomerization is similarly based on nucleophilic attack by a free thiol group on a disulfide bond to form a new disulfide, but this time, a transient mixed disulfide is formed, which in turn undergoes nucleophilic attack by another free thiol group (see Fig. 3B). In intra- or intermolecular isomerization reactions, no net changes occur in the number of disulfide bonds or free thiol groups at any stage of the reaction. Note that this is also true of oxidation reactions, in which a disulfide bond–containing species (e.g., GSSG or PDI) transfers its disulfide bond to the folding protein (see earlier). The same overall reaction can be called an oxidation reaction or an isomerization reaction, depending on the viewpoint used. Usually the net effect on the folding protein is used to determine which terminology to adopt, but this can cause confusion, for example, when examining in vivo routes for oxidation of PDI (see Section VIII).


Although we said earlier that isomerization is based on a nucleophilic attack by a thiol group on a disulfide bond, this is not usually the case in practice. Whereas thiol groups are nucleophiles, thiolate anions (i.e., the –S− species) are much more potent nucleophiles. Given this, it is important to consider factors that change the ionization state of cysteine thiols (i.e., factors that modulate the pK a of these groups). The pK a of a chemical moiety is defined as the pH at which 50% of the species is in the protonated state, and so 50% is in the deprotonated state (see Fig. 4A). The proportion of the moiety in the protonated/deprotonated state at any pH can be linked to the pK a value by the Henderson–Hasselbalch equation. The typical pK a of cysteine thiols in proteins is often cited as being around 8.3; however, considerable variations occur in this depending on the local microenvironment of the thiol group. Because buried unpaired charged groups are thermodynamically unfavorable, a buried cysteine side chain would be relatively more stable in the thiol form, and so the pK a would be higher than average. Local electrostatics also play a major role in modulating pK a values. For example, a positive charge adjacent to the thiol group would provide a stabilizing electrostatic attraction between this charged species and the deprotonated thiolate state, stabilizing the thiolate form and lowering the pK a value for the cysteine (see Fig. 4B). Similarly, adjacent negative charges would result in electrostatic repulsion with the thiolate and would destabilize its formation, resulting in an increase in pK a. The pK a of the thiol group in GSH is 8.75, higher than normal value cited for cysteine thiol groups in proteins and peptides because of the local negative charge density on this tripeptide.

The kinetics of noncatalyzed folding pathways of disulfide bond–containing proteins are probably determined by a combination of the pK a values of individual cysteines in the folding protein, along with accessibility and the constraints put on the juxtaposition of pairs of cysteine residues by the formation of regular secondary structural elements. Because the pK a values of the cysteine residues will depend on the conformation of the folding intermediates, which cannot easily be determined, this cannot be easily verified. However, it is noteworthy that the nonnative disulfide bonds observed by Creighton and co-workers (43) in the refolding pathway of BPTI (43) all contain cysteine residues with adjacent positively charged residues (Lys15 adjacent to Cys14, and Arg39 adjacent to Cys 38). A low pK a value for these cysteines may be responsible for the rapid intramolecular rearrangements that occurred during quenching (see Section VI).
Because PDI is a catalyst of thiol-disulfide isomerization, we would expect the pK a of at least one of its reactive thiols to be lower than average to increase its nucleophilicity for the first step of intermolecular-based thiol-disulfide isomerization. This is indeed the case, with the pK a of the N-terminal active-site cysteine being <5.5 (see Section V). The fundamental basis of the structure of the catalytic domains of PDI seems to be attaining the correct pK a values for the two active-site cysteines at different steps of the catalytic process (see Sections V and VII). However, before we look at PDI itself, we must consider first one more important aspect of thiol-disulfide chemistry, reduction potential.
IV. The Reduction Potential of PDI
The term “redox potential” is used variably in the field of catalyzed thiol-disulfide exchange. The term itself is imprecise, with reduction potential (Er 0) or oxidation potential (Eo 0) (the tendency of a chemical species to either gain or lose electrons when it is subject to change by the addition of another chemical species) being the more-accurate terms. A chemical species with a higher reduction potential will have a tendency to gain electrons from a species with a lower reduction potential (i.e., to become reduced by oxidizing the lower-reduction-potential species). Reduction potentials are measured in volts, or more usually for biochemical systems in millivolts, defined relative to a standard hydrogen electrode. More usually, the standard reduction potential (Er 0) rather than the reduction potential is reported. This is the reduction potential under standard temperature, pressure, and concentration. However, the standard reduction potential implies that [H+] = 1.0 M (equivalent to pH 0), which is clearly inappropriate for biological systems. Therefore, the most commonly used system in biochemistry is the biochemical standard reduction potential (Er 0′, often written just as E0′), which is defined at [H+] = 10−7 M (i.e., pH 7.0). In the rest of this review, when we use the phrase “reduction potential,” we will mean the biochemical standard reduction potential. Measuring Er 0′ is problematic; for many biochemical systems, the reduction potential is measured indirectly by measuring the relative reduction potential against a species with defined Er 0′. Usually in the case of thiol-disulfide exchange systems, the relative reduction potential is defined against glutathione (see Fig. 5A; for example, see refs. 103, 111, and 129). However, where the protein thiol-glutathione mixed disulfide is a longer-lasting species, direct protein–protein redox equilibria (see Fig. 5B) (346) is a more accurate method to determine relative reduction potential. The currently accepted reduction potentials of various species, including a range of thioredoxin-superfamily members, such as PDI, are shown in Fig. 6.

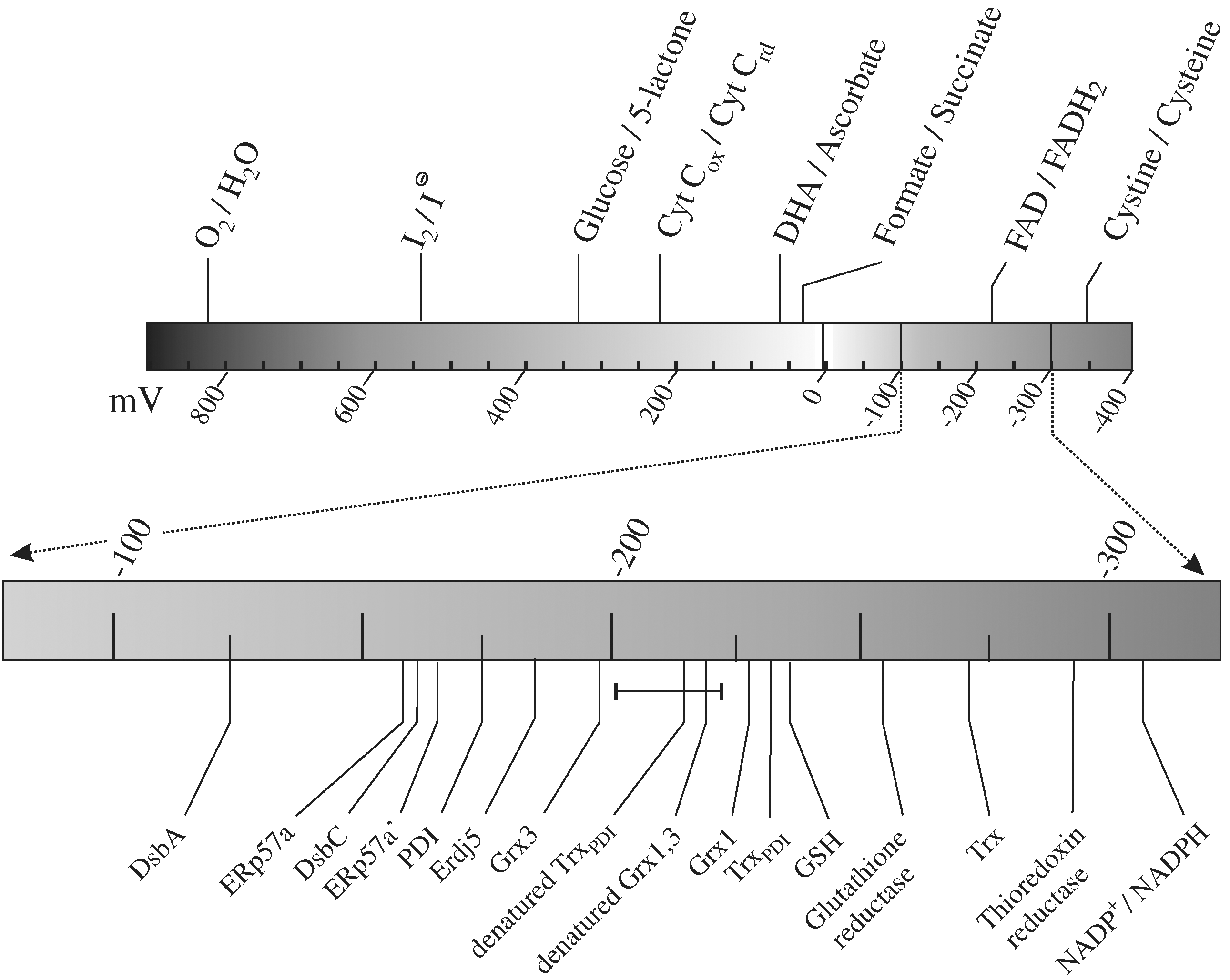
A number of problems and misunderstandings are associated with the reduction potential of PDI. First, because reduction potentials are negative numbers for thiol-disulfide exchange reactions, the terminology can become confusing. For example, a species with a reduction potential of −180 mV (e.g., PDI) (189) has a higher reduction potential than a species with a reduction potential of around −220 mV (e.g., denatured proteins and peptides) (for example, see refs. 275 and 346) and so will be reduced by it. In addition, when talking about the reduction potential of a protein oxidant such as PDI, it can be easy to become confused about which reaction is being discussed. Combining these two issues, PDI (Er 0′ = −180 mV) has a higher reducing potential than denatured proteins (Er 0′ ≈ −220 mV), which means it will be preferentially reduced, and the equilibrium shown later will tend to the right (i.e., the higher reducing potential of PDI implies that it will act as a protein dithiol-disulfide oxidant toward the denatured protein).
Second, the reduction potential is a thermodynamic property; it measures the potential direction of the system, but does not tell us anything about the kinetics of the system. The reduction potential of NADPH is −315 mV, whereas that of glutathione is −240 mV (254, 346). This implies that, based on the thermodynamics of the system, NADPH should spontaneously reduce GSSG. However, the reaction is kinetically very slow unless catalyzed, for example, by glutathione reductase. Two enzymes that catalyze thiol-disulfide exchange reactions may have the same reduction potential but have very different kinetics or specificities or both.
Third, the reduction potential of a species cannot be seen in isolation. By itself, the number is meaningless; it must always be seen in the context of reaction with another species (i.e., a comparison of reduction potentials). Reaction schemes are often erroneously simplified to consider only two species, ignoring contributions from other species present. In vivo, the reduction potential of the cytoplasm for thiol-disulfide exchange reactions is primarily defined by the major redox species present, reduced glutathione (and the NADPH produced by the pentose phosphate pathway to keep glutathione reduced). Similarly, the reduction potential of the ER is defined primarily by the ratio of the major redox species present, GSH and GSSG (20, 130) and, in particular, the [GSH]2/[GSSG] ratio, assuming limited glutathione-protein mixed disulfides, although all redox species present in the ER contribute.
Fourth, and linked to the previous point, the reduction potential is an equilibrium measurement, implying both that all thiol-disulfide redox reactions are equilibrium reactions (i.e., they proceed in both directions) and that the relative concentrations of the species present plays a major role in the net reaction. For example, the cytoplasmic enzyme thioredoxin has the lowest reduction potential of the thioredoxin-superfamily members (i.e., it is the most reducing enzyme). Thermodynamically, it reduces disulfide bonds in proteins, and this is its primary physiological role in vivo (see ref. 182 for a review). The reduction potential of thioredoxin is −270 mV (168), 30 mV lower than that of glutathione (254, 346) and circa 50 mV lower than that of denatured proteins and peptides (for example, see refs. 275 and 346). However, in vitro, it is able to catalyze the oxidation of dithiols to disulfides in folding proteins by using GSSG as the net electron acceptor (see Fig. 7A; 190). Thus all thioredoxin-superfamily members have the potential to act as catalysts of protein disulfide bond formation, reduction, and isomerization (see Fig. 7). However, the relative efficiencies of these will vary; in part dependent on the reduction potential of their active sites, but also dependent on their kinetic properties and on their physiological redox environment.

Fifth, the reduction potential should not be seen as being an immutable value. What is cited is the standard reduction potential. The actual reduction potential of a system varies with biophysical conditions, including temperature and, for systems such as ours that include a proton, the pH of the solution. The reduction potential of enzymes, such as PDI, will also depend on the conformation of the protein. Intrinsic conformational changes in PDI (see Section VII) or conformational changes due to substrate binding or due to changes in the biophysical conditions can change the reduction potential of the active sites.
So what determines the reduction potential of PDI? Because the reduction potential is a measurement of the relative stability of the reduced (dithiol) and oxidized (disulfide) states of the active site, factors that stabilize the disulfide state will lower the reduction potential and will make the enzyme a better reductant: the enzyme is stabilized by having the active-site disulfide, and so it is preferential to take it from nonnative protein substrates (i.e., to reduce them). Similarly, factors that stabilize the dithiol state will increase the reduction potential and will make the enzyme a better oxidant—the enzyme is destabilized by having the active-site disulfide, and so it is preferential to donate it to a nonnative protein substrate (i.e., to oxidize it).
Relatively little work has been done on determining the factors that determine the reduction potential of PDI. However, a considerable amount of work has been done on other thioredoxin-superfamily members, particularly thioredoxin and DsbA (for examples, see refs. 95, 103, 129, 168, 191, 220, and 345), and it is generally assumed that the same factors determine the reduction potential of PDI. Two main factors influence the reduction potential of thioredoxin-superfamily members, the pK a of the N-terminal cysteine thiol and the effects of the two intervening residues between the active-site cysteines on stabilizing/destabilizing the disulfide state. Factors that reduce the pK a of the N-terminal cysteine will favor the formation of the thiolate, which in turn favors the formation of the dithiol state; the net effect is to increase the reduction potential of the active site (i.e., to make the enzyme more oxidizing). Factors that modulate the pK a of the N-terminal cysteine of PDI are discussed in more detail later (see Section VII), but it is widely accepted that the pK a of the N-terminal cysteine of PDI lies between that of the more-oxidizing DsbA (pK a, 3.3 to 3.5) (103, 129, 220) and that of the more-reducing thioredoxin (pK a, 6.3 to 7.1) (69, 79). As mentioned earlier, the pH of the solution changes the reduction potential of thioredoxin-superfamily members by altering either the thiol-thiolate equilibria for the active-site cysteines (36) or the conformation of the protein, including at extremes of pH by denaturation.
The role of the other amino acids in the CXXC active site motif is twofold. First, these amino acids are juxtaposed to the active-site cysteines and so can have a major effect on the pK a values of the active-site thiols. Second, they can directly affect the stability of the disulfide state and its relative stability to the reduced state. For example, the active site of DsbA contains the motif CPHC. The histidine, also found in the active sites of PDI, is thought to modulate the pK a of the N-terminal cysteine (see refs. 117 and 165 for effects on PDI), whereas the proline introduces strain into the disulfide state, destabilizing it. The net effect of these two residues is reported to make the CXXC motif into a rheostat (36), although their function goes beyond this (245), because they must at least also form part of the substrate interaction site.
The reduction potential of the active sites of PDI is about −180 mV (189). The reduction potential of the glutathione present in the ER depends on the [GSH]2/[GSSG] ratio. An agreement exists in the field that the ratio of [GSH] to [GSSG] in the ER is lower than in the cytoplasm (i.e., that the ER is a more-oxidizing redox environment), and a consensus that the ratio is 3:1 (20, 130); however, this has recently been disputed (65). The absolute concentration of glutathione in the ER is unknown, but it has been estimated to be around 9 mM (20), and this is consistent with typically cited values for total cellular glutathione concentration. Because the reduction potential of glutathione depends on the [GSH]2 to [GSSG] ratio, it is critical to know the absolute concentrations of the two species and not just their relative ratios. However, a ratio of 3:1 and a total glutathione concentration of 10 mM would give a reduction potential for ER glutathione of −191 mV (see Fig. 8). This value is sufficiently oxidizing to allow the formation of disulfide bonds because Er 0′ ≈ −220 mV for denatured protein thiol/disulfides. In addition, this value is around the optimal value found for native disulfide bond formation in vitro (see Section VI). These concentrations are also sufficiently high for glutathione to act as a redox buffer (see Section VIII). In addition, this reduction potential for the glutathione redox buffer present in the ER would imply that at equilibrium in the ER, we would expect the active sites of PDI to be present in both the oxidized and reduced states, allowing PDI to act as a catalyst of protein dithiol-disulfide oxidation and isomerization reactions. PDI is found in this mixed state (8; see also see Section IX).

The understanding of the reduction potential(s) of PDI helps us to understand the physiological relevance of the in vitro and in vivo reactions that PDI can catalyze. However, before examining these, it is worthwhile to look at what is known about the domain architecture and structure of PDI.
V. The Architecture and Structure of PDI
The mature form of human PDI is 491 amino acids. Like most proteins larger than a couple of hundred amino acids, PDI has a multidomain structure. PDI is currently recognized as having four distinct domains,

No published structure exists for full-length mammalian PDI, despite decades of trials from multiple groups. The structures of the isolated
One problem associated with the study of catalyzed disulfide bond formation is the poor use of nomenclature and the concomitant cross-correlations between different proteins that may be part of the same family but may not have the same physiological function. For example, PDI-family members, especially ERp57 (see Section XI), are sometimes referred to as PDI, despite clear physiological differences. Similarly, PDIs from different species are often bundled together without full consideration of the potential differences in sequence, structure, or function. For example, the most highly studied PDIs are bovine PDI, human PDI, and the yeast S. cerevisiae PDI1 gene product Pdi1p. Although bovine and human PDI are very similar (94.9% identity), human PDI and yeast Pdi1p are not, although they share the same overall domain architecture. Pdi1p is 14 (or 22 for the longer variant found in some S. cerevisiae strains) amino acids longer than human PDI. Furthermore, human PDI is not N-glycosylated, whereas Pdi1p is heavily glycosylated. Overall, the percentage identity between the two mature proteins is reasonably high, given the species difference at 29.1%. However, whereas the catalytic domains show 40.9% (
Another confusion that arises is connected with the numbering of the protein sequence. A significant amount of experimental work was done on PDI-family members before the full amino acid sequence was known or before the cleavable signal sequences were defined. Therefore at least two sets of numberings exist, those based on the full-length protein and those based on the mature protein. Unfortunately, because of differences of opinion on the length of the signal sequence, some family members have different numbering systems even for the mature protein. Because the length of the signal sequence has not been experimentally determined for most PDI-family members, we will use full-length protein numbering throughout this review. For those who wish to convert to the mature numbering, the signal sequence of human PDI is 17 amino acids in length.
Given the high degree of sequence homology between the catalytic domains of PDI and thioredoxin, it is perhaps unsurprising that they share very similar structure. In addition to being the founding member of the thioredoxin superfamily of proteins, the structure of thioredoxin forms the foundation of a protein fold known as the thioredoxin fold. This fold is found in a wide range of proteins including thioredoxin, DsbA, PDI, glutathione peroxidase, glutathione S-transferase, arsenate reductase, calsequestrin, and circadian oscillation regulator, many of which are involved in thiol metabolism or use thiol-based chemistry. The thioredoxin fold is defined as a three-layer α–β–α structure with two α-helices packing onto one side of a four stranded β-sheet, of which strand three is antiparallel to the others, and with an additional α-helix packing onto the other side of the β-sheet. In thioredoxin, the structure is β–α–β–α–β–β–α. However, α3 is sometimes missing and, allowing for this, and for circular permutations of the fold, Qi and Grishin identified a significant number of other thioredoxin-fold–containing proteins and protein families, including tubulin C-terminal domain, cytidine deaminase, chorismate mutase, RNA 3
Although the thioredoxin fold is defined by the core three α-helices and four β-strands, this is not the structure of thioredoxin. Instead, thioredoxins, including human thioredoxin (80) and Escherichia coli thioredoxin (124, 147), show a β–α–β–α–β–α–β–β–α structure (i.e., they have an additional β–α before the thioredoxin-fold). This fact causes some problems connected with nomenclature. For example, the active site of thioredoxin lies at the N-terminus of the first α-helix of the thioredoxin fold (α1), but this is the second α-helix of thioredoxin (the first α-helix is α0). In this review, we use the standard nomenclature in the field (i.e., that the active site of PDI lies at the N-terminus of α2), and the phrase “thioredoxin-like” fold to describe structures with a β–α–β–α–β–α–β–β–α structure.
As stated, the active site of thioredoxin and of the catalytic domains of PDI lie at the N-terminus of the α2 helix, with the N-terminal active-site cysteine being at the N-terminus of the helix, and the C-terminal active-site cysteine having limited solvent exposure. This arrangement is important in determining the pK
a of the active-site cysteine residues. The pK
a of the N-terminal active-site cysteine must be low both to have a sufficiently high reduction potential so as to act efficiently in protein dithiol oxidation (see Section IV) and to have efficient kinetics for the initial steps of catalysis of thiol-disulfide exchange (see Section III). The pK
a of the N-terminal cysteine of the catalytic domains of PDI has been reported to be in the range 4.4 to 6.7 (114, 165, 260), considerably lower than the normal pK
a of a protein cysteine thiol. This low pK
a results from multiple effects. In part, it results from the N-terminal active-site cysteine being located at the N-terminus of an α-helix. α-Helices have a permanent dipole associated with them, with the N-terminus of the helix having a positive dipole moment, whereas the C-terminus has a negative dipole moment. This leads to a decrease in the thiol pK
a of up to 1.6 pH units for cysteine residues at the N-terminus of α-helices in model peptides (164). This by itself its not sufficient to explain the abnormally low pK
a value of Cys 53 and Cys 397 of PDI, because thioredoxin has a similar structure, and the pK
a of the N-terminal active-site cysteine of thioredoxin is usually cited as being 7.1 (69). Other electrostatic effects must be around the active site of PDI. The primary effector is probably the histidine residues, His 55 and His 399, which lie between the active-site cysteines. However, other charged residues are within 7 Å of the active-site cysteines in the NMR structure of the
The requirements for the pK a of the C-terminal active-site cysteine residue, Cys 56 and Cys 400 in PDI, are more complex. However, for much of the catalytic cycle of protein dithiol oxidation or isomerization, this group must be in the protonated thiol state, so these cysteines require a higher than average pK a value. The pK a of this group is so high that it is difficult to measure, without competition from protein denaturation, but general agreement in the field indicates that it is >10, and it has a calculated value of 12.8 (178). However, at one step of the catalytic cycle for protein dithiol oxidation, this group must be a thiolate, and a conformational change within the catalytic domain lowers the pK a of this group to <7 (see Section VII).
Other structural features that are conserved between other thioredoxin-superfamily members and the catalytic domains of PDI include (a) a conserved proline residue in the middle of α2, Pro 61, and Pro 405 in PDI, which introduces a kink into the helical structure and allows it to wrap around the core β-sheet; (b) a cis-proline residue, Pro 100 and Pro 441 in PDI, found at the start of β4 and juxtaposed to the active site. This residue, forming part of what was known as the “Egypt motif,” as the sequence is GYPT in the
Whereas the
No multidomain structures are available for human or any other mammalian PDI. However, in the last few years, crystal structures have been reported for yeast Pdi1p (297, 298) and the human PDI-family members ERp57 (in complex with tapasin; PDB: 3f8u) (66), ERp29 (a structure that contains only one thioredoxin-like domain; PDB: 2qc7) (16) and ERp44 (PDB: 2r2j) (326), along with structures for the
With structural information at hand, it is time to move on to examining the reactions that PDI can catalyze and the methods used to characterize its mechanisms of action, starting with in vitro analysis.
VI. In vitro Thiol-Disulfide Exchange Reactions of PDI
A long and sometimes contradictory history is associated with the study of the reactions that PDI is able to catalyze. This is best presented as a summary of the reactions PDI is able to catalyze in vitro, in line with Section III, the assays used to examine these activities, and then the roles of individual domains in these processes.
PDI is oxidized by molecular oxygen relatively slowly, and therefore, it should not be categorized as an oxidase (296). Therefore, all of the reactions catalyzed by PDI are thiol-disulfide exchange reactions. However, it is important to distinguish at least three categories of reactions that PDI can catalyze (see Fig. 10).
Oxidation reactions in which a protein or peptide substrate dithiol is oxidized to the disulfide state, with the concomitant loss of an active-site disulfide from PDI. To complete the catalytic cycle, PDI must be reoxidized. In vitro, GSSG is usually used as the terminal electron acceptor. This generates GSH and results in a change in the reduction potential and redox-buffering capacity of the buffer. Care must be taken to ensure that sufficient GSSG is present to complete the reaction and that the change in reduction potential during the reaction does not significantly affect the folding efficiency or yield. If no GSSG or equivalent electron acceptor is present, PDI is also able to reduce a disulfide bond in one nonnative protein molecule to form a disulfide in another nonnative protein. Reduction reactions in which a protein- or peptide-substrate disulfide is reduced to the dithiol state, with the concomitant gain of an active-site disulfide in PDI. To complete the catalytic cycle, PDI must be reduced. In vitro, GSH or DTT is often used as the electron donor. Again, this results in a change in the reduction potential and redox-buffering capacity of the buffer. Isomerization reactions in which the disulfides and thiols in a protein or peptide substrate are rearranged to give a different disulfide-bonding pattern. In direct isomerization, no net change exists in the redox state of the active site of PDI, and so no other redox reagents are required.
All of these reactions can involve intra- or intermolecular substrate disulfides, with the focus to date being on the former. In addition, certain reactions also have specialist names (e.g., the reduction of a protein or peptide mixed disulfide with glutathione by using GSH as the electron donor to generate GSSG and a protein thiol is known as a deglutathionylation reaction). Similarly, the formation of a protein or peptide mixed disulfide with glutathione is known as a glutathionylation reaction.
A. Assays for PDI-like activity
Multiple different assays have been used to determine the activities of PDI, all of which have advantages and disadvantages. A consensus is found in the field that it is better to use “natural” substrates (i.e., those based on proteins and peptides) rather than artificial ones, although the use of extrinsic fluorophores can give highly sensitive systems (251). However, only rarely is a physiological substrate used. Instead, a fairly narrow range of “well-behaved” proteins and peptides are used. It should also be remembered that in vitro, we usually deal with refolding of proteins in dilute solutions with PDI present in catalytic amounts, whereas in vivo, folding occurs co-translationally at high protein concentrations and with PDI present in very high concentrations and probably in excess over substrate. The in vitro systems may give a distorted picture of the physiological functions of the enzyme. However, given the dearth of methods for dissecting mechanisms of action in natural systems, the only realistic approach is to combine in vitro and in vivo data, accepting that both may be prone to artifact.
Some in vitro assays are based on the gain of activity of a substrate protein. Here the classic example is the use of a ribonuclease; usually RNase A. RNase A is a one-domain protein that contains four disulfide bonds in the native state. The RNase assay comes in many different formats. The starting material can be (a) reduced RNase, to give an assay that looks primarily at oxidation; or (b) “scrambled” RNase, in which the protein has been reduced and allowed to oxidize under denaturing conditions, which produces a set of proteins in which the disulfide bonds are predominantly nonnative, to give an assay that looks primarily at isomerization, or (c) glutathionylated RNase T1 (261, 262), in which all of the cysteines in the protein are glutathionylated at the start, in an assay that looks at isomerization, including the deglutathionylation subsystem. Assays that are based on the gain of activity of a substrate protein are widely used, in part because of their relative simplicity. The major advantage that drove their development was their sensitivity, allowing detection after only a very small percentage of the refolding protein had reached the active state. However, a number of problems are associated with them. The first problem is that the assay does not directly measure disulfide-bond formation in the substrate protein. A gain of activity does not directly correlate with disulfide-bond formation because many folding intermediates may have biological activity, with the degree of activity depending on the structure of individual folding intermediates and being protein dependent. In addition, for mechanistic studies, the biophysical conditions of the system are often varied, and care must be taken that any changes observed relate to changes in the activity of PDI and not to changes in activity or stability of the folded substrate or of its folding intermediates.
The second problem relates to the complexity of the material: 764 different disulfide-bonded states are possible for RNase A. Whereas only a small fraction of these may be experienced during refolding, each intermediate is in effect a different substrate for PDI with different requirements for catalysis, and a global assay, such as the regain of activity, will miss many of the subtleties. This problem is exacerbated for “scrambled” RNase in that the starting material itself is not homogeneous (and shows considerable batch-to-batch variation).
The third problem is related to assaying the activity of RNase. Essentially, two different assays are used: (a) the hydrolysis of RNA (102, 133, 173, 288), which uses a natural substrate but which requires very small changes in absorbance to be accurately measured with time, necessitating the use of a good double-beam spectrometer; or (b) the hydrolysis of cCMP (193, 322, 323, 340), an assay that, for detailed kinetics, requires the accurate measurement of very high absorbance values. The cCMP variant has also been used in a continuous variant of a PDI assay. This is based on taking the first derivative of the absorbance with respect to time and correcting for the depletion of cCMP and product inhibition of RNase by CMP (193, 340). Because of these problems with the assays, often a less-comprehensive measurement is used: either the use of measuring the “lag-phases” (the time taken before activity of the substrate enzyme starts to appear), or of measuring the initial rate of appearance of RNase activity after the “lag-phase,” sometimes based on just two time points. Although these methods can be used to measure the relative order of activity (i.e., protein X is more active than protein Y), they are poor methods to determine PDI activity quantitatively, given the complexity of the system and the heterogeneity of the starting material in the “scrambled” RNase assay. A recent addition to this category of assays is the use of riboflavin-binding protein (250), a protein that contains nine disulfide bonds. Quenching of riboflavin fluorescence on binding by functional riboflavin-binding protein allows continuous monitoring of the system, even at high protein concentrations, making it potentially a very powerful assay. However, as with most assays in this category, it is a measure of gain of some function and not necessarily a measure of gain of the native state, and so care must be taken in interpretation of the results.
A second category of assays is based on changes in the biophysical properties of a substrate. An early assay in this category was the insulin-reduction assay (202, 224). In this assay, insulin reduction by GSH or DTT results in the aggregation of the B chains of insulin. This aggregation event is followed by changes in light scattering, usually by monitoring the apparent increase in absorbance at ∼600 nm. Again, this assay is very useful to determine the relative order of activity of different catalysts, but it is a poor method for quantification. Because the assay measures aggregation properties of the product, it is also very sensitive to changes in biophysical conditions. However, this method forms the basis of a proposed high-throughput screen for PDI inhibitors (278). A more quantitative version of this assay is the coupled insulin-reduction assay (176, 244, 245). In this assay, the GSSG formed by the reduction of insulin by GSH is reduced by glutathione reductase, with the concomitant oxidation of NADPH and a decrease in absorbance at 340 nm. This assay is quantitative, but only up to time points at which insulin aggregation starts to contribute to the apparent changes in absorbance at 340 nm due to light scattering. A similar assay is also possible by using thioredoxin reductase in place of the GSH and glutathione reductase, because PDI is a substrate for thioredoxin reductase (190). Two additional drawbacks of the insulin-reduction–based assays are that (a) many other enzymes show significant activity in this assay; and (b) the assay looks at the reduction of a substrate disulfide that is neither the main physiological function of PDI (see Section X) nor the main defining reaction of the enzyme.
Other assays that fall into this category are peptide-based thiol-disulfide exchange assays in which an associated change in fluorescence occurs. The first of these published was an oxidation assay in which disulfide-bond formation is accompanied by a change in intrinsic fluorescence of a tryptophan residue in the substrate peptide (260). This is a quantitative assay, and the designed peptide allows a wide range of biophysical conditions to be used, but being based on intrinsic fluorescence, it has limitations, especially because the change in signal is only ∼18%. This assay also formed the basis for similar assays to examine glutathionylation and deglutathionylation activity (235). Subsequently Winther and co-workers (335) introduced a series of substrate peptides with a fluorescent aminobenzoic acid residue and a nitrotyrosine quencher, which on reduction (37, 335) or oxidation (37) show very significant changes in fluorescence, with spectral parameters that are not affected by the intrinsic fluorescence of proteins. Another recent addition is a peptide based on tachyplesin I that allows examination of isomerization reactions in a homogeneous substrate via donor-quencher–based fluorescence assays (151). This very useful and welcome assay does, however, have complications relating to the properties of the peptide, including its adsorption to cuvettes.
A third grouping of assays is based on quenching dithiol-disulfide exchange reactions and then subsequently monitoring the disulfide bond state of the substrate. These quenching-based assays have the disadvantage that they are considerably more time consuming than the other assays presented here, and they are always discontinuous. However, they have the potential to provide much more detailed kinetic analysis of even complex systems. Although various peptides and proteins have been used, the classic substrate in this type of assay is BPTI (44). BPTI is a small one-domain protein that contains three disulfide bonds. Its use has the advantages that the folding pathway has been extensively characterized (but see later) and that all of the folding intermediates are soluble. These assays are based on quenching or freezing the dithiol-disulfide exchange reaction at discrete time points. Quenchers fall into two categories: acidification and chemical modification (see Fig. 11). Acidification, the reduction of the pH of the solution by the addition of acid, has two advantages: it is very rapid, and it results in denaturation of the enzyme and the substrate. However, it also has disadvantages. Specifically, it does not permanently quench thiol-disulfide exchange reactions, it only slows them (see Fig. 11A). For example, PDI has residual activity under the most commonly used acid-quenching conditions (A.-K. Lappi and L.W. Ruddock, unpublished results). In contrast, chemical modification of the thiol groups will permanently stop thiol-disulfide exchange reactions. Typical chemical quenchers include iodoacetamide, iodoacetate, and maleimides such as N-ethyl maleimide (NEM), but any thiol-reactive compound can potentially be used. However, care must be taken in the choice of chemical modifier used. For example, thiosulfonates such as S-methyl methanethiosulfonate have been used as quenching reagents (132, 187, 233); however, these reagents can result in a change of the thiol-disulfide state of the system (145). Similarly any iodo-based compounds must be high grade, because any contaminating iodine can catalyze disulfide-bond formation. The use of chemical quenchers has one major disadvantage: the reactions of thiol groups with these compounds are nucleophilic reactions, which are in direct kinetic competition with the nucleophilic reactions involved in thiol-disulfide exchange by the same thiol groups (see Fig. 11B). For surface-exposed thiol groups, this problem can be minimized by the use of a large excess of quenching reagent. Iodoacetamide is often used as a quencher at molar concentrations. For PDI, the surface-exposed N-terminal active-site cysteine has a second-order rate constant of 11 M−1s−1 for reacting with iodoacetate when in the thiolate state, and circa 7 M−1s−1 at pH 7 (114). This implies a half-time for reaction at pH 7 of around 0.1 s when 1 M iodoacetamide is used. This time-scale is fast enough to trap most thiol-disulfide exchange reactions. However, the pK a of this thiol is unusually low and, assuming a similar second-order rate constant with a typical cysteine thiol pK a of 8.3, the half time for quenching under the same conditions becomes 1.3 s. In addition, buried thiols in folding proteins may be poorly accessible to chemical quenchers and may continue to undergo intramolecular thiol-disulfide exchange reactions rather than reacting with the chemical quencher. This is a serious problem. For example, in BPTI with chemical quenchers, Creighton and co-workers (43) saw major populations of nonnative two-disulfide bonded states, whereas Weissman and Kim (330, 331) saw predominantly only native-like two-disulfide states by using acid quenching. The difference is probably due to the inaccessibility of buried thiol groups to chemical quenchers (see also 42, 332). Similarly mixed disulfides between PDI and substrates or glutathione are very difficult to trap, as the C-terminal active site cysteine is very inaccessible to chemical quenchers (see Fig. 11) (53; A.-K. Lappi and L.W. Ruddock, unpublished observations).

After the quenching, reaction analysis of the thiol-disulfide state of the protein is often done by reverse-phase HPLC or by mass spectrometric methods. Mass spectrometric methods are more rapid. However, they give information only on the populations of different disulfide-bonded states (e.g., no disulfides vs. one disulfide vs. two disulfides). They do not give information on subpopulations (e.g., for folding intermediates that contain two disulfide bonds, what proportion of these are native). In contrast, under ideal conditions, reverse-phase HPLC allows the separation and relative quantification of all of the different species present in the sample. Although it is a much more powerful technique, it requires extensive optimization and identification of the different eluting species, something that is often complicated by the presence of one or more species co-eluting.
Given the narrow range of substrates used to assay PDI-like activity, it is possible that a significant bias exists in the data set obtained to date, especially for PDI-family members that are not as promiscuous as PDI.
B. Domain contribution to thiol-disulfide exchange activity
Because PDI has two catalytic domains, along with two domains that lack active sites but that may contribute to the overall catalytic activity of PDI, it is useful to dissect the molecule to examine the roles of each domain in isolation and in combination. Similarly, the use of mutants, either in full-length PDI or in domain constructs, allows a dissection of mechanisms of action. However, in both cases, it is essential to ensure that the structure of the construct is not compromised.
The minimal unit required for catalysis of thiol-disulfide exchange reactions is usually considered to be the isolated
In a landmark study, Darby and co-workers (56) dissected out the contributions of domains to the relative activities of PDI by using a range of substrates including two different kinetically trapped nonnative disulfide-containing BPTI-refolding intermediates, a 28-amino-acid peptide based on the sequence of BPTI, and the insulin-reduction assay (56). This study, and others (162, 325), showed (a) that deletion of the
For oxidative protein folding in vitro, the rate-limiting steps are thiol-disulfide exchange reactions in folding intermediates that contain substantial regular secondary structure. For example, for some two-disulfide BPTI-folding intermediates, the noncatalyzed half-times to reach the native state in a glutathione buffer are measured in hours or even days. It is for these reactions involving quasi-native intermediates that PDI would be expected to have the greatest catalytic effects. The relative contribution of PDI catalysis to oxidation, reduction, and isomerization is difficult to gauge and is probably substrate dependent, but it is clear that PDI is efficient at catalyzing all three thiol-disulfide exchange reactions. Although PDI is able to catalyze all thiol-disulfide exchange reactions, it is reactions in late-stage folding intermediates that are the rate-limiting steps in oxidative folding in vitro. How efficient is it at catalysis? Values from the literature for which a defined transition is looked at can be examined for evidence. For example, for BPTI refolding at pH 7.3 in a buffer containing 2.0 mM GSH and 0.5 mM GSSG, the acceleration for transitions were 3,500- to 6,000-fold (330). For the oxidation of a kinetically trapped state of RTEM-1 β-lactamase, catalysis was 500-fold faster than the noncatalyzed GSSG reaction at pH 8.0 (321). Similarly, when using 0.5 mM GSSG as the electron acceptor for oxidation of a simple peptide substrate at pH 7.0, a circa eightfold difference in rate of refolding was found in the presence and absence of 0.74 μM PDI (260), which, given the relative concentrations, implies that PDI was >5,000-fold more efficient at introducing the disulfide bond into the peptide than was GSSG. As a final example, at pH 7.3, a larger and unrelated peptide substrate PDI, with GSSG as the net electron acceptor, was ∼100- to 120-fold faster than GSSG at introducing a disulfide bond (54). It should be noted that both the catalyzed and noncatalyzed reactions show a very strong pH dependence, and that the major effect on catalysis is the unusually low pK a of the active-site cysteine of PDI, which allows it to be an efficient nucleophile at physiological pH values. At pH 7, the proportion of N-terminal active-site cysteine that is in the thiolate state (assuming a pK a of 5.1) is 98.8%, whereas the proportion of a normal protein or peptide cysteine in the thiolate state (assuming a pK a of 8.3) is 4.8%. This 20-fold difference certainly contributes to the catalytic activity of PDI at pH 7. At higher pH values, the proportion of the N-terminal active-site cysteine in the thiolate state cannot significantly increase, whereas that of a normal cysteine can and does. As the pH increases above 7.3, the difference between the rates of the catalyzed and uncatalyzed reactions decreases. For this reason, and for reasons connected to the structure and stability of PDI and of folding intermediates, the study of catalysis should be done, wherever possible, either at pH 7.3 (physiological pH) or at pH 7 (the pH for determining standard biochemical reduction potential).
Very little work has been reported on the substrate specificity of catalysis of thiol-disulfide exchange by PDI. In a landmark article, Westphal and co-workers (335) screened a library of random peptides and found and characterized 13 different peptides whose reduction could be catalyzed by PDI and two that could not. Although this is a small dataset, a preference for small amino or imino acids existed before the cysteine to be reduced (Ser, Ala, Gly and Pro were found in 10 of 13 positive peptides), and a preference for basic amino acids at a position two amino acids after the cysteine (His, Arg, Lys were found in seven of 13 positive peptides), which may be linked to modulation of the pK a of one or more of the cysteines in the system. Substrates that could not be reduced by PDI contained multiple aromatic groups in positions adjacent to the peptide cysteine, suggesting that steric hindrance was the problem.
C. Substrate binding by PDI
To be able to catalyze thiol-disulfide exchange reactions in folding proteins, PDI must be able to bind them. Initial studies on catalysis showed that the K
M of PDI for peptide substrates was <3 μM (54). To identify substrate-binding sites directly, a cross-linking–based approach has been used, in which a radiolabeled peptide is added to PDI, usually in a lysate to minimize potential artifacts. This approach first identified PDI as being equivalent to the glycosylation-site binding peptide (97), as it was able to bind to a radiolabeled peptide Asn-Lys-Thr, although from subsequent analysis of PDI specificity, it is possible that the actual groups bound were the aromatic labels and not the tripeptide itself. This same probe was used to locate the interaction site in PDI (223). The interaction site was found to be in a 26-amino-acid fragment that included part of the last helix of the
To date, the specificities of binding by PDI or of individual domains of PDI have not been determined. However, the specificity of substrate binding by the other PDI-family members PDIp (259) and ERp29 (16) have both been shown to have preferences for specific patterns of hydrophobic amino acids (see Section XI): Such a requirement for hydrophobic residues for substrate binding by the
D. Molecular chaperone and antichaperone activity
In addition to its ability to catalyze thiol-disulfide exchange reactions, PDI has also been suggested to have molecular chaperone activity (324). This is manifested during folding of proteins that contain disulfide bonds (242, 339, 342) and the ability to assist in the refolding of proteins with no disulfide bonds (30, 282). This activity does not depend on the catalytic domain active sites and is inhibited by the presence of a peptide substrate for the
In addition to a chaperone-like activity, PDI has also been reported to have an antichaperone activity (242), which is inhibited by the
VII. Conformational Change in PDI
Protein structures are dynamic in nature. All proteins have structures that constantly undergo small “breathing” motions, and many undergo larger conformational changes, such as induced-fit substrate binding, that are directly linked to their function. One of the early models for PDI was that the four domains formed “beads on a string” (i.e., that they were linked by highly flexible regions). Such an arrangement would allow PDI the flexibility to bind to and act on its very wide range of protein substrates. This model slowly lost favor, as it was realized that the interdomain linker regions between the
The active site of PDI, and other thioredoxin-superfamily members, exists in two redox states, the reduced dithiol state and the oxidized disulfide state, as well as in transient mixed disulfides. The oxidized and reduced species are able to catalyze different reactions, oxidation of protein substrate dithiols, and isomerization of protein substrate disulfides, respectively. The different redox states of PDI must interact with different protein substrates, or the same protein substrate at different stages of the folding pathway. In addition, PDI must interact with Ero1 (see Section VIII) only when it is in the reduced state. Very little is known about conformational differences between reduced and oxidized PDI. For other superfamily members, such as thioredoxin or DsbA, small, but distinct conformational differences exist between the oxidized and reduced states (106, 123). These are centered mainly on the active-site region, as would be expected, but at least in DsbA, they are also transmitted to the farthest point in the protein. Preliminary NMR studies have revealed that the
The second conformational change that occurs in PDI is linked to the paradox of the pK
a of the C-terminal active-site cysteine. For catalysis of oxidation of a substrate protein to occur, the pK
a of the C-terminal active-site Cys must be high, so as to prevent the reverse reaction from occurring (see Fig. 12). However, in one of the steps of reoxidation of PDI, this cysteine must be in the thiolate state (see Fig. 12B). This implies that a significant change in the pK
a of the C-terminal active-site Cys (Cys 56 and Cys 400) must occur during the catalytic cycle of PDI. The pK
a value of chemical moieties varies depending on their local environment (see Section III). In the NMR structure of the
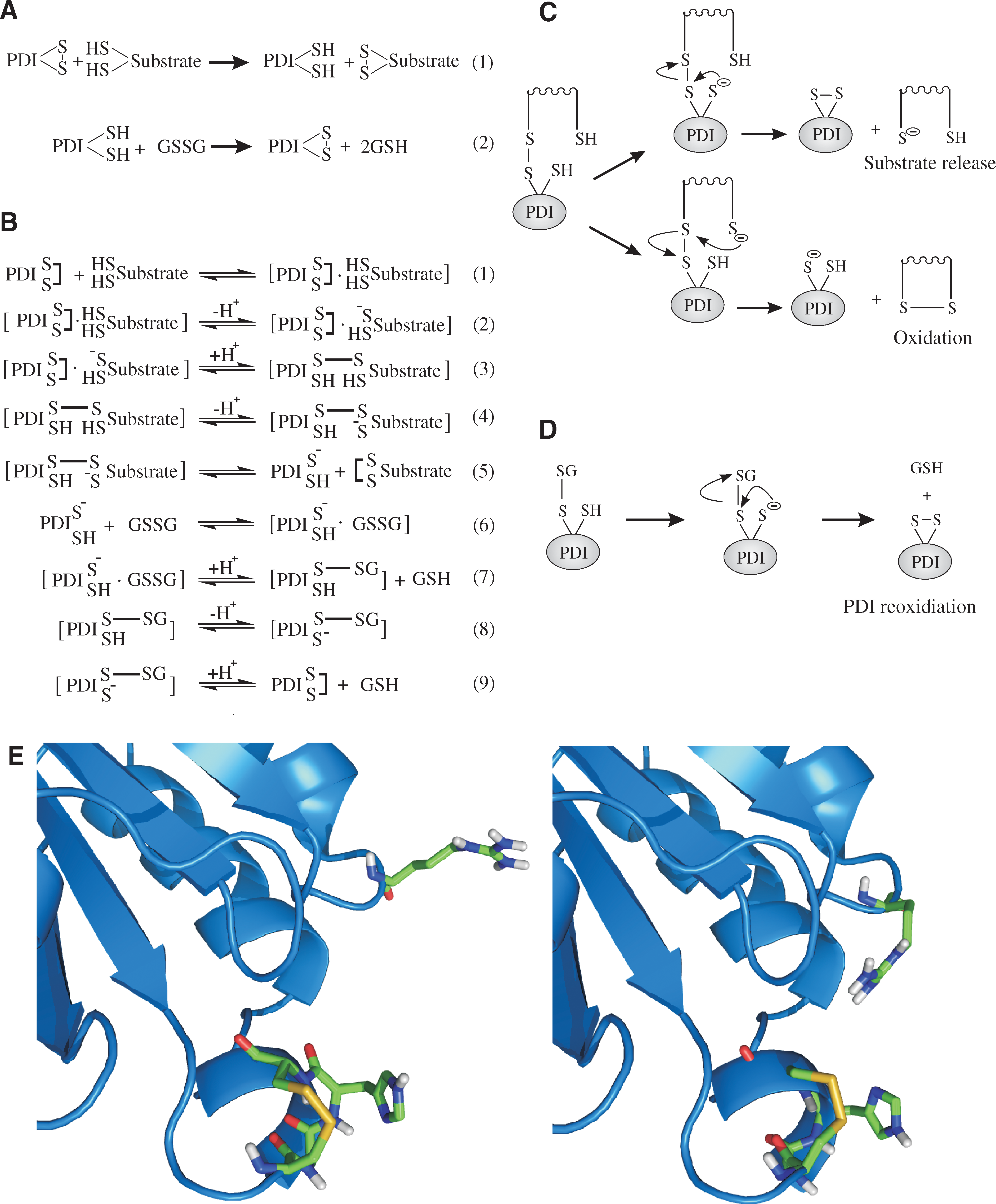
Other human PDI-family members similarly saw a significant reduction in activity on mutation of the analogous arginine residue. In addition to being linked to the reoxidation cycle of PDI, the movement of arginine 120 is also potentially linked to the release of kinetically trapped mixed-disulfide species between PDI and substrate proteins during isomerization (see Fig. 12D). This dual function implies a balance between the kinetics of oxidation (which require a fast deprotonation of Cys 56) and the kinetics of isomerization (which require a more-stable mixed disulfide between PDI and the substrate protein to be formed). However, this has yet to be shown experimentally. The positively charged amino acid, usually an arginine, is conserved in many catalytically active PDI-family members (71).
The third conformational change reported to occur in human PDI is linked to the substrate-binding site in the

One of the most important aspects of PDI function, rarely considered, is the ability of this enzyme to access buried disulfides and thiols in substrate proteins, and so the need to induce conformational change in the folding substrate. As discussed earlier, folding proteins are not in their most thermodynamically favored state, and it should be relatively easy to induce conformational change in what is essentially an unstable state. We recently identified a second conformational change in the catalytic domains of PDI, which contributes significantly to the ability of PDI to induce conformational change in a folding substrate (A.-R. Karala, M.F. Lensink, A.H. Juffer, and L.W. Ruddock, unpublished observations) and which we believe is conserved across most of the PDI family. However, clearly other factors are at work, and an intermolecular conformational change in the structure of PDI (i.e., conformational change within one or more domains of PDI or an interdomain conformational change) would be a simple method for inducing conformational change in substrate proteins, especially if they were bound to all three substrate-binding sites in the

Although in vitro analysis of PDI allows us to determine potential mechanisms of action of the protein, it must always be viewed as an artificial system, so correlations with the in vivo activities of PDI are essential. Before examining these in vivo activities, we must first examine in more detail the potential mechanisms of oxidation and reduction of PDI in vivo and the in vivo redox state of PDI.
VIII. Oxidation and Reduction of PDI in vivo
Over the past decade, a revolution has occurred in the perception of the physiologically relevant mechanisms for PDI oxidation in vivo. Prior to 1998, the consensus in the field was that the active sites of PDI were oxidized in vivo by reaction with GSSG (i.e., by the same mechanisms by which PDI was oxidized in vitro). This viewpoint was overthrown by the discovery and subsequent characterization of the Ero1 subfamily of sulfhydryl oxidases. This change in viewpoint went so far that the GSSG present in the ER came to be regarded as a nonfunctional byproduct of the natural pathway for disulfide-bond formation and that any reactions of GSSG in vivo were either irrelevant or even possibly deleterious to the system. The actual physiologically relevant mechanisms for in vivo oxidation of PDI probably lie between these two extremes.
A. Ero1-based oxidation
The Ero1 family was first reported as a gene product in the yeast S. cerevisiae that was required for the formation of disulfide bonds in the ER (84, 239). The yeast protein, Ero1p, is a highly glycosylated, cysteine-rich, membrane-associated enzyme that is essential for S. cerevisiae viability and for which homologues in a wide range of eukaryotic organisms were immediately identified. Overexpression of Ero1p gives increased resistance to the addition of the reductant DTT. It was subsequently reported that Ero1p is essential for the oxidation of Pdi1p and that Ero1p and an N-terminal active-site cysteine of Pdi1p form a mixed disulfide in vivo, implying that Ero1p directly oxidizes Pdi1p (85). Two distinct Ero1-family members, Ero1α and Ero1β, are found in mammals, and both are able to complement several phenotypic traits associated with the functional loss of Ero1p in S. cerevisiae (29, 230). It is still unclear why we need two Ero1-family members. However, Ero1α expression is induced by hypoxia (98) and ER stress (198), whereas Ero1β expression is induced by stress resulting from an accumulation of unfolded proteins in the ER (230). Ero1α was subsequently shown to form a mixed disulfide complex with wild-type PDI in vivo (21). Because this complex was observed only 1 h after the start of the pulse-chase, it was concluded that the Ero1α–PDI complexes were the intermediates in the oxidation of PDI by Ero1α (see Fig. 15) and not the alternate possibility that they represented Ero1α being a folding substrate for PDI. Both Ero1α and Ero1β were then shown to be involved in the oxidation of PDI, but not of the PDI-family member ERp57 (209). Ero1 family members have a large number of cysteine residues, and many of these were found to be essential for the folding and function of Ero1p (86) and Ero1α (21). These cysteines form a number of distinct disulfide-bonded states in vivo, states that were subsequently found to be regulatory (see later). The next big step forward was the publication by Tu and co-workers (302) that identified Ero1p as a FAD-binding protein and showed that Ero1p plus PDI and FAD allowed the effective in vitro reconstitution of a pathway for oxidative-folding protein by using RNaseA as a substrate. Mutagenesis studies of Ero1α and Ero1p (23, 272), aided in part by the crystal structure of Ero1p (104), revealed a complex pathway of intra- and intermolecular disulfide-bond transfer between the FAD cofactor and PDI or Pdi1p. Furthermore, a strong preference exists for Ero1p to oxidize the active site of the

B. GSSG-based oxidation
The primary redox buffer in the ER is thought to be based on the oxidized and reduced states of glutathione. Whereas the cytoplasm is a highly reducing environment with <1% of the glutathione present in the oxidized GSSG state, the ER is more oxidizing, with ∼25% of the ER-resident glutathione thought to be present as GSSG (20, 130). In vitro GSSG is able to oxidize the active site of PDI efficiently (see Section VI), and for many years, it was thought to be the primary oxidant of PDI in vivo. However, whereas Ero1p is an essential gene product in yeast (84, 239), Gsh1p, the first enzyme in glutathione biosynthesis, is not (226). This led to the idea that GSSG may not be directly involved in native disulfide-bond formation in the ER, but rather that its production is competing with disulfide-bond formation in folding proteins (see ref. 33 for a recent review on the role of glutathione in disulfide-bond formation). The evidence for this comes from several indirect sources that are linked, not to the function of GSSG, but to the function of GSH. GSH is a reductant; in vitro, it is able to reduce disulfide bonds in folding proteins and the active-site disulfide in PDI, and it is essential for the isomerization reactions that many proteins require to reach the native disulfide-bonded state (see Section VI). A similar function would be expected in vivo, as the ER probably contains millimolar concentrations of GSH (20, 130). The first evidence for a role of GSH in vivo was reported from S. cerevisiae. Here the production of cellular GSSG was shown to be linked to Ero1p activity (50), most likely through the reduction of disulfides in folding proteins or in PDI-family members (i.e., the direct oxidation of GSH by Ero1p is limited (303). Furthermore, in a screen for suppression of the growth defect of the ero1-1 temperature-sensitive Ero1p mutant, from 21,000 transposon insertions, all nine positive mutants were linked to insertions in the gene for Gsh1p (50) (i.e., inhibition of glutathione biosynthesis rescued the viability of a strain harboring a mutated Ero1p). Furthermore, the maturation of a widely used model protein for monitoring disulfide-bond formation, carboxypeptidase Y, showed a strong defect in the ero1-1 mutant, but in the wild-type strain and in the ero1-1 gsh1 double-mutant, maturation was complete by the first time point of the assay. These results were interpreted to mean that GSH acts as a competitor with protein thiols for Ero1p, with the GSSG produced being secreted from the ER. However, these results do not show that GSSG has no function in the ER during disulfide-bond formation, especially because the disulfide bond in GSSG may be transferred directly, or indirectly via a PDI-family member, to a folding protein. In mammalian systems, cytoplasmic GSH has been shown to limit disulfide-bond formation in the ER of HeLa cells; specifically, it was required to maintain PDI and Ero1α in their physiological redox states (214). Reducing the level of cellular glutathione in CHO cells resulted in acceleration of oxidative folding, after treatment with the reductant DTT, but not in the rate of native disulfide-bond formation (32). These results suggest that glutathione is directly linked to the reductive or isomerization pathway or both in vivo. Glutathione has also been shown to reduce directly the PDI-family member ERp57 in vivo (135), and presumably, PDI is reduced via the same direct mechanism, as well as indirectly via reduction–oxidation cycles with folding proteins. This is required to keep at least part of the population of PDI, or other PDI-family members, in the dithiol state required to catalyze thiol-disulfide isomerization reactions (see Sections VI and IX).
How do the in vivo GSSG data correlate with data from in vitro kinetics? The second-order rate constant for oxidation of the active site of the GSSG is not an oxidase or a net oxidant. The oxidation of the active site of PDI by GSSG is a net isomerization reaction; with the formation of the disulfide bond in PDI being accompanied by the reduction of GSSG to GSH. Thus, GSSG cannot be the net oxidant in the system for the production of disulfide bonds, but it can be an intermediary. The oxidant for converting GSH to GSSG is the subject of much debate in the literature, but two widely acknowledged routes are the reduction of (nonnative) disulfide bonds in folding proteins and the reduction of the active site of PDI-family members. In both of these cases, the pathways can be traced back to Ero1, consistent with the published link between Ero1 activity and cellular GSSG levels (50). Although the absolute concentration of GSSG and GSH in the ER lumen is not known, the in vivo ratio between these species and estimates of their concentration (20, 130) are consistent with the optimal concentrations of these species required for efficient native disulfide-bond formation in vitro (see Section VI). Furthermore, no reports exist of naturally occurring variations in this ratio with varying production of secreted disulfide-bonded proteins. This implies that the ratio is probably carefully regulated and does not solely arise with the production of GSSG as a byproduct of native disulfide-bond formation. Ero1-family members are membrane-associated proteins. Although disulfide-bond formation occurs co-translationally, we suppose that many of the reactions connected with native disulfide-bond formation of soluble proteins happen in the ER lumen. This would either require two pools of PDI, one associated with co-translation disulfide-bond formation and one with subsequent formation of native disulfide bonds, or for PDI to cycle back between the substrate in the lumen and Ero1 on the membrane. Such a requirement for PDI cycling seems inefficient, especially given the very high intracellular protein concentrations, which reduce the rate of diffusion. It would be more efficient to use the glutathione redox buffer to change the redox state of PDI in the lumen as required.
In vitro, the rate-limiting steps associated with native disulfide-bond formation are late-stage events in substrates with a high degree of native-like secondary structure (see Section VI). If this is mirrored in vivo, and it is likely to be, then in vivo oxidative events could have their efficiency significantly reduced without an overall effect on the rates or yields of native disulfide-bond formation.
Given these arguments, it is possible that both GSSG and GSH are involved in the physiological oxidation, reduction, and isomerization events in native disulfide-bond formation. If so, this would primarily be via a PDI-family member, because the rates of their direct reactions with folding substrate proteins are low [e.g., whereas the second-order rate constant for GSSG reacting with reduced PDI is 188 M−1s−1, the initial oxidation of reduced BPTI by GSSG is only 7.3 M−1s−1 (146)]. This difference is probably determined primarily by the 20-fold difference in the proportion of the thiolate state of the nucleophilic cysteines in PDI and an unfolded protein (see Section VI). In addition, glutathione could serve multiple other purposes in the ER: (a) It could act as a redox buffer, buffering the ER against both oxidizing and reducing agents; (b) It could act as a reservoir of both oxidizing and reducing potential, allowing the cells to react rapidly to changes in the levels of production of disulfide bond–containing proteins; (c) Via the oxidation and reduction of PDI family members, it could regulate the activity of Ero1 (see Fig. 15B). Under resting conditions, the activity of Ero1 and the reduction potential of the glutathione buffer would be intimately interlinked via PDI-family members. If the rate of production of secreted disulfide bond–containing proteins increased, the initial effects would be a net decrease in GSSG concentrations, which in turn would result in the activation of Ero1 (via PDI) until redox homeostasis was resumed. Similarly, if the rate of production of secreted disulfide bond–containing proteins decreased, the initial effects would be a transient net increase in GSSG concentrations, which would lead to the increased net inactivation of Ero1 (via PDI) until redox homeostasis was resumed. Although cells lacking glutathione would be able to survive and to produce disulfide-bonded proteins via Ero1–PDI alone, they would lack the ability to adapt smoothly and rapidly to changes in disulfide-bond protein production and to other changes in redox conditions. This would be especially important when considering the redox conditions in the microenvironment around the membrane-associated Ero1, which would oscillate more wildly than the bulk ER. The finding that a major fraction of the glutathione in the ER may be in mixed disulfides with proteins (20) supports the hypothesis of a direct role for GSSG in native disulfide-bond formation; however, this article described microsomal preparations, and the validity of the results for the ER have been questioned (65).
C. Peroxide-based oxidation
Other systems may also result in oxidation of PDI in vivo. In vitro sulfhydryl oxidases, including Ero1-family members, make one molecule of hydrogen peroxide per disulfide bond. Although it has yet to be shown that hydrogen peroxide is similarly generated during disulfide-bond formation in vivo, the assumption in the field is that it is produced. This peroxide generation has been widely viewed as a harmful byproduct, such that the formation of native disulfide bonds in folding proteins is thought to cause oxidative stress in cells (116, 304). However, peroxide is an oxidant that can lead to the formation of disulfide bonds from cysteine thiols via the formation of a sulphenic acid intermediate (see Fig. 16A). The exogenous addition of hydrogen peroxide to cells results in the widespread formation of both protein sulphenic acids (27, 34) and protein disulfides (48, 267). It is possible that any peroxide generated by Ero1 during disulfide-bond formation in vivo is used in the formation of disulfide bonds. Recently, in vitro data supporting this hypothesis were published (146), with Karala and co-workers showing that peroxide added directly to folding BPTI or generated in situ by the action of glucose oxidase and glucose (see Fig. 16B), was sufficient to result in the efficient formation of a natively folded protein containing three disulfide bonds. At pH 7, peroxide-mediated oxidation was much faster than that mediated by a glutathione redox buffer. Furthermore, as long as the peroxide was not present in large excess, no detectable formation of dead-end cysteine sulphenic acid species and minimal side reactions, such as methionine oxidation, were seen. Peroxide was also able to oxidize the active site of PDI, or less efficiently to oxidize GSH to GSSG. It was also speculated that the peroxide generated may be able to oxidize the regulatory disulfides in Ero1, providing an efficient negative-regulation feedback mechanism. In vivo data supporting the notion that the peroxide generated during disulfide-bond formation may be involved in disulfide-bond formation is limited; however, two supporting pieces of evidence exist. First, two ER-resident human glutathione peroxidases have been reported as part of a study on KDEL variants for ER localization (252). Because glutathione peroxidases catalyze the peroxide-mediated oxidation of GSH (see Fig. 16C) their existence in the ER implies a functional role for peroxide in disulfide-bond formation, especially when combined with an observed in vivo interaction of these proteins with Ero1α (V.D. Nguyen, H.I. Alanen, and L.W. Ruddock, unpublished data). Second, in a landmark study by Malhotra and co-workers (197), it was shown that the overproduction of a protein containing eight disulfide bonds did not result in oxidative stress in cell culture or in mice, whereas the overproduction of a protein that misfolded in the ER did result in oxidative stress. Disulfide-bond formation in the ER per se does not result in oxidative stress in vivo. These results imply that very efficient cellular mechanisms must exist either for the disposal of peroxide generated by Ero1 during disulfide-bond formation in vivo or for productive utilization of this peroxide. It should also be remembered that other processes in the ER generate peroxide (for example, the last step of ascorbate biogenesis by

D. Other oxidation systems
Ascorbate also features in another possible route for PDI oxidation in vivo. In the 1960s and early 1970s, dehydroascorbate (DHA) was used as the net oxidant in disulfide bond formation in vitro (see refs. 100, 277, 283, and 317 as examples). DHA is one of the oxidized forms of ascorbate formed during its function as a cellular antioxidant (see Fig. 17A) (184). DHA is unstable in aqueous solutions, undergoing a hydrolysis reaction with a reported half time of ∼100 min at 20°C, pH 7 (26). DHA is also able to react with thiols to form disulfide bonds and ascorbate (see Fig. 17B), although the uncatalyzed reaction with cellular thiols such as glutathione is thought to be slow (338). PDI and glutaredoxin (a thioredoxin-superfamily member whose primary function is thought to be the catalysis of deglutathionylation reactions) are regarded as dehydroascorbate reductases (76, 333, 341). The initial step in the reaction is the reduction of DHA to ascorbate, resulting in the concomitant oxidation of the active site of PDI (see Fig. 17C). In vitro, the oxidized PDI is reduced by GSH to form GSSG, but oxidized PDI can also introduce disulfide bonds into folding proteins. The potential function of DHA as an oxidant of PDI in vitro and in vivo has largely been overlooked for the past 40 years. One group has repeatedly proposed an in vivo role for DHA in disulfide-bond formation (for examples, see refs. 14, 15, 45 –47, 219, and 289), but this has been largely ignored by the field, in part because of the lack of supporting data and the gaps in the model proposed [for example, the identity of the metalloenzyme required to oxidize ascorbate in the ER microsomal preparation (14)], but also in part probably owing to the timing of this work against the proven in vivo and in vitro role of Ero1 in PDI oxidation (see earlier). However, this area deserves further investigation and recognition.

Other potential oxidation systems have been mentioned in the literature. One for which some supporting experimental evidence exists is based on vitamin K. The term vitamin K covers a collection of related compounds (see Fig. 18A). One of these, vitamin K3, otherwise known as menadione, is routinely used as a strong oxidant in the study of the effects of overoxidation on disulfide-bond formation. The dione structure of menadione is similar to that found in DHA, which also acts as an electron acceptor in disulfide-bond formation in vitro (see earlier). One of the physiological functions of vitamin K is to play a role in the biogenesis of γ-carboxyglutamate–containing proteins. This reaction is a two-step process (see Fig. 18B). The reduced form of vitamin K, vitamin K hydroquinone, is used by the vitamin K–dependent γ-carboxylase as a cofactor during γ-carboxylation. The vitamin K 2,3-epoxide formed from this reaction is then reduced back to vitamin K hydroquinone by the action of vitamin K 2,3-epoxide reductase (VKOR) (227, 255). In vitro, the electron acceptor in this process is DTT. It was proposed that in vivo thioredoxin played the role of electron acceptor (139), but it was subsequently shown that the thioredoxin–thioredoxin reductase combination was unable to act in this manner in intact microsomes (241). Recently, PDI was shown to be in a complex with VKOR (320), so γ-carboxylation is probably linked to disulfide-bond formation in the ER.

Other evidence for a role of vitamin K in disulfide-bond formation is forthcoming from prokaryotic systems. Very recently Dutton and co-workers (68) showed in a landmark article that a bacterial homologue of VKOR may functionally replace the transmembrane protein DsbB (which plays a role in disulfide-bond formation in the periplasm similar to that Ero1 plays in the ER). In addition, they found that most bacteria that lack a DsbB homologue, and that would be predicted to make disulfide bonds, have a VKOR homologue, and, in some cases, this VKOR homologue was found fused to a DsbA homologue. Although no evidence was presented that menaquinone, rather than another quinone, was the electron acceptor in this system, it strongly suggests a direct role for vitamin K in disulfide-bond formation in at least some prokaryotes.
Under what conditions could vitamin K play a major role in disulfide-bond formation in eukaryotes? Menaquinone (vitamin K2) is synthesized by bacteria and used for anaerobic respiration (see ref. 306 for a review). For example, in E. coli, a shift occurs in the quinone intermediate used in respiration from ubiquinone under aerobic growth to menaquinone under anaerobic growth by using fumarate as the electron acceptor. It would, therefore, be tempting to speculate that vitamin K may play a role as a cofactor in a process for disulfide-bond formation under hypoxic or anoxic conditions or both. This has yet to be examined experimentally.
E. Reduction of PDI
Three possible sources exist for the formation of the dithiol state of the active site of PDI. The first is the catalysis of oxidation of protein substrates, during which the active site converts from the disulfide to the dithiol state. The second option is reduction of the active-site disulfide by GSH. In vitro, GSH is able to reduce the active sites of the isolated
IX. The Redox State of PDI in vivo
The redox state of the active sites of PDI in the ER is another subject on which considerable differences of opinion are held. Some of these differences may be in the method or reagents used; some, due to real differences in the redox state of PDI in different cell types or under different conditions; but some are undoubtedly due to the technical challenges involved in obtaining an accurate picture of this highly dynamic system.
The active sites of PDI can exist in four potential redox states (see Fig. 19A); both active sites in the dithiol reduced state, both in the oxidized disulfide state, with the

The method for determining the redox state of the active sites and trapping mixed disulfides in vivo is essentially the same as that in vitro. A thiol-reactive reagent is added to the system and reacts with the active-site cysteines in the reduced or mixed disulfide states, but not in the disulfide state. To trap the thiol-disulfide state, in vitro reagents such as iodoacetamide, iodoacetic acid, and N-ethylmaleimide are often used, and these are also used for cell-based systems, although only N-ethylmaleimide is membrane permeable. Acid quenching is also frequently used, often in combination with subsequent neutralization and reaction with a chemical quencher. Because these reagents do not result in significant changes in molecular weight, other maleimides, such as 4-acetamido-4
Determining the redox state of PDI or trapping PDI-mixed disulfides in vivo by direct chemical trapping is extremely technically challenging. The first consideration is the ability to pass a thiol-reactive reagent across multiple cellular membranes into the ER and at high enough concentrations to quench the system rapidly. Although the exact concentration of reactive thiol groups in the ER is not known, and will vary depending on cell type and the amount of disulfide-containing proteins that it is producing, from a combination of the glutathione and thiol groups on proteins, the minimum estimate of ER thiol concentration must be on the order of 20 mM. Any thiol-reactive reagent must be present in excess of this. In vitro reagents, such as iodoacetamide, are often used at concentrations as high as 1 M, but even this is insufficient to prevent disulfide rearrangement (see Section VI). In vivo reagents are often used at much lower concentrations, typically 10–50 mM, including sometimes at concentrations below the concentration of free thiols in the system. This reduces the chance of obtaining physiologically relevant information.
The second consideration is the kinetics of the system. Accurately to determine the redox state of PDI in vitro, any quenching reaction must happen on a faster time scale than thiol-disulfide exchange. In vitro, and presumably in vivo, thiol-disulfide exchange reactions occur on the second time scale. For example, the nucleophilic attack of the N-terminal active-site cysteine of PDI on GSSG, with GSSG approaching the best estimates of physiological concentrations, has a half-time of <1.5 s (see Section VIII). Similarly, the nucleophilic attack by the C-terminal cysteine on the PDI–glutathione mixed disulfide has a half-time of ∼2.6 s in vitro at pH 7 (146). By comparison the half-time for the reaction of the N-terminal active-site cysteines (a highly reactive thiol group) with 20 mM iodoacetamide at physiological pH would be ∼3 s.
The third consideration is the accessibility of the C-terminal active-site cysteine of PDI. In the structure of the
The most accurate systems are probably those based on acid quenching of thiol-disulfide exchange reactions followed by rapid processing, addition of a large excess of chemical quenching reagent, and neutralization of the pH to allow the quenching reaction to occur. In a recent study, Appenzeller-Herzog and Ellgaard (8) were able to show for the first time that all four possible thiol/disulfide redox states of PDI exist in vivo. Furthermore, these states in relative ratios are consistent with the expected ratio from the reduction potential of PDI and the possible reduction potential of the ER, as defined by glutathione (see Section IV).
In addition to the problems associated with determining the redox state of PDI in vivo, considerable confusion exists in the field regarding the significance of the in vivo redox state of PDI and how this relates to the function of the protein. For example, in a highly simplified scheme, PDI can be considered to be predominantly in the reduced state or predominantly in the oxidized state. The reduced state of PDI is linked to its ability to catalyze isomerization or disulfide reduction reactions, whereas the oxidized state is linked to its ability to introduce disulfide bonds into substrate proteins. It is, therefore, easy to fall into the trap of saying that if, for example, PDI exists predominantly in the reduced state, that the primary function of PDI in vivo is to act as an isomerase. This is, however, misleading without an understanding of the kinetic steps in the various processes that result in this steady-state measurement and especially without knowing which steps are rate limiting in vivo (see Fig. 20). Even if the in vivo redox state can be determined accurately, the existence of one predominant redox state does not directly, by itself, give information on function. However, vital information on the interconnectedness of in vivo pathways and function can be obtained from examining how the in vivo redox state of PDI changes as a function of manipulating the environment of the cell (for example, overexpressing an Ero1-family member or a substrate protein) (10).

X. In vivo Thiol-Disulfide Exchange Reactions of PDI
A direct role for PDI in catalyzing thiol-disulfide exchange reactions in vivo was elegantly shown in two landmark articles. First, it was shown that defective co-translational formation of disulfide bonds exist in γ-gliadin in PDI-deficient microsomes, a defect that could be reversed by the addition of purified PDI (28). Second, the formation of mixed disulfides between PDI, and the PDI-family member ERp57, with folding proteins was observed in living cells (213). Despite these studies, relatively little work has been done on determining the nature of the reactions that PDI catalyses in vivo, in part because of the complexity of mammalian systems and the lack of a viable PDI-knockout strain. In contrast, a considerable number of apparently contradictory reports exist on the essential function of Pdi1p, and an examination of these can be used to gain some idea of the in vivo function of PDI.
Pdi1p has been recognized as an essential gene product since it was first identified by multiple groups in 1991 (74, 175, 293). Although now a common consensus agrees that the essential function of Pdi1p is related to its function in native disulfide-bond formation, considerable disagreement concerns whether this represents its ability to oxidize protein substrates or its ability to isomerize protein substrates (35, 173, 174, 280, 340). The truth is that both are physiological functions and that trying to define the “essential” function can be misleading. However, because this forms a significant section of the literature, it should be examined.
Because Ero1p is an essential gene product (84, 239), which appears to be an inefficient electron acceptor from GSH (272) or from folding substrate proteins, any disruption of the ability of Pdi1p to interact with Ero1p, or to transfer the disulfide bond from Ero1p to either substrate proteins or glutathione, would be expected to be lethal. The ability to act as an oxidant, or, more specifically, to act as a transfer molecule between Ero1p and folding proteins, is almost certainly a physiological function of Pdi1p (and PDI). This can be used as an argument that it is the essential function of the protein. Similarly, in vitro at physiological pH, Pdi1p (and PDI) has significant catalytic activity as an oxidant; however, by far the biggest fold difference between the catalyzed and uncatalyzed rate is for isomerization reactions, with some in vitro uncatalyzed reactions having half-times measured in days (see Section VI). The isomerization of nonnative disulfide bonds is almost certainly a physiological function of Pdi1p (and PDI).
So how do we explain the results that show that either oxidation or isomerization are not essential in vivo functions of Pdi1p? These conclusions were reached on evidence derived from studies using different constructs to rescue the viability of a PDI1-null or disrupted strain. Three distinct classes of construct have been used, active-site mutants of Pdi1p, domain constructs of Pdi1p and PDI, and the use of other thioredoxin-superfamily members.
The initial work reported was based on truncations and mutations of Pdi1p (174). This showed that truncations of Pdi1p from the C-terminus down to the equivalent of an
This analysis leaves us with another question. How are constructs such as Eug1p or CGPS thioredoxin able to support the viability of a PDI1-null or disrupted strain if the construct replacing Pdi1p lacks the C-terminal active-site cysteine required for the flow of electrons from folding proteins to Ero1p? Two answers to this question are possible. First, potential mechanisms exist by which a one-thiol thioredoxin-superfamily member can act as a catalyst of substrate protein dithiol-disulfide oxidation in vivo (see Fig. 21). However, these have not been demonstrated in vitro. Second, Pdi1p does not exist in isolation in the ER. For example, it is possible that under physiological in vivo conditions, Ero1p would be able to directly oxidize GSH to GSSG, although it does not do this efficiently in vitro (272). Pdi1p is also only one member of the yeast PDI family, and it has been shown that another family member, Mpd2p, is essential for Pdi1p with its C-terminal active-site cysteines mutated to replace endogenous Pdi1p functionally (225). Because a C-terminal active-site mutant of Mpd2p has been found to be in a mixed disulfide with Ero1p (85), the most likely explanation is that Ero1p is able to use Mpd2p as well as Pdi1p to transfer disulfide bonds to folding proteins.

Is PDI essential? Whereas reports of knockout cells and animals have been found for other ER-resident protein-folding catalysts and molecular chaperones (for example, 61, 96, 207), no such report exists for a knockout of PDI. Given the primary role of PDI, such an omission probably implies that such knockouts are not viable. Furthermore, PDI knockdown studies using RNAi show that efficient knockdown is linked to cytotoxic effects (231). Both of these suggest that PDI is an essential gene product, and, given the similarities between PDI and Pdi1p, it is likely that the physiological function is twofold: to act as an isomerase and as a direct, or indirect, transfer molecule between Ero1α and folding proteins (i.e., to act as a protein dithiol-sulfide oxidant).
To date, very few natural substrates for PDI have been reported (12, 64, 213, 257, 312), although the assumption from the promiscuity of the enzyme in vitro and its apparently essential nature in vivo is that a very significant number of proteins that fold in the ER will require PDI for disulfide-bond formation. A number of studies implicating PDI-family members in thiol-disulfide exchange reactions, especially outside the ER, are based heavily on the use of inhibitors. However, care must be taken in the interpretation of in vivo results based solely on the use of inhibitors; thiol-modifying agents will alter thiol-disulfide exchange reactions irrespective of whether PDI is catalyzing the reaction, and a strong argument can be made that bacitracin, the one “selective” inhibitor of PDI, is neither an effective inhibitor nor selective for PDI (110).
XI. Other PDI-Family Members
Although PDI was, for a long time, considered to be the sole enzymatic catalyst of thiol-disulfide exchange reactions in the ER, it has been clear for more than two decades that it is just part of an ever-growing family, a family named after the archetypal and most abundant member, the PDI family.
The physiological functions of the PDI-family members are far from well defined. For many of them, it is possible to state reactions that they can catalyze, but whether they actually catalyze these reactions under normal physiological conditions is unclear. What is clear is that different organisms have different families of PDI, some of which may be connected in function, such as human PDI and yeast Pdi1p, but many of which are unique to a subset of organisms. Because each organism has a unique set of PDIs, some focus must exist. In this review, the focus is on mammalian PDI-family members and, in particular, on human PDI-family members. However, it is worth saying a few words first about the PDI-family members found in S. cerevisiae, as they are among the best characterized as a grouping from a single organism.
Saccharomyces cerevisiae has four soluble PDI-family members, Pdi1p, Eug1p (291), Mpd1p (294), and Mpd2p (292), along with the transmembrane protein Eps1p (327), all of which are N-glycosylated, unlike PDI, which is not. Any of these family members can rescue the viability of a PDI1-null or disrupted strain when highly overexpressed. However, in a landmark study, Nørgaard and co-workers (225) found that the ability of different family members to rescue viability was, in part, dependent on the presence of low endogenous levels of one or more other family members. This implies that the different yeast PDI-family members are not functionally interchangeable.
Eug1p shares the same overall domain architecture as Pdi1p and PDI and shows 42.5% identity over the region
Mpd1p and Mpd2p are small, two-domain PDI-family members with active-site sequences WCGHC and CQHC, respectively. Mpd1p associates with Cne1p (153), the yeast homologue of calnexin (compare with ERp57 later) and is the only yeast PDI-family member that can rescue strains deleted of any of the other family members when placed under the control of the PDI1 promoter (225). The crystal structure of Mpd1p was recently solved to 2.0-Å resolution (318). This structure revealed that Mpd1p comprises one catalytic and one noncatalytic domain, both with thioredoxin-like folds. The relative orientation of these domains is significantly different from that found in other multidomain PDI-family member structures solved to date. Relatively little is known about Mpd2p. However, the CQHA active-site mutant of Mpd2p forms a mixed disulfide with Ero1p (85). Furthermore, mutation of the active sites of Pdi1p to CGHS restricted the ability of the gene to rescue a PDI1-null strain to systems in which Mpd2p was present (225). These two results imply that, in this strain, Mpd2p acts as the direct or indirect transfer molecule between Ero1p and folding proteins when Pdi1p is unable to act in this way (see Section X).
Eps1p is a large transmembrane PDI-family member. It has been implicated in ER quality control (327, 328) and may play a role in presentation of misfolded proteins to the degradation machinery. However, the precise role of Eps1p in this process is unclear, and it should be noted that homologues of Eps1p can be found only in Saccharomycetales.
Whereas S. cerevisiae has only five PDI-family members, humans currently have 20 defined PDI-family members. These are defined by similarity to PDI and localization in the ER rather than by physiological function. All of the human PDI-family members therefore contain at least one domain that is similar to one of the four domains of PDI (see Fig. 22). Most contain at least one catalytic domain, but several have either no catalytic domain or have lost one or both of the active-site cysteines in

As per PDI, many PDI-family members have been implicated in a wide range of physiological functions, but here we concentrate on those related to protein folding and in particular to the catalysis of thiol-disulfide exchange. We start the examination of other human PDI-family members with the two that show the same architecture as PDI (i.e.,
A. ERp57
ERp57 (122), (also known as ERp60, ERp61, GRP57, GRP58, HIP-70, and Q-2) was initially identified as a phosphoinositide-specific phospholipase C (22). Despite this inauspicious start, ERp57 rapidly became one of the most-reported human PDI-family members. It is very similar in length and domain architecture to PDI, with the most striking difference being that whereas the
ERp57 has been assigned an even greater range of functions outside the ER than has PDI itself. These fall outside of the range of this review, but again, it is unclear how ERp57 moves to other cellular compartments, although it is the only PDI-family member that has a C-terminal motif that is inefficient at ER retrieval (252). In addition, these functions are often assigned to ERp57 based on general thiol-reactive reagents or from the use of the peptide “PDI-specific inhibitor” bacitracin at concentrations of up to 7 mg/ml (see 110 for a discussion on bacitracin as a PDI inhibitor).
ERp57 is involved in MHC class I folding and forms part of the MHC I peptide-loading complex along with calreticulin, TAP, and tapasin (232), although the exact function of ERp57 in this complex is the subject of significant discussion in the literature. Tapasin is a potential substrate for ERp57, and it forms a very stable mixed-disulfide complex with ERp57 (233). Very recently, the crystal structure of ERp57 and tapasin, linked by a disulfide, has been solved to 2.6-Å resolution (66). The structure shows extensive contacts between the two molecules, which the authors suggest accounts for the inactivation of the escape pathway (see earlier). Given the supposed specificity of the ERp57–tapasin complex, it is surprising that all of the contact residues in ERp57 are conserved in PDI and ERp72.
ERp57 is most associated with the oxidative folding of glycoproteins (see ref. 258 for a review of the quality control cycle for glycoprotein folding). The involvement of ERp57 in the folding of glycoproteins was first demonstrated by chemical cross-linking of ERp57 to nascent glycoproteins in microsomes (229) and further substantiated in a subsequent study in living cells (213). It was subsequently shown that ERp57 interacts with the ER lectins calnexin and calreticulin (228), and detailed analysis of the ERp57 engagement with viral glycoproteins indicated that ERp57 acts co-translationally with these lectins (212). Both calreticulin and calnexin contain a remarkable armlike domain called the P-domain. ERp57 binds to the P-domain with a K
d of 26 μM (166) and 10 μM (91) for calnexin and calreticulin P-domains, respectively. The ERp57 interaction site has been mapped with NMR to near the tip of this highly acidic and proline-rich arm (91). By a combination of a cross-comparison with PDI and mutagenesis studies, a binding site in the
As is the case for other ER housekeeping enzymes, the deletion of ERp57 caused embryonic lethality in mice (96), suggesting the importance of this enzyme in the early development and differentiation of embryonic cells. In contrast, ERp57-deficient cell lines are viable and show no gross cellular defects. Furthermore, examination of protein-folding pathways in these cells did not show a global folding perturbation, with only a few proteins showing significant changes in folding and with ERp72 being recruited to assist in the folding of orphan ERp57 substrates (279). ERp57 has been shown to associate by mixed disulfide bonds with proteins that traverse the secretory pathway. Examples include ER-resident proteins, plasma surface-membrane proteins, secretory hormones, and endocytosed viral proteins. One intriguing example was shown in a study to dissect the stepwise entry and uncoating program of Simian Virus 40, a DNA virus that enters the cell through the endocytic vascular system (268). In this study, it was shown that ERp57 functions in a calreticulin/calnexin-independent manner, to isomerize disulfide bonds in the viral capsid, which allows viral access to the cytoplasm. In another elegant study, Jessop and co-workers (136) trapped ERp57-substrate mixed disulfides by using a C-terminal active-site cysteine mutant and thereby identified a range of ERp57 substrates in vivo. These included integrins, laminin, collagen, clusterin, and others. Although this technique does not trap all possible substrates (see ref. 109 for discussion), it does offer a unique insight into the possible range of substrates that a PDI-family member may have. In a follow-up study, this substrate specificity of ERp57 was shown to be determined primarily by its interaction with calnexin and calreticulin (137).
B. PDIp
Human PDIp is one of the few PDI-family members that show very-specific tissue distribution. Its name derives from the fact that it was initially reported to be expressed only in the acinar cells of the pancreas (62, 63), although current database analysis implies expression in both the pancreas and the brain, with potentially lower levels also found in organs adjacent to the pancreas, such as the stomach and bladder. PDIp shares the same domain architecture as PDI, and the
The unusual tissue distribution of PDIp led to the speculation that it evolved to fold a subset of pancreatic enzymes that PDI was unable to (i.e., that it would have a different range of substrate specificities). In microsomes, PDIp is able to bind peptides derived from a range of proteins including prolactin, procecropin, glycophorin C, pro-α-factor (72, 319), and in vitro, to peptides from pancreatic digestive enzymes, including pancreatic lipase, pancreatic α-amylase, cholesterol esterase, colipase, pancreatic trypsin inhibitor, and somatostatin (159). This binding can be inhibited by a wide variety of estrogens and xeno-estrogens (159) and other hydroxyaryl-containing compounds (155), which partially mimic the natural tyrosine and tryptophan recognition motifs used by PDIp to bind peptides (259).
The physiological function of PDIp is still unclear, but two potential pathological roles have been reported. First, PDIp was the only one of eight PDI-family members tested to be upregulated in a cellular experimental model for Parkinson disease, and it was found in Lewy bodies from the brains of postmortem human Parkinson disease subjects (39). It is unclear whether this association is a net effect of the disease state or contributes in some way to it. Second, in an animal model for type 1 diabetes with a loss of function of autoimmune regulator, the major targets of autoimmune destruction were acinar cells, and this was linked to the production of an autoantibody against PDIp (222).
C. ERp72
ERp72, also known as CaBP2, was one of the first PDI-family members reported (203), but it currently remains the poor cousin of ERp57, with which it shares considerable sequence homology. The domain organization and active-site chemistry suggest that it can catalyze thiol-disulfide exchange reactions similar to those of PDI. ERp72 has been shown to have such activity in vitro (1, 167, 192, 263), and it can complement lethality of yeast deficient in Pdi1p in vivo (108). The sequence homology between ERp72 and ERp57 extends to the
ERp72 forms a complex with PDI, P5, ERdj3, BiP, CypB, HSP40, GRP94, GRP170, UDP-glucosyltransferase, and SDF2-L1, both in the presence (where the complex also includes ERdj3) and in the absence of an immunoglobulin heavy chain (208). ERp72 has also been reported in association with a number of wild-type or mutant proteins including thrombospondin (170), thyroglobulin (206), and matrilin-3 (41), and presumably, this is linked to its ability to catalyze thiol-disulfide exchange. Reducing the levels of ERp72 by siRNA enhanced retrotranslocation of cholera toxin (81) and boosted SV40 infectivity (268), the former by an unknown mechanism, and the latter, probably linked to a lowered capacity to reduce disulfide bonds.
D. P5
P5, also known as ERp5 and CaBP1, is, like ERp72, one of the less well studied and known PDI-family members despite being the third one identified, after PDI and ERp72 (192). Like ERp72, its active-site chemistry in the two catalytic domains implies that P5 should be able to catalyze thiol-disulfide exchange reactions, and indeed, in vitro, it can (4, 152, 167, 192, 263). P5 is the smallest of the family members with both catalytic and noncatalytic
E. ERp44
Human ERp44 was first reported in 2002 (6). It is 408 amino acids in length, with homologues being found across metazoans. The gene is localized to chromosome 9, and transcription is under an ER stress-response element (343), CCAACN9CCCAG, which is similar to that of PDI, CCAACN9CCCCG. The protein is highly expressed in secretory tissues and during B-cell and adipocyte differentiation (329). Although the protein has an efficient ER-retention motif (252), the endogenous protein is localized mainly to the ER-Golgi intermediate compartment and to the cis-Golgi, as visualized by immunofluorescence (7, 329). However, overexpressed ERp44 largely localizes to the ER.
The crystal structure of ERp44 was recently solved to 2.6-Å resolution (326). The three domains (
ERp44 can be immunoprecipitated with a number of proteins that reside in the ER, including Ero1α and β (5, 6) and inositol 1,4,5-triphosphate receptor type I (119), and also with proteins that mature and oligomerize throughout the secretory pathway, including adiponectin (329), SUMF1/FGE (83, 199), and IgM (7). Some of these complexes can be isolated without and with reducing agents, implying that at least some of the interactions between ERp44 and other proteins are stable and are not limited only to the formation of a mixed disulfide with the unique CRFS active site. The data currently available are at times contradictory, but the simplest interpretation for the role of ERp44 is that it plays a role in either a late-stage, thiol-dependent, quality-control system or trafficking or a role in late-stage oligomerization reactions or both. The interaction of ERp44 with inositol 1,4,5-triphosphate receptor type I plays a role in the regulation of cellular calcium homeostasis (119), although further characterization of this system is required.
F. ERp29
ERp29, also known as ERp28, is a two-domain protein that lacks a catalytic
Rat ERp29 has been purified from a natural source, and, consistent with it lacking an
The role of ERp29 in thyroglobulin processing was first postulated by the identification of ERp29 as part of a folding complex that also included the molecular chaperones BiP and GRP94 (266). Overexpression of ERp29 increases thyroglobulin secretion, whereas RNAi-mediated silencing of ERp29 led to the attenuation of thyroglobulin export (19). What is surprising about the role of ERp29 is that it appears to be secreted into the media with thyroglobulin. The authors concluded that ERp29 functions as an escort “chaperone” that accompanies thyroglobulin all the way through the secretory pathway, although how a KEEL-containing protein can escape the ER-retrieval machinery is currently unknown.
Apart from the assigned “escort” role of ERp29, a second distinct function has been discovered related to protein conformational remodeling before “retrotranslocation,” a function that is exploited by polyomaviruses. Polyomaviruses, such as SV40, are non-enveloped viruses that require direct interaction between viral coat proteins and the biological membrane to be internalized across the ER membrane to initiate infection (300). ERp29 is reported to alter the conformation of the VP1 major structural-coat protein, exposing the C-terminal arm of V1 and triggering viral membrane binding (196). The altered conformation of VP1 exposes the internal-coat protein VP2 and triggers perforation of the membrane (246). The C-terminal domain of ERp29 is responsible for this function (248). Surprisingly, bacterially expressed ERp29 is unable to have the same effect (196), and therefore, either an unknown posttranslational modification of ERp29 is required, or ERp29 may work in concert with one of the other six proteins in the active ERp29-enriched fractions. As yet, no reports exist of a similar function for ERp29 on potential endogenous substrates, either secreted or ER-resident proteins.
G. ERdj5
The gene for ERdj5 (49), also known as JPDI (127), is located on human chromosome 2 and encodes for a 793-amino-acid protein. It is unique among human PDI-family members in many ways. It has five recognized domains (see later), four of which are
Although ERdj5 is currently recognized as having five domains, an undefined stretch of 219 amino acids remains between the first and second catalytic domains; this is approximately the size of two thioredoxin-like domains. This region contains six cysteine residues, which are conserved between the human, mouse, and Caenorhabditis elegans proteins (49). The position and spacing of four of these are equivalent to the structural disulfides found in the catalytic domains of ERdj5 and other PDI-family members. Probably two more thioredoxin-like domains exist in ERdj5, bringing the total number of domains to seven, six of which are thioredoxin like. The second of these domains contains no other cysteine residues, and so should be considered as
ERdj5 is among the many genes upregulated in response to overexpression of a disease-causing and folding-incompetent mutant of surfactant protein C and among the few genes that has three unfolded protein response elements in its promoter region (67). This is consistent with induction of expression of ERdj5 by stress-promoting compounds such as thapsigargin, DTT, tunicamycin, or fenretinide (40, 49). Recently, two groups independently published details of the role of ERdj5 in the degradation of misfolded ER proteins (67, 307). This activity required both the J domain and the redox activity of the catalytic domains, and so, ERdj5 provides the missing link that exhibits the prerequisite reductase activity for ER-associated degradation. However, it still remains to be elucidated whether GSH provides the direct source of electrons required for reduction, or whether an ER-resident protein similar to the cytoplasmic thioredoxin reductase reduces ERdj5 directly.
H. The transmembrane PDI-family members
Whereas S. cerevisiae has only one transmembrane PDI-family member, humans probably have five, all shorter in length than Eps1p. To date, three of these have been published, TMX (202), TMX2 (205), and TMX3 (111), with TMX4 appearing in reviews (9, 71). The ER localization of TMX2, TMX4, and TMX5 has not yet been reported in the literature, but all three have putative type I transmembrane protein ER-localization signals. TMX2 is unusual, in that the active site in the
I. Other PDI-family members
PDIr was one of the first PDI-family members identified more than a decade ago (115), and yet the physiological function of the protein is still unclear. Like PDI, PDIr is a four-domain protein, but with the three C-terminal domains being
ERp27 is, like ERp29, a small human PDI-family member that lacks a catalytic domain. It can bind to peptides and nonnative proteins via its
ERp18, also known as ERp19, ERp16, and hLTP19, was originally reported independently by two groups in 2003 (3, 160). It is the smallest of the human PDI-family members, comprising a single, slightly extended, catalytic domain. The active site of ERp18 is unusual, having a CGAC motif, with a reduction potential of −165 mV (134) and being able to catalyze thiol-disulfide exchange reactions, although with relatively slow kinetics (3, 134). The kinetics of catalysis of peptide oxidation by ERp18, by using GSSG as the electron acceptor, is unusual, in that the rate was limited by oxidation of the peptide and not subsequent oxidation of the active site by GSSG to complete the catalytic cycle (3). The physiological function is still unknown.
ERp46 (160), also known as endoPDI (287), was reported independently by two groups in 2003. ERp46 also has a unique domain architecture with three catalytic domains linked by long spacers, 22 and 30 amino acids, respectively, with the linker between the first and second domains being very rich in proline and glutamic acid. Mouse ERp46 can functionally replace Pdi1p (160) and has been reported to play a role in protecting cells against hypoxia (287). ERp46, along with the PDI-family members ERp57 and ERp29, was downregulated in fructose-fed (insulin-resistant) hamsters, whereas PDI itself was upregulated (215), implying differential regulation and function.
Like PDIp, PDILT (309) is unusual in that it shows a highly specific tissue distribution, being expressed primarily in the testes after puberty and being the first PDI-family member to show developmental control (310). Whereas it has the same overall domain architecture as PDI, PDILT has the unusual active-site motifs SSKQS and WSKKC. PDILT forms mixed disulfide complexes with a variety of ER proteins, including Ero1α (309), and forms a complex with calmegin (310), the testis-specific homologue of calnexin that is required for sperm fertility (131). Again the physiological significance of this interaction is unclear at the moment.
hAG-2 and hAG-3 are the latest members added to the PDI family (236), with both having recently been shown to be located in the ER (252). Both are small single-domain proteins that show sequence homology with ERp18, although they have very different active-site motifs, WCGAC (ERp18), ECPHS (hAG-2), and DCQYS (hAG-3). Despite this, the crystal structure of human hAG-3 (M. Salin, V.D. Nguyen, R. Wierenga, and L.W. Ruddock, unpublished observations) shows a high degree of structural homology with human ERp18. hAG-2 and hAG-3 have been suggested to be molecular markers or potential therapeutic targets for hormone-responsive breast tumor (78), or both, but few clues exist as to their physiological function.
XII. Conclusions
These are exciting times in the PDI field. After years with limited structural information to guide studies, we now have the publication of six structures in the past year, increasing the number of structures of full-length human PDI-family members fourfold. Some details from these new structures could have been predicted beforehand, but many are new, including new concepts relating to the bind–release cycle of PDI and novel orientations of thioredoxin folds. We have the information available to make significant progress in structure–function relations, in determining the mechanisms of action of this remarkable enzyme, and perhaps in using this information to increase the efficiency and yields of in vitro refolding or cell factories or both for the production of therapeutic or industrially relevant proteins. However, the clear need exists for a greater degree of correlation between in vitro and in vivo results and a wider acknowledgment that both may be prone to artifact. We also need new, preferably physiological substrates, to assay activity, as well as to use existing assays in a more-appropriate and quantitative manner, including greater consideration of mimicking physiologically relevant conditions. We need to distinguish whether observed changes in the “catalytic activity” are due to changes in K m or changes in k cat (or due to significant changes in structure). We must acknowledge that even the simplest oxidation reaction that PDI catalyzes has four distinct covalent steps, which we need to differentiate. We must consider more carefully issues relating to heterogeneity in substrates and substrate specificities and affinities for different proteins and for different intermediates of the same protein. PDI was the first protein-folding catalyst to be reported. Much has been learned about it, but much more is still unknown.
Footnotes
Acknowledgments
We thank Robert Freedman and Christian Appenzeller-Herzog for useful discussion.
Abbreviations Used
Reviewing Editors: Kawngseog Ahn, Adam Benham, David Craik, Lars Ellgaard, David Ferrari, and Salvador Ventura