
Abstract
The therapeutic response to traumatic brain injury (TBI) still lacks a strategy to treat its acute and chronic phases. Given that TBI affects the lives of 1.5 million people a year, and the management of its early stages has significant effects on outcome, identifying a druggable target to mitigate damage could protect the cognitive and motor function of patients. Given the value of rescuing cells during TBI and of the health of mitochondria in preventing cell death, it is important to explore means of preventing mitochondrial stress. There is a growing body of evidence that identifies a role for androgens (i.e., testosterone and its derivative, dihydrotestosterone) in both this TBI pathology and managing mitochondrial stress. Androgen signaling is involved in regulating gene expression of several proteins that interact with key mitochondrial pathways, and since TBI alters androgen signaling in time-specific stages, it appears to be a promising target for a druggable intervention. In this review, we discuss the physiopathological events underlying TBI pathology, focusing especially on the impact inflammatory cascade has on disrupting cell function and the integrity of mitochondrion. Finally, we propose that the administration of androgens might be considered a promising pharmacological approach to alleviate inflammation and mitochondria impairment post-TBI.
Introduction
Traumatic brain injury (TBI) due to traffic accidents and falls is one of the leading causes of death in the world. Currently, it is believed that TBI causes ∼1.5 million hospitalizations with ∼75,000 deaths per year, the most affected being adults between 19 and 60 years of age. However, an exponential increase has been observed in the elderly population (+65 years old) with some type of brain trauma, whose population is the most vulnerable due to advanced age. The fact TBI affects young people implicates a huge economic and financial impact for a country, as it does not only affect people at productive age but also their relatives who lose their jobs to become full-time lifelong caregivers, which generates an increased financial burden on a country's welfare program. Despite the scientific and technological advances in recent years, there is still no effective therapy to minimize the damage caused by TBI.
TBI is a systemic, heterogeneous, and multifactorial pathology, characterized by acute and chronic phases. First there is an acute abrupt inflammatory response, which involves the destruction of the cerebral vessels leading to hemorrhage, blood–brain barrier (BBB) breakdown with consequent infiltration of neutrophils, leukocytes and monocytes, edema and axonal trauma that produce the massive death of neurons, and glial cells due to mechanical trauma. In response, dying neurons release damage-associated molecular patterns (DAMPs). This promotes microglia to release proinflammatory type 1 interferons,1,2 IL-1β, 3 IL6,4,5 TNF-α, 6 CCL2, 7 and chemokines CXCL10, CXCL6, 8 RANTES/CCL5, and MIP-1β or CCL49 in the first 24–72 h post-TBI. This continues with an expansion of brain involvement due to the activation of oxidative cascades that not only affect neurons but also astrocytes and microglia, leading to long-term motor and cognitive damage. In the chronic phase of the pathology, generally from day 7 after trauma, there is a latent chronic inflammation, with a sustained activation of microglia that can last for years, affecting the processes of remodeling, recovery, and reorganization of the brain neuronal circuits, leaving the patients with serious motor and cognitive sequelae. At cellular level, during both the acute and chronic phases, there is a severe compromise of mitochondrial integrity, loss of adenosine triphosphate (ATP) production, concomitant with the activation of mitochondrial-dependent cell death cascades such as apoptosis. In dealing with current therapies against TBI, the therapeutic management in each phase is different, and one of the failures in the treatment of this pathology is the lack of complete knowledge of the cellular and molecular processes occurring in each phase to be able to address the problem with more specificity and precision.
An important feature of the pathophysiological mechanism of TBI is the neuroendocrine disruption in the brain. First and the more evident aspect is an altered expression of androgen receptor (AR) TBI. 10 Indeed, not only are the plasma and brain levels of gonadal hormones affected,11,12 other hormones such as follicle-stimulating hormone, growth hormone, and pituitary hormone 13 are also found to be significantly reduced after a TBI event, for which the magnitude of their levels are inversely correlated with trauma intensity and severity. Considering that the brain is an endocrine organ, it can be speculated that brain-synthetized hormones (i.e., neurosteroidogenesis) are particularly vulnerable and may become a potential complication in chronic post-TBI. It should be noted TBI male patients present lower blood levels of testosterone (T), 14 a similar outcome observed in the brain where its levels reduce up to 72 h but increase by week 2 as opposed to dihydrotestosterone (DHT), a T derivative product of 5α-reductase, whose levels are significantly attenuated. 15 Whether or not these neurosteroid levels are correlated with better prognosis or improved outcome overall, this is still controversial with some studies showing distinct outcome when T levels positively correlate with Glasgow Coma Scale, 14 suggesting that further studies are warranted to precisely link these changes to the degree of protection. Apart from these inconsistencies, the benefits of androgens (T and DHT) to stimulate neuroprotective signaling in TBI is well known by modulating the response of both glial cells and neurons to lesion, including at cellular level the mitochondrial modulation of redox reactions, inflammation, and apoptosis.16–20 Given the importance of androgens toward mitochondria well-being in this review, we highlight the main aspects of TBI pathology and discuss how T and DHT may have benefits as therapy to counteract inflammation and mitochondria impairment in TBI.
Acute and Chronic Inflammation Post-TBI
Neuroinflammation is a characteristic hallmark of TBI, where several brain cells, namely astrocytes and microglia, work together to control mechanical and pro-oxidative damage at site of injury that may generate an innate immune response that, in most cases, provoke an exaggerated response leading to more severe damage to neurons. Astrocytes bordering the injury become activated, as characterized by an increase in the expression of glial fibrillary protein (GFAP) and vimentin,21,22 also reflected by an augment in cell body and number of prolongations/ramifications. This early reaction is thought to be an immediate protective mechanism against mechanical damage, with the aim of limiting the expansion of damaged tissue through the formation of a dense glial scar. Glial scarring has long been considered to having a double-edged role that in one side it favors the containment of the lesion but eventually may inhibit axonal regeneration due to the excessive release of proteoglycans and inhibitory proteins that affect the formation of new axonal cones (for full review please see Filous and Silver 23 ). Furthermore, the release of cytokines and chemokines by activated astrocytes surrounding the injury likely promotes the activation of neighboring microglia, in addition to contributing to the attraction of immune cells from the periphery, favoring the rise of local inflammation.
The first 48 h postinjury is critical and marked by an exponential increase in the release of proinflammatory molecules, DAMPs, which continue with an initially acute but then chronic activation of glial cells, occurring in parallel with the infiltration of immune cells. On acute onset, both astrocytes and the brain-resident immune cells, microglia, surrounding the lesion wound, release an excessive amount of inflammatory cytokines that, concomitantly with the formation of reactive oxygen species and nitrogen oxygen species (ROS/NOS), can lead to massive death of neurons, which, inevitably, generate motor and cognitive sequelae with time of injury. Of these cytokines, IL6 is significantly upregulated in TBI and considered a biomarker of the severity of the lesion.24,25 The rapid increase in cytokine levels starts after 1 h post-TBI, with IL6, IL-1β, and TNF-α being the main ones expressed in the first few hours, including lasting up to 12 months after injury. 26 Not only are these cytokines implicated in the pathophysiological mechanism of TBI, but also other chemokines such as CXCL12, CXCL4,26,27 CXCL10,25,28 CCL2 and CCR28 that can not only increase the local inflammatory process at the site of injury, but also serve as chemical mediators that attract monocytes, neutrophils, T cells, macrophages, and leukocytes. Once reaching the brain tissue due to the loss of permeability of the BBB, these cells release more proinflammatory molecules that cause even more damage to the already injured tissue, leading to a perpetuation of the activation of glial cells.
More recently there is a growing interest in the role played by complement proteins in the secondary injury in TBI. The complement system comprising >30 distinct proteins is part of the innate immune system, and along with adaptive immunity both are responsible for the protection of the brain against pathogens that may eventually enter this organ. Interestingly, M1 microglia, an activated state, express C1q and C3, which seems to participate in or contribute to their reactive morphology. A rise on C1q and C3d and C3b levels has been observed in the area surrounding the injury in TBI patients, 29 which is in accordance with their levels in cerebral spine fluid, 30 suggesting the facilitation of neuroinflammatory signaling with involvement of these proteins. Apart from C1 and C3 anaphylatoxins, C5a has also been a target for further investigation in TBI, with studies showing that genetic deletion of C5 or administration of antagonists against its receptor, C5aR, attenuates secondary damage by reducing the infiltration of immune cells (i.e., leucocytes) and improving behavior outcome in injured animals.31,32 The fact that the expression of these complement proteins does not occur homogeneously and continues over time post-TBI, some having a peak in hours while others after days, suggests the importance of identifying the right timing of their levels, whether up or down, for a more precise pharmacological intervention.
Mitochondria response to TBI
Chronically orchestrated inflammation affects cell's ability to maintain its physiological functions. Mitochondria are one of the most affected organelles that lose their integrity and become unable to cope with physiological redox reactions, oxidative phosphorylation for ATP production, and maintaining cellular well-being overall. It is well known and widely accepted that all cells are extremely dependent on mitochondria for their correct functioning, especially considering that the brain has a high energy demand and little capacity to store energy make this organelle vital for pharmacological strategies that can protect them against TBI.
There are pathological events occurring within mitochondria that are pivotal to developing a better understanding of the damaging effects of TBI on a cellular level. Along with the endoplasmic reticulum (ERe), mitochondria play a key role in regulating cytosolic Ca+2 levels, which is an essential intracellular mechanism to control the dynamics of mitochondrial metabolism and oxygen consumption, 33 as this ion can be considered as an allosteric activator for some Krebs cycle catabolic enzymes such as pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase. 34 However, as TBI greatly affects these dynamics by interfering with the ability of the ERe and mitochondria to uptake Ca+2, this ion begins to accumulate intracellularly, leading to the activation of kinases and downstream enzymes capable of degrading membrane phospholipids, and affect kinase proteins regulating cell survival such as extracellular-signal regulated kinase (ERK) and mitogen-activated protein kinase (MAPK). 35 More specifically, this impaired mitochondrial regulation of Ca+2 promotes a disruption of the fusion (MFN1/2 and OPA1) and fission (Fis1/Drp1) processes, two critical mechanisms that play an essential role in maintaining mitochondrial homeostasis and quality control. It has been observed, for example, that acute TBI increases fission over mitochondrial fusion, 36 causing mitochondria fragmentation, which may be correlated with mitophagy and a significant decrease in endogenous ATP production. Not only that, but also the massive Ca+2 influx to mitochondria 37 causes an uncoupling of the electron transport chain located in the inner mitochondrial membrane (IMM), leading to a reduction in its capacity to accept and donate electrons forming a concentration gradient between the mitochondrial matrix and the mitochondrial intermembrane space. Inevitably, this has three immediate consequences: (1) a major reduction in protonmotive force; (2) this favors a greater production of oxidative stress by increasing free electrons, which are easily trapped by oxygen; and (3) increased levels of ROS are responsible for oxidizing mitochondrial membrane phospholipids such as cardiolipin that are essential to maintain the selectivity and permeability of the IMM. 38 The importance of cardiolipin for mitochondrial integrity is observed when there is a TBI, where this IMM-localized phospholipid is oxidized due to the peroxidase activity of cytochrome C (Cytc). 39 This promotes the cytosolic translocation of Cytc, one of the mitochondria-dependent mechanisms for the activation of apoptosis, programmed cell death, since once in the cytosol Cytc activates caspase proteins (i.e., caspase 3), leading to downstream triggering of the other apoptotic proteins (i.e., caspases 7 and 9). 40
Neurons and glial cells have developed amazing intracellular mechanisms to counteract, or at least minimize, the secondary damage caused by TBI. A protein expressed by these cells with great involvement in the pathophysiological mechanism of TBI is neuroglobin (Ngb), a protein highly regulated by androgens, especially T 19 (Fig. 1). Belonging to the globin family, similar to myoglobin and its more famous relative, hemoglobin, Ngb with 151 amino acids has a hexacoordinated group in place of the pentacoordinate—as seen with myoglobin and hemoglobin—favoring the union of oxygen with iron groups of the porphyrins. In a slightly different way, in the case of Ngb, His64 and His96 residues form the sixth hem-coordinated link with iron, which when oxygen binds Ngb it causes a displacement of both residues to generate the oxygenated form of this protein. Despite this evidence, the dissociation of Ngb by oxygen seems to be low and, instead of binding O2 with high affinity, this protein may serve as a sensor for this gas, especially in those hypoxic conditions similar to what occurs in the TBI with destruction of vessels and arteries due to mechanical trauma. Furthermore, Ngb also binds to ROS and NOS, 41 suggesting an antioxidant role, in addition to the fact that it interacts with proteins belonging to mitochondrial complexes III, for example, cytochrome c1. 42 Indeed, Ngb has the ability to sequester Cytc, with studies showing a physical interaction, avoiding its translocation to the cytosol in case of mitochondrial damage, thus assuming an antiapoptotic role. 43 It should be noted that Ngb levels reduce post-TBI, meaning that this protein could be considered as a target for future pharmacological strategies with potential clinical interest for the treatment of patients suffering from TBI. Nevertheless, Ngb inability to cross plasma membranes, hence not able to cross the BBB, makes its clinical use of little interest, and some ways to circumvent this is the application of encapsulation methods for its efficient and optimal delivery to the brain.

Androgenic regulation of neuroglobin signaling. Upon binding to testosterone, the AR is internalized to nucleus to stimulate transcriptional regulation of Ngb. Another upstream cascade that may upregulate this protein is through PI3k/AKT and p38/MAPK. Ngb is then transported to mitochondria where it interacts with various complexes of the electron transport chain (C1–C4), stimulating metabolism, oxidative phosphorylation, and regulating ROS-mediated inflammation possibly through the p32/C1qbp complex. Ngb protects CL from oxidation, prevents cytosolic translocation of Cytc, inhibits apoptosis, and also interacts with the mitochondrial transition pore formed by VDAC and ANT. Ngb interacts very intrinsically with cyc1 in complex III, and may also with the ETFDH/Q/ETFA complex, favoring electron transport and promoting ATP synthesis. At ATP synthase (AS), Ngb may bind to ATP5F1D, favoring ATP synthesis. This suggests that Ngb-inducing actions of testosterone are essential to maintain these organelles' stability and integrity under pathological conditions. ANT, adenine nucleotide translocator; AR, androgen receptor; AT, ATP synthase; ATP5F1D, ATP synthase F1 subunit delta; C1, mitochondrial complex I; C2, mitochondrial complex II; C3, mitochondrial complex III; C4, mitochondrial complex IV; CL, cardiolipin; Cytc, cytochrome C; cytc1, cytochrome c1 subunit; ETFA, electron transfer flavoprotein subunit alpha; IMM, inner mitochondrial membrane; MM, mitochondrial matrix; Ngb, neuroglobin; OMM, outer mitochondrial membrane; p32/C1qbp, complement C1q binding protein complexed with p32; p38/MAPK, p38, and mitogen-activated protein kinase; PI3K/AKT, phosphoinositide 3-kinases/protein kinase B; ROS, reactive oxygen species; T, testosterone; VDAC, voltage-dependent anion channel.
Involvement of Androgen Signaling in TBI
There are two isoforms of the AR, called AR-A (87 kDa) and AR-B (110 kDa). Isoform A is a minority and is due to a translation of the AR-B messenger from Met188, which produces a protein with the N-terminal region truncated and that could have less transcriptional activity. Isoform B corresponds to the complete coding sequence of the AR.44–46
Regulation of gene transcription is the primary mechanism of action of the AR. Upon binding to T or DHT, the AR dimer binds to a specific DNA sequence known as the androgen response element. 47 It has been revealed that the AR, apart from having genomic actions, may also have effects independent of its role as a transcription factor,48,49 interacting with components of signaling pathways initiated in the plasma membrane that induce, among other effects, rapid modifications in ion transport. Regulation by the AR of signal translation cascades in the cytoplasm can indirectly affect transcriptional activity through phosphorylation of other transcription factors.50,51 For instance, physical interaction of T with GPRC6A, a membrane AR, has been reported, 52 which may lead to a rapid activation of downstream signaling mediated by ERK and Ca+2 as messenger. 53 Being regarded as a zinc transporter, Slc39a9 (or ZIP9) has been predicted to be a target of androgens, in particular T, resulting in a myriad of actions ranging from ERK1/2, CREB, and ATF1 activation, upregulation of apoptotic proteins (i.e., Bax, p53, and caspase 3), and increase in intracellular zinc.54–59 These mixed effects may rely on cellular type, for example, it has been seen that the activation of ZIP9 in prostate cancer cells is associated with an increase in migration and invasiveness, 60 or perhaps be dependent on the cellular response to the concentration of T administered such as the induction of apoptotic mechanisms in ovarian cancer cells and breast cancer.56,57
Androgen signaling in the brain is severely altered in TBI. For example, AR expression is notably augmented in microglia at 72 h, decreasing afterward by day 28 after lesion. 61 Contrasting results have been observed with proteome data analysis revealing an important reduction of 48% and 42% on the expression of AR at 8 and 24 h, respectively, whereas Slc39a9 is reduced by ∼20% at 72 h post-TBI rats. 62 Using zebra finch subjected to unilateral brain injury, Mehos et al. show decreased AR messenger RNA levels at 48 h after damage. 63 Whether AR is up- or downregulated after injury, this indicates that the timing and animal model are two variables to be considered when studying its transient expression in TBI. Because of these discrepancies, one can speculate that due to reduced brain 15 and blood levels of T and DHT as observed in TBI, 64 brain cells may overexpress the AR to compensate the low bioavailability of its substrates or perhaps its reduced expression is coupled to low T, and DHT being not able to trigger a transcriptional regulation of AR. Another possible explanation is that since TBI dysregulates the brain synthesis of neurosteroids, their higher levels might be different and, in some cases, correlated with detrimental rather than protective outcome. Indeed, important to note is that not always the levels of steroids in blood correlate with those in the brain,65,66 this being a possible confounding factor when analyzing their peripheral versus brain levels. Collectively, these studies suggest three important outcomes: (1) the brain is an endocrine organ severely affected by lower production of local androgens, this impairs the androgenic regulation of mitochondria metabolism, and redox enzymatic reactions due to limited availability of T and DHT; (2) the reduction of T and DHT levels is associated with the development of other diseases (i.e., neurodegenerative diseases), being their levels in some cases inversely correlated with prognosis in TBI; and (3) exogenous supplementation with T might reduce TBI burden, facilitating a faster recovery and better overall outcome.
Actions of Androgens on the Attenuation of TBI Pathology
Mitochondrion is largely an androgen-regulated organelle. The fact that it is considered a converging point between the activation of survival and cell death mechanisms due to inflammatory stimulation demonstrates the importance it has for the post-TBI period. Bearing in mind that endocrine dysfunction occurs in TBI, which can again lead to dysregulation of metabolic and oxidative processes, the current evidence indicating the presence of ARs within the mitochondria may suggest this organelle as a full target of androgens. Next, we discuss the neuroprotective role of androgens in the setting of a traumatic damage to the brain.
Regulation of gliosis by androgens
One of the first studies to report that T reduces reactive gliosis was described by Garcia-Estrada et al. 67 In this study, they found that GFAP-positive astrocytes located at 0–500 μm from injury are significantly reduced in both cortex and hippocampus when rats are given 250 μg of T at 24, 48, and 72 h after a penetrating brain injury, with this approach also consistent with a significant reduction of proliferating (BrdU-positive) astrocytes close to the wound. 67 These findings were of clinical interest at that time, and many other studies that have tried to further determine how T, or its metabolite DHT, can regulate reactive glia after an injury have been carried out. Confirming the previous evidence, not only T but also DHT at 1 mg/kg was able to reduce reactive microglia in male rats subjected to a penetrating brain lesion, 21 possibly this effect may be directly related to the anti-inflammatory effects of these compounds. In line with this, pretreated BV2 microglial cells with 10 nM of DHT followed by 100 ng/mL of lipopolysaccharide (LPS), an inductor of neuroinflammation, show lower levels of IL6, TNF-α, IL-1β, iNOS, and COX-2, likely by regulating the TLR4/NFκB/MAPK/p38 signaling pathways. These findings are confirmed when studying primary microglia under the same inflammatory stimuli. 68 In the absence of T, animals castrated for 15 days and given 5 mg/kg DHT for 15 days and then injected with LPS have significantly lower levels of reactive microglia and astroglia in both cortex and hippocampus, thus confirming the ability of DHT and T to modulate neuroinflammation and lowering glia activation with LPS. 68
Since males seem to display more severe brain damage than females after a TBI event,69,70 to delve into these sex differences over the neuroprotective actions of T and DHT in brain trauma, male and female mice can be used to explore how each sex differentially responds to damage and what mechanisms underlie this. One of the recent attempts was the use of mice subjected to controlled cortical impact (CCI) whose outcome was monitored up to 72 h after lesion. 71 Although there was no significant sex difference with respect to GFAP expression in the ipsilateral hemisphere up to 72 h in TBI animals, there seems to have a sex-dependent effect on the lesion volume. Furthermore, it appears that these effects may be regulated by the endogenous production of hormones in the brain. For example, the animals were treated with either finasteride, an inhibitor of 5α-reductase, the enzyme that converts T to DHT, thus favoring the aromatization of T to estradiol, or letrozole, an aromatase inhibitor, the enzyme converting T to estradiol. Finasteride-treated male mice show a slight rise on the number of GFAP-positive astrocytes, and although this was not observed in the other sex, female mice given letrozole present significant levels of reactive astrocytes compared with vehicle-treated counterparts. It is also possible that the mechanism underlying this could be related to the brain levels of neurotrophins and their receptors, which are found significantly attenuated in TBI. For instance, the lesion itself appears to decrease TrkB and TrkC in both sexes, but the levels of both normal growth factor (NGF) and its receptor, TrkA, are significantly decreased (compared with vehicle) in males treated with finasteride, 71 suggesting that blocking endogenous DHT synthesis causes a disruption of NGF-TrkA as trophic signaling affecting neuronal fate, inflammation, and brain development. 72
Finally, tibolone, a synthetic steroid with estrogenic, androgenic, and progestogenic properties, may be used for drug repurposing in TBI. 17 For instance, tibolone is prescribed for the treatment of osteoporosis and climacteric symptoms in postmenopausal women. 73 After administration, it is rapidly metabolized in the body into 3α-OH tibolone and 3β-OH-tibolone that mediate their actions through estrogen receptors, whereas δ4-tibolone has preference for androgen and progesterone receptors.74–76 One of the first studies to show the actions of tibolone in TBI found an important reduction in the number of GFAP-positive astrocytes, and an attenuation of both the number and the reactive phenotype of microglia along with improvement in neuronal survival in animals after a stab wound injury. 77 Although tibolone has anti-inflammatory, antioxidant, and antiapoptotic effects,78–83 it is still unclear which of its metabolites may be mediating these actions in TBI, and further studies are warranted to explore which enzymes responsible for tibolone metabolism are being regulated in the face of brain injury. If this is confirmed in future studies, it could largely explain whether these benefits of this prodrug are being triggered by its metabolites with estrogenic, progestogenic, or androgenic activity.
Regulation of TBI-induced mitochondria impairment and inflammation by androgens
The mitochondrial network is a critical compartment for cell survival and development, due to its great function such as the proportion of cellular energy. It is a major player in the production of reactive species and regulates apoptotic cell death.18,84–86 Traumatic brain damage directly affects mitochondrial function 87 by interfering with the electron transport chain and contributing to the loss of mitochondrial membrane potential (ΔΨm) and the generation of ATP.
The fact the AR possesses an amino acid sequence for importation into mitochondria 88 suggests an intrinsic relationship between androgens and functions carried out by this organelle. Interestingly, the absence of androgens in male rats due to orchiectomy leads to lower expression of ND1 and ND5, two mitochondrial complex I subunits, leading to an important reduction of this complex's activity, all these cellular processes being ameliorated upon exogenous supplementation of T. 89 Furthermore, T has also been shown to upregulate mitochondrial complex V, 90 boosting ATP production and improving mitochondria integrity and metabolism. In contrast to these previous observations, ectopic expression of AR has some deleterious effects by attenuating the mitochondrial respiratory complexes, whereas an augmented activity of these complexes is noted when the AR is blocked in prostate cells. 88
Animals treated with T and subjected to CCI show a considerable improvement in ΔΨm, O2 consumption, oxidative phosphorylation, and a reduced level of hydrogen peroxide possibly due to an upregulation of the mitochondrial SOD2. 16 Similar evidence has been observed in glial cells pretreated with tibolone and subjected to metabolic damage, showing that this compound, in addition to positively stimulating the expression of Ngb, cardiolipin, and ΔΨm, is also able to attenuate the expression of 8-hydroxy-2′-deoxyguanosine, a marker of oxidative DNA damage, and 4-hydroxynonenal, a marker of lipid peroxidation.81,82 Indeed, tibolone reduces 3-nitrotyrosine, 82 a marker of nitrated proteins, in which the incorporation of the nitro group into the phenolic ring of tyrosine produces changes in certain properties of the amino acid. 91 The immediate consequences of tyrosine nitration are (1) no effects on the function of the protein, (2) a loss of function occurs, and (3) a gain of function is observed. An example is the inactivation of MnSOD by nitration of a key tyrosine residue, 92 which could increase the endogenous production of free radicals at the mitochondrial level. Finally, due to the increased production of nitric oxide in TBI, 93 this may inevitably inhibit the antioxidant enzymes glutathione peroxidase 94 and Cytc oxidase within mitochondria, 95 thus altering the mitochondrial respiratory chain, leading to attenuated ATP production.
To better understand how T protects mitochondria in TBI, some proteins known to be targets of this hormone and involved in TBI pathology can be retrieved from public databases for functional assessment. A mitochondrial cluster can be drawn, showing that these proteins are mainly related to the control of apoptosis, neuronal death, and cellular stress response (Fig. 2). For instance, by downregulating caspase 3 and Bax while increasing Bcl-2 family in TBI mice T16,96 and DHT,97,98 both are able to negatively regulate mitochondrial-dependent apoptotic signaling. Not only do T and DHT control the expression of proteins responsible for programmed cell death, thus favoring cell survival, but also upregulate AKT1, confirming their prosurvival properties that can be beneficial during post-TBI chronic inflammation. Quite intriguingly, another protein with possible involvement in the protective mechanism of T is the antiapoptotic member of the BCL2 family, MCL1, which has been sought to be involved in steroidogenesis, its blockade promotes an important reduction in the expression of P450 SCC (Cyp11a1) and steroidogenic acute regulatory protein, 99 and consequently leading to a decrease in precursors for the endogenous synthesis of T. Finally, androgen-deprived animals have higher levels of tumor protein p53, 100 a master regulator of DNA damage and repair with close participation in senescence and apoptosis, suggesting a link between endogenous androgens and processes regulating cell fate applicable to TBI. More recently, our group has discovered that the catalytic subunit of telomerase (TERT) has a key role in not only telomeres maintenance, but also that upon cellular damage it migrates to the cytosol to activate signaling pathways that can be essential for cell survival. It should be noted that its inhibition alters the protective actions of tibolone over astrocytic cells upon metabolic dysfunction, 80 clearly demonstrating TERT participation in neuroprotective mechanisms. Given that tibolone is metabolized into three different metabolites, it is yet to be explored whether these observed effects are due to its conversion to δ4-tibolone, with a potential perspective to regulate the AR.
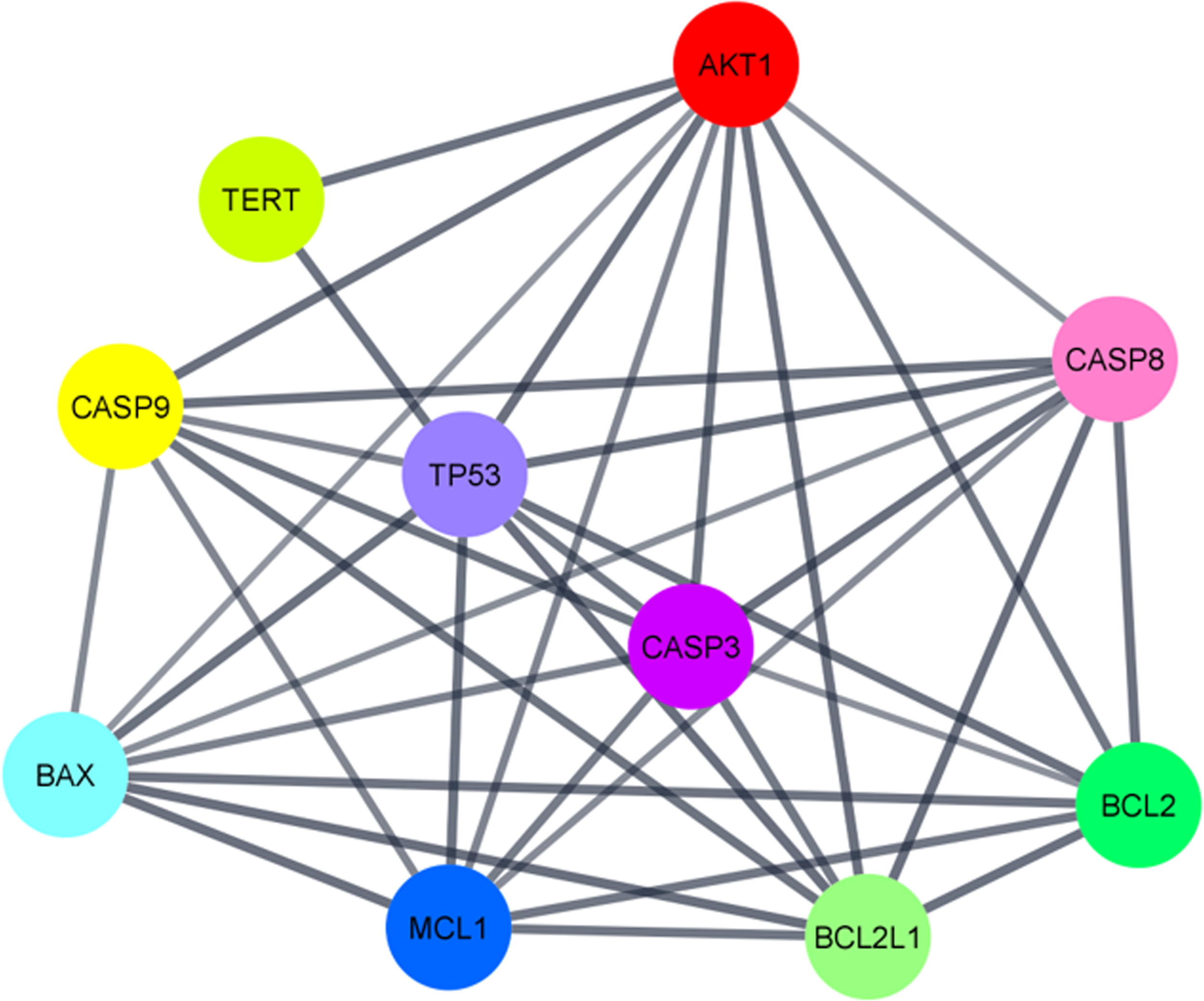
Mitochondrial subcluster of proteins regulated by testosterone as potential druggable targets in traumatic brain injury pathology.
Conclusions
The therapeutic strategy for TBI still lacks a means to manage the immediate inflammatory response that influences an individual's recovery. This is worrying as early intervention post-TBI has repeatedly been emphasized as critical to outcome, as it can mediate the extent of the damage to the brain and, therefore, protect the faculties of patients. However, although the value of an early intervention is apparent, current treatments are primarily in education, psychological, or rehabilitative assistance for patients and the data have mixed views of the efficacy of these treatments. As such, in this review, we have outlined the importance of considering mitochondrial dysfunction in preventing neuronal cell death as well as identifying important protective agents such as T and DHT by targeting mitochondrial proteins (i.e., Ngb), which have potential as druggable therapeutic targets. TBI also dysregulates the neuroendocrine system, suppressing the action of androgens. This coincides with evidence showing that men have worse outcome to TBI compared with women. We suggest that given the role of androgen signaling in regulating gliosis and protecting mitochondrial function under stress, targeting this system has potential as an early interceptive therapy in the acute stages of TBI.
Footnotes
Authors' Contributions
A.J.M. and G.E.B. wrote the article. Both authors agree with the final article, submission, and publication.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
No funding was received for this article.