
Abstract
Diabetic foot disease is a major health problem, which affects 15% of the 200 million patients with diabetes worldwide. Diminished peripheral blood flow and decreased local neovascularization are critical factors that contribute to the delayed or nonhealing wounds in these patients. The correction of impaired local angiogenesis may be a key component in developing therapeutic protocols for treating chronic wounds of the lower extremity and diabetic foot ulcers. Endothelial progenitor cells (EPCs) are the key cellular effectors of postnatal neovascularization and play a central role in wound healing, but their circulating and wound-level numbers are decreased in diabetes, implicating an abnormality in EPC mobilization and homing mechanisms. The deficiency in EPC mobilization is presumably due to impairment of eNOS-NO cascade in bone marrow (BM). Hyperoxia, induced by a clinically relevant hyperbaric oxygen therapy (HBO) protocol, can significantly enhance the mobilization of EPCs from the BM into peripheral blood. However, increased circulating EPCs failed to reach to wound tissues. This is partly a result of downregulated production of SDF-1α in local wound lesions with diabetes. Administration of exogenous SDF-1α into wounds reversed the EPC homing impairment and, with hyperoxia, synergistically enhanced EPC mobilization, homing, neovascularization, and wound healing. Antioxid. Redox Signal. 10, 1869–1882.
Chronic Wounds of the Lower Extremity and Diabetic Foot Ulcers
Diabetes mellitus is increasing in incidence and represents a major health problem for the 21st century. Indeed, the total number of diabetic patients has been projected to increase from 171 million in 2000 to 366 million in 2030 (135). The annual incidence of foot ulcers among people with diabetes has been variously estimated at between 1% and 4.1%, and the annual incidence of amputation is 0.21–1.37% (9).
The pathophysiology of diabetic foot ulcers and delayed healing has been well described. Contributing factors include progressive development of a sensory, vasomotor, and autonomic neuropathy, leading to loss of protective sensation; joint and bone deformities that increase plantar foot pressure; and alterations in autoregulation of dermal blood flow. Diabetic patients show earlier development and progression of lower extremity peripheral arterial occlusive disease (PAD), with a predilection for blockages in the trifurcation level of vessels just distal to the knee.
The healing of a wound requires a well-orchestrated integration of the complex biologic and molecular events of cell migration, cell proliferation, and extracellular matrix (ECM) deposition. Normal wound healing requires proper circulation, nutrition, immune status, and avoidance of negative mechanical forces. The process usually takes 3–14 days to complete and has three phases: inflammation, proliferation, and remodeling with wound contraction (11, 30, 117) (Fig. 1). During the inflammatory phase, neutrophils and macrophages appear in the wounded area to phagocytize bacteria and debris. A functioning immune system and adequate supply of growth factors are necessary in this phase of wound healing. In the proliferative phase, fibroblasts produce a collagen matrix, new blood vessels invade the forming granulation tissue, and epidermal cells migrate across the wound surface to close the breach. During the remodeling phase, fibroblasts reorganize the collagen matrix and ultimately assume a myofibroblast phenotype to effect connective tissue compaction and wound contraction. Wounds gain ∼80% of their final strength in the first 3 weeks of normal wound healing through collagen deposition, remodeling, and wound contraction (117). When any of the components of the wound healing process is compromised, healing may be delayed. Chronic wounds are those that have failed to follow this sequence and do not achieve a sustained anatomic and functional result.
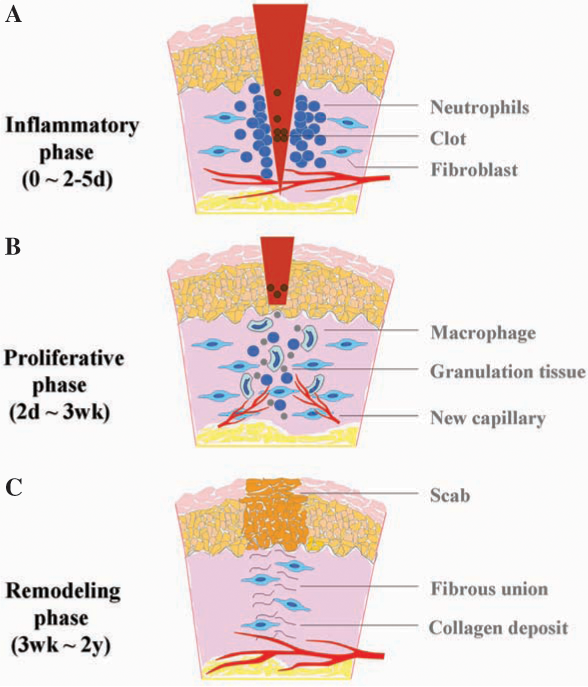
In diabetes, myriad factors, including intrinsic factors (neuropathy, vascular problems, other complicating systemic effects due to diabetes) and extrinsic factors (wound infection, callus formation, and excessive pressure to the site) can impair wound healing. It is well known that peripheral vascular disease (macroangiopathy), along with diabetic neuropathy, plays a major role in diabetic foot ulceration. About 20% of diabetic lower extremity ulcers have arterial flow insufficiency as their primary etiology, ∼50% will have primary diabetic neuropathy, and ∼30% will have both conditions (105). Even after correction of large blood vessel dysfunction by open surgical or endovascular revascularization, only ∼47% of patients will heal in a span of 20 weeks with standardized treatment, including glycemic control, debridement of necrotic tissue, control of infection, use of moist dressings, protection from pressure or trauma related to ambulation, and adjuvant HBO therapy (2, 54, 58). Recently, microangiopathy has also been implicated in the pathogenesis of diabetic foot ulcers (67, 91). Some of these vascular complications in diabetes, as well as the healing defects, have been associated with a decrease in number and function of circulating endothelial progenitor cells (EPCs) (22, 59, 76, 77, 125, 132). In addition, impaired host responses to infection and other cellular dysfunctions contribute to the refractory nature of diabetic wounds.
Despite the existence of standard protocols and the adoption of novel biologic and cell therapies for the treatment of diabetic foot ulcers (15, 114), the effectiveness is limited, and the amputation rate remains high. Given the morbidity and mortality of amputations (2-year survival rate of 50–60%) (121), it is imperative to develop better therapies to treat diabetic ulcers. The development of novel, more efficacious modalities of treatment would have tremendous benefit to both individual patients and society.
Neovascularization and EPCs
Restoring blood flow to the site of injured tissue is a prerequisite for mounting a successful repair response. It is now well established that an essential part of normal healing for full-thickness cutaneous wounds is the formation of new blood vessels within the provisional wound matrix, which is referred to as granulation tissue. New blood vessel growth occurs in both physiologic and pathologic conditions. Until recently, neovascularization was thought to arise solely from preexisting vessels (angiogenesis). However, recent studies have proven that EPCs can contribute to neovascularization (vasculogenesis) as well (102). In 1997, Asahara et al. (5) first identified circulating EPCs contributing to neovascularization. Since then, increasing evidence has suggested that BM-derived EPCs can functionally contribute to neovascularization during wound healing, limb ischemia (52, 81, 122), postmyocardial infarction (65, 93, 94), endothelialization of vascular grafts (56, 115), atherosclerosis (109), retinal and lymphoid organ neovascularization (34, 95), vascularization during neonatal growth (140), and tumor growth (79, 106). It has been estimated that EPCs contribute up to 25% of endothelial cells in newly formed vessels in animal models (125).
Although reports have been conflicting regarding the importance of EPCs in neovascularization (33), a substantial amount of study in both animals and humans has provided strong evidence for the role of BM-derived EPCs in neovascularization (66). In particular, a very recent study showed that notwithstanding low numbers of EPC, recruitment of these EPCs is pivotal for the progression of avascular micrometastatic tumors to lethal macrometastatic ones (26). The lack of consensus may be due in part to heterogeneous phenotypic definitions of EPCs. In this aspect, two recent reports (89, 139) showed that the subpopulations of EPCs with distinct phenotypes give rise to different outcomes, indicating the importance of defining the correct EPCs for such studies. Overall, however, these researchers clearly demonstrated a significant contribution of EPCs to neovascularization. Other factors, such as types of ischemic models or tumors [in which the profiles of chemoattractant factors may vary (107)], time frame of the experiment, and method of in situ endothelial cell identification, may also contribute to some of the reported discrepancy. In general, it is now well accepted that both adjacent preexisting blood vessels and recruitment of BM-derived EPCs participate in tissue vascularization.
Multiple markers have been used for characterization of EPCs and matured endothelial cells (ECs). Platelet endothelial cell-adhesion molecule-1 (PECAM-1/CD31), vascular endothelial growth factor receptor 2 (VEGFR2, also known as KDR or Flk1), von Willebrand factor (vWF), and vascular endothelial cadherin (VE-cadherin) are commonly used for identification of endothelial cells (28, 44, 92, 101). The ability to take up Dil-labeled acetylated low-density lipoprotein (Dil-Ac-LDL) (5, 28) has been used as a marker for identification of endothelial precursors. CD34 is a marker for both endothelial and hematopoietic progenitor cells. In humans, EPCs (but not mature endothelial cells) express AC133 (CD133), a stem cell marker with as-yet-unrecognized functions (108). Purified populations of CD133+VEGFR2+ EPCs proliferate in vitro in an anchorage-independent manner and can be induced to differentiate into mature adherent CD133−VEGFR2+ endothelial cells. Expression of various markers is a dynamic process. It is likely that different markers are present on EPCs and matured endothelial cells at various points along their differentiation cascade from immature progenitors to mature endothelial cells. Expression of CD133 will be turned off with the maturation of endothelial cells. The corresponding marker for CD133 in murine EPCs has not yet been substantiated. Murine EPCs are characterized as Sca-1+/c-Kit+/Lin−/VEGFR2+/CXCR4+/Tie2+ (100). With maturation, expression of stem cell marker Sca-1 and hematopoietic marker c-Kit is turned off.
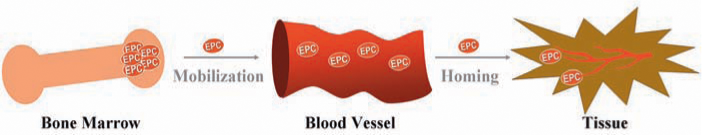
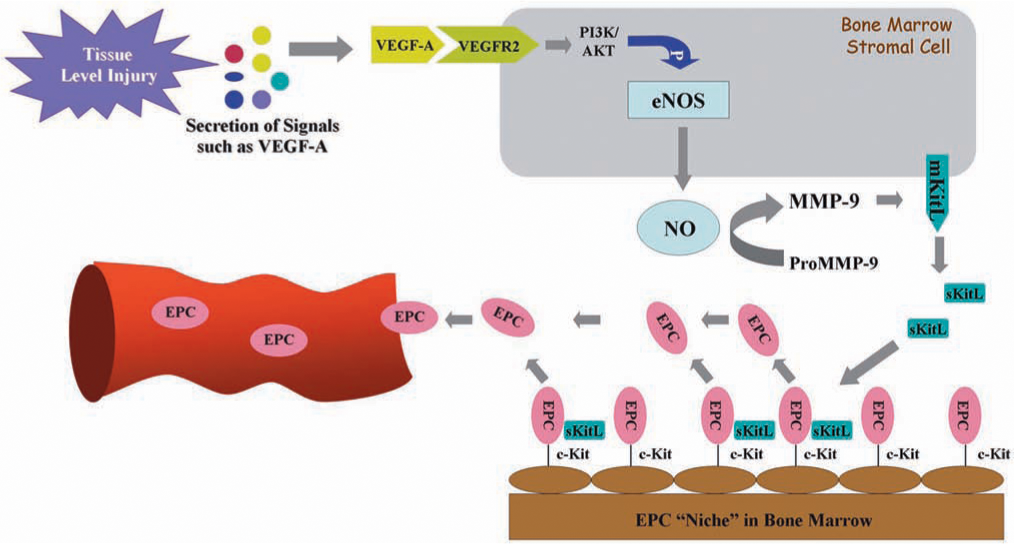
EPCs are embedded in a microenvironment (niche) of bone marrow and can be mobilized to the circulation (55). Under normal conditions, the number of circulating EPCs is relatively small but is increased in response to trauma or ischemia, which mobilizes these cells from the bone marrow and allows them to proliferate (29). The recruitment of EPCs from the bone marrow and circulation to homing sites of neovascularization is subject to regulation by many factors, including chemokines and growth factors (Fig. 2). The precise mechanism of EPC mobilization is not entirely elucidated and is still under investigation. Many chemokines and cytokines trigger stem/progenitor cell release by induction of matrix metallopeptidase 9 (MMP-9) in bone marrow. Nitric oxide (NO)-mediated signaling pathways have been previously proposed to be essential for EPC mobilization (3, 39, 40) (Fig. 3). By using vascular endothelial growth factor-A (VEGF-A) as a proximal stimulus, Aicher et al. (3, 4) demonstrated that endothelial nitric oxide synthase (eNOS) becomes activated in bone marrow stroma; NO then S-nitrosylates by paracrine mechanisms and activates MMP-9, which releases the stem cell–active cytokine, soluble Kit ligand. This agent shifts endothelial progenitor and hematopoietic stem cells from a quiescent to a proliferative niche and stimulates rapid stem cell mobilization to the peripheral blood (3, 39, 40, 90, 100, 116). It has been demonstrated that in the setting of trauma and ischemia, systemic VEGF-A levels increase, with a time course that mirrors the increase in circulating BM-derived EPCs. In addition, recent studies have suggested that EPCs may promote local neovascularization by secreting angiogenic growth factors in a paracrine manner. Transplantation of EPCs into ischemic tissues may emerge as a promising approach in the therapy of diseases associated with blood vessel disorders.
Impairment of EPC and eNOS Activity in Diabetes
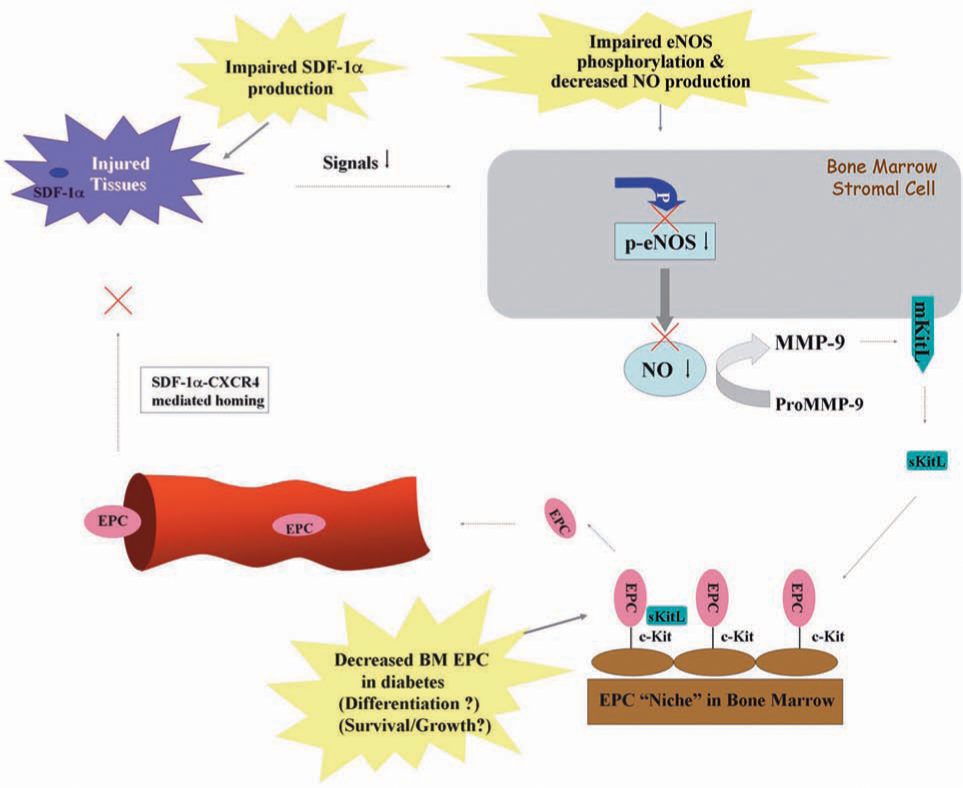
The number and function of EPCs can be affected in diabetes mellitus. Reduced levels and impaired function of EPCs have been described in both type 1 and type 2 diabetic patients (77, 125). EPC recruitment for re-endothelialization after vascular injury is also impaired in diabetes (48). These alterations are likely to be involved in the pathogenesis of vascular disease in diabetes (22). Increasing evidence suggests that wound-healing mechanisms, in both the bone marrow and within the peripheral wound, are compromised by diabetes as a result of BM-derived EPC impairments (22, 59, 76, 77, 125, 132). Although cytokines such as granulocyte colony-stimulating factor (G-CSF) and growth factors such as VEGF-A can induce the release of progenitor cells from the bone marrow, the nonspecific effects on release of other white cells and platelets or the leaky-capillary effect has made these factors unsuitable to treat diabetic patients with nonhealing chronic wounds (24, 57, 72, 74, 130). eNOS is of paramount importance for the regulation of mobilization and function of EPCs (3, 133); however, evidence concerning the effect of eNOS deficiency on diabetic EPCs is not available. We have hypothesized that eNOS function is impaired in diabetics, thus preventing these cells from reaching the wound site in significant numbers. Data from our laboratory confirmed that the levels of biologically active phosphorylated eNOS protein were decreased in diabetic mice, although no changes in the amount of total eNOS protein were observed (25). In the experimental diabetic mouse model, it was found that the EPC and HSC populations remain constant, whereas the mesenchymal stromal cell and lymphocyte populations demonstrate a slight decrease (by ∼27% and 26%, respectively). Thus, it is possible that diabetes-induced changes in mesenchymal stromal cell and lymphocyte populations in the BM might be responsible for the observed downregulation of BM eNOS activation. However, the precise mechanism related to the impaired eNOS in diabetes mellitus remains unclear. Also, it is unknown whether NO-mediated signaling pathways are altered in the human diabetic bone marrow.
Enhancement of Mobilization of BM-Derived EPCs by Hyperoxia
Adult bone marrow is a rich reservoir of pluripotent stem cells and various lineage progenitor cells (37, 128). Emigration of stem/progenitor cells (in particular, hematopoietic progenitor cells) from the bone marrow is generally thought to occur after a period of cell proliferation within the marrow niche (19, 97, 136). However, the emerging evidence also indicates a rapid mobilization of progenitor/stem cells from bone marrow, which suggests that cell proliferation is not always necessary. In mice, infusion of soluble Kit ligand triggers mobilization of CD34+ cells in 1 h (90). A fourfold elevation in circulating progenitor cells occurred within 10 min when human volunteers were subjected to highly strenuous exercise (104). It was shown that a specialized microenvironment exists in the marrow where stem/progenitor cells exhibit different propensities for proliferation and mobilization and where MMP-9 activity mediates BM-derived EPC release (39, 40, 100). From our findings (25, 31, 128) and reports by Nakamura et al. (90) and Rehman et al. (104), it appears that a subpopulation of BM-derived EPCs exists within specialized bone marrow niches that are poised for rapid release to the circulation.
Oxygen plays a key role in wound healing. Low oxygen tension (hypoxia) around the wound is one of several critical factors that mutually enhance the progression of a chronic ulcer, whereas a plentiful supply of oxygen is essential for a variety of the healing processes. Oxygen tension is positively correlated with collagen production (46, 51, 112), bacterial killing (17, 42, 61), epithelialization (131), and angiogenesis (62, 63). All these components of wound healing increase greatly in well-oxygenated wounds. Of the available oxygen-delivery methods, HBO is noninvasive and appears to be the most potent choice. Systemic hyperoxia induced by HBO is a treatment approved by the United States Food and Drug Administration (FDA) as a safe, adjunctive therapy to stimulate wound healing in diabetic patients. Patients typically receive ≥20 treatments with pure oxygen at 2.0–2.4 atmospheres absolute (ATA), once or twice daily. Oxygen is transported by the blood in two different ways: (a) chemically bound to hemoglobin in erythrocytes, and (b) physically dissolved in plasma according to Henry's law. This law states that the degree to which a gas enters into physical solution in body fluids is directly proportional to the partial pressure of the gas to which the fluid is exposed (7). HBO treatment is known to accelerate healing in ischemic (43, 46) and refractory diabetic wounds (2, 8, 141).
Despite the beneficial actions of HBO therapy on chronic, nonhealing wounds, the molecular mechanisms of the action are incompletely understood. NO has been shown to play a central role in the bone marrow mobilization and release of EPCs (3). Based on the fact that the generation of NO results in EPC release from the BM and HBO has been shown to up-regulate NO production in cerebral cortex tissue, perivascular pulmonary tissue, and neutrophils via stimulation of NOS (126, 127, 129), it is likely that HBO benefits wound healing via NOS-NO cascade-mediated mobilization of progenitor cells from the bone marrow. Recent investigations from our laboratory and collaborators confirmed this possibility and demonstrated that EPC mobilization into circulation is triggered by HBO through induction of bone marrow NO, with resulting enhancement in ischemic limb perfusion and wound healing (31, 128). With ischemic and diabetic murine models, we recently determined that hyperoxia, induced by a clinically relevant HBO protocol, increases NO levels within femoral bone marrow, accelerates the spontaneous revascularization of surgically induced hindlimb ischemia, and increases the number of BM-derived EPCs in circulation and within cutaneous hindlimb ischemic incisional wounds and diabetic excisional wounds (25). These effects appear to be specific to the release of BM-derived EPCs, but not lymphocytes, and responsive to the cytokine milieu of the wound. In the ischemic and diabetic murine models that were used, therapeutic wound-healing effects of increased BM-derived EPC mobilization into circulation and recruitment into wounds were observed in association with enhancement of neovascularization of the wounds and spontaneous recovery of hindlimb perfusion.
Hyperoxia and the NOS-NO Cascade
NO is a small, pleiotropic, free radical that was discovered as the endothelium-derived relaxing factor responsible for the maintenance of vascular tone (47). Because of its highly hydrophobic characteristics (73), NO is a unique messenger in that it is produced in one cell and diffuses into adjacent target cells to activate cytosolic guanylate cyclase–bound heme to generate the NO-heme adduct of guanylate cyclase (84). NO plays a significant role in the intracellular signaling process in cardiovascular as well as in other systems.
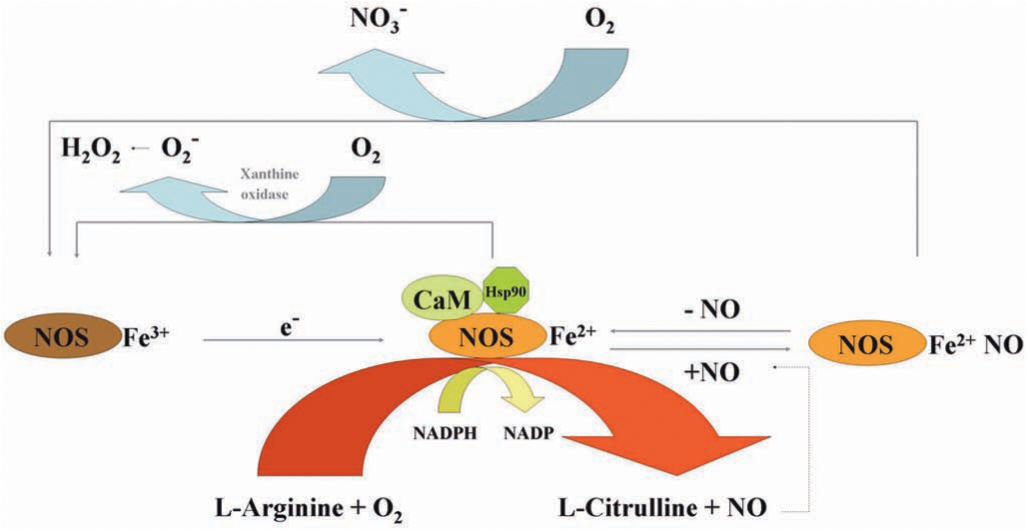
Production of NO is controlled by NOSs, which catalyze a stepwise oxidation of
eNOS is selectively expressed in vascular endothelial cells or surrounding stromal cells and therefore has been a focus of attention in vascular biology. eNOS plays an essential role in endothelial cell proliferation and is a central mediator of several endothelium growth stimulators, such as VEGF-A.
Among the four NOS enzymes, the activity of iNOS is controlled at the level of gene transcription, whereas the activities of nNOS and eNOS are controlled by intracellular calcium/calmodulin, several different phosphorylation mechanisms, and by binding of the molecular chaperone heat-shock protein 90 (HSP90) (13, 23, 27). It is believed that mtNOS is a constitutively active eNOS-like isoform (68). Studies with three major NOS enzymes (except mtNOS in limited studies) in vitro have shown that enzyme activity is influenced by the redox state and specifically by O2 tension. Elevated O2 tension influences NOS activity by hastening conversion of ferrous heme back to the native ferric conformation (Fig. 4).
In diabetic mice, the hyperoxia-induced increase of NO in BM was attenuated, likely as a result of impaired eNOS phosphorylation. However, with the induction of hyperoxic conditions, other NOS isoenzymes appear to compensate, leading to NO increases in the BM that are substantial and sufficient to reverse the defect in EPC mobilization in our diabetic mouse model.
An intriguing paradox exists. Based on our findings and the reported effects of hypoxia, it appears that both hyperoxia and hypoxia can result in NOS activation and a subsequent increase of NO level in BM and induce EPC release (25, 100, 124). The reasons for this seemingly paradoxic phenomenon are unclear. One explanation for this response involves the potential existence of an oxygen-tension sensor, triggered by any perturbation in the oxygen levels that results in NO production and EPC release. Another potential explanation is that both high and low local oxygen concentrations act via distinct mechanisms that ultimately converge on NOS activation (Fig. 5). Ischemic wounds signal for hypoxia by hypoxia-inducible factor (HIF)-dependent mechanisms. A functional HIF response requires stabilization of the α-subunit (e.g., HIF-1α), during hypoxia, and dimerization with HIF-1β, to drive target gene activation (60). Intriguingly, high concentrations of NO stabilize HIF-1α and thus mimic a hypoxic response under normoxia. Thus, it is likely that hyperoxia activates NOS activity and increases the BM NO level, which can result in HIF-1α stabilization. If this be the case, it supports a concept that both hyperoxia- and hypoxia-induced upstream biochemical events converge at the level of HIF-1α, and this oxygen-tension sensor will trigger a common signal to mobilize BM EPCs. Whatever the reason, it is clear that with hypoxia, systemic effects of VEGF-A via VEGFR2 mediate NOS activation, whereas with hyperoxia, NO levels increase within minutes, and the effects are quickly reversible on withdrawal of the stimulus, suggesting a direct activation of NOS by a change in oxygen tension. Further studies are needed more specifically to identify the underlying mechanisms.
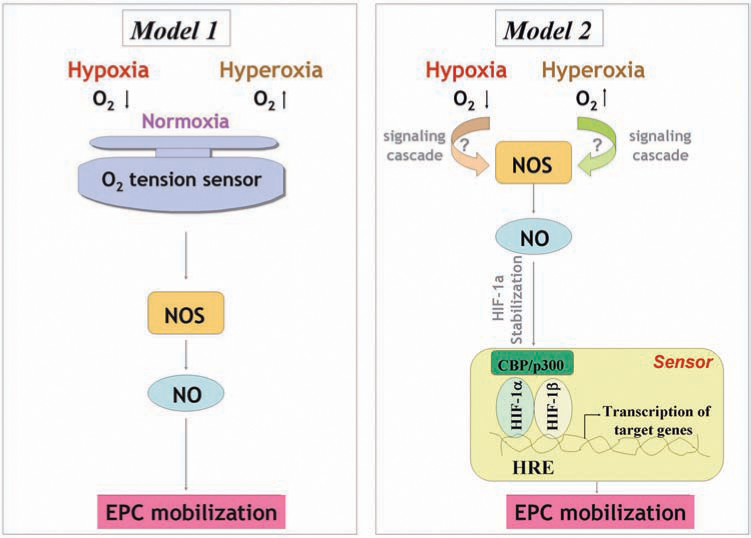
One of the advantages of HBO-induced EPC mobilization is that HBO specifically stimulates BM EPCs to be released into circulation without having a significant impact on the inflammatory cell numbers in circulation. In contrast, although G-CSF and other chemokines are able to increase circulating EPCs (99), an associated increase in leukocytes results in inflammation with the potential for enhanced acute coronary events. It, therefore, raises questions about the safety and clinical utility of these chemokines (53, 87). Thus, HBO appears to be a safe method in driving EPC mobilization.
Conversely, it should be pointed out that HBO-mediated enhancement of wound healing is multifactorial. HBO may have local tissue effects unrelated to EPC release that also enhance wound healing in selective wound environments. One of these effects may include increased tissue-level release of angiogenic factors such as VEGF-A (111, 113).
Oxygen-Dependent Redox-Sensitive Signaling Processes
It is known that hyperoxia induces the generation of excessive reactive oxygen species (ROS), including superoxide anions (O2 −), hydroxyl radicals (OH−), and hydrogen peroxide (H2O2) (Fig. 6). ROS can affect the activity of signaling pathways by direct modification of regulatory proteins via disulphide bond formation, nitrosylation, carbonylation, or glutathionylation (21). ROS and the ROS-generating NADPH oxidases play important roles as signaling molecules in the vasculature (12, 41). Whether and how hyperoxia-induced ROS participate in the regulation of BM EPC mobilization remains unknown.
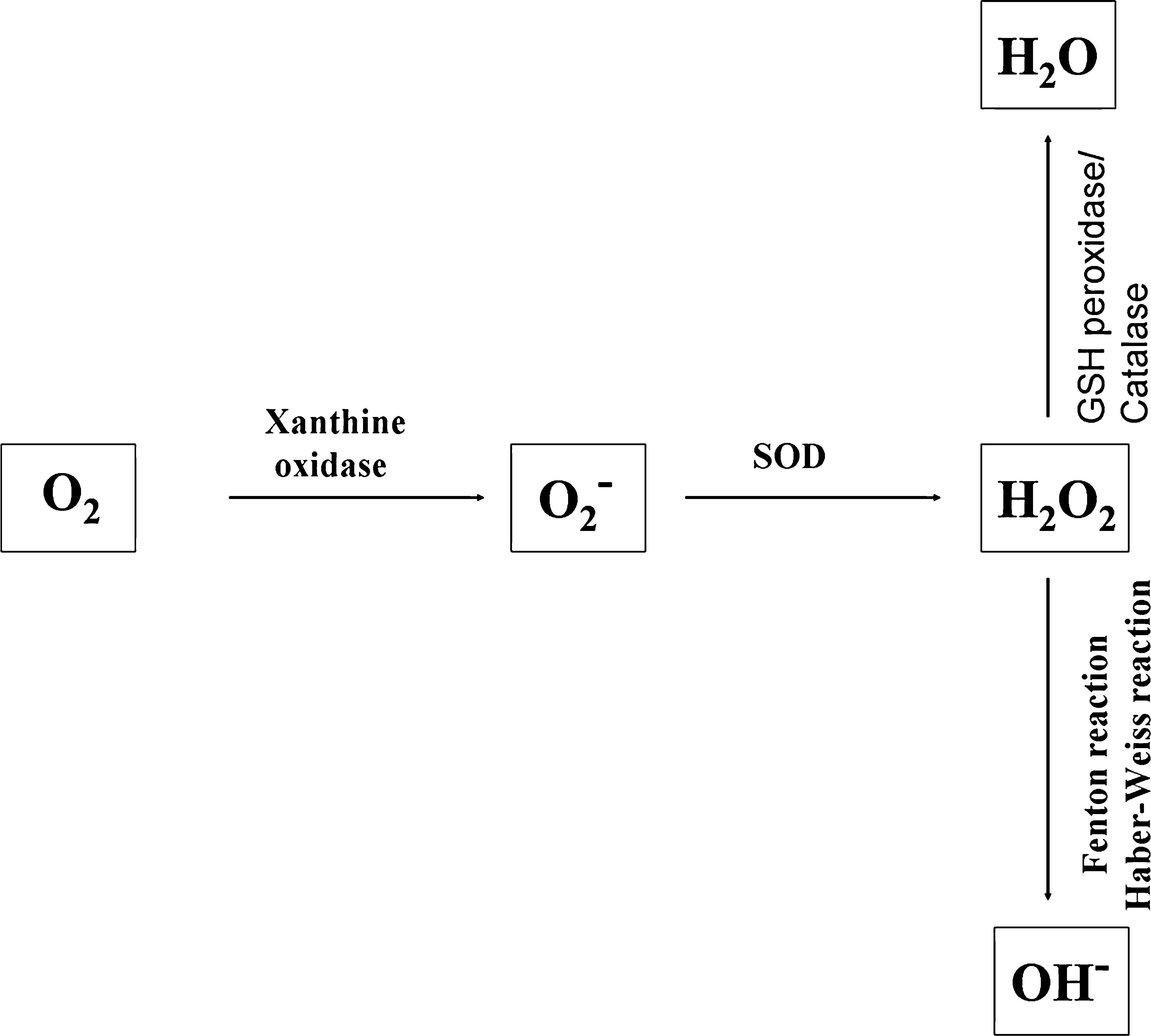
Although direct studies are lacking, several lines of evidence suggest that ROS may be involved in the control of EPC mobilization through modulating cell adhesion, regulating cytoskeleton and cell motility, and inducing VEGF-A production. For instance, it has been reported that ROS are capable of inducing conformational changes in integrins to change their binding affinity and function (35). In addition, ROS are important regulators of the actin cytoskeletal dynamics and cellular motility (86). Given the knowledge that the detachment of EPC from BM niche requires alteration of cell adhesion, and actin cytoskeleton is necessary for cell motility, it would be of interest to investigate whether hyperoxia–ROS–integrin (or other adhesion molecules) and hyperoxia–ROS interaction with the cell actin cytoskeleton are involved in this process. Moreover, at micromolar concentrations, H2O2 induces VEGF-A expression (111). Signaling studies identified a cascade comprising Ras–Raf–MEK1–ERK1/2 as the main pathway mediating H2O2-induced VEGF-A transcription (110). Besides, H2O2 also induces the expression by VEGFR2 by a NF-κB–dependent pathway (32). The consequence of VEGF-A–VEGFR2 interactions in the activation of bone marrow NOS, producing NO, which then stimulates MMP-9, enabling release of soluble Kit-ligand and liberating progenitor cells from the marrow, has been well-established (3, 38, (39). It may be interesting to understand whether BM EPCs respond to ROS in the same way as the matured endothelial cells in terms of VEGF-A and VEGFR2 induction, or which cell population(s) is the target(s) of the ROS and responsible for the activation of NOS–NO–MMP9-soluble Kit ligand cascade in the BM microenvironment.
Recruitment/Homing of EPCs to Diabetic Wound Tissues
The contribution of BM EPCs to neovascularization in wounds results from a multistep process. It involves sensing the ischemia signal from the remote tissue, releasing EPCs from the BM niche into circulation, homing of circulating EPCs to the target tissues, the integration of the EPCs into blood vessels, and the in situ differentiation/maturation of EPCs into matured, functional ECs. Our data demonstrate that hyperoxia selectively enhances EPC release, resulting in a small but significant improvement in diabetic wound healing, yet not having a significant impact on wound EPC homing (25). If EPCs are mobilized into circulation but fail to reach the injured tissue, the clinical usefulness of the HBO treatment becomes suboptimal. This may explain the variable clinical effects on wound healing reported with HBO treatment alone. Our findings suggest that homing factor(s) that controls recruitment of circulating EPCs into wounds may be decreased with diabetes. Therefore, a full understanding of impaired homing mechanisms in diabetic wounds is crucial for enhancing EPC recruitment and engraftment.
The process of EPC homing to sites of ischemia includes detachment from the BM niche, rolling into blood vessels and traveling within the circulation, adhesion to the endothelial cell monolayers, and incorporation into neovessels. Recent studies support the idea that EPC and progenitor cells use adhesion molecules for homing to sites of neovascularization, similar to the adhesion molecules engaged by leukocytes for recruitment to sites of inflammation (49, 71, 78). Chemokines play critical roles in the regulation of this trafficking of circulating EPCs from the bloodstream to ischemic tissues. Many of these factors are chemoattractants. Stromal cell–derived factor (SDF)-1α is the predominant chemokine that is upregulated in ischemic tissue and acts as a homing signal for EPCs (69). Inhibition of the SDF-1α–CXCR4 axis partially blocks the homing of progenitor/stem cells to the ischemic myocardium (1). Likewise, suppression of CXCR4 by anti-CXCR4 neutralizing antibodies significantly reduced SDF-1α–induced adhesion of EPC to endothelial cell mono-layers, the migration of EPCs in vitro (16), and the in vivo homing of myeloid EPCs to the ischemic limb in a model of hindlimb ischemia (134).
Although the role of SDF-1α in EPC homing to ischemic tissue is known, the effect of SDF-1α expression at the tissue level in diabetic wounds had not been previously studied. We recently demonstrated that the local concentration of SDF-1α in a diabetic wound is significantly decreased, and epithelial cells and myofibroblasts appeared to be responsible for the downregulation of SDF-1α in diabetic wounds (25) (Fig. 7). Moreover, the exogenous administration of SDF-1α to wounds of diabetic mice increases the wound-level EPC recruitment. More important, in combination with HBO therapy, which brings an increased amount of circulating EPCs, raising SDF-1α levels by local injection of recombinant SDF-1α protein significantly enhances EPC recruitment to wound tissues and improves wound healing in this diabetic animal model (Fig. 8).
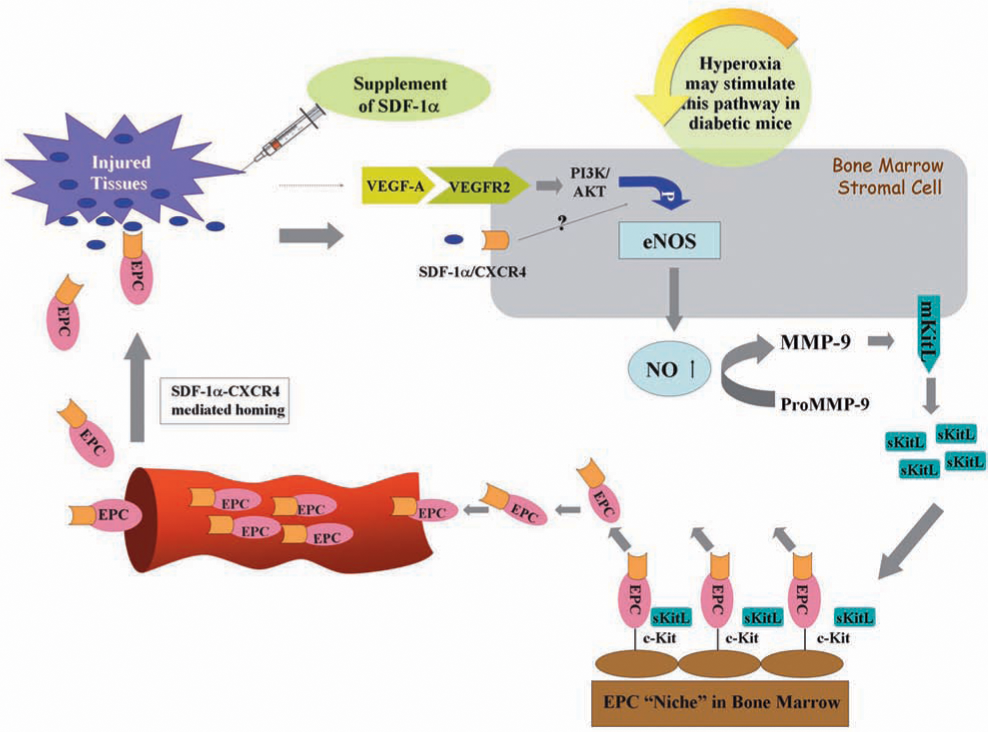
In addition to the SDF-1α/CXCR4 axis and VEGF, which is ischemia induced and well known for its role in recruiting EPCs, other factors may be potentially involved in the regulation of EPC homing to diabetic wounds but have not been studied. For instance, high-mobility group box-1 (HMGB-1) is a nuclear protein that is released extracellularly on activation of cells by inflammatory cytokines and during cell necrosis and acts as a chemoattractant for inflammatory cells, stem cells, and EPCs in vitro and in vivo (18, 96). IL-8 is an inflammatory chemokine that is able to stimulate angiogenesis (88). Local injection of IL-8 in the nonischemic myocardium increased the recruitment of CD34+ cells (64). Neutralizing anti-IL-8/Gro-α antibodies or antibodies against the IL-8 receptors, CXCR1 or CXCR2, reduced CD34+ cell-mediated improvement of neovascularization, establishing a role for CXC-chemokines (IL-8/Gro-α) for homing and neovascularization improvement by CD34+ cells. In addition, blocking CXCR2 inhibited the incorporation of human EPCs expressing CXCR2 at sites of arterial injury (45). Other elements, such as the integrin α4β1–VCAM pair, may also mediate EPC homing (50). It would be worthwhile to test whether these factors are impaired in chronic diabetic wounds and are responsible for mediating EPC homing. The identification of critical homing factor(s) will enhance the efficiency and specificity of EPC therapies and increase the efficacy of EPC-mediated neovascularization in diabetic wounds.
Summary and Perspective
Cell-based therapy is a promising therapeutic option for treating patients with diabetic, nonhealing wounds. Of various different types of stem or progenitor cells, the EPC is one type of cell that has been moved from experimental models to clinical trials. EPC has thus far been tested in patients with acute and chronic ischemic disease, and outcomes are very encouraging (6, 119). A few properties, such as its endogenous, BM-derived character, ability to home to sites of pathologic entities, and relative stability in terms of lineage specification in culture, which allows genetic and epigenetic manipulation, make EPC an ideal cell candidate to be tested in cell-based therapeutic applications for ischemic disorders.
The efficacy of cell therapies to augment neovascularization and healing depends not only on the sufficient amount of circulating EPCs, but also on the efficient recruitment of these cells to the target tissue. This is not surprising, as the contribution of EPC to neovascularization is a coordinated sequence of multiple steps, and each step is controlled by a specific mechanism(s). Several of these mechanisms are impaired in diabetes. Thus, a combination of therapeutic approaches to target individual impairments and to correct diabetes-related EPC deficits will likely synergize and lead to a more-successful treatment outcome for diabetic wounds.
The identification of hyperoxia as an efficient tool in the mobilization of BM-derived EPCs is of great significance, as HBO treatment is an FDA-approved, safe method. The biologic effect of hyperoxia on mobilizing EPCs is evident and somehow paradoxically similar to that produced by hypoxia. Although further study is required to elucidate the molecular and cellular mechanism underlying the biologic effect of HBO, from the therapeutic point of view, HBO is an acceptable approach and has actually been applied in the treatment of diabetic wounds with varying degrees of success. Understanding the limitations of HBO in modulating only the EPC mobilization step but not the homing of the increased circulating EPCs to the target tissue provides useful guidance for the development of new clinical protocols to treat diabetic wounds. In addition, development of more efficient and specific treatments targeting the eNOS-NO pathway, which may synergize with the HBO effects, would be a plus. Moreover, in addition to SDF-1α, other strategies for modifying the wound environment, such as PDGF-BB (118), fibroblasts delivered in an absorbable mesh (83), or in type 1 collagen (14), which, at least partially, function directly or indirectly through accelerating recruitment of EPCs, are under investigation. These strategies could be combined with HBO therapy, thus synergistically influencing EPC effects by targeting both homing and mobilization steps. Furthermore, elucidation of the mechanisms underlying the decreased number of BM EPCs in diabetes and development of a solution to correct this impairment and to increase the BM EPC number could provide a scientific basis for the establishment of combined treatment protocols. A better understanding of the molecular and cellular etiologies of nonhealing, diabetic wounds in combination with development of efficient approaches for correcting EPC deficits and functional impairments will eventually result in the development of clinically efficient and feasible therapies that prevent wound progression, eliminate amputations, and promote rapid healing in patients with diabetes.
Footnotes
Abbreviations
ATA, atmospheres absolute; BM, bone marrow; CaM, calmodulin; CXCR4, chemokine receptor 4; Dil-Ac-LDL, Dil-labeled acetylated low-density lipoprotein; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; ECM, extracellular matrix; G-CSF, granulocyte colony-stimulating factor; HSP90, heat-shock protein 90; HMGB-1, high-mobility group box-1; H2O2, hydrogen peroxide; OH−, hydroxyl radical; HIF, hypoxia inducible factor; HBO, hyperbaric oxygen; MMP-9, matrix metallopeptidase 9; NO, nitric oxide; O2, oxygen; PAD, peripheral arterial occlusive disease;:PECAM-1/CD31, platelet endothelial cell–adhesion molecule-1; ROS, reactive oxygen species; SDF, stromal cell–derived factor; O2 −, superoxide anion; FDA, United States Food and Drug Administration; VEGF, vascular endothelial growth factor; VE-cadherin, vascular endothelial cadherin; VEGFR2, vascular endothelial growth factor receptor 2; vWF, von Willebrand factor.