
Abstract
Loss of redox homeostasis and formation of excessive free radicals play an important role in the pathogenesis of kidney disease and hypertension. Free radicals such as reactive oxygen species (ROS) are necessary in physiologic processes. However, loss of redox homeostasis contributes to proinflammatory and profibrotic pathways in the kidney, which in turn lead to reduced vascular compliance and proteinuria. The kidney is susceptible to the influence of various extracellular and intracellular cues, including the renin–angiotensin–aldosterone system (RAAS), hyperglycemia, lipid peroxidation, inflammatory cytokines, and growth factors. Redox control of kidney function is a dynamic process with reversible pro– and anti-free radical processes. The imbalance of redox homeostasis within the kidney is integral in hypertension and the progression of kidney disease. An emerging paradigm exists for renal redox contribution to hypertension. Antioxid. Redox Signal. 11, 2047–2089.
Redox Control of Cellular Function: How Is It Achieved?
Pathologic Role of ROS in Hypertension
NAD(P)H Oxidase Inhibition for the Treatment of Hypertension: Promises and Limitations
I. Introduction
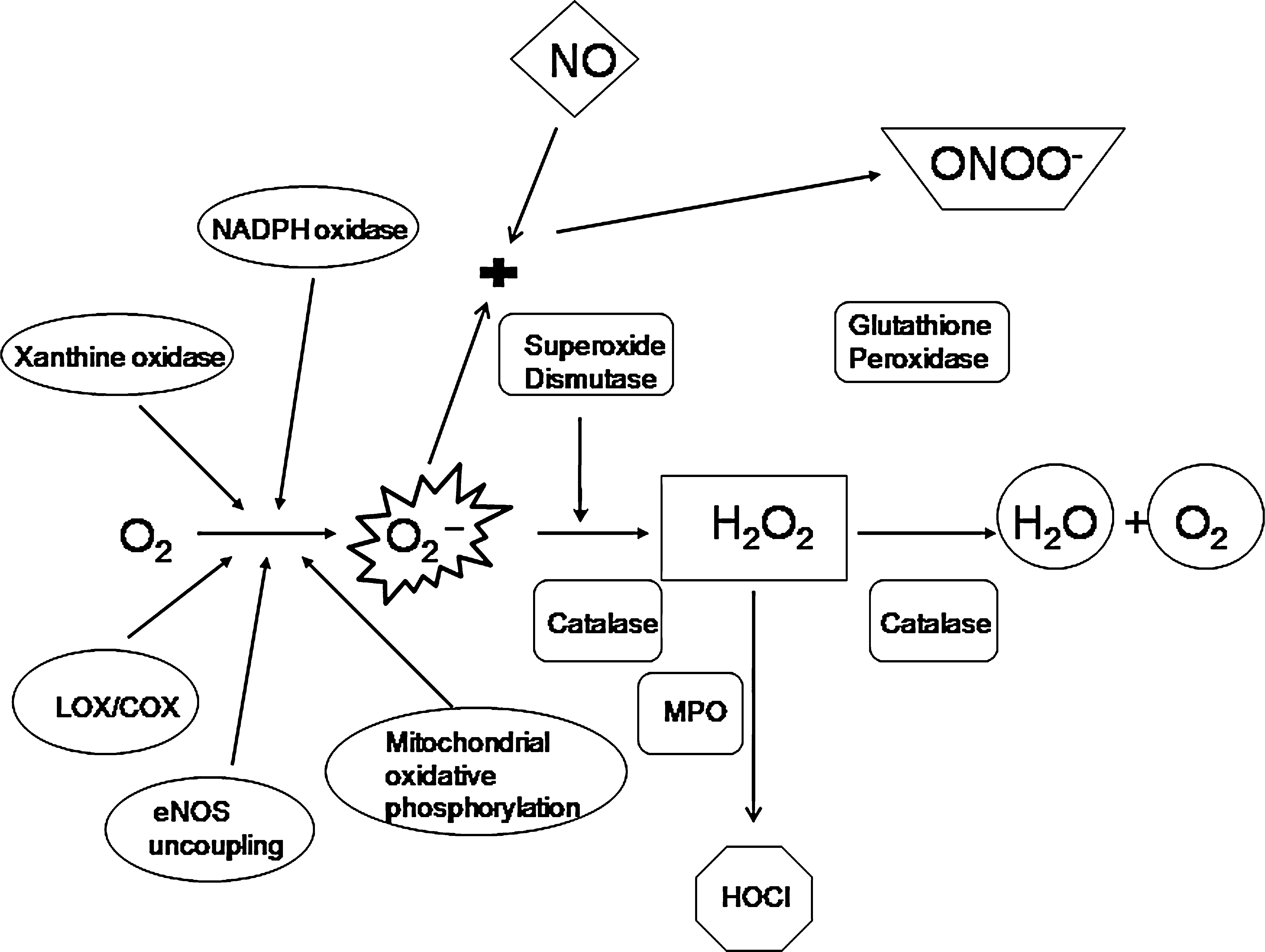
O2 •−, superoxide anion; 1O2, singlet oxygen; 3O2, ozone; •OH, hydroxyl anion; ClO−, chlorite ion; HOCl−, hypochlorite anion; ONOO−, peroxynitrite; NO+, nitrosonium ion; NO−, nitroxyl anion; oxLDL, oxidized LDL; •LOO, lipid peroxyl; •LO, lipid alkoxyl.
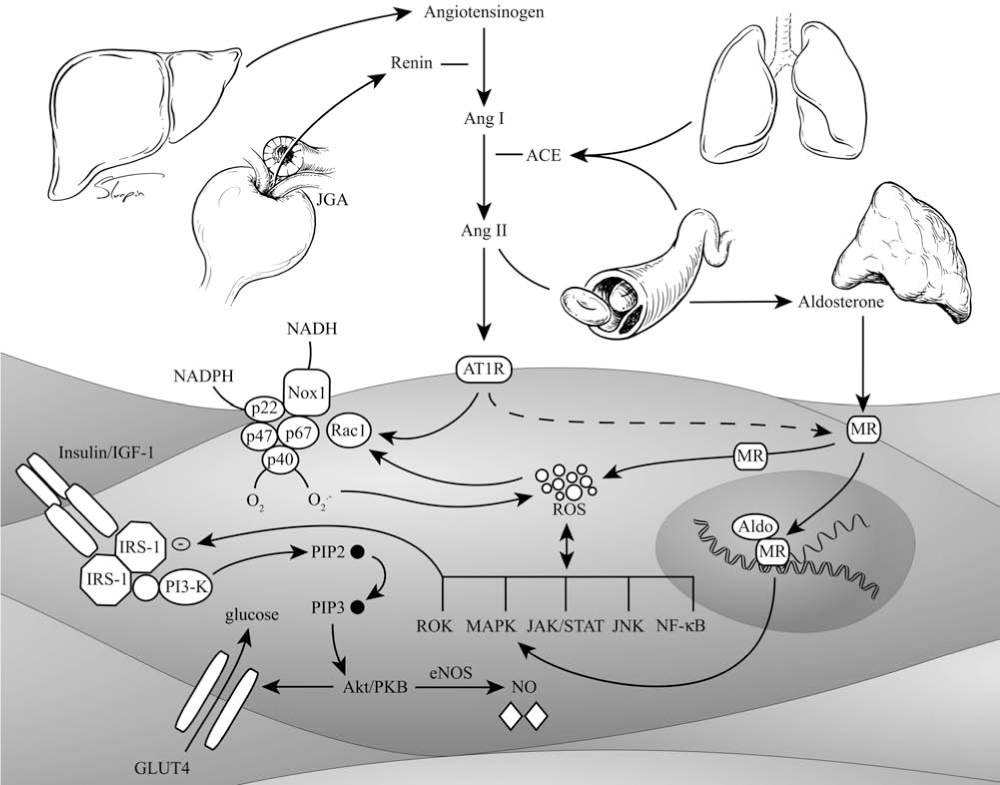
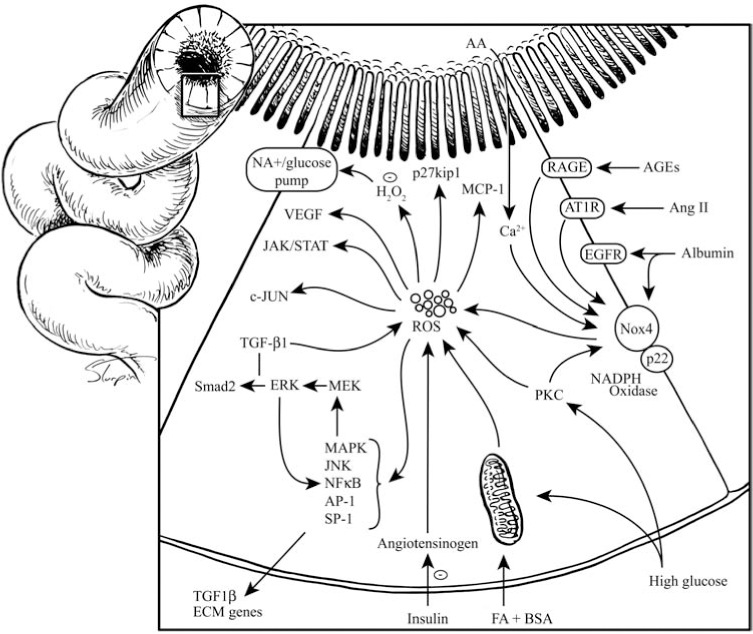
Increases in the chronic disease burden have led to identification of several important modifiers of the cellular redox mechanisms. Important among the modifiers are Ang II, aldosterone, hyperglycemia/hyperinsulinemia, modified lipids, high salt, and the peripheral nervous systems, including sympathetic and dopaminergic systems (Fig. 2). Modification of cellular redox mechanisms results in the abnormal production of O2 ·− and hydrogen peroxide (H2O2), which are mediators of many downstream signaling pathways, such as transcription factors, tyrosine kinases/phosphatases, ion channels, mitogenic factors, and cytokines. Through these signaling pathways, ROS have distinct functional effects in the kidney and various renal cellular mechanisms, including tubular sodium transport, tubuloglomerular feedback, medullary blood flow, cell migration and growth, hypertrophy, expression of inflammatory and extracellular matrix genes, and apoptosis. The effects of ROS generated within various components of the kidney ultimately depend on the locally generated concentrations and the balance of pro- and antioxidant pathways.
The purpose of this review is to describe the important mechanisms that contribute to generation of oxidative stress and hypertension in the kidney and the extrarenal vascular contribution to hypertension. Further, we review pathways with antioxidant properties that support a link with hypertension. Last, we discuss ways to reduce oxidative stress–mediated renal injury.
II. Redox Control of Cellular Function: How Is It Achieved?
A. Free radical contribution to redox control of hypertension
Free radicals can broadly be subdivided into ROS and reactive nitrogen species (RNS) (Table 1). These are generated for specific cellular processes: second messengers in cell-cycle progression, smooth muscle relaxation and inhibition of platelet adhesion, cell growth and differentiation, phagocytosis, production of cytokines [e.g., interleukin-2 (IL-2)], stimulation of hemoxygenase-1 (HO-1), activation of transcription factors [e.g., nuclear factor-kappa B (NF-κB)], insulin signaling, and others. In the kidney, ROS are involved in erythropoiesis, sodium handling, and fluid homeostasis, as discussed in greater detail in later sections.
The predominant form of ROS is the O2 ·− (Fig. 1 and Table 1). In the mitochondria, most of the O2 ·− is generated by leakage from the electron-transport chain (nonenzymatically produced by semiubiquinone) and the Krebs cycle (278). This constitutes 2% of all electron-transport chain by-products; the remaining 98% goes toward producing O2 and H2O. In overall quantitative terms, mitochondrial electron-transport chain–generated O2 ·− may be the most important source (278). O2 ·− also is produced by metabolic oxidases, including nicotinamide adenine dinucleotide phosphate [NAD(P)H] oxidase, xanthine oxidase (XO), P-450 monooxygenase, lipooxygenase (LOX), and cyclooxygenase (COX) (278). Superoxide dismutase (SOD) converts O2 ·− into H2O2, which is detoxified into H2O by either glutathione peroxidase (GPx) or catalase. H2O2 can oxidize chloride to form the reactive HOCl− in cells that express myeloperoxidase (MPO). HOCl− can react with O2 ·− to form OH. HOCl− can further react with H2O2 to produce singlet oxygen (1O2). Excess O2 ·− also reduces transition metal ions such as Fe3+ and Cu2+, the reduced forms of which, in turn, can react with H2O2 to produce ·OH (Fenton and Haber–Weiss reactions, respectively). ·OH is the strongest of the oxidant species and reacts indiscriminately with nucleic acids, lipids, and proteins. No detoxification system is known for OH; therefore, scavenging OH is a critical antioxidant process.
The NO radical (NO·) is produced by oxidation of one of the terminal nitrogen atoms of
B. Clinical contribution to redox control of hypertension
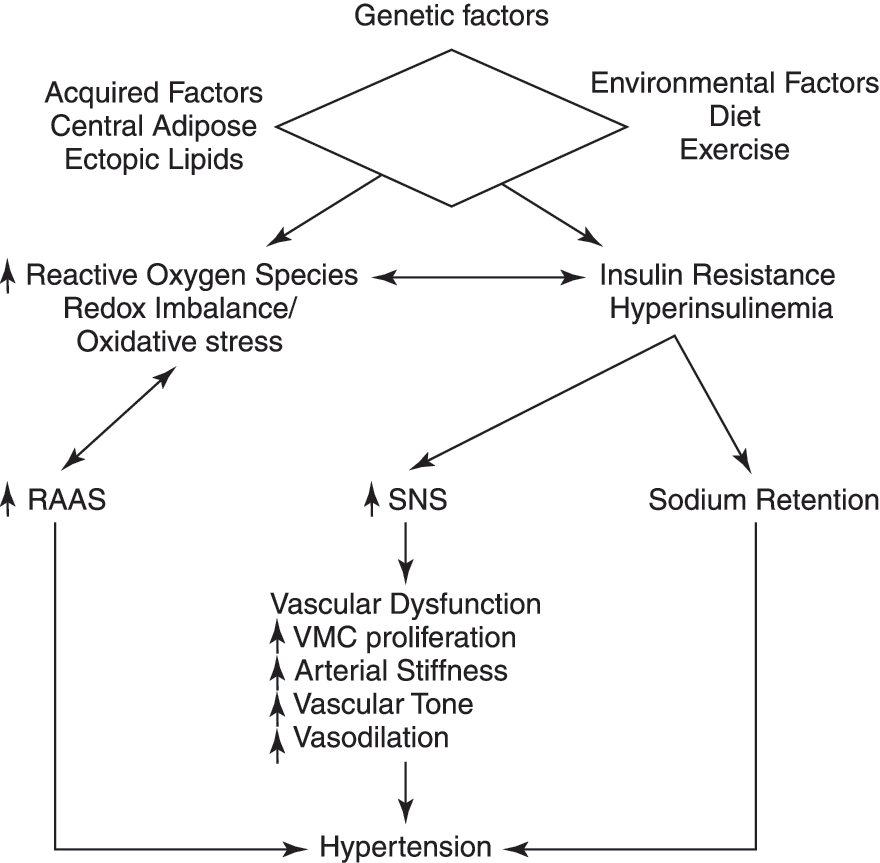
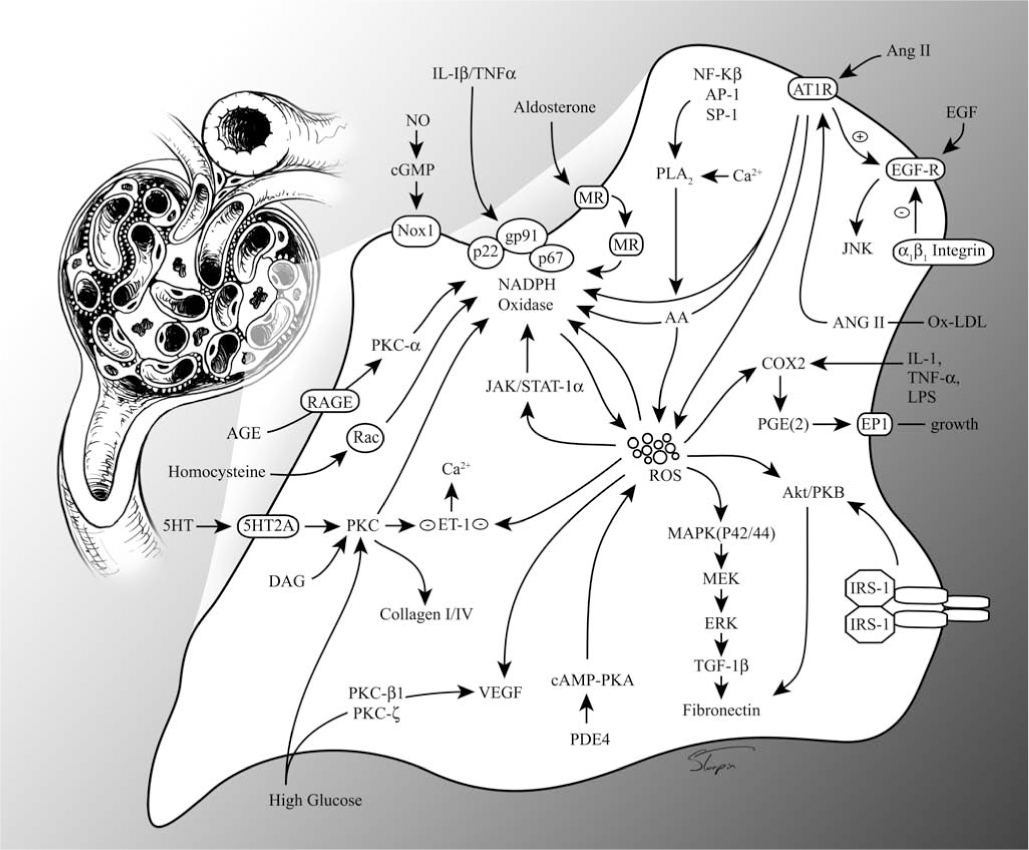
Environmental and cellular cues that increase production of ROS include activation of the renin–angiotensin–aldosterone system with elevated levels of angiotensin II (Ang II) and aldosterone, lipids, insulin resistance with subsequent hyperinsulinemia and hyperglycemia with formation of advanced glycation end-products (AGEs), as well as mechanical shear forces (Figs. 2 and 3). These ROS-generating cues are present at increased levels in certain populations genetically predisposed to hypertension and renal disease (64). Classically this has been observed in individuals that display obesity, insulin resistance, hypertension, and other components that compose the cardiometabolic syndrome [e.g., any pathophysiologic state characterized by elevated plasma lipids, of the modified (ox-LDL) or unmodified type, are associated with generation of ROS, redox imbalance, and oxidative stress and are known to contribute to hypertension (Fig. 4)]. The cholesterol lowering achieved after short-term use of an HMG-CoA reductase inhibitor (statin) therapy is associated with a decrease in ONOO−-mediated oxidative stress and an improvement in large-artery distensibility. Indeed, the use of statins alone or in combination with angiotensin-converting enzyme inhibitors (ACE inhibitors) improved antiatherosclerotic endothelial expression of quotient Q, which includes the NAD(P)H oxidase subunit gp91 phox (Nox2) expression.
In addition to ox-LDL, hyperglycemia by itself can lead to oxidative stress and hypertension in normal subjects and in people with diabetes (26). In healthy human subjects, administration of methacholine in euglycemia resulted in decreased endothelium-dependent vasodilation after the cohort was subjected to 6 h of acute hyperglycemia (10). Concurrent administration of the antioxidant vitamin C abrogated hyperglycemia-induced changes. The authors postulated that hyperglycemia inhibits endothelium-dependent vasodilation through preferential production of O2 ·− over NO, and vitamin C scavenges the extracellular oxygen-derived free radicals. Furthermore, the work of other investigators on hyperglycemia has led to the hypothesis that oxidative stress in this condition is a result of imbalances between oxidant-sensitive mechanisms and NO availability (39). Oxidative stress is attenuated by free radical scavengers that indirectly increase NO bioavailability (10, 323). Other pathways through which hyperglycemia may promote O2 ·− production are glucose autooxidation, advanced glycation end products (AGE) signaling, abnormal arachidonic acid (AA) metabolism and its coupling to cyclooxygenase catalysis, protein kinase C activation, depletion of BH4, and wall shear stress (10, 26, 242, 301, 323). As mentioned, hyperglycemia contributes to the formation of AGEs, examples of which include pentosidine, carboxymethyllysine, carboxyethyllysine, and argpyrimidine. These have been shown to have direct effects on oxidative stress via binding to the receptor for AGE (RAGE) (229, 290). Furthermore, blockade of RAGE resulted in abrogation of oxidative stress. This evidence is derived largely from small animal investigations and clinical trials using aminoguanidine (a.k.a., pimagedine) in Aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION-II) and Alt-711 (a.k.a., alagebrium) in phase 2b Systolic Pressure Efficacy and Safety Trial of Alagebrium (SPECTRA) (7, 69). In the insulin-resistant state, hyperglycemia has been shown to have deleterious effects on vascular fluid dynamics, thereby contributing to hypertension.
C. Prooxidant enzymes and pathways
1. NAD(P)H oxidase
The enzyme complex better known as NAD(P)H oxidase is now well recognized as a major source of ROS production in vascular and renal cells (91, 248). More important, it is the enzyme complex responsible for generating Ang II-mediated O2 ·− in the vast majority of cardiovascular diseases including hypertension (278). The structure and function of this enzyme complex have been extensively reviewed elsewhere (6). In brief, the phagocyte enzyme complex is made up of both membrane and cytosolic components. The membrane complex consists of two subunits, gp91phox (phox stands for phagocyte oxidase), a 91-kDa protein, and p22phox, a 22-kDa protein, which together form a heterodimer, flavocytochrome b558 (154). The membrane complex is inactive under normal cell-resting conditions (an exception may be Nox4, a 66-kDa gp91phox homologue expressed in nonphagocytic cells and other Noxes expressed in nonphagocytic cells). Cytosolic components include p47phox, or its homologue NoxO1 (expressed in nonphagocytic cells), which is considered an organizer subunit, as it is strategically phosphorylated when activated. Under unstimulated conditions, the bis-SH3 domain (contains tandem repeats of SH3) is not available for binding to the proline-rich region (PRR) of p22phox because of being buried in an autoinhibitory region (AIR). On stimulation, serine phosphorylation of the AIR occurs, uncovering the bis-SH3 domain for interaction with the membrane complex cyt b558 (specifically PRR of p22phox). An additional PX domain facilitates binding to the membrane phosphatidylinositol groups. p67phox (67-kDa protein) and p40phox (40-kDa protein) are the other cytosolic components with PRR binding and activation domains (ADs). The AD in p67phox facilitates transfer of hydride ion from NAD(P)H to FAD on interaction with small G-protein Rac through the tetratricopeptide domain (TPR) of p67phox. The Rac proteins themselves exist in an inactive state when bound to GDP; however, phosphorylation leads to activated Rac-GTP and translocation to the NAD(P)H membrane complex. p40phox binds to p67phox through its counterpart phox/Bem1 (PB1) and facilitates the assembly of p47 phox−p67phox at the membrane through an additional lipid PX binding to the membrane. The final transfer of an electron to form O2 ·−is facilitated by two heme groups present within the NAD(P)H multisubunit complex.
Several mutations have been identified in the phagocytic NAD(P)H oxidase subunits, leading to a condition known as chronic granulomatous disease (CGD). This disease condition is characterized by the inability of the organism to kill engulfed bacteria by production of O2 ·− and other free radicals. Whereas mutations in gp91 phox are inherited as an X-linked trait, mutations in other subunits are inherited as autosomal recessive conditions. So far, no mutation has been identified in the human p40phox gene, contributing to chronic granulomatous disease (CGD); however, p40 phox knockout mice exhibit CGD defects similar to those of other subunit knockouts (60). Importantly, characterization of a mutation in the phox homology domain of the NAD(P)H oxidase component p40 phox identified a mechanism for negative regulation of O2 ·− production (30).
The nonphagocytic NAD(P)H oxidase differs substantially from its phagocytic counterpart (Table 2). It is constitutively active under resting conditions, which means all subunits must be in their active conformation and together in the cell. Li et al. (182) showed that the nonphagocytic enzyme complex resides mostly in a perinuclear pattern in endothelial cells. Whereas the phagocytic NAD(P)H oxidase can generate millimolar quantities of O2 ·− under stimulation, the nonphagocytic counterpart produces only micromolar quantities. It is now believed that the constitutively active NAD(P)H oxidase present in nonphagocytic cells is the original NAD(P)H oxidase, and the highly regulated, damaging version active only in respiratory bursts is the mutated and evolved enzyme (Table 2).
NAD(P)H oxidase is regulated by agonist-induced activation and antagonist-induced suppression. Agonist-induced mechanisms include upregulation of the p47 phox subunit by Ang II, tumor necrosis factor (TNF-α), and pure pressure with helium gas, among other stimuli (141, 180, 378). This was corroborated in p47 phox knockout mice, which demonstrate complete loss of agonist stimulation. Antagonists including NO are capable of suppressing NAD(P)H oxidase activity, illustrated best in mesangial cells by the downregulation of Nox1 (255).
Importantly, feedback mechanisms may be involved in the maintenance of low, constitutively active NAD(P)H oxidase-mediated ROS during physiologic processes. An example of such a regulation is the increased turnover of ubiquinated Rac1 in the presence of excess H2O2 and decreased turnover of Rac1 in the presence of NAD(P)H oxidase inhibitors like diphenyliodonium (DPI) (169). Conversely, exogenous exposure of smooth muscle cells (SMCs) or fibroblasts to H2O2 activates NAD(P)H oxidase to generate more superoxide anion, displaying a feed-forward process (185). The feed-forward mechanism may play a role in NAD(P)H oxidase–dependent oxidative stress in a variety of disease processes, including insulin resistance, hypertension, and kidney disease.
The roles of NAD(P)H oxidase in mediation of redox homeostasis and hypertension are reviewed in later sections.
2. Xanthine oxidase (XO)
XO was first proposed to play an important role in generating vascular oxidative stress in the hearts of chronically ethanol-treated rats (239). Since then, several groups have focused on the role of this enzyme in generating oxidative stress and endothelial dysfunction. Data from double transgenic rats (dTRs) harboring both human renin and angiotensinogen genes suggests endothelial function of renal arterial rings was impaired (213). Moreover, relaxation was improved with superoxide scavenger SOD and oxopurinol, an XO inhibitor. Blockade of the angiotensin-receptor type 1 (AT1R) normalized blood pressure, oxidative stress measures, endothelial dysfunction, and the contractile responses. This supports another mechanism by which the RAAS and elevated Ang II contribute to endothelial dysfunction, redox and oxidative stress in the vasculature. However, conflicting reports exist on the function of XO in redox control of the kidney.
As discussed earlier, hypercholesterolemia is an important risk factor for the progression of kidney disease and contributes to redox imbalance and oxidative stress, potentially causing endothelial dysfunction via XO. To elucidate a contribution of XO activity and redox control of kidney function, investigators incorporated a pig model of hypercholesterolemia to evaluate renal hemodynamics (45). While infusing oxypurinol into the kidneys, they measured renal hemodynamics before and after endothelium-dependent (acetylcholine, ACh) and -independent (sodium nitroprusside) challenge. Hypercholesterolemic pigs demonstrated elevated oxidative stress, higher plasma uric acid, lower urinary xanthine, and greater renal XO expression compared with controls. Inhibition of XO with oxypurinol significantly improved the blunted responses to ACh of cortical perfusion, renal blood flow, and glomerular filtration rate, restored medullary perfusion, and, in addition, improved the blunted cortical perfusion response to sodium nitroprusside, further supporting a role for XO in redox control of the kidney. Alternatively, XO levels have been shown to be down-regulated in the 5/6 nephrectomized rats, as well as a rat model of mesangioproliferative anti-Thy 1.1 GN (glomerulonephritis) (76). NAD(P)H oxidase–dependent increases in ROS production were observed, but XO levels were not changed in these animal models. Furthermore, another study in Dahl salt-sensitive (DSS) rats showed an increase in NAD(P)H and mitochondrial sources of ROS and no change in XO levels (320).
The contribution of XO to renal redox control of hypertension may be more apparent as it relates to uric acid. Uric acid, an end product of the xanthine oxidase pathway, has been proposed both to have antioxidant and oxidant properties (145, 311). The antioxidant property of uric acid may relate to its ability to scavenge free radicals, including O2 ·−, ·OH, and singlet oxygen, and to increase the levels of extracellular SOD (ecSOD), thereby preventing endothelial NOS (eNOS) uncoupling and endothelial dysfunction (121, 145). Although elevated levels of uric acid may confer protective antioxidant effects on the vasculature and the kidneys, the correlation with cardiovascular disease risk is not readily apparent (145). Interestingly, elevated serum uric acid levels have been proposed as a marker for cardiovascular dysfunction and renal disease (145, 149, 152, 311). Administration of uricase inhibitor, oxonic acid (OA), has been used extensively to study the detrimental effects of increased uric acid levels. Experimentally increased uric acid levels result in accelerated renal-disease progression by variably increasing systolic BP, afferent glomerular arteriolopathy, oxidative stress, and endothelial dysfunction in different rat models, and administration of allopurinol reversed these deleterious effects (149, 158). In addition, reduction of BP with enalapril and potentiation of NO with NOS substrate L-arginine also variably improved renal function (209, 283). However, the role of uric acid as a risk factor for kidney disease is largely unknown (145). Whereas it appears that reducing uric acid levels in early kidney disease may be useful, established kidney disease is less responsive to treatment with uric acid–reducing agents, as other factors including salt sensitivity of kidneys may predominate. Moreover, clinical trials, including the Modified Diet in Renal Disease (MDRD) study, have failed to show association of uric acid levels with kidney-disease progression. Interestingly, the GREek Atorvastatin and Coronary heart disease (CHD) Evaluation (GREACE) and LIFE studies reopened a fierce debate on the benefits of uric acid–level monitoring in a subset of patients with more-severe CVD risk (311).
3. Lipooxygenases (LOX) and cyclooxygenases (COX)
LOX and COX produce oxidative stress through NAD(P)H oxidase–dependent and –independent pathways (29, 195). Ang II treatment of rat aortic smooth muscle cells induced 5-lipoxygenase (5-LOX), resulting in an increased production of leukotriene B(4) [LTB(4)]. LTB(4) stimulated NAD(P)H oxidase and downstream IL-6 transcripts through generation of ROS. Reversal of oxidative stress through inhibition of LOX and COX via AT1R blockade as well as NSAIDS/aspirin has been shown to modulate kidney function (83, 339). Further, COX-2 expression was increased in N(omega)-nitro-
Prostaglandins (PGs), products of COX, have important physiologic roles in maintaining vascular tone, salt and fluid homeostasis, and renin release. In the kidneys, salt deprivation activates Ang II, which in turn inhibits renin production once homeostasis is achieved. One pathway that has been proposed to participate in this feedback loop is COX-2–mediated suppression of renin in the juxtaglomerular apparatus (JGA) (382). This is a long-term effect. However, before homeostasis is achieved, salt deprivation due to insufficient salt intake or diuretic adminstration contributes to increases in COX-2 expression in the cortical part of the thick ascending loop of Henle (cTALH) and JGA, leading to increase in renin release and subsequent activation of the RAAS. Thus, PGs contribute to the tubuloglomerular-feedback (TGF) response to low-salt delivery to macula densa by increasing Ang II levels and by preventing Ang II from decreasing the glomerular filtration rate (GFR). In animal models, blockade of the AT1R and ACE inhibition increase the expression of COX-2 in the macula densa. Furthermore, AT1A inhibition potentiated Ang II actions via AT2 receptors, resulting in further increases in COX-2 expression.
Interestingly, some investigators propose that COX-2 expression may be regulated in response to local chloride concentrations and not sodium concentrations (109), whereas others propose that it is the sodium load regulating COX-2 expression (170). In the renal medulla, COX-2 is regulated in a manner opposite that of the cortex. COX-2 expression decreases with salt deprivation and increases with high-salt diets to promote adaptation of medullary interstitial cells to hypertonic stress (34). Use of an NO donor S-nitroso-N-acetylpenicillamine (SNAP) in primary cultures of rabbit cTALH leads to increased expression of COX-2 in the cTALH (35) through NF-κB and p38MAPK pathways. COX-2 is expressed in the macula densa, cTALH, medullary interstitial cells, and podocytes.
In addition to the normal physiologic role of COX-2, its role in increasing oxidative stress, inflammation, hypertension, and proteinuria has generated considerable interest. Several animal models that demonstrate hyperfiltration also demonstrate increased COX-2 expression; including the subtotal renal-ablation model, the streptozotocin-induced diabetes model, and the diabetes plus deoxycorticosterone acetate (DOCA) salt-hypertension model (34). Long-term treatment with COX-2 inhibitors in these models significantly decreased proteinuria and reduced extracellular matrix deposition, as indicated by decreases in immunoreactive fibronectin expression and mesangial matrix expansion. These studies exemplify the complex role of COX-2.
4. P450 monooxygenase and mitochondrial respiratory chain enzymes (I-IV)
Although P450 monooxygenase has been proposed to play a role in the redox control of kidney function, little evidence exists of this enzyme in this role (1, 262). Importantly, the P4502E1 isoform has been proposed to be activated by ·OH and, by positive feedback, acts as an iron donator and for further production of ·OH through the Fenton reaction. In addition, this isoform was found at five-to eightfold higher levels in kidneys of streptozotocin-treated rats. Importantly, the mitochondrial respiratory chain enzymes (I-IV) have been proposed to play important physiologic and pathologic roles in the vasculature and the kidney. Complex I and III are the major sites of superoxide production, which is an unavoidable byproduct of ATP generation in the mitochondria. Mitochondrial ROS have also been proposed to play important roles in cellular signaling processes. Autosomal recessive mutations in the coenzyme Q(10) protein leads to glomerular damage via decreased activity of complex II + III in abnormal mitochondrions in podocytes (54). Blockade of mitochondrial complex I leads to lack of ureteric bud-branching morphogenesis in response to high glucose (383). Hyperglycemia induces mitochondrial electron-transport chain enzymes in cultured human mesangial cells to increase ROS production, leading to NF-κB activation and COX-2 protein expression (162). The renal outer medulla is considered to be the major site for mitochondrial respiratory chain enzyme–mediated ROS production and, together with nicotinamide adenine nucleotide (NADH) oxidase (not NADPH oxidase), has been implicated in regulation of medullary blood flow (MBF) and sodium excretion (387). DETC (a SOD inhibitor) decreased MBF and increased sodium retention and hypertension, whereas the SOD mimetic, tempol, had the opposite effects, demonstrating that O2 ·− is vasconstrictive and antinatriuretic. In addition to unmasking NO-induced vasodilation, tempol has other effects on O2 ·−. Among the proposed mechanisms are reduction in intracellular Ca2+ concentrations, thereby keeping the vascular smooth muscle cells relaxed and preventing inhibition of prostacycline (PGI2).
Importantly, several lines of evidence indicate that hypertension may be associated with mitochondrial dysfunction through mechanisms involving increased ROS production and RAAS activation (253, 326). Losartan attenuates renal mitochondrial dysfunction in SHRs and decreases proteinuria (48). Pretreatment of streptozotocin-induced diabetic rats with losartan was associated with lower mitochondrial H2O2 production, kidney structural damage, and proteinuria (47). The benefits of losartan were shown to be beyond its blood pressure–lowering effects, as treatment with amlodipine also lowered BP but did not have the improvement in mitochondrial dysfunction (47). Interestingly, Ang II has been shown to stimulate mitochondrial H2O2 production, leading to endothelial dysfunction via a PKC-dependent pathway; however, p22 phox inhibition did abrogate mitochondrial H2O2 production, underscoring the role of NAD(P)H oxidase enzyme once again (56). Furthermore, a study by Kimura et al. (160) demonstrated that abrogation of mitochondrial ROS did not result in a decrease in the vasoconstriction induced by prolonged Ang II inhibition, although other benefits, including reduction in MAP kinase activity, were apparent. In spite of this conflicting report, the consensus points to an important role for mitochondria-generated ROS and hypertension via the mediation of RAAS and other cellular cues.
D. Antioxidant enzymes and pathways
O2 ·− free radicals are scavenged efficiently by cellular antioxidants when their normal functions (e.g., cellular signaling) are over (Fig. 1). Antioxidants, including SOD, convert O2 ·− to H2O2, which is then converted to O2 and H2O by catalase and glutathione peroxidase (GPx). Under certain conditions of excess O2 ·− or depleted SOD and catalase enzymes, the thioredoxin (TRX)/glutaredoxin (GRX) system comes to the rescue of cells under attack from oxidants (15, 207, 302, 374). The TRX and GRX systems have been extensively reviewed elsewhere (15). In brief, the TRX system consists of cytosolic TRX1/TRXR1 and mitochondrial TRX2/TRXR2. Mammalian TRXs are homodimeric selenoenzymes containing an FAD and a penultimate COOH-terminal selenocysteine residue in their Gly-Cys-SeCys-Gly active site. Cytosolic TRX1 plays important roles as electron donors to enzymes forming disulfide bonds and exert redox control of transcription factors such as NF-κB. Mitochondrial TRX2 are essential for embryonic development and actively respiring cells. The GRX system consists of the glutathione redox couple (GSH/GSSG), glutathione reductase, and GRX. Humans have three types of GRXs; cytosolic GRX1 is an electron donor for enzymes forming disulfide bonds similar to cytosolic TRX1; mitochondrial GRX catalyzes the reversible glutathionylation of mitochondrial complex I. This modification of two critical thiol groups regulates the production of O2 ·− by the complex, and knockout of yeast mitochondrial GRX5 (yGRX5) led to constitutive oxidative damage (15, 276). The TRX/GRX enzyme systems are especially important in protecting against ischemia/reperfusion injury after myocardial infarction, atherosclerotic plaque rupture, left ventricular hypertrophy in congestive heart failure, and kidney oxidative stress. In SHRs and SHRSP rats, TRX levels were lower in kidneys by both immunohistochemistry/Western blots and real time PCR, when compared with those in normal WKY rats (319). Of note, whereas the roles of TRX/GRX systems are better understood in the cardiovascular disease, their role in the kidney is just emerging.
NO is another biomolecule with antioxidant functions, and decreased NO is associated with increased oxidative stress and hypertension (3, 23, 111, 118, 167). NO bioavailability has been shown to be decreased in advanced atherosclerotic lesions in humans attributed to lower expression of eNOS (361). A lack of substrate or cofactors for eNOS can lead to decreased bioavailability of NO (257). In addition, loss of endothelial pertussis toxin–sensitive G-protein function in atherosclerotic porcine coronary arteries showed that alterations of cellular signaling led to eNOS not being appropriately activated (300). Furthermore, accelerated NO degradation by ROS occurs because of kinetics favoring this reaction over scavenging of ROS by SOD (111). Overexpression of NO via delivery of neuronal NOS (nNOS) results in improvement in oxidative stress and hypertension through improved parasympathetic nerve transmission (116). However, NO is a complex molecule, and uncoupling of eNOS due to reduced levels of tetrahydrobiopterin (BH4) can lead to increased generation of O2 ·−; the interaction of NO and O2 ·− can lead to production of other oxidants including peroxynitrite (ONOO−). NO has important roles in the kidney, including regulation of medullary perfusion and pressure natriuresis, tubuloglomerular feedback, tubular sodium transport, and modulation of renal sympathetic nerves (224, 358). These roles of NO are discussed in further detail in section IVC.
E. Role of ROS in physiologic processes
Chronic inflammation and ROS have been implicated in the progression of insulin resistance, cardiovascular and kidney disease, and age-related diseases (Fig. 4). ROS serve a dual role not only in enabling immune cells to kill invading pathogens but also essential mediators of inflammatory signaling. This latter role is not limited to inflammation, but it is now known that these species serve as essential mediators of a wide array of signaling mechanisms, such as cell-cycle progression, growth, and proliferation, as well as insulin signaling and others. In addition to the contributions from metabolic oxidases such as NAD(P)H oxidase and xanthine oxidase, ROS are produced as by-products of mitochondrial aerobic metabolism, with ∼2% of oxygen (O2) being converted to O2 ·− at any given time. Excess ROS, redox imbalance, and oxidative stress are thought to be the main cause underlying the aging processes and chronic diseases, as their electrophilic character allows them to oxidize cell constituents such as proteins, lipids, and DNA. The paradoxic nature of ROS can be resolved by understanding that only excessive production of these radicals results in damage, whereas their roles as mediators of cell signaling are temporally and spatially controlled. As an example, ROS are known to oxidize/reduce cysteine residues within proteins, a mechanism particularly active with MAPK, protein tyrosine phosphatases (PTP), protein tyrosine kinase (PTK), transcription factors, and even other enzymes being redox regulated via their cysteine residues. This redox regulation allows the cell to activate/inhibit signaling proteins and hence dynamically to change gene expression according to external stimuli. NF-κB is a redox-regulated transcription factor involved in the activation of inflammatory signaling, and its constitutive activation may underlie its role as a long-term inflammatory mediator in the pathogenesis of age-related diseases.
ROS are involved in routine cell functions including (a) host defense, (b) cellular signaling, (c) gene expression, (d) cellular death and senescence, (e) regulation of cell growth, (f) oxygen sensing, (g) biosynthesis and protein crosslinking, (h) regulation of cellular redox potential, (i) reduction of metal ions, (j) regulation of matrix metalloproteinases, (k) angiogenesis, and (l) cross-talk with the nitric oxide system (11).
III. Pathologic Role of ROS in Hypertension
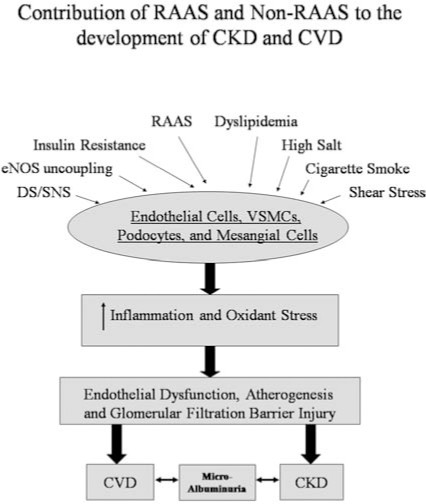
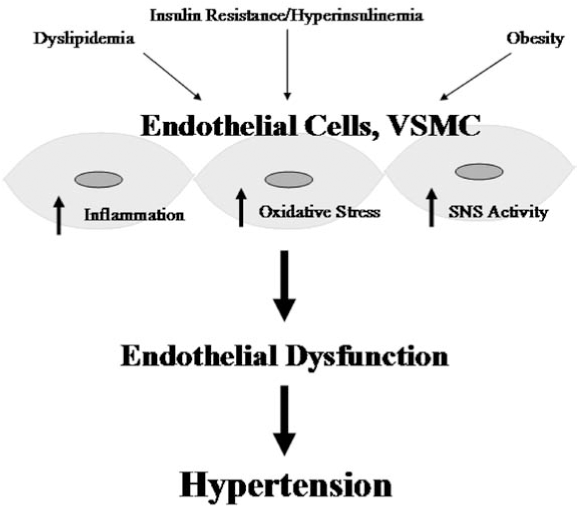
In general, increased production of ROS can lead to the development of insulin resistance, hypertension, dyslipidemia, cardiovascular disease (ischemic heart injury, congestive heart failure), acute and chronic kidney disease (precipitating factors for these include ischemic renal injury, hypertension, nephritis, obstructive nephropathy, glomerular damage, and rhabdomyolysis), obesity, cancers, and shock (sepsis, etc.) (Fig. 3). ROS can cause hypertension, as demonstrated by several lines of evidence (359). Rats administered lead in drinking water can generate O2 ·− and ·OH in blood vessels, which ultimately results in hypertension. Scavenging of ROS with vitamin E or ·OH scavenger dimethylthiourea prevented hypertension in these rats. It was further shown that development of oxidative stress can precede hypertension in the SHRs. Our laboratory also observed this phenomenon of oxidative stress preceding hypertension in Ren2 transgenic rats (unpublished observations). Interestingly, deletion of ecSOD results in oxidative stress and hypertension, whereas gene transfer to SHRs ameliorates hypertension. We and others also observed that tempol fails to “treat hypertension” (61, 358), although it has multiple beneficial effects, including reduction of oxidative stress, improvement in insulin sensitivity, reduction of myocardial remodeling, etc. This illustrates the complex relation between oxidative stress and hypertension and requires further clarification through studies. ROS can be generated by RAAS and non-RAAS–mediated mechanisms, and this section focuses on extrarenal ROS in the control of systemic and renal hypertension (Figs. 2, 3, and 5).
A. Non–RAAS-mediated oxidative stress in hypertension
1. High intravascular pressure
Elevated blood pressure, or hypertension, is characterized by increased hydrostatic pressure within the kidney arterial/arteriolar and intraglomerular systems. High pressure induces O2 ·− production in isolated arteries via protein kinase C (PKC)-dependent activation of NAD(P)H oxidase. In this experimental model system, reduction of high intraluminal pressure (Pi) with SOD, DPI, apocynin, and protein kinase C inhibitors (chelerythrine or staurosporin) or the removal of calcium during high-Pi treatment prevented the increases in O2 ·− production, whereas inhibition of the RAAS had no effect. The authors concluded that high Pi itself elicits arterial O2 ·− production, most likely by PKC-dependent activation of NAD(P)H oxidase, thus providing a potential explanation for the presence of oxidative stress and endothelial dysfunction in various forms of hypertension and the vasculo-protective effect of antihypertensive agents with different mechanisms of action (332). However, in subsequent in vivo studies, the same authors showed a partial effect of Ang II inhibitors and proposed that in vivo, a local RAAS-mediated control of redox homeostasis may exist. In a separate group of experiments in an aortic-coarctation animal model, hypertension per se was postulated to be enough to cause upregulation of NAD(P)H oxidase subunits above the coarctation (340).
2. Shear stress
As described in the previous sections, mechanical forces, comprising both unidirectional laminar and oscillatory shear, have been postulated to lead to alterations in the redox state in the kidney, resulting in differential expression of genes (247). Vascular studies suggest laminar shear stress to be beneficial to intimal health by upregulating eNOS and NO production, whereas oscillatory shear results in increased production of ROS via NAD(P)H oxidase and XO (49, 212). Increased ROS production then leads to a familiar cascade of events, leading to inflammation, growth, fibrosis, and scarring in the intimal portion of the vasculature, which ultimately contributes to kidney disease and hypertension.
Oscillatory shear stress, present at sites where atherosclerosis develops, appears to be a potent stimulus of O2 ·− production (110). Atherosclerotic lesions are found opposite vascular flow dividers at sites of low shear stress and oscillatory flow (49, 306). Continuous oscillatory shear stress leads to a sustained activation of prooxidant processes resulting in redox-sensitive gene expression in human endothelial cells. Steady laminar shear stress initially activates these processes but appears to induce compensatory antioxidant defenses. It is speculated that differences in the endothelial redox state, orchestrated by different regimens of shear stress, may contribute to the focal nature of atherosclerosis.
Although the majority of the evidence for oscillatory shear stress stems from the vascular investigations, limited evidence does exist in the kidneys (70). Podocytes have been shown to be sensitive to fluid shear forces in vitro, and mechanical stretch has been proposed as a mechanism that can lead to glomerulosclerosis. Specifically, in the presence of hypertension, NO may work in the kidney by inhibiting both mesangial cell hypertrophy and hyperplasia (260). The efferent arteriole at the transition of the intraglomerular segment to the segment that passes through the extraglomerular mesangium has a conspicuously narrow portion with endothelial cells protruding into the vessel lumen. In addition, this segment is prominent for the expression of nNOS. Therefore, it has been proposed that this segment acts as a specific shear-stress receptor (58, 59, 123).
Collectively, these data suggest that elevated blood pressure and increased hydrostatic forces within the kidney contribute to excess ROS, redox imbalance, and oxidative stress mediated by several factors, including oscillatory shear and NO. Abrogation of high blood pressure is through a common Ca2+/PKC/p47 phox -mediated pathway in the kidney.
3. Lipids
This class of molecules is important physiologically for nutrition, energy, membrane integrity, cellular signaling, binding, and transport of important minerals and vitamins. However, excessive amounts of lipids or modification of these lipids (i.e., dyslipidemia) directly contribute to altered redox homeostasis, oxidative stress, and hypertension (105, 241). Epidemiologic studies, such as the Helsinki Heart Study and Physician Health Study, demonstrate that higher levels of the LDL/HDL ratio (>4.4) and higher cholesterol levels are associated with rapidly deteriorating kidney function and hypertension, respectively (288). Further, high levels of cholesterol and LDL-C (e.g., ox-LDL) were predictors of risk for development of CKD in the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) study (21).
Animal models for dyslipidemia-induced renal disease include genetic models (zucker obese and SHRs), diet-induced model (rat, guinea pig, and rabbit), and secondary hyperlipidemia model (DSS rat, mass-reduction kidney model). Rats fed a high-fat diet develop obesity, insulin resistance, and hypertension via an increase in NAD(P)H oxidase activity in the kidney, as seen in other animal models (273), and this contributes to endothelial dysfunction via oxidant degradation of NO (112).
In addition, impaired redox homeostasis can result in the formation of ox-LDL, which further potentiates renal injury. A proatherosclerotic, vicious cycle of NAD(P)H oxidase–dependent ROS formation with an augmented generation and uptake of ox-LDL, which then causes further potentiation of oxidative stress by ox-LDL itself, has been proposed (277). ox-LDL promotes glomerulosclerosis, similar to the progression of atherosclerosis: both are characterized by the presence of LDL and ox-LDL; infiltration of monocytes/macrophages; and overexpression of adhesion molecules (e.g., monocyte chemotactic proteins, growth factors, and cytokines) within the lesions. Both LDL receptors, which bind native LDL, and scavenger receptors (SR-AI), receptors for ox-LDL and acetylated LDL, are expressed in glomerular epithelial and mesangial cells. LDL stimulates DNA synthesis and cell proliferation, whereas ox-LDL is cytotoxic and induces apoptosis. In other experiments, the expression of ox-LDL receptor LOX-1, was increased in experimental hypertensive glomerulosclerosis (227). DSS rats, when salt loaded with 0.8% salt, showed evidence of impaired renal function and histologic glomerulosclerotic changes, along with markedly elevated levels of LOX-1. These deleterious effects were ameliorated when these animals were treated with a calcium channel blocker.
Clinically, hyperlipidemia is an independent risk factor for cardiovascular diseases [JNC VII, (36)]. Substantial evidence from both human and animal studies now indicates that reduction of cholesterol levels has beneficial effects on blood-pressure regulation and kidney function. HMG-CoA reductase inhibitors (statins) have been used to reduce cholesterol. Statins have also been proposed to have pleiotropic effects, actions independent of their cholesterol-lowering mechanism. Evidence for non–HMG-CoA effects now includes inhibition of NF-κB, a nuclear transcription factor, decrease in geranylation and translocation of Rac1 to the membrane, and even transcriptional regulation of NAD(P)H oxidase subunits, including Nox4, p22 phox , and Nox2 in the kidney (71, 353).
4. Eicosanoids
In addition to LDL and ox-LDL, small lipids such as eicosanoids are implicated in the pathogenesis of glomerular injury and hypertension. COX-derived prostanoids are integral in preserving renal function, vascular fluid homeostasis, and blood pressure. Kidney cortical COX2-derived prostanoids such as PGI2 and PGE2 preserve blood pressure and renal function in the volume-contracted state, whereas medulla-derived prostanoids appear to have an antihypertensive effect in individuals challenged with a high salt diet.
5-LOX-derived leukotrienes are involved in inflammatory glomerular injury. LOX product 12-hydroxyeicosatetraenoic acid (12-HETE) is associated with pathogenesis of hypertension and may mediate Ang II- and transforming growth factor (TGF-β)–induced mesangial cell abnormalities observed in diabetic kidney disease. P450 hydroxylase–derived 20-HETE is a potent vasoconstrictor and is involved in the pathogenesis of hypertension. P450 epoxygenase–derived epoxyeicosatrienoic acids (EETs) have both vasodilator and natriuretic effects. Blockade of EET formation is associated with salt-sensitive hypertension. Ceramide has also been demonstrated to be an important signaling molecule, which is involved in pathogenesis of acute kidney injury caused by ischemia/reperfusion and toxic insults. 8-Isoprostaglandin F2α (8-ISO) is formed nonenzymatically from the attack of the O2 ·− radical on arachidonic acid and is considered a marker of kidney oxidative stress in vivo (293). These pathways provide compelling targets for pharmacologic intervention in renal redox control of hypertension.
5. High salt
Humans have evolved to function in a low-salt environment. The high-salt content of modern diets contributes to hypertension, and reduction in dietary sodium intake to <2.4 g/day has been shown to reduce the severity of hypertension by 2–8 mm Hg, on average [JNC-VII (36)]. Recent data suggest a more nontraditional role for salt in promoting hypertension. Dobrian et al. (55, 228) demonstrated that salt loading leads to hypertension via NAD(P)H-dependent mechanisms that can also exacerbate renal injury and proteinuria in obese spontaneously hypertensive rats. Salt loading has also been shown to increase oxidative stress and blood pressure when the animals are treated for longer intervals and to induce p47phox and gp91phox in rat kidney cortices (201). Proposed mechanisms include paradoxic mineralocorticoid receptor (MR) activation of NAD(P)H oxidase and oxidative stress. Another study in DSS rats demonstrated that MR blockade reduced salt-induced hypertension by increasing endothelium-derived relaxing factors, inhibiting RAAS components, and decreasing oxidative stress (9).
6. Cigarette smoke
Smoking is an independent risk factor for hypertension, cardiovascular disease, and chronic kidney disease (350). In a Texas cohort with hypertension but no evidence of end-stage renal disease (ESRD), smoking was independently associated with worsening of GFR after patients were monitored prospectively (264). Furthermore, in patients with diabetic nephropathy (DN) undergoing therapy with an ACE inhibitor for blood pressure control, smokers had worse plasma creatinine levels after 7 years of follow-up (37). Smoking has been shown to induce oxidative stress (330) and also has been found to be a strong predictor for the extent of endothelial injury in patients with hypertensive kidney injury (350). Subjects who smoke heavily and for a long time, as well as passive smokers, seem to be particularly exposed to endothelial damage.
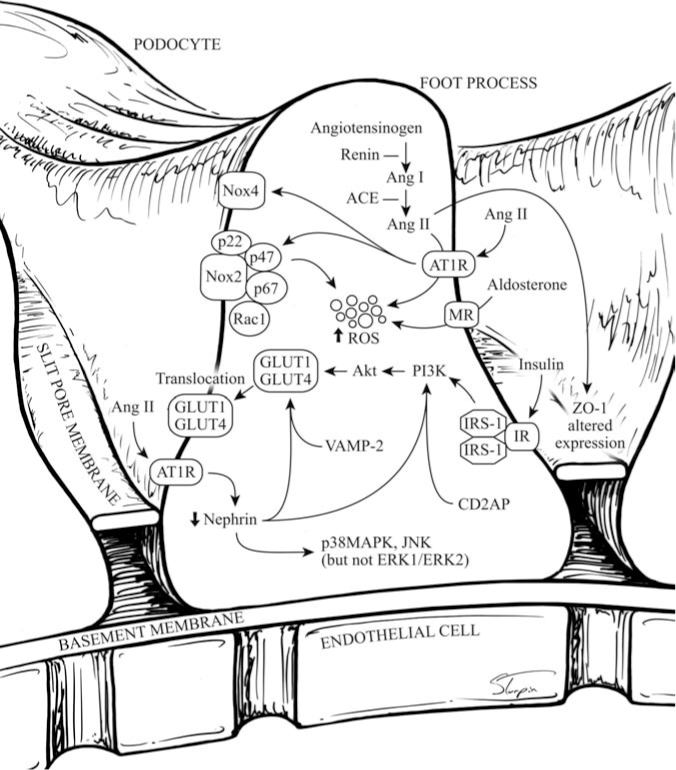
7. Insulin resistance/hyperinsulinemia
Insulin resistance is a prominent feature of the cardiometabolic syndrome (CMS) and is the precursor to type 2 diabetes and links between obesity, hypertension, and renal disease (Figs. 4 and 5). The metabolic dysregulation observed in the development of insulin-resistance/hyperinsulinemia is proposed to result from oxidative stress: disruption of phosphatidyl inositol 3–kinase (PI3-kinase) and Akt signaling through serine/threonine phosphorylation, reduced and impaired glucose transporter 4 (GLUT4) function (286). Insulin resistance has been shown to induce oxidative stress via generation of excessive superoxide anion/H2O2 and decreased catalase synthesis. This is part of a feed-forward mechanism that results in chronic conditions of oxidative stress. Oxidative stress then leads to modulation of vascular endothelial function, smooth muscle contractility, and organ function (Fig. 5). As described in detail elsewhere, the acute sodium-retaining action of insulin at the level of the kidney is one of the unifying mechanisms that connect insulin resistance with renal redox control of hypertension. Indeed, this mechanism appears to play a role in the development of salt sensitivity. Recent data support that oxidative stress can also be involved in this association, as ROS influence sodium handling in the kidney. In particular, increased free radical generation is suggested to promote primary salt retention. One of the mechanisms possibly involved in this action is the modulation of renal cortical and medullary microcirculation. In isolated renal afferent arterioles, O2 ·− can lead to vasoconstriction, inhibited by a mimetic of SOD, the enzyme responsible for O2 ·− degradation. Furthermore, infusion of an SOD inhibitor in the medullary interstitium resulted in vasoconstriction accompanied with decreases in urine flow and sodium excretion, whereas infusion of an SOD mimetic opposed these alterations.
8. eNOS uncoupling
eNOS is responsible for the production of NO, a potent vasodilator with an important role in preserving endothelial function. Superoxide-producing enzymes, such as NAD(P)H oxidase and xanthine oxidase, promote NO degradation as they generate a variety of ROS. For example, in hypercholesterolemic rabbits with impaired endothelial relaxation, NO bioavailability was decreased because of oxidation of NO into vasoinactive nitrates/nitrites, and treatment with SOD reversed endothelial dysfunction (225). Other animal models also demonstrate increased degradation of NO as the cause of endothelial dysfunction and hypertension (192). eNOS is a cytochrome p450 reductase–like enzyme that catalyzes flavin-mediated electron transport from the electron donor NAD(P)H to a prosthetic heme group. eNOS uncoupling is a phenomenon that results when deficiency of
9. Dopaminergic system (DS)/sympathetic nervous system
Dopamine is produced locally in the proximal tubule cells and, after binding to D1 and D5 receptors, inhibits sodium absorption, thereby promoting natriuresis (381). Downstream signaling is mediated through G-protein subunits that are activated by G protein–coupled receptor kinase type 4 (GRK-4). Activating variants of GRK-4, along with abnormal dopamine production, leads to uncoupling of the receptors from their G-protein effector complexes. Both people with essential hypertension and animal models, including DSS rats and SHRs, have been shown to harbor these variants. Overexpression of GRK-4 in mice contributes to hypertension, probably by allowing the unopposed action of Ang II, whereas GRK-4 blockade ameliorates hypertension in rat genetic models. The dopaminergic system has been extensively reviewed by Zeng et al. (381), and readers are advised to refer to their article for further details.
An extensive body of literature links the sympathetic nervous system (SNS) and hypertension (2, 191, 205, 279, 285). NO has been shown to play a central role in offsetting oxidative stress/SNS–mediated increase in arterial pressures (118). The NO-cGMP pathway is known to suppress cardiac norepinephrine (NE) release, and oxidative stress can upset this protective relation. A recent study demonstrated that restoring NO levels via adenovirus-mediated nNOS in SHRs improved cardiac sympathetic neurotransmission and ameliorated hypertension (179). In another recent study in the mesenteric arterial bed from SHRs, NE levels were shown to be increased, whereas neuropeptide Y (NPY) levels were reduced (196). N-Acetylcysteine (NAC) reversed these levels and restored NO modulation of SNS activity, demonstrating the importance of antioxidants in maintenance of redox states and normal vascular function. In the DOCA salt-sensitive rats, NAD(P)H oxidase subunits are differentially regulated in the sympathetic (celiac ganglia) and sensory (dorsal root ganglia) nervous systems. The authors postulate that this may lead to vasomotor imbalances and vasoconstriction in the splanchnic beds (24).
It has been shown that modulation of the SNS through various mechanisms contributes to the control of hypertension. In salt-loaded DSS rats, administration of intracere-broventricular tempol (SOD-mimetic) and DPI reversed increased systemic arterial pressure, SNS activity, and heart rate (73). Furthermore, inhibition of Rac1-derived ROS in nucleus tractus solitarius decreased blood pressure and heart rate in stroke-prone SHRs (236). The antioxidant adrenomedullin was shown to inhibit SNS-induced hypertension in salt-loaded mice (74). Overexpression of inducible NO synthase in the rostral ventrolateral medulla causes hypertension and sympathoexcitation via an increase in oxidative stress (161). Increased ROS in the rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone SHRs (163). ET-1 was shown to be important in O2 ·− production in the sympathetic neurons of DOCA-salt hypertensive rats, and this was mediated by upregulation of ET (B) receptors (46). Renal sympathetic nerves respond to tempol by ameliorating elevation of high blood pressures in SHRs by decreasing oxidative stress (304). Longterm antioxidant treatment improves sympathetic functions and the β-adrenergic pathway in the SHRs (80). Collectively, these experiments emphasize the role of the SNS in mediating hypertension via dysregulation of redox balances in the neurons and the vasculature.
B. Role of the RAAS in oxidative stress and hypertension
The contribution of the RAAS to redox homeostasis has generated extraordinary interest since the discovery of renin >100 years ago. Initially, renin was discovered in kidney extracts by Tigersted and Bergman in 1898, and its role later was explained by Harry Goldblatt, in 1934 (8). It was not until 1939 that hypertensin was discovered by Braun-Menendez, and later in 1958, it was renamed angiotensin by Braun-Menendez and Page (19, 20). Subsequent experiments, including those done by Sir Arthur Guyton, elucidated the physiologic implications of the RAAS and highlighted the importance of the kidney in blood pressure control (97 –104, 151). Several decades of hard work ushered us into the 1980s, when Ang II was established as a potent vasoconstrictor that, when inhibited, would lead to improvements in hypertension. Ang II has other important biologic functions, including growth and neovascularization, regulation of glomerular filtration, and tubular transport.
The role of Ang II in the development of kidneys is supported by evidence from deletion of the angiotensinogen gene in mice (Agt−/−). The consequent defective kidney organogenesis is lethal within 10 days of birth (53, 308). Angiotensinogen is expressed mainly in the kidney tubules of mice by embryonic day 17, and its expression declines shortly after birth. Similarly, mice with targeted mutations in ACE, AT1R genes, and in both renin genes, have nearly identical phenotypes characterized by poor survival to weaning, low blood pressure, and abnormal kidney structure. Their kidneys are thickened with hypercellular arterial walls, interstitial fibrosis, inflammation, papillary atrophy, and tubular dilation. Angiotensinogen-overexpression models (transgenic mice expressing the rat angiotensinogen gene alone in the liver and brain, TGM[rAOGEN]123) and (transgenic mice expressing the human angiotensinogen gene alone and treated with human renin bolus, or double transgenic mice expressing both human renin and human angiotensinogen), manifest hypertension (159, 375, 376). Over-expression of angiotensinogen and elevated levels of Ang II can lead to kidney disease by causing growth, inflammation, and fibrosis.
Ang II classically exerts its effects through AT1R and AT2R binding. AT1R binding has been shown to lead to many of the deleterious effects and vasoconstrictor actions of Ang II. Alternatively, AT2R binding leads to some beneficial effects, including reduction of blood pressure. In addition, many other angiotensin receptor–binding effects of Ang II exist, and it is now widely recognized to have not only endocrine effects but also exocrine, paracrine, and autocrine effects. Ligand-receptor binding also leads to G protein versus non–G protein–mediated effects. G protein–mediated effects may be mediated through phospholipase C with formation of 1,4,5-inositol and DAG, whereas non–G protein–mediated effects are through stimulation of tyrosine kinases. Both of these pathways eventually contribute to activation of components of NAD(P)H oxidase/other metabolic oxidases and generation of O2 ·− and other free radicals.
Some effects of Ang II are direct; however, a number of profibrotic effects are mediated through stimulation of aldosterone. In addition, aldosterone itself has certain actions that are independent of Ang II stimulation. For example, aldosterone acts through AT1R to mediate vascular oxidative stress by increasing mRNA levels of reduced nicotinamide adenine dinucleotide phosphate oxidase components; this effect was abolished by selective MR antagonism, whereas AT1R blockade and tempol decreased only the p47 phox mRNA level but not that of p22 phox or gp91 phox (122). In the same study, MR antagonism uniformly abrogated aldosterone-mediated stimulation of proinflammatory genes, whereas AT1R blockade and tempol had gene-specific effects. The authors concluded that both Ang II–dependent and Ang II–independent pathways are involved in the mechanisms leading to the development of hypertension, vascular inflammation, and oxidative stress induced by aldosterone.
Last, renin itself has been proposed to have proinflammatory effects through binding to the renin receptor on target tissues. Direct inhibition of renin with aliskerin has been shown to have beneficial effects on blood pressure, inflammation, fibrosis, and end-organ damage (155, 243, 256, 333). A recent study,
1. Ang II, ROS, and systemic hypertension
During the late 1980s and early 1990s, evidence for Ang II–mediated ROS production and its role in hypertension began to emerge. Landmark studies demonstrated that O2 ·− production underlies the pathogenesis of hypertension, and this was attributed primarily to Ang II, whereas other catecholamines failed to stimulate production of ROS (176, 233). Others showed that treatment of rats with Ang II infusions led to increases in ROS production and elevated blood pressure, which was effectively reduced by treatment with SOD, catalase, glutathione peroxidase, and dimethyl sulfoxide (DMSO) (363). A direct link was then established between Ang II–impaired vasomotor tone resulting in hypertension and increasing vascular O2 ·− production via membrane NAD(P)H oxidase activation (261).
2. Ang II stimulation of NAD(P)H oxidase and hypertension
Vascular and renal NAD(P)H oxidase is a multicomponent enzyme complex implicated in the pathogenesis of hypertension. In addition, NAD(P)H oxidase requires Rac1/Rac2 and Rap1 for activation in some cellular systems. These activating small-molecular-weight G proteins have been demonstrated in some studies to affect O2 ·− production and hypertension (115, 240, 355). The vascular and renal NAD(P)H oxidases share several characteristics with the multicomponent enzyme complex, as described in neutrophils in an earlier section (6). The active enzyme complex is composed of one of the Noxs plus or minus p22 phox . p47 phox , p67 phox , and p40 phox , or one of their isoforms, completes the subunit architecture. However, organizational and functional differences are found between nonphagocytic and phagocytic NAD(P)H oxidase, as illustrated in Table 2, including the perinuclear location of all of the subunits. This facilitates rapid recruitment of subunits to generate ROS for signaling purposes.
In the vasculature, the endothelial as well as adventitial NAD(P)H oxidase is composed of gp91 phox (Nox2) and p22 phox , as well as p47 phox and p67 phox and the G protein Rac1 (88, 245). It is now established that NAD(P)H oxidase expressed in endothelial cells is an important source of free oxygen radical generation in the arterial wall (147). In contrast, smooth muscle cells either lack Nox2 or have several orders of magnitude lower expression of this subunit when compared with Nox2 homologues such as Nox1 and Nox4 (174). In vitro studies in vascular smooth muscle cells (VSMCs) support Ang II–stimulated Nox1 expression in a protein kinase C (PKC)-dependent fashion (245). Use of PKC inhibitor GF109203X efficiently inhibited PKC activity, decreased Nox1 basal expression, and abrogated Ang II-induced upregulation of Nox1 expression. Anti-sense Nox1 mRNA completely inhibited Ang II-induced O2 ·− production, supporting a role for Nox1 in redox signaling in vascular smooth muscle cells.
NAD(P)H oxidase, as well as other metabolic oxidases such as xanthine oxidase and mitochondrial respiratory chain enzymes (I–IV), are functional in the kidney (278). However, we and others have identified NAD(P)H oxidase–induced ROS production as a primary initiator of oxidative stress within the kidney (183, 353, 355, 356). Various cells in the kidney have now been shown to possess a fully functional NAD(P)H oxidase system, which produces O2 ·− free radicals under stimulation by cues such as Ang II, agonists that bind to D1-like receptors, and to H+ fluxes (28, 89, 146, 184, 259, 353, 377, 384) (Figs. 6 through 9). Nox4 is expressed at high levels in kidney and other Noxs, including Nox1, Nox2, and Nox-regulatory subunits, are expressed at lower but quantitatively significant levels (4, 78, 146, 214, 244, 259, 303) making Nox enzymes attractive candidates for the origin of renal ROS, including the relatively high levels of H2O2 seen in urine (172). No consensus appears to exist for the corticomedullary differences between the different subunits, as the literature has data from different animals undergoing different treatments. Indeed, a few studies demonstrated no or very little expression of the regulatory subunits in the medulla. However, one report shows that, in SHRs, the expression of Nox1, Nox2, and p67 phox is higher in the renal medulla as compared with the cortex, and the expression of Nox4 and p22 phox is higher in the cortex than in the medulla (291). Per this group, the expression of p47 is about the same in the cortex versus medulla. The mesangial cells are unique in that they express Nox1, Nox4, and p22 phox , p47 phox , and p67 phox , but not Nox2 (86, 127, 146, 164, 214, 223, 371, 380) (Fig. 6). This may be biologically important, as some data support the fact that mesangial cells may be similar to VSMCs, which also lack Nox2 (expressed at very low levels) (Stockand et al.). In mesangial cells, NO inhibits the expression of Nox1, suggesting cross-talk between NO and O2 ·−-generating systems (244). p22 phox , p47 phox , and p67 phox are all expressed in the PCT, DCT, CCD, and the macula densa cells (12, 72, 107, 139, 148, 342, 366) (Fig. 6). Nox2 expression has been shown in the PCT and CCD; Nox4, in the PCT and DCT; whereas Nox1 expression is confined to the PCT, once again demonstrating differential regulation of redox control within the cortex. In situ hybridization experiments initially localized Nox4 mRNA expression to the renal cortex, whereas the medulla showed much lower expression (244, 303). However, immunohistochemical studies also demonstrate Nox4 expression in distal portions of the human nephron, and Nox4 mRNA has been demonstrated in medullary collecting ducts (244, 303). Moreover, as mentioned earlier, the data for Nox4 were obtained from different animal groups. The Nox4 isoform of NAD(P)H oxidase is unique in that it also can use NADH as the substrate for generating O2 ·− (173). This may be relevant to the kidneys, as the cortex and outer medulla preferentially use NADH as a substrate and express Nox4 at high levels in these regions (387).
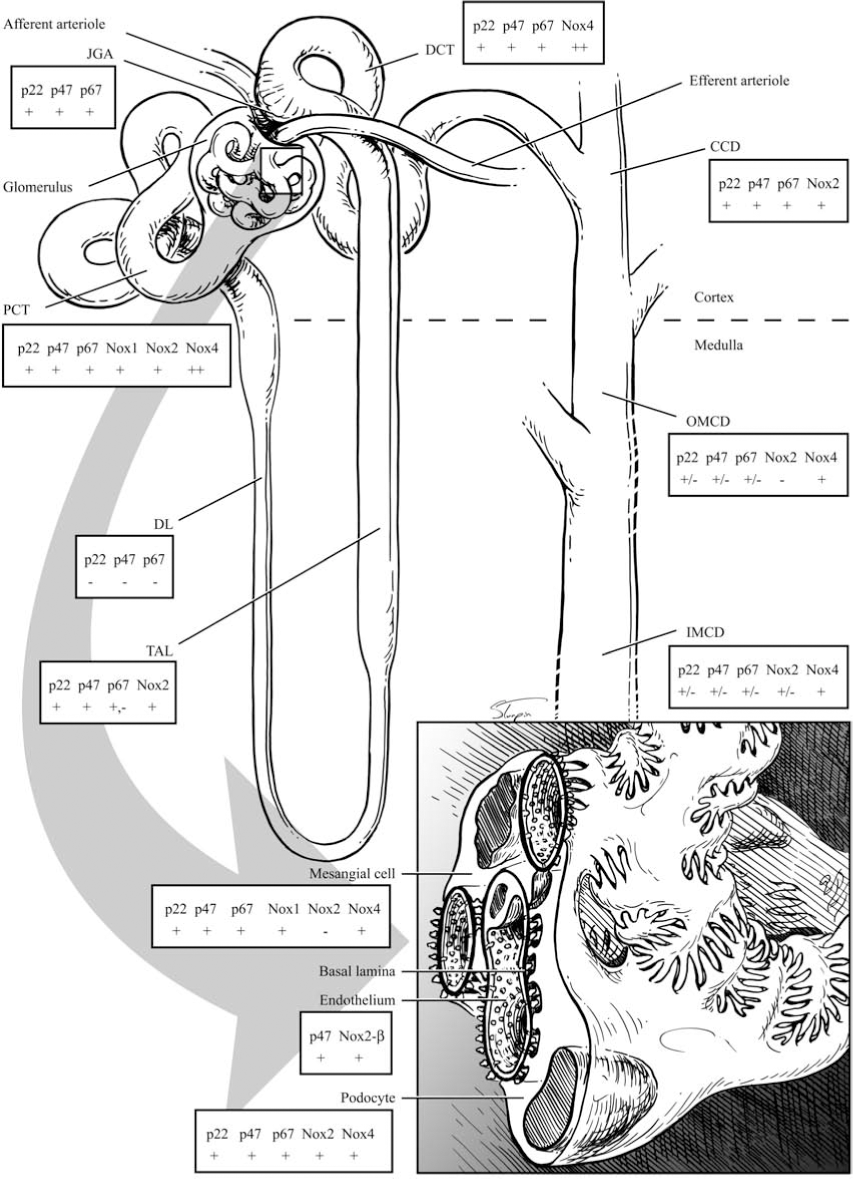
The main physiologic function of Nox4 is largely unknown. Nox4 may serve varying roles, depending on the tissue where it is expressed and its function as an ROS producer. The expression pattern of Nox4 in the kidney is consistent with several renal-specific functions. For example, by virtue of close proximity to EPO expression, Nox4 might regulate EPO production in the kidney. The major site for EPO synthesis in adults is generally believed to be the peritubular fibroblast-like cells, although EPO receptors have been demonstrated in mesangial cells, PTCs, and the glomerulus (208, 238, 297). As mentioned before, EPO synthesis is ramped up in response to hypoxia (with proline hydroxylases as main sensors) and anemia. Interestingly, recent observations suggest that Nox4 may regulate EPO synthesis through HIF-dependent pathways [hypoxia-inducible factor-2α (HIF-2α)] and HIF-independent pathways (H2O2-sensitive target GATA-2) (131; 202). Finally, SOD3, which is expressed in PTC, has been proposed to play a role in EPO production (314).
Another putative functional role in host defense has been proposed in the kidney for Nox4, by virtue of expression in the inner medulla, which is known to be a hypoxic environment (244). ROS produced by Nox4 could potentially have microbicidal effects on the urine, and this is supported by evidence of high H2O2 in the urine (338).
Most important, Nox4 is integral in the development of the renal complications of insulin resistance and hypertension. Increased expression of Nox4 and p22 phox and accumulation of 8-hydroydeoxyguanosine (8-OHdG), marker for ROS-induced DNA damage, in the kidney of streptozotocin-induced diabetic rats was reversed after treatment with insulin for 2 weeks (63). Another group working with the same animal model showed that infusing antisense oligonucleotides for 2 weeks, targeting Nox4 mRNA, was effective in reducing ROS in cortical and glomerular homogenates, organelle hypertrophy, and fibronectin expression, which are characteristic markers of diabetic kidney disease. One putative pathway proposed to increase Nox4 expression has been the Rho/Rho-kinase pathway, and this was inhibited by administration of fasudil (Rho-kinase inhibitor) and statins (81). Angiotensin-(1-7) [Ang-(1-7)] attenuated Nox4 expression, decreased NAD(P)H oxidase activity, and improved proteinuria in streptozotocin-treated SHRs (diabetic SHRs) (14). These data suggest a signaling role for Nox4-derived ROS in the development of diabetic kidney disease (84). We now discuss the role of each subunit as it relates to Ang II–mediated hypertension.
a. p22phox and hypertension
One of the first subunits to be implicated in redox control of hypertension was p22 phox . This 22-kDa protein was initially thought to be confined to the plasma membranes of cells; however, data now show that p22 phox , along with Nox2, may localize to the perinuclear area (181). Expression of p22 phox mRNA was reported to increase in Ang II–infused hypertensive rats (75). Impaired endothelium-dependent vascular relaxation observed in this animal model was reversed in part by increasing vascular SOD levels, suggesting a crucial role for vascular (O2 ·−) in endothelial dysfunction. Additionally, a polymorphism of p22 phox , which alters an amino acid in the potential heme-binding sites, has been demonstrated to be more frequent in control subjects compared with patients with coronary artery disease (135). Overexpression of p22 phox , selectively in smooth muscle of transgenic (Tg) mice [Tg(p22smc)], potentiated Ang II–induced hypertrophy of the aorta and led to hypertension (346). Interestingly, mice not stimulated with Ang II did not manifest either increases in aortic-wall thickness or blood pressure, emphasizing the importance of the stimulated enzyme in hypertension when compared with the role of constitutive enzyme in normal processes, such as cellular signaling. Further, overexpression of p22phox led to chronic oxidative stress caused by excessive H2O2 production (175). This evoked a compensatory response involving increased eNOS expression and NO production. NO in turn increased eSOD protein expression and counterbalanced increased ROS production, leading to the maintenance of normal vascular function and hemodynamics. So far, p22 phox -specific inhibitors are not available; however, siRNA induced knockdown of p22phox gene expression in SD rats, led to decrease in renal NAD(P)H oxidase activity, expression of Nox proteins and oxidative stress, and a slight decrease in blood pressure during an Ang II slow-pressor response (217). With the recent identification of a naturally existing p22 phox knockout mouse (nmf333 mouse strain), the contribution of this subunit to redox regulation of blood pressure can be studied (232). Collectively, these reports support the hypothesis that the alteration of p22 phox expression might modulate ROS levels in the vasculature and influence the progression of vascular diseases (203).
b. gp91phox (Nox2) and hypertension
gp91 phox (Nox2) was among the first to be discovered to cause a phagocytic burst to kill engulfed bacteria. The expression of nonphagocytic gp91 phox , as described earlier, is confined to the endothelial cells and adventitia of the blood vessels; surprisingly, it is not detected in the VSMCs of large arteries; however, its isoform Nox2 is expressed in the VSMCs (173, 174, 312). In contrast, gp91 phox expression was knocked down by antisense oligos in resistance arteries from humans, and this led to abrogation of ROS production, whereas Nox1 expression was not detected here (327). The role of gp91 phox (Nox2) in hypertension was described by Morawietz et al. (219), who showed that tenfold-elevated mRNA levels in the aorta were associated with threefold elevated levels of O2 ·− anion production in stroke-prone SHRs (SHR15) (219). In SHR15 rats, a decreased response to ACh, NO-donor (S-nitroso-N-acetyl-d, l-penicillamine), and organic nitrate (glyceryl trinitrate) was found, compared with an age-matched wild-type control, Wistar rats (WIS15). NO stimulates soluble guanylate cyclase (sGC) and sGC β(1)-subunit proteins (heterodimers), which are involved in the formation of cGMP, which is an important vasodilator. Expression of guanylate cyclase enzyme complex was downregulated in both aorta and lungs of SHR15. This article was among the first to suggest that hypertension is a manifestation of an imbalance between excess vascular ROS and an impaired NO signal-transduction pathway. In another model of hypertension, Ang II–infused SD rats, gp91 phox expression was increased threefold at both mRNA and protein levels when the infusion was continued for 1 week. They also showed that NAD(P)H oxidase–induced superoxide production may trigger NOS III uncoupling, leading to impaired NO/cGMP signaling and to endothelial dysfunction in this animal model. However, as noted earlier, gp91 phox has been documented to be absent in VSMCs from large arteries but present in resistance arteries in humans.
c. p47phox and hypertension
The identification of p47 phox in addition to p67 phox and p22 phox in the vasculature, macula densa, distal convoluted tubule, cortical collecting duct, and outer and inner medullary collecting ducts provides further evidence for redox-mediated gene expression in these cells (18). The kidney of SHRs expresses genes for all the main components of phagocyte NAD(P)H oxidase and gp91 phox homologues Nox4 and Nox1. Expression of p47 phox was higher in the kidney cortices of SHRs when compared with age-matched and normotensive Wistar Kyoto rats (WKYs) (28). Furthermore, the prominent increase of p47 phox mRNA and protein expression observed in 4-week-old SHRs in the vasculature, macula densa, and distal nephron demonstrates that oxidative stress precedes development of hypertension. Mechanical stretch of wild-type VSMCs resulted in ROS formation and p47 phox translocation to the plasma membrane followed by an increase in Nox1 transcripts (92). In addition, Ang II–stimulated p47 phox translocation through c-Src phosphorylation and enhanced activity of NAD(P)H oxidase in human VSMCs (328). Interestingly, it has been proposed that p47 phox deficiency may paradoxically lead to activation of the RAAS through ROS-independent mechanisms, and that p47 phox may not be a major player under non–agonist-stimulated conditions (93). However, it should be borne in mind that the majority of studies in animal models of increased oxidative stress and hypertension have shown an increase in p47 phox expression and translocation to the membrane. So far, overexpression studies with p47 phox have not been published per our review of the literature, and it would be interesting to see the effects on oxidative stress, hypertension, and end-organ damage.
In genetically hypertensive rats, p47 phox protein and NAD(P)H oxidase activity was increased in the media of endothelium-denuded arteries, and it was shown that this resulted in the maintenance of spontaneous aortic tone in the absence of vasoconstrictors. Importantly, the increase in aortic tone was mitigated by antioxidants specific to the NAD(P)H oxidase pathway (193). When fibroblasts from hypertensive and normotensive individuals were compared for Ang II–induced stimulated expression of extracellular signal-regulated kinase (ERK1/2), expression of p47 phox , NAD(P)H oxidase activity, and extracellular O2 ·− was found to be higher in hypertensive subjects (246). Further, treatment of phenylephrine-contracted aortic rings with Ang II resulted in decreased relaxation in response to ACh and increased O2 − by dihydroethidium (DHE) staining. Treatment of these aortic rings with quercetin/isorhamnitin or SOD/apocynin decreased p47 phox expression and endothelial dysfunction (282).
d. p67phox and hypertension
The role of p67 phox in oxidative stress and hypertension was described by Pagano et al. (245) in their landmark article by establishing a link between Ang II stimulation and enhanced generation of ROS in the aortic adventitial fibroblasts. Importantly, increases in p67phox gene transcription may be responsible for mediating the increase in NAD(P)H oxidase activity as the increase in mRNA levels for this subunit preceded the peak in NAD(P)H oxidase activity (38).
The role for p67 phox in hypertension was best illustrated recently with nebivolol, a β-receptor blocker with both vasodilator and antioxidant properties. Ang II–infused male Wistar rats were treated with either nebivolol or metoprolol, and translocation of p67 phox and Rac1 was assessed in isolated heart membranes (240). Whereas metoprolol had no effect, nebivolol inhibited Rac1 and p67 phox association at the membranes, decreased NAD(P)H oxidase activity and superoxide anion production, and prevented NOS III uncoupling. The authors attributed the beneficial effects of nebivolol on Ang II–induced endothelial dysfunction to its ability to modulate NAD(P)H oxidase activity.
e. p40phox and hypertension
Relatively scant data are available on the role of p40 phox in hypertension. Its expression has been demonstrated in the aorta and renal cortex (136, 345). In DSS rats, increased expression of p40 phox in the kidney cortices, along with p47 phox and gp91 phox , was associated with decreased renal function and increased blood pressure. Adrenomedullin, a multifunctional vasodilator peptide, has been shown to have renoprotective effects. Combined treatment with adrenomedullin and omapatrilat of DSS rats resulted in abrogation of an increase in expression of p40 phox when compared with that in nontreated DSS rats (137).
IV. Kidney Redox Function and Hypertension
A. ROS in normal kidney physiology
ROS are produced at constitutive levels in nonphagocytic cells (e.g., glomerular cells and tubular epithelial cells) for preservation of routine cellular physiology. However, derangements in their production can lead to loss of redox homeostasis and oxidative stress in the kidney. Ultimately, the balance between ROS and reactive nitrogen species (RNS) is required to preserve antiapoptotic mechanisms (284).
Formation of ROS is evident in many areas of the kidney, predominantly in the renal cortices, whereas the medulla can be susceptible to hypoxia and less ROS production under physiologic conditions (11, 386) (Fig. 6). This is important to note in regard to the normal function of the medulla, including sodium and fluid homeostasis by inhibiting tubular ion-transport activity. Hypoxia-inducible factor-1 (HIF-1) is perhaps the most strategically important transcription factor involved in proper functioning of the medulla under hypoxic conditions. HIF-1 consists of two subunits, HIF-1α and HIF1-Iβ, the former being the O2-sensitive part of the complex. It is involved in the transcription of many O2-sensitive genes, including HO, erythropoietin (EPO), vascular endothelial growth factor (VEGF), glucose transporters, glycolytic enzymes, inducible NO synthase (iNOS), and transferrin. ROS have been shown to alter adenosine production, NO bioavailability, arachidonic acid metabolism, and Na/K/2Cl cotransporter activity in the short term, thereby regulating TGF and renal blood flow (RBF). Ultimately, this leads to control of renal cortical and medullary oxygenation.
In addition, ROS may have long-term effects on renal blood flow and oxygenation through the modulation of HIF-1α gene expression. Increased ROS, such as in the TALH, may lead to degradation of HIF-1α through prolyl-4-hydroxylase–mediated hydroxylation. Degradation of HIF-1α will lead to decrease in transcription of hypoxia-sensitive genes and lead to perturbation of medullary function, ultimately resulting in sodium and fluid retention. ROS-stimulated increases in response to Ang II, aldosterone, and chemokines, can be classified into various categories:
Alteration of cell fate: (a) augmentation of epithelial-mesenchymal transition through activation of MAPK pathways; (b) induction of mesangial cell apoptosis; (c) promotion of cellular hypertrophy through activation of ERK1/ERK2 and through p27Kip1-dependent cell-cycle arrest;
Regulation of renal blood flow: ROS interact with NO to cause limitation of its relaxation effect on afferent arterioles. On the basis of studies involving Nox2-deficient mice and apocynin, Nox enzymes are a likely source of ROS involved in regulation of renal blood flow.
Regulation of gene expression: Nox-dependent oxidative activation of transcription factors. NF-κB/c-jun leads to increased expression of renal target genes [monocyte chemoattractant protein-1 (MCP-1), transforming growth factor-β1 (TGF-β1), phospholipase A2 (PLA2), and COX-2].
B. RAAS in the kidney
Most of the circulating/local factors implicated in vascular ROS production are also important players in contributing to kidney ROS production. Non–RAAS-mediated factors important in ROS production include hyperinsulinemia, AGEs, mechanical shear stress, cigarette smoke, hypertension, lipids, and high salt. Their role in kidney redox modulation was described earlier. Ultimately, many of these pathways were recently shown to influence the RAAS.
In this section, we focus on RAAS-mediated mechanisms for ROS production in the kidney, which are currently the subject of intense research. Many reasons exist for this interest. However, the important ones include existence of identifiable human disease from overexpression/mutations/polymorphisms in the renin, angiotensinogen, ACE, AT1R, aldosterone, and MR genes (Table 3). This is exemplified in a Dutch cohort with insulin-dependent diabetes mellitus (IDDM)-induced incipient diabetic kidney disease (336). Within this population, the T-allele of the AGT-M235T polymorphism is associated with increases in proteinuria, and the CC-genotype of the AT1-A1166C polymorphism is associated with hypertension. In addition, mutations in other components of RAAS pathways are associated with hypertension. Several animal models with genetic contributions to hypertension exist, including SHRs; DSS rats; Ren2 transgenic rat [TG(mRen2)27, harbors extra copies of mouse renin gene on Sprague–Dawley background]; PAC140/160 X hAGT mice (PAC140/160 containing the human renin gene and >100 kb of genomic flanking regions, bred with hAGT-containing multiple copies of human angiotensinogen gene at its native locus); and double transgenic rats [(dTR), 17.6 kb of human genomic DNA containing the renin gene TGR(hREN) bred with 16.3 kb of human genomic DNA containing the angiotensinogen gene TGR(hAOGEN)] (213, 307). Elegant studies in these and other animal models have contributed extensively to our knowledge of the RAAS and its tissue affects including the kidneys. Furthermore, the ability to manipulate both primary and immortal cell lines easily has given rise to an explosion of data on RAAS-mediated signaling in monolayers and, in turn, its application to the milieu in vivo. Last, the discovery of compounds to suppress the RAAS at each step of the pathway has yielded opportunities to dissect the contribution of each component toward redox control and hypertension.
M235T, methionine-to-threonine substitution; G1051A, guanine to adenine; ACE I/D, insertion/deletion polymorphism; A1116C, adenine to cytosine; CYP11B, gene for 11-β hydroxylase enzyme; T344C, thymine to cytosine; S810L, serine to leucine; Y618H, tyrosine to histidine; C825T, cytosine to thymine; R65L, arginine to lysine; A142V/A486V, alanine to valine.
1. RAAS expression in developing and adult kidneys
Among varying roles of Ang II, including sodium homeostasis and regulation of aldosterone synthesis, the involvement in renal organogenesis is compelling (252). Angiotensinogen mRNA, among other components of the RAAS, was detected by days 30 to 35 of gestation in the mesonephros of human embryos, whereas AT1R and AT2R were detected by day 24 (294). In the human metanephros, ACE was expressed in proximal tubule cells/cortical collecting ducts (PTC/CCD), angiotensinogen in PTC at 8 weeks, AT1R in glomeruli and PTC, and AT2R in the undifferentiated mesenchyme (252). It is known the RAAS is upregulated during renal development and in the perinatal period (365); the AT1R is expressed at high levels postpartum, whereas AT2R expression is higher in the kidney prepartum. Evidence now suggests that Ang II stimulates growth of tubular cells in vitro and induces synthesis of collagen IV, mediated through the AT1R (367). Complete knockouts of AT1R results in lack of development of the renal pelvis and ureteral peristaltic movement (216); the phenotype is mimicked by wild-type mice with one ureter ligated (obstructive uropathy). Interestingly, gene targeting of angiotensinogen/ACE produces a more severe phenotype when compared with AT-receptor targeting. The reason could very well be the effects on non–AT-receptor–mediated pathways, such as aldosterone-mediated MR activation, or it could even be AT2 receptor–mediated signaling (364). As a corollary, consumption of ACE inhibitors during pregnancy has been shown to result in kidney malformation, mainly through defective angiogenesis/tubulogenesis. Thus, Ang II and the RAAS contribute to normal growth and development of the nephron through an interplay of AT1R- and AT2R-mediated effects and has additional roles in cell-cycle progression (369).
Components of the RAAS expressed in the adult kidney include renin, angiotensinogen, ACE, aldosterone-sensitive epithelial sodium channels (ENaC), and the AT1R and AT2R (134, 188, 364, 365). Renin is traditionally thought to be expressed in the juxtaglomerular apparatus (JGA) and secreted into blood vessels for its endocrine, exocrine, and paracrine functions. However, it has been shown that ACE inhibition increases the staining of renin by recruiting more cells farther along the afferent arterioles by the JGA (82). Further, recent data also identified renin expression along other parts of the nephron, such as the proximal tubules and collecting ducts (165). Angiotensinogen expression was thought to originate from the liver; however, evidence now exists for strong angiotensinogen expression in kidney cortices, proximal tubules, distal convoluted tubules, and renal endosomes (132, 134). It is also expressed weakly in the glomerular endothelial cells (165). ACE was thought to originate in the pulmonary epithelium; however, ACE also is expressed abundantly on the brush-border membrane of the PTC, in addition to renal endosomes (132). Furthermore, ACE activity has been detected throughout the nephron by others (25).
Aldosterone in the kidney has traditionally been thought to be active only in the collecting ducts. However, amiloride-sensitive ENaC is expressed in renal distal convoluted tubules (DCT), connecting tubules (CNT), cortical collecting ducts (CCD), and outer medullary collecting ducts (OMCD), but not in the inner medullary collecting ducts (IMCD), consistent with tighter regulation of sodium transport in the cortex and outer medulla (57). AT1R is expressed most prominently in the interlobular arteries and tubulointerstitial fibrous regions surrounding interlobular arteries and glomeruli, followed in decreasing order by glomeruli and cortical tubules (296, 329). Among the tubular cells, the proximal tubule brush-border and basolateral membranes (204, 295, 329), distal tubules, and cortical and medullary collecting ducts exhibited specific immunoreactivity. Glomerular staining for AT1R was observed in mesangial cells and podocytes. Macula densa cells stained positively as well (113). AT2R expression has been confined to the interlobular arteries (204).
2. RAAS-mediated redox mechanisms
The RAAS plays a pivotal role in regulating physiologic and pathophysiologic processes in the kidney. Although different components of the RAAS, such as renin, aldosterone, and various Ang fragments, can initiate renal function impairment on their own, Ang II is the primary effector of this system. Sufficient and convincing evidence supports Ang II as a key contributor to progression of kidney disease by stimulating growth, hypertrophy, oxidative stress, inflammation, and fibrosis within the kidney (Fig. 3). Many of the same downstream pathways that are effective in the vasculature, as described earlier, have been identified in the kidney (e.g., PKC/Ca2+ signaling, IP3/DAG signaling, MAPK/TGF-β1 signaling, IGF-1/IRS-1 signaling, AT1R/G-protein signaling, and NADPH oxidase/ROS signaling) (Fig. 2).
Most of the known physiologic and pathophysiologic effects of Ang II are transduced via the AT1R, a 359-amino acid (aa) protein that belongs to the seven-membrane superfamily of G protein–associated receptors (43). A minority of the effects of Ang II effects are also transduced through AT2R a 363-aa protein whose gene was mapped to the X chromosome (166). After the binding of Ang II to AT1R, a series of signaling cascades is activated. Although traditionally divided into G protein– and non–G protein–related signaling, various interactions between these subgroups of Ang II–induced signaling pathways make a strict distinction difficult. An example of a G protein–dependent pathway is activation of phospholipase C (PLC) with the subsequent production of inositol 1,4,5-phosphate (IP3) and diacylglycerol (DAG). IP3 then stimulates Ca2+-mediated activation of PKC, which ultimately leads to activation of NAD(P)H oxidase and ROS generation. Non–G protein–dependent pathways induced by Ang II are phosphorylated and activate various tyrosine kinases. An example of this is activation of epidermal growth factor receptor (EGFR), which then leads to activation of transcription factor c-jun, which could in turn induce ROS formation through NAD(P)H oxidase.
Sufficient evidence supports the notion that Ang II promotes ROS formation via NAD(P)H oxidase activation by direct stimulatory effects (stimulation of the AT1R); studies on inhibition of the RAAS have contributed to this evidence as well (27, 141, 343, 348, 349, 355). Renal afferent arterioles, when infused with Ang II at a slow pressor rate, exhibit renal cortical NAD(P)H-stimulated O2 ·− generation and lipid peroxidation in addition to impaired ACh-induced endothelium-dependent relaxation and enhanced contractile responses to Ang II. The arterioles have a profound downregulation of the mRNA for AT1R but an upregulation of the p22 phox component of NAD(P)H oxidase (343). In addition, Ang II infusion in rats led to increased expression of NAD(P)H oxidase subunits, an effect abrogated with blockade of the AT1R. Moreover, administration of AT2R inhibitor PD-123,319 resulted in worsening of Ang II–mediated oxidative stress, as AT2R is known to mediate the beneficial effects of Ang II stimulation (27).
The Ren2 transgenic rat [(mRen2)27] is a unique model for investigating the influence of Ang II and the RAAS on redox control of the kidney. Elevated tissue RAAS has been shown to increase oxidative stress in these animals, contributing to insulin resistance, hypertension, and proteinuria. Existing work in the heart, aorta, and kidney established Ang II as a key mediator of tissue inflammation, oxidative stress, and remodeling (75, 174, 218, 261, 287, 334, 346, 347, 351 –355). Work from our laboratory and others indicated a role for Ang II production of ROS in the kidney by stimulating NAD(P)H oxidase and subunits, including p47 phox , p67 phox , and Rac1 (355). ROS, in turn, can contribute to stimulation of proinflammatory and profibrotic pathways in the vasculature and kidney.
Although the role of circulating RAAS cannot be minimized, renal RAAS activation has been demonstrated through various paradigms to contribute to the development and progression of hypertension (43, 52, 94, 97, 99, 101, 151, 165). Importantly, renal AT1R activation has been shown to influence systemic hypertension compared with systemic contributions of the AT1R. Investigators used a kidney cross-transplantation technique to compare four groups of mice: a wild-type group containing transplanted kidneys from wild-type donors expressing AT1A receptor (AT1A in mouse is most homologous to AT1 in humans) in the kidney transplant and in all systemic tissues; a systemic AT1AR KO group transplanted with kidney expressing AT1A; a kidney AT1AR KO group expressing AT1A in all tissues except for kidney; and a total KO group with no expression of AT1A (43). Blood-pressure measurements between the groups suggested that systemic and kidney AT1A made equal contributions to baseline blood pressure. However, when mice were stimulated with Ang II, the systemic KO group had pressures similar to those of the wild-type group, suggesting that hypertension was mediated primarily through kidney AT1AR and not through systemic AT1AR. Additional deleterious effects of kidney AT1A activation include decreased sodium excretion, weight gain, cardiac hypertrophy, and atrial natriuretic peptide and brain natriuretic peptide stimulation, further demonstrating the important role of kidney AT1A in cardiovascular pathology.
The glomerulus is the primary part of the nephron involved in various functions, including blood pressure regulation, filtration of nutrients and waste products and sodium/water homeostasis. Increases in pressure (PGC) within the glomerular capillary network leads to several effects including increases in blood pressure and damage to the filtration barrier leading to proteinuria. Damages to the filtration barrier have been demonstrated in the transgenic rat TG(mRen2)27 (Ren2) at the podocyte foot process/slit membrane (355). The Ren2 rat overexpresses the mouse renin transgene and demonstrates elevated tissue Ang II, including a local renal tissue RAAS. In this model, we and others have observed increases in local components of the RAAS with subsequent increases in NAD(P)H oxidase and ROS formation. A significant filtration barrier, as well as proximal tubule remodeling, suggests a direct link between activation of the RAAS, NAD(P)H oxidase, ROS, and renal remodeling. Further, we have observed that blockade of the AT1R or administration of SOD mimetic, tempol, decreased the levels of NAD(P)H oxidase activity, subunit expression, and markers of oxidative stress (350, 353, 356).
3. Methods for detecting ROS in the laboratory and clinic
Several markers for detecting ROS have been described, and they are important read-outs for measuring Ang II–mediated effects on NAD(P)H oxidase. Some of these have been developed to measure kidney-mediated excretion of ROS-modified compounds. For example, O2 ·− interacts with esterified or free arachidonate to yield a family of isoprostanes that includes 8-ISO. The steady-state excretion of 8-ISO reflects oxidative stress, and subpressor doses of Ang II are known to increase excretion of 8-ISO (274). LPO breakdown products participate in the formation of adducts with proteins, resulting in the formation of malondialdehyde (MDA) and 4-hydroxynonenal-lysine (4-HNE). Levels of these compounds can also easily be measured to quantify the levels of endothelial dysfunction and oxidative stress. Other methods include 3-nitrotyrosine and carbonylation by using immunoassays. Medtronik has a machine called Oxi-med that measures oxidative stress levels in capillary blood. It uses only 20 μl of blood and can give a readout in 60 seconds. However, it cannot distinguish one oxidative stress from another (i.e., circulating vs. tissue-generated free radicals). Blood oxygen level-dependent MRI (BOLD-MRI) is a technique in which deoxyhemoglobin is used as an endogenous contrast agent for the noninvasive assessment of tissue-oxygen bioavailability. This technique has been used to assess the correlation to oxidative stress markers including H2O2, hsp27, F2-isoprostanes, total NO, and total antioxidant property (TAOP).
C. Nephron handling of ROS and hypertension: redox control of renal function
Each kidney is made up of 1 to 2 million nephrons, which are the functional units of the kidney filtration system (Fig. 6). The kidney is further organized into the outer cortex, which has most of the glomerular corpuscles and convoluted parts of the tubules. The medulla consists of the straight parts of the tubules and the major portion of collecting ducts and is relatively hypoxic when compared with the cortex. Although nephrons constitute <0.5% of the body mass, they garner 25% of the body's blood supply. This allows the efficient delivery of water and nutrients to the nephrons for filtration of waste and absorption of essential components. However, this also affords circulating biologic effectors (i.e., Ang II, AGEs, chemokines, growth factors, immune complexes, and toxins) greater access to the kidney structures, especially the nephrons. The nephrons also regulate salt and fluid homeostasis through different segmental receptors and effectors. This tight control of whole-body salt and fluid homeostasis is partly exerted through differential pressures in the afferent and efferent arterioles. The RAAS system is uniquely positioned to play a major role in kidney function as described. This last decade has seen an immense amount of interest concerning RAAS-mediated signaling in the nephron and its supporting cellular mechanisms. Central to the signaling pathways is the ubiquitous O2 ·− generated by NAD(P)H oxidase, as described earlier. Although the nephron is a single continuous structural unit, it is functionally divided into several highly specialized units. It is only natural that functional specialization be achieved through differential expression of genes and activation of unique pathways. This is exemplified by selective expression of NAD(P)H oxidase subunits in different segments of the nephron (Fig. 6). Furthermore, this allows the various nephron segments to be regulated by redox mechanisms that are specific to that cell type. For example, renal cortical blood flow is regulated mainly by O2 ·− generated by NAD(P)H oxidase, whereas the outer medullary blood flow (MBF) is regulated by O2 ·− from both NAD(P)H oxidase and mitochondrial enzymes. SOD inhibition with diethyldithiocarbamate markedly decreased renal MBF and sodium excretion, whereas tempol produced the opposite effect (387). Last, each nephron segment responds uniquely to hypertension.
The mechanisms whereby ROS formation results in morphologic lesions and in modifications of glomerular permeability, blood flow, and filtration rate have been inferred from in vitro studies. They involve direct and indirect injury to resident cells (mesangiolysis) and glomerular basement membrane (in concert with metalloproteases) and alteration of both the release and binding of vasoactive substances, such as bioactive lipids (e.g., prostaglandin E2, prostacyclin, thromboxane), cytokines (e.g., tumor necrosis factor α), and possibly endothelium-derived relaxing factor (NO). The importance of such processes appears to be modulated by the intrinsic antioxidant defenses of the glomeruli. Further, the kidney is particularly rich in polyunsaturated fatty acids, which constitute an abundant source of ROS.
The SOD mimetic, tempol, has been used extensively to demonstrate the beneficial effects of ROS inactivation and modulation of systemic blood pressure (358). In the SHR, tempol decreased both BP and renal vascular resistance (RVR) through NO-mediated mechanisms, whereas short-term infusions had, in addition, NO-independent effects, likely through inhibition of renal sympathetic nerve activity (373). Reductions in RVR and BP lead to increased diuresis and natriuresis, even when simultaneous NOS blockade is instituted (199). Simultaneous treatment of mice with tempol and Ang II for 2 weeks results in decreased excretion of 8-ISO, prevention of hypertension, and increase in RVR (153). Either direct vasoconstriction of afferent arteriole or indirect enhancement of TGF by ROS is blocked by tempol. H2O2 has biphasic actions on renal afferent arterioles, an initial vasoconstricting action followed later by a vasodilatory action (292). High-dose Ang II treatment of rats for 2 weeks leads to salt sensitivity and an increase in blood pressure in the succeeding weeks via a persistent inflammatory cell infiltrate in the renal cortex that leads to oxidative stress (194).
1. Redox control of kidney function
a. Tubuloglomerular feedback and role of ROS in macula densa
Tubuloglomerular feedback is classically thought to be mediated by NaCl concentrations in the distal tubule and is detected through the furosemide-sensitive Na/K/2Cl transporter (13). The macula densa cells are uniquely positioned to detect the changes in NaCl concentrations, which then direct the afferent and efferent arteriole to either decrease or increase GFR through changes in arteriolar resistance. Both O2 ·− and NO are generated in the macula densa cells and afferent arterioles (358). All of the components of NAD(P)H oxidase, including p22 phox , Nox2, p47 phox , and p67 phox are expressed in the macula densa cells (28). The NOS type 1 or nNOS expression is limited to the macula densa cells, whereas NOS type 2 or eNOS is expressed in the afferent arterioles (358). Although the roles of superoxide and NO in mediating macula densa and afferent arteriolar redox function have been extensively described, the efferent arteriolar oxidative stress has also been investigated (268). NO from nNOS in macula densa, decreases TGF in response to increased NaCl concentrations (140, 269, 360). Use of the NOS-specific inhibitor 7-nitroindazole (7-NI) confirms that this isoform that mediates TGF in rats. This ensures that, in the face of increased total body sodium and volume expansion, sodium is removed from the body efficiently. Moreover, increased renal vascular resistance and systemic blood pressure may result in lesser delivery of sodium to the nephrons, resulting in sustained volume expansion and elevated pressures. NO can dilate the afferent arteriole, decreasing RVR and improving GFR to maintain sodium excretion at high levels (358). nNOS inhibitor L-NAME can increase TGF in high-salt conditions but does not affect TGF in low-salt conditions. O2 ·− can increase the TGF response by quenching NO to form ONOO− anions (267, 358). NO decreases NaCl absorption in TAL by inactivating the luminal Na/K/2Cl cotransporter and the Na/H exchanger through generation of cGMP. A higher concentration of NO also inhibits basolateral Na/K-ATPase. Therefore, one mechanism by which O2 ·−can mediate increased afferent arteriole tone is by inactivating NO in the JGA (267). Ang II, products of AA pathway, and adenosine can increase TGF by downregulating nNOS in the macula densa cells (268). TGF is blunted by ACE inhibitors and absent in AT1A receptor–deficient mice. SOD can decrease TGF by inhibition of superoxide anion, which may have both direct potentiation effects on TGF and indirect effects through formation of ONOO− anions.
b. Medullary perfusion and renal hemodynamics
NO has a central role in the regulation of renal blood flow via its modulation of renal vascular resistance, which is known to be lower than that in most other organs (198). Administration of NOS inhibitors nitro-
c. Pressure natriuresis
NO mediates the pressure-natriuresis response that is so central to the salt- and water-balance function of the kidney. Increase in intravascular volume leads to increase in blood pressure, resulting in increased delivery of sodium to the distal tubules. This results in TGF; however, NO dilates the afferent arterioles, leading to maintenance of natriuresis and reduction of sodium load and return of blood pressure to normal (200). Blockade of NO with inhibitors impaired pressure-natriuresis and led to a shift of the curve to the right and higher blood pressure (281). Mechanisms for NO-mediated pressure-natriuresis are poorly understood, although some suggest it may be through decreased tubular sodium reabsorption through an amiloride-sensitive transport process (197).
d. Tubular sodium transport
The overall effect of NO on the tubules is to decrease tubular sodium reabsorption. In the proximal tubules, it decreases apical Na/H exchange and Na/K-ATPase activity (187, 275). In the TAL, eNOS generates NO that inhibits sodium resorption (254). In the CCD, NO inhibits the amiloride-sensitive sodium channel (eNAC) and vasopressin-mediated osmotic pump activity through cGMP and cAMP (77, 310). Surprisingly, no studies have been published on NO in the distal tubule. Moreover, the direct role of NO in decreasing absorption of sodium is unclear, as many of the studies have been done in isolated single-nephron types of experiments and not in the intact kidney.
e. Renal sympathetic nerves
The renal sympathetic nerves are important for the maintenance of renal blood flow and sodium homeostasis. However, derangement in sympathetic nerve function can lead to increase in sodium retention and blood pressure. Interestingly, NO has bidirectional roles in mediating or modulating renal sympathetic nerve action. In the proximal tubules, it may mediate the sodium-retaining effects of sympathetic nerves (370), whereas it may blunt the neural effects in isolated perfused rat kidney and in the renal medulla (62, 266).
In the following section, we describe redox mechanisms as known in special cell types in the kidney. It should be noted that some of the pathways described are not directly related to hypertension but are included to elucidate redox mechanisms that are relevant to kidney function and pathophysiology.
2. Nephron components and their contribution to ROS and hypertension
a. ROS in the glomeruli/podocytes
The area of ROS-mediated damage to the glomerular-filtration barrier is still in its nascent stages. However, several compelling areas of work have emerged (Fig. 7). All of the components of RAS are expressed in the podocytes and are functional (188). Ang II is a critical determinant of glomerular function involving both pressure-mediated and pressure-independent effects that are insufficiently understood. To tease out the non-hemodynamic effects of RAAS signaling, the human AT1R (hAT1) gene that was driven by a nephrin promoter was used to make a transgenic rat (TGR) model, which overexpressed hAT1 at twice the endogenous levels in podocytes (124). In this model, male transgenic rats aged 8–15 weeks developed albuminuria, and this was independent of blood-pressure effects when compared with nontransgenic littermates. Roughly in the same period as the development of albuminuria, structural changes in the podocytes were seen, including pseudocyst formation, podocyte effacement, and detachment, leading to focal segmental glomerulosclerosis (FSGS). This elegant rat transgenic model showed the blood pressure-independent effects of RAAS signaling in podocyte pathology and glomerular filtration barrier injury when compared with the Ren2 rat that displays blood pressure-dependent effects (Fig. 10). The data in hAT1 rats correlates with our data in Ren2 rats and provides direct evidence that increased AT1R signaling in podocytes leads to protein leakage and structural podocyte damage and subsequent renal injury (124).
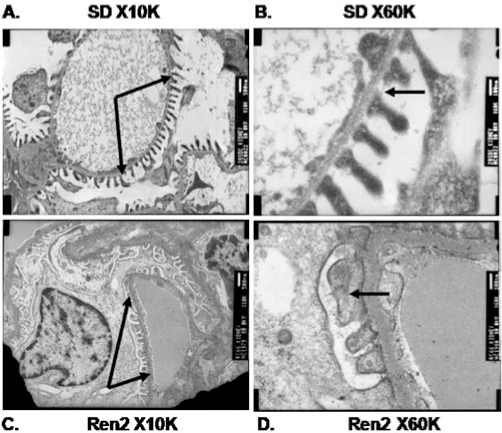
In addition to Ang II–mediated oxidative stress and filtration barrier injury, aldosterone has been implicated in the kidney oxidative stress, inflammation, fibrosis, and sclerosis. Shibata et al. (299) demonstrated that aldosterone infusions and high-salt diet, when administered to uninephrectomized SD rats, led to glomerulosclerosis, hypertension, and kidney damage. This pathophysiology was mediated through elevations in oxidative stress, as demonstrated by increased ROS production/NAD(P)H oxidase activity, increased NAD(P)H oxidase subunit protein levels, decreased nephrin and podocin expression, and increased aldosterone effector kinase (Sgk1) activity. Administration of MR blockade ameliorated the harmful effects of aldosterone administration. It should be noted, however, that levels of the other components of the RAAS were not measured, notably Ang II or the AT1R. This is important, as non–(MR)-mediated affects of aldosterone have been demonstrated by other authors (129, 258, 289). In their next work, these authors did use an MR blocker + AT1R blockade to demonstrate the additive effects in lowering blood pressure, oxidative stress, TGF-β levels, and kidney damage.
LPO by ROS is one of the mechanisms by which glomerular filtration barrier injury and proteinuria can be induced. In the passive Heymann nephritis model of human membranous nephropathy, subepithelial immune complex deposition and complement activation leads to adduct formation between LPO breakdown products and proteins [e.g., malondialdehyde (MDA) and 4-hydroxynonenal-lysine (234)]. MDA adducts localized to cytoplasmic vesicles and cell membranes of glomerular epithelial cells, to the GBM, and to the immune deposits, modify type IV collagen, and pretreatment with the antioxidant probucol reduced proteinuria. The authors concluded that LPO alters glomerular permselectivity by damaging cell membranes and modifying type IV collagen.
Although the kidneys are not a major storage site for glucose, insulin is still needed to transport glucose into the cells for local us in energy production. Recent work demonstrated that insulin initiated translocation of GLUT1/GLUT4 to the podocyte membrane, and this is mediated by nephrin, the slit-diaphragm protein with exclusivity of expression in the podocytes (40, 41). It also was shown that the COOH-terminal of nephrin binds to vesicular SNARE protein, vesicle-associated membrane protein 2 (VAMP-2), and facilitates translocation of GLUT proteins. Conditionally immortalized podocytes from nephrin-mutation-harboring humans and in vitro knockout of nephrin from podocytes both demonstrated decreased insulin response (40, 280). Most important, stable nephrin transfection restored insulin signaling in podocytes carrying a nephrin mutation. Another group showed that nephrin and CD2-associated protein (CD2AP) bind to PI3-K and initiate phosphorylation of Akt (128). Akt target proteins are 14-3-3 adaptor proteins and Bad proapoptotic protein; binding of latter to the former prevents apoptosis of podocytes. It is well known that Akt facilitates GLUT translocation to the cell membranes from work in endothelial cells and VSMCs; however, this has not been specifically demonstrated in podocytes. A substantial body of work exists on the Ang II disruption of Akt signaling via ROS generation in the cardiovascular system, leading to insulin resistance and hypertension. Although it is not known how ROS affects insulin signaling in the podocytes, the demonstration of this important signaling pathway warrants further investigation.
b. ROS and the glomerular basement membrane (GBM)
The GBM, which is composed primarily of type IV collagen, laminin, nidogen, and heparin sulfate proteoglycans, has traditionally been thought to be an important mediator of filtration-barrier integrity. The onus for the maintenance of GBM falls on glomerular endothelial cells and podocytes. Some forms of hereditary glomerulonephritis are characterized by defects in the primary components of the GBM, and these include Alport syndrome, familial benign hematuria (type IV collagen), and Pierson syndrome (laminin) (96). Others are a consequence of overdeposition, and these include the nail-patella syndrome (type III collagen, an interstitial collagen) and fibronectin nephropathy (fibronectin) (96). A body of work demonstrates the importance of GBM in modulation of protein loss in the urine of animal models; however, very limited data exist on the role of oxidative stress in mediating GBM injury.
In the Ren2 rat, which is characterized by hypertension, insulin resistance, oxidative stress, and proteinuria, the glomerular filtration barrier injury includes thinning of the GBM when compared with nontransgenic SD controls (355). ROS have been implicated in the production of glomerular damage in PHN, an experimental form of membranous nephropathy (MN) with proteinuria (156, 235). Functional activity of the glomerular ROS-generating system was demonstrated exclusively in proteinuric PHN, in which H2O2 was found in the highest concentration within the GBM (235). These findings provide evidence that in rats with PHN and proteinuria, the glomerular endothelial cells express and externalize Noxs that generate ROS, which could then lead to GBM damage. In the same animal model, complement-mediated attack on glomerular epithelial cells caused upregulation of NAD(P)H oxidase expression and secretion of ROS to the GBM (156). Formation of LPO adducts on type IV collagen leads to structural changes within the GBM and, importantly, potent LPO-antagonist probucol markedly reduced proteinuria. The authors postulated that accumulation of lipoproteins within subepithelial immune deposits and generation of oxidative stress was akin to atherosclerotic lesions and ox-LDL.
Ang II has been shown to inhibit heparin sulfate proteoglycan (HSPG) production and to stimulate TGF-β production in cultured human mesangial cells, and recent evidence suggests that ACE inhibition preserved HSPGs in the GBM of rats with established doxorubicin (Adriamycin)-induced nephropathy (335, 344). In vitro studies with podocytes suggest that Ang II is able to modulate the production of HSPG (22). Because HSPG plays a fundamental role in the permselectivity of the GBM, these results thus may explain at least partially the antiproteinuric effects of inhibitors of the RAAS in patients with diabetic kidney disease.
c. ROS and the mesangium
ROS are produced by the renal mesangium and also by the inflammatory bone marrow-derived cells invading the renal tissue. Regardless of their origin, ROS have been shown to degrade the GBM and alter the glomerular and tubular cell functions. The mesangium is the best-characterized location in the kidney in terms of ROS and its generation of oxidative stress (146, 259). These cells have been shown to respond to multiple cues in vivo, and subsequently, use of several of these as exogenous cues was made in vitro, and their interactions were fairly well described (Fig. 8). Prominent among these are Ang II, high glucose/AGEs, aldosterone, LDL, cytokines, growth factors, and NO. Ang II has been shown to activate ERK1/ERK2 and Akt/protein kinase B (PKB) through ROS, and this effect is mimicked by AA (84 –87). The ERK1/ERK2 pathway is important in the generation of fibronectin, and Akt is a known mediator of protein synthesis, a hallmark of mesangial cell hypertrophy. Use of phospholipase A2 (PLA2) inhibitors, mepacholine or aristocholic acid, abolished AA-mediated increases in ROS. Furthermore, PLA2 inhibitors also suppressed AA-mediated activation of ERK1/ERK2 and Akt. Increased ROS production was attributed to Nox4 and Rac1 in mesangial cells. Anti-sense to Nox4 abrogated ROS production, which in turn decreased both ERK1/ERK2 stimulation and Akt activation. Dominant-negative Rac1 also produced similar results demonstrating the importance of Ang II–mediated Rac1/NAD(P)H oxidase activation and ROS generation in modulation of mesangial cell function. Last, the same group of investigators showed that Akt is involved in the induction of fibronectin expression, and this was abrogated by NAC and anti-sense to p22 phox (16).
Transactivation of EGFR by Ang II via ROS and subsequent activation of c-Jun N-terminal kinase (JNK) led to mesangial cell proliferation (51). LDL has been shown to stimulate intracellular Ang II production that in turn leads to activation of AT1R and the ROS cascade, leading to mesangial cell hypertrophy (249). Aldosterone, another member of the RAAS pathway, has been implicated in the generation of oxidative stress via NAD(P)H oxidase–mediated ROS generation, and eplerenone completely abolished membrane translocation of p47 phox and p67 phox in rat mesangial cells (214). AGEs bind to RAGE and activate PKC-α, which in turn activates NAD(P)H oxidase. PKC-α inhibition with RO-32-0432 reverses NAD(P)H activation (324).
High glucose (e.g., hyperglycemia) as occurs in diabetes, stimulates NAD(P)H oxidase-mediated ROS generation, which is inhibited by DPI and apocynin and mitochondrial electron-transfer chain complex 1 inhibitor (rotenone) in mesangial cells (178). High glucose also acts through PKC to modulate collagen I/IV, and antisense against p22 phox reversed an increase in collagen mRNA expression, whereas antisense to p22 phox /p47 phox and tempol abrogated ROS generation and collagen IV protein expression (371). Furthermore, PKC-ë mediates high glucose–induced stimulation of NAD(P)H oxidase and F-actin disassembly; this is reversed with antisense RNAi to p22 phox and p47 phox (171). Last, PKC-ξ and PKC-β1 mediate high glucose–induced VEGF expression, and this increase was reversed with tempol, PKC-ξ, and PKC-β1 inhibition (372).
Cytokines are important modulators of mesangial cell function. 5-Hydroxytryptamine (5-HT), through its receptor 5-HT2A, induced MAPK/MEK via ROS production, which in turn leads to stimulation of TGF-β (90). Interleukin-1 (IL-1), TNF-α, and lipopolysaccharide (LPS) induced COX-2 expression, a generator of proinflammatory prostanoids, and blockade of NAD(P)H oxidase abrogated TNF-α-mediated effects (67). Homocysteine was shown to stimulate NAD(P)H-dependent ROS production through Rac1, and this was mediated by vav2, a guanine nucleotide exchange factor (GEF). Si-RNA to Rac1 and vav2 were shown to attenuate ROS production (379).
Last, antioxidants and inhibitors of ROS production have been studied in mesangial cells. Integrin α1-null mice demonstrate increased ROS production via amplified Rac1 expression and translocation to the membrane and increased deposition of collagen. Importantly, integrin α1β1 was shown to downregulate EGFR via inhibition of Rac1 (32). Insulin treatment of streptozotocin-induced diabetic rats ameliorated NAD(P)H oxidase–induced ROS production and diabetic nephropathy (63). NO was shown to downregulate Nox1-mediated ROS production in mesangial cells via c-GMP signaling, demonstrating yet again the cross-talk between pro- and antioxidant pathways (255).
d. ROS and the tubule
Proximal tubule origin of ROS formation and downstream signaling pathways is an area of active research, and many investigators believe that tubulointerstitial processes are the major contributors to hypertensive and diabetic kidney disease (Fig. 9). All of the components of RAS are functional in the PTC, and this was demonstrated in SV40 transformed rat primary culture (317). In regard to the PTC, Ang II similarly generates ROS through NAD(P)H oxidase, which is abrogated by AT1R blockade, DPI, and tiron (108). Ang II was shown to increase the expression of p27Kip1, an inhibitor of G1-phase cyclin/cyclin-dependent kinase complexes via ROS-dependent mechanisms, which led to tubular hypertrophy. Elevated intrarenal Ang II leads to proteinuria, stimulation of cytokines, and growth factors, which ultimately results in tubular hypertrophy and scarring, as evidenced in cross sections of tubules from transgenic Ren2 rats (unpublished observations, our laboratory; Fig. 11). TUNEL staining for apoptosis of tubular epithelial cells showed marked increase of this process in the Ren2 rats when compared with the normal SD rats, and apoptosis was ameliorated by treatment of rats with AT1R blockade (unpublished observations, our laboratory; Fig. 12). Ang II–induced hypertrophy of cultured murine proximal tubular cells was shown to be mediated by endogenous TGF-β (368). Ang II also stimulates VEGF expression and PTC modulation, both via transcriptional and translational mechanisms (65, 272). The translational increase in VEGF expression is brief (detected at 5 min and lasts 45 min) and is mediated by activation of PI3-kinase and its downstream target Akt. Transcriptional control of VEGF expression is more sustained and has been shown to occur through AT1R and AT2R in glomerular epithelial cells. Finally, Ang II–mediated increases in VEGF translation require ROS as NAC and DPI; both inhibited VEGF protein expression (66). Chemokines such as MCP-1 have been implicated in proximal tubule origins of kidney disease (72, 318). Ang II induced MCP-1 expression via increased ROS generation, and this was largely abrogated by the ROS inhibitor, NAC, and partially by AT1R and AT2R blockade (318). Interestingly, these investigators also showed that ROS-mediated MCP-1 stimulation was more prominent in PTCs when compared with MCs. In DSS rats fed a high-salt diet, NAD(P)H oxidase and MCP-1 expression was increased in response to albuminuria, and NAC blocked p47 phox subunit and MCP-1 expression (72). In contrast, exposure of isolated PTCs to fatty acid–conjugated albumin induced mitochondrial ROS production that was reversed with rotenone and not by NAD(P)H oxidase or XO inhibitors (139). Further to elucidate the importance of redox mechanisms in the kidney, pericytes of intact vasa recta (VR) were compared with pericytes of VR with disrupted endothelium for Ang II-mediated superoxide production (221). Interestingly, Ang II did not increase superoxide production in either scenario; however, addition of an NO scavenger led to increased superoxide production in endothelium-denuded vessels, suggesting crosstalk between different cell types, which is inhibited by antioxidants (NO).
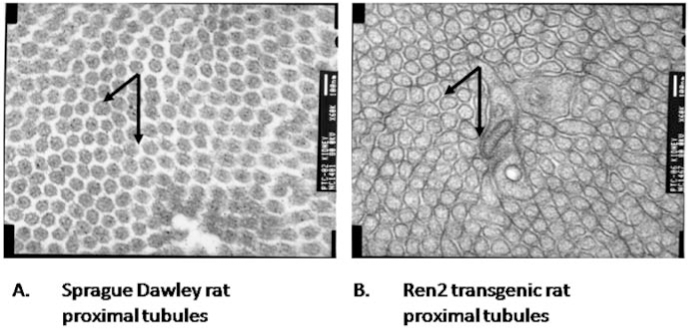
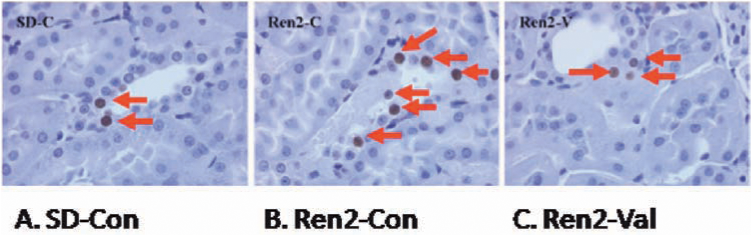
Increased albumin leak through the glomerular capillaries in hypertensive and diabetic kidney diseases results in heightened absorption into PTCs, leading to activation of proinflammatory and fibrotic pathways, as well as ROS generation. In vitro treatment of PTC with albumin leads to activation of the JAK/STAT pathway and inflammation, cell differentiation, fibrosis, and apoptosis (230). Furthermore, albumin-treated PTCs demonstrate reduced levels of antioxidants catalase and GPx, and levels were brought back to normal with antioxidant (NAC) treatment. Albumin activated NF-κB in PTCs via H2O2 generated through the PKC system (222). Both antioxidants dimethyl-thiourea and pyrrolidine dithiocarbamate prevented H2O2 generation and activation of NF-κB. In addition, DPI and rotenone also abolished NF-κB activation, demonstrating that albumin stimulation of NAD(P)H oxidase leads to PKC activation and ROS/H2O2 generation, which in turn leads to NF-κB–dependent inflammatory signals. Finally, albumin has been shown to activate ERK1/ERK2 by phosphorylation of EGFR, and this was reversed with AG-1478, an EGFR kinase inhibitor (265). It was further apparent that in PTCs, albumin acts through EGFR to stimulate NAD(P)H oxidase and ROS production. ROS then activate NF-κB, which then ultimately leads to activation of ERK1/ERK2 pathway.
High glucose levels (e.g., hyperglycemia) are characteristic of diabetic kidney disease. As described for mesangial cell signaling, high glucose results in activation of PKC-mediated signaling in the PTCs, resulting in generation of ROS via NAD(P)H oxidase and mitochondrial electron-transfer chain complex 1 enzymes (178). ROS in turn ramp up the expression of TGF-β1 and transcription factors, leading to modulation of extracellular matrix (ECM) genes. ROS-mediated stimulation of TGF-β1 was blocked by inhibition of PKC, NAD(P)H oxidase, and mitochondrial complex 1. High glucose by stimulating H2O2 also suppresses Na+/glucose cotransporter activity of PTCs, and this was reversed by aminoguanidine, DPI, and rotenone (106). Importantly, this model also revealed increased activity of catalase and decreased glutathione (GSH) levels, consistent with increased oxidative stress. Concomitant insulin administration to PTCs subject to high glucose was shown to suppress the angiotensinogen gene expression (126). Moreover, antisense to angiotensinogen in this model abrogated high glucose-induced expression of angiotensinogen and abrogation of oxidative stress. AGEs bind to RAGE and stimulate NAD(P)H oxidase to generate oxidative stress (142, 220, 324).
V. NAD(P)H Oxidase Inhibition for the Treatment of Hypertension: Promises and Limitations
To summarize the current status of understanding, NAD(P)H oxidase is an important contributor to redox-mediated hypertension and structural damage in the kidney. In the context of therapeutic strategies, several inhibitors have been used to suppress NAD(P)H oxidase. Some of these are specific to NAD(P)H oxidase, whereas others are more general inhibitors of ROS production. Relatively specific inhibitors of NAD(P)H oxidase include apocynin, DPI, gp91ds, and gp91ds-tat. The nonspecific inhibitors include PKC inhibitors (chelerythrine and staurosporin), statins, ACE inhibitors, ARBs, and antioxidants. In this section, we mainly focus on newer, less-studied NAD(P)H oxidase inhibitors. In addition, where relevant, limitations of the widely used inhibitors are discussed. The better-recognized inhibitors such as ACE inhibitors were previously addressed over the course of this review.
A. NAD(P)H oxidase–specific inhibitors
1. Apocynin
Apocynin (4-hydroxy-3-methoxy-acetophenone) has been the favorite NAD(P)H oxidase inhibitor for the past several years. It has been shown to decrease NAD(P)H oxidase–mediated ROS generation quite effectively. However, the exact mechanism of inhibition still remains elusive. It has been suggested that apocynin may inhibit the translocation of p47 phox to the membrane, thereby preventing the assembly of an active enzyme complex (309). Others have suggested that apocynin is most effective in myeloid cells expressing myeloperoxidase, as this enzyme is critical in activating apocynin (309). If this were true, then apocynin should not be active in endothelial cells or, for that matter, in any cell type deficient in myeloperoxidase. Some have speculated the existence of similar peroxidases in myeloperoxidase-deficient cells; however, no such enzyme system has been demonstrated (270). The debate over the use of apocynin as an NAD(P)H oxidase–specific inhibitor was fueled further with the demonstration of nonspecific effects of this compound on superoxide anion generation via non-NAD(P)H metabolic oxidases (120). A few studies even showed that apocynin stimulates ROS production in nonphagocytic cells and increases expression of oxidative stress markers in glial cells (271, 341). Last, human trials with apocynin and hypertension have been lacking. Apocynin is commercially available.
2. DPI
Diphenyleneiodonium (DPI) is an inhibitor of flavin-containing enzymes and can be used as a NAD(P)H oxidase–specific inhibitor. Use of this compound has demonstrated abrogation of NAD(P)H oxidase–generated superoxide anion and amelioration of vasoconstriction, endothelial dysfunction, activation of proinflammatory and profibrotic pathways, kidney dysfunction, and hypertension. However, DPI can also inhibit any flavin-containing enzyme and is a potent inhibitor of mitochondrial respiratory-chain enzymes, NO synthase, etc. Furthermore, it can cause hypoglycemia via inhibition of gluconeogenesis, limiting its clinical usefulness (125). This has led to the search for more-specific inhibitors for NAD(P)H oxidase.
3. Neopterin/phenylarsine oxide
The oxidized form of neopterin (α-amino hydroxypteridine derivative), and a precursor of biopterin, has been demonstrated in the liver, neuroendocrine tissues, and macrophages. It has been proposed to be a direct inhibitor of the phagocyte NAD(P)H oxidase system (168). However, others have documented activity against XO (11). In addition, it may increase the generation of singlet oxygen, nitric oxide, and the hydroxyl radical. Furthermore, studies have linked elevated levels of neopterin to coronary artery disease and hypertension and have proposed neopterin as a marker for heightened risk of these diseases (5, 150). In light of these conflicting reports, neopterin has failed to take off as a useful NAD(P)H oxidase inhibitor for treating hypertension. Neopterin is commercially available. Phenylarsine oxide (trivalent arsenic compound, PAO) was first shown to inhibit NAD(P)H oxidase in phagocytes via covalent binding to thiol groups, reversibly and completely (177). Later, its use was extended to other cell lines and ex vivo studies (331). However, one important caveat to the general applicability of this compound is its inability to prevent ROS generation from the fully assembled NAD(P)H oxidase enzyme, as it cannot reach the thiol groups. Moreover, it cannot prevent ROS generation from cytosolic NAD(P)H oxidase, as it cannot penetrate the cell membrane. This is a major drawback of this compound, as we now know that nonphagocytic NAD(P)H oxidase is active in the cytosol itself. Some studies have found utility for this compound in modulating Ang II signaling through its anti-tyrosine phosphatase activity (138, 186). Tyrosine phosphatases play an important role in the internalization of AT1R and Ang II into PTCs (186). Others have demonstrated activation of eNOS, leading to improvement in endothelial dysfunction (68). The latter effects may have some utility in modulating hypertension. PAO is commercially available.
4. Phycobilins
Phycobilins including phycocyanobilin (PCB), phycochromobilin, and phycoerythrobilin have been found to have NAD(P)H oxidase–inhibiting activity after they have been converted to phycorubins by biliverdin reductase. Phycorubins are similar in activity to bilirubin, which is ubiquitously found in mammalian systems and actively inhibits the translocation of p47 phox to the plasma membrane (144, 210). The obvious advantage of using compounds that are converted to bilirubin is their oral bioavailability. However, phycobilins are not easily commercially available [patent for PCB is pending (210)]. Meanwhile, a Japanese company has bioengineered Escherichia coli to make PCB in large amounts, potentially opening doors for further research (210). Spirulina (cyanobacteria) makes large amounts of PCB, and ingestion of these bacteria may provide physiologically relevant doses to inhibit NAD(P)H oxidase until other palatable ways of delivering PCB are available (210). Although a few rodent studies have borne out the promise of phycobilins, no human studies have been done to prove their utility in vivo. Moreover, it must be established with certainty that bilirubin is a specific NAD(P)H oxidase inhibitor and not just an antioxidant.
5. gp91ds/gp91ds-tat
The quest for NAD(P)H subunit–specific inhibitors led to the construction of a chimeric gp91 phox protein. Initial work led to the discovery of gp91-ds (gp91 phox docking sequence) based on neutrophil cell-free assays and peptide phage display library searches (50). gp91ds is capable of inhibiting only 80% of phagocyte NAD(P)H oxidase, further fueling searches for more-potent and more-specific inhibitors for this enzyme. The chimeric protein, gp91ds-tat, consists of nine amino acids of the HIV virus coat protein (tat) fused to nine amino acids of the p47 phox interacting domain of gp91 phox . When this chimeric protein was co-infused with Ang II intraperitoneally into mice, the authors saw abrogation of NAD(P)H oxidase activity in aortic rings, along with a sustained reduction in blood pressure that lasted at least a week (270). The complete inhibition of vascular superoxide production in aortic rings indicates that gp9ds-tat is capable of inhibiting all Noxes. This may or may not be useful, depending on the goal of the studies (i.e., if the goal is to inhibit Nox1 exclusively, then the study will be hampered by nonspecific inhibition of Nox2 and/or Nox4). Interestingly, the amount of gp91ds-tat needed to inhibit vascular NAD(P)H oxidase (50 μM) only resulted in a 24% inhibition of phagocyte NAD(P)H oxidase, likely preserving the phagocytic role of this enzyme (270). Last, use of gp91ds-tat could overcome the potentially detrimental compensatory and developmental issues associated with whole-organism knockouts when compared with tissue-specific delivery of this compound after birth.
Subsequent studies have shown that gp91ds-tat inhibits the dipsogenic response and increases blood pressure in response to Ang II treatment in WKY rats, possibly by inhibiting the neuronal chronotropic effects of Ang II (315). Smooth muscle hypertrophy, which is a common characteristic of hypertension, was reduced by gene transfer of gp91ds into the adventitia (189). gp91ds-tat also has been effective in attenuating some aspects of hypertension in salt-sensitive models. In the Dahl salt-sensitive rat model, gp91ds-tat ameliorates endothelial dysfunction by reducing vascular superoxide and peroxynitrite formation, but it has no significant effect on blood pressure (385). This may be due to the difference in the animal model (DSS rat is a low-renin hypertension model). Another study showed a similar result, in which the BP was not affected by gp91ds-tat (190). This has been attributed to the use of 10-fold lower dilution of gp91ds-tat than that used in the original study by Rey et al. (270).
In conclusion, although gp91ds-tat is useful in animals, its role in humans is limited by the fact that it can be administered only IV (it is a peptide and subject to digestive enzymes) and the potential to generate an immune response after long-term use. Further pharmacokinetic studies in vivo will probably answer these vital questions. gp91ds-tat is available commercially through companies such as Biosynthesis, Lewisville, TX, or Agrisera AB, Vannas, Sweden. However, the compound must be custom made. In addition, custom-made constructs can be availed through Tufts University Core Facility, Boston, MA, or Molecular Biology Core Facility at MCG, Augusta, GA, or similar such facilities.
6. PR-39
PR-39 is a proline-arginine–rich peptide, initially discovered in pig intestinal cells and later in phagocytes (298). It is a potent inhibitor of the assembly of NAD(P)H oxidase components p47 phox and p22 phox via competitive binding to the SH3 domain of p47 phox . Although its role in preventing ischemia/reperfusion injury has been relatively well characterized, the utility of this compound in modulating hypertension is unknown (130). Furthermore, PR-39 is unique in that it has dual roles in cells, as a mediator of respiratory burst in phagocytes and as a suppressor of NAD(P)H oxidase activity in conditions of oxidative stress (298). It is unknown how the administration of this compound will affect these multiple roles in vivo. Because of its inherent property of binding to SH3 homology domains, it could potentially bind to any protein with such a domain, and last, PR-39 is a peptide as well, limiting its oral bioavailability and general use.
7. VAS2870, S17834, and AEBSF
VAS2870 (3-bezyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo(4,5-d)pyrimidine] was discovered via a high-throughput screening assay specific for NAD(P)H oxidase activity at Vasopharm Biotech. Its unique property is that it can inhibit agonist-induced NAD(P)H activity but not the basal activity of this enzyme complex, suggesting specificity within the NAD(P)H oxidase family [e.g., Nox1 activation by PDGF is inhibited, but Nox4 activity at basal level in VSMCs is unaffected (322)]. However, a screening for Nox specificity has not been conducted. Furthermore, in vivo pharmacokinetics, bioavailability, and efficacy are unknown. S17834 [6,8-diallyl 5,7-dihydroxy 2-(2-allyl 3-hy-droxy 4-methoxyphenyl)1-H benzo(b)pyran-4-one], a synthetic polyphenol originally evaluated in the treatment of chronic venous insufficiency due to its regulation of adhesion molecule expression, has been shown to inhibit NAD(P)H oxidase activity in endothelial cells. It is orally bioavailable, does not scavenge superoxide or inhibit the XO system, and is not toxic to animals. However, its mechanism of NAD(P)H oxidase inhibition remains unknown, as well as its potential usefulness in humans. AEBSF (aminoethyl benzenesulfonofluoride, Pefabloc) has been shown to inhibit NAD(P)H oxidase activity in vitro and in cell-free systems by blocking the association between p47 phox and membrane components. However, this compound can also irreversibly inhibit serine proteases, limiting its usefulness in humans.
8. siRNAs
Small interference RNA (siRNA) molecules have found widespread use in biology because of their ease of use and relative specificity for the knockdown of any potential gene of choice. It is no surprise that siRNAs have been used by several investigators to demonstrate the role of NAD(P)H oxidase subunits in redox control of kidney function and hypertension (84, 327, 371). Some of the studies using siRNAs were described previously in the review. However, these moieties have several limitations, including poor oral bioavailability, immunogenic potential, and lack of any meaningful human in vivo data. In addition, concerns exist for off-target effects [i.e., on nontarget RNAs (337)]. Sirna Therapeutics, a subsidiary of Merck, is running trials of siRNA for age-related macular degeneration, and the results from this clinical trial should guide us further about the usefulness and pitfalls of siRNA.
9. Monoclonal antibodies
Monoclonal antibodies have become mainstays in the treatment of cancer, rheumatologic, and other immune-mediated diseases. However, the development of antibodies that are active in vivo against NAD(P)H oxidase is lagging far behind. Several mouse antibodies against gp91 phox and p22 phox have been developed in individual laboratories with variable inhibition of NAD(P)H oxidase activity in vitro (321). Polyclonal and monoclonal antibodies against these and other subunits are commercially available from Upstate Signaling (now Millipore, Billerica, MA) and Santa Cruz Biotechnology, Santa Cruz, CA. However, these antibodies are variable in their specificity and potency. Many data have been published about these antibodies, but the question of specificity does pose a problem. Furthermore, a few of the better antibodies are limited to the laboratories where they were developed, and they have not been tested in vivo. The utility of monoclonal antibodies in treating hypertension via inhibition of NAD(P)H subunits may appear promising, but we will have to wait several years before their in vivo efficacy can be adequately demonstrated.
B. Nonspecific NAD(P)H Oxidase Inhibitors
1. PKC inhibitors
Activation of the PKC pathway leads to phosphorylation of p47 phox and its membrane translocation. Inhibition of PKC with calphostin C, chelerythrine, and selective PKC-β inhibitor LY333531 (ruboxistaurin mesylate, RBX) ameliorated oxidative stress in the 2K1C model of renovascular hypertension, Ang II–infused rats, and in humans with endothelial dysfunction, respectively (362). However, as is obvious, PKC has roles in multiple pathways involving cell permeability, gene transcription, and other cellular responses, limiting its use to very special circumstances (i.e., diabetic macrovascular complications for which suppression of multiple pathways may be desirable). Furthermore, adverse drug reactions, including dyspepsia and increase in creatinine phosphokinase, have been reported with RBX (211). Calphostin C and chelerythrine remain to be tested in humans. Both calphostin C and chelerythrine are commercially available.
2. Antioxidants
Some compounds act by scavenging superoxide anion produced by NAD(P)H oxidase and other metabolic oxidases, thereby reducing oxidative stress. Ebselen [2,-phenyl-1,2-benzisoselenazol-3(2H)-one, PZ 51, DR3305] acts by mimicking GSH peroxidases and reacting with the peroxynitrite moiety. In addition, it can indiscriminately inhibit lipooxygenases, NO synthases, NAD(P)H oxidase, protein kinase C, and H+/K+-ATPase (250). Ebselen is orally bioavailable and has low toxicity, as the selenium molecule is restricted to the ring structure. This compound is most useful in mitigating the effects of ischemia and has found use in poststroke models. A few studies have shown the beneficial effects of ebselen in inhibiting endothelial dysfunction and vascular remodeling, but most have shown no blood-pressure effects (175, 313). Ebselen is readily available commercially. Other antioxidants include tempol and vitamins C and E; their role in redox control of cell function was discussed previously in the review. An important limitation to the use of tempol is that its efficacy has not been formally tested in humans. Although vitamins C and E showed promise in reducing endothelial dysfunction in type I diabetes and even improved blood pressures in isolated studies, phase III clinical trials failed to replicate these data. This led to questions regarding the timing of interventions, study population selection, and other confounding factors. However, as is evident from recent publications, the quest for the perfect antioxidant continues.
3. Statins, ACE inhibitors/ARBs, and aldosterone antagonists
These agents were discussed in some measure over the course of the review; however, a few important observations must be made. All of these drugs have the ability either to inhibit multiple pathways (statins) or to inhibit pathways that are involved in multiple cellular functions (ACE inhibitor/ARBs). Both of these mechanisms are due to pleiotropic effects of these drugs that may not be specific to NAD(P)H oxidase activity.
NAD(P)H oxidase activation has been shown to be a major contributor to redox control of kidney function and hypertension. Modulation of this enzyme pathway has been shown to have important salutary effects on endothelial dysfunction, end-organ damage, and blood pressure regulation. NAD(P)H oxidase inhibitors that are more specific, having greater bioavailability, are less toxic, and ultimately are more beneficial in treating kidney dysfunction and hypertension are needed.
VI. Future Perspectives/Conclusions
The role of redox mechanisms in normal functioning of the kidney and control of hypertension is complex. Myriad cytosolic factors and cellular and extracellular signaling pathways work in concert for seamless control of cellular mechanisms and the kidney as a whole. Dysregulation of these intracellular processes contributes to an imbalance in redox homeostasis and oxidative stress. Although the structure and function of the kidney is very well defined by the nephron, the nephron itself has numerous cell types. Furthermore, extracellular cues/stimuli from the surrounding environment and from circulating humoral factors also contribute to kidney function and control of blood pressure. The RAAS, among other pathways, is integral in salt and fluid homeostasis, and its intrarenal control of hypertension is more lucid. Our understanding of vascular and renal NAD(P)H oxidase is expanding in many of the RAAS-mediated and non–RAAS-mediated effects, both of which lead to increased ROS formation, oxidative stress, redox imbalance, and hypertension. However, a common final pathway for renal control of hypertension is still elusive.
Small-animal and genetic investigations have expanded our understanding of human disease processes, including hypertension. The incorporation of various cell lines further contributed to understanding basic signaling pathways as they contribute to hypertension. However, our understanding of signaling pathways among the numerous cells within the kidney and how they contribute to renal function is variable at best.
Modification of the RAAS-mediated oxidative stress in the vasculature and kidney has provided our best clinical understanding of renal redox control of hypertension. However, suppression of other mechanisms may also be a feasible strategy in redox control of hypertension. For example, smoking cessation is a preventable cause of oxidative stress, hypertension, and renal disease. Control of diabetes and weight loss for obese and cardiometabolic syndrome patients can result in a decrease in insulin resistance and hyperinsulinemia. Intriguingly, in some studies, although oxidative stress was mitigated with antioxidants, hypertension still persisted, implying that mechanisms other than oxidative stress are at play. Future studies directed to answering some of the questions raised will help us better to understand the redox control of kidney function and thereby help understand the process of developing essential hypertension.
Footnotes
Acknowledgments
Dr. Sowers' research is supported by NIH (R01 HL73101-01A1) and the Veterans Affairs Merit System (0018), and Dr. Whaley-Connell is supported by the Missouri Kidney Program and Veterans Affairs VISN 15. We thank Yongzhong Wei, M.D., Ph.D. (Research Assistant Professor in the Department of Internal Medicine, University of Missouri-Columbia School of Medicine) for contributing the figure on TUNEL staining in the proximal tubule cells. We also thank Stacy Turpin, M.S., CMI (Medical Illustrator, Department of Surgery, Applied Research, University of Missouri-Columbia School of Medicine) for her excellent illustrations.
Abbreviations
AA, arachidonic acid; ACE, Angiotensin-converting enzyme; Ach, Acetylcholine; AGE, advanced glycation end product; ANG, angiotensin; ARB, angiotensin-receptor blocker; ATR, angiotensin receptor; BH4, tetrahydrobiopterin; CCD, cortical collecting duct; c-GMP, cyclic guanosine monophosphate; COX, cyclooxygenase; DPI, diphenyleneiodonium; DS, dopaminergic system; DSS, Dahl salt-sensitive rat; EGFR, epidermal growth factor receptor; ENaC, epithelial sodium channel; EP, PGE receptor; EPO, erythropoetin; ERK, extracellular regulated protein kinase; GBM, glomerular basement membrane; GLUT, glucose transporter; GPx, glutathione peroxidase; GRK-4, G protein-coupled receptor kinase type 4; GSH, glutathione; HSPG, heparin sulfate proteoglycan; IGF, insulin growth factor; IMCD, inner medullary collecting duct; IL, interleukin; IR, insulin receptor; IRS, insulin-receptor substrate; JAK, Janus activated kinase; JGA, juxtaglomerular apparatus; JNK, c-jun N-terminal kinase; LDL, low-density lipoprotein; LOX, lipooxygenase; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MCP, monocyte chemoattractant protein; MR, mineralocorticoid receptor; NAC, N-acetylcysteine; NAD(P)H nicotinamide adenine dinucleotide oxidase, phosphate dehydrogenase; NF-κB, nuclear factor-κB; NO, nitric oxide; NOS, nitric oxide synthase; OMCD, outer medullary collecting duct; PGE/PGI, prostaglandin/prostacyclin; PI3-K, phosphoinositol 3-kinase; PKC, protein kinase C; PLA, phospholipase A; PTC, proximal tubule cell; RAAS, renin–angiotensin–aldosterone system; RNS, reactive nitrogen species; ROS, reactive oxygen species; SHRs, spontaneously hypertensive rats; SOD, superoxide dismutase; SNS, sympathetic nervous system; TGF, transforming growth factor; TNF, tumor necrosis factor; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling; VEGF, vascular endothelial growth factor; VSMC, vascular smooth muscle cell; and XO, xanthine oxidase.
Reviewing Editors: Tohru Fukai, John Hoidal, Reiko Inagi, Edgar Jaimes, and Rhian Touyz