
Abstract
Introduction:
The development of in vitro models that can link multiple organs and provide toxicokinetic data is important for rapid screening and risk assessment of chemicals and drugs. The primary objective of this study was to evaluate a new meso-scale MPS system that links multiple organs together via a simulated vasculature system.
Methods:
The MPS system used in this study is comprised of three organ compartments linked by a simulated vasculature system driven by a micro syringe pump. The portion of simulated vasculature inside each organ chamber is a semipermeable membrane, which allows small molecules to be exchanged by osmotic diffusion. To evaluate this model, acrylamide (AA) was selected as a relevant chemical of interest to the FDA. A three-organ system was prepared with human intestine (EpiIntestinal), liver [primary human hepatocytes (PHH)], and kidney (HK-2) cells/tissues. AA was administered to the apical surface of the intestinal chamber (0.3, 0.5, 1.0 mg) to simulate an oral exposure. Samples were collected over multiple time points for analysis of AA by LC/MS/MS.
Results:
The kinetic curves showed absorption, distribution, and elimination phases. Following the 72-hour time point, the tissue/cells from each chamber were collected for cytotoxicity measurements. A 30–40% loss of mitochondrial function in the intestinal tissue was observed, along with a limited amount of cell death. In the liver chamber, there was a marked dose-dependent reduction in intracellular ATP and glutathione (GSH). Finally, in the kidney, there was an increase in β-N-acetylglucosaminidase (NAG) and kidney injury marker-1 (KIM-1) activity indicating renal toxicity.
Conclusions:
These cytotoxicity findings match those reported in rodents following AA treatments and are similar to those used in this study. These findings indicate that the new multi-organ MPS can be used as a rapid screen to generate kinetic and cytotoxicity data.
Introduction
During the past 15 years or more there has been a global awareness and push to reduce, refine, and replace the use of animals for evaluating the safety of drugs and chemicals and to change the paradigm by which safety evaluation of drugs and chemicals is approached. A seminal report published by the National Academy of Sciences (2007) began the global paradigm shift by asserting that the future of toxicity testing should be less focused on whole animal testing and more focused on in vitro methods that identify biological processes linked to adverse effects using human cells and tissues. 1 In the European Union, the reduction of animal use began in earnest with the European Commission’s Cosmetic Directive banning the use of animals for the testing of cosmetic products or ingredients in 2009. The use of animals for more complex tests for cosmetics, such as repeated dose, reproductive toxicity, and toxicokinetics, was banned in 2013. In 2007 the U.S. Environmental Protection Agency began phase I of ToxCast, a large chemical screening project that employed multiple in vitro methods to collect toxicity data on more than 300 compounds. 2 The U.S. Food and Drug Administration (FDA) has also started to focus on new alternative methods (NAMs). The FDA Modernization Act allows agency scientists to review and incorporate NAMs data into their safety assessment process. Over the years the data from these projects as well as many others has paved the way for next generation risk assessment (NGRA). NGRA is exposure-focused, hypothesis-driven risk assessment that integrates chemical information from in silico, in chemico, and in vitro approaches without relying on animal data. 3 The development, validation, and acceptance of in vitro tests that replace animals for dermal and ocular irritation and dermal sensitization have been successful, resulting in several OECD test guidelines. A key challenge now is the development of NAMs that can address the question of complex testing assays (e.g., repeated dose, reproductive, and toxicokinetic) and systemic exposure.
To investigate more complex types of exposure scenarios and the toxicity associated with them, the development of NAMs that can recapitulate the in vivo organ-to-organ environment is required. The idea of putting organ models into a microenvironment was first presented by Dr. Linda Griffith with her groundbreaking work with organ-on-a-chip technology.4,5 Early models successfully showed that chemical toxicity in models where cells are exposed to simulated flow and possess active contractile activity (e.g., lung) can be significantly different from a static cell system and can change the toxicity profile of chemicals. 4 These organ-on-a-chip models also presented several challenges. The PDMS plastics use possessed a high degree of non-specific binding sites, making detection of applied materials difficult. 6 The ability to use many different types of cells and tissues has not been easy, and linking chips that mimic different organs together has also been challenging. This, combined with extremely small volumes of fluid and tissue mass, has made the use of these models difficult for the development of pharmacokinetic and toxicokinetic data sets.
In vitro organ-linked information is essential for building physiologically based pharmacokinetic (PBPK) models that can be used to predict exposure in humans and extrapolate in vitro safety data to in vivo effects (IVIVE). The development of kinetic datasets along with organ toxicity via relevant routes of exposure have remained elusive but are key to developing our ability to link the exposure organ (intestine, skin, and lung) to key systemic organs such as liver and kidney. More importantly, communication between the organ models cannot be a simple transfer of all culture medium in one organ chamber to the next organ chamber. A more physiologically relevant approach would be to simulate the in vivo reality by creating biological barriers that mimic those known to be present in vivo. When intracellular, intravascular, and interstitial compartments are part of the in vitro model the system itself can be parameterized and described mathematically, getting us one step closer to a complex in vitro PBPK model.
An area of need for the FDA is the ability to rapidly screen representative groups of chemicals associated with toxicity without the use of animals. Initially, the objective would be to find an in vitro system that could provide human-based information on organ toxicity at relevant exposure concentrations that could be used to identify potential hazards.
Therefore, the primary objective of the work presented here was to evaluate the applicability of a meso-scale human-based integrated organ MPS platform with simulated blood flow to determine if the system could be used to rapidly identify a potential chemical hazard and to demonstrate that kinetic parameters for chemical movement could be developed in an in vitro integrated organ model provided that basic biological barriers are present in the system. The test material used for this case study was acrylamide (AA).
AA was selected for this case study because it has been found in many different types of fried and baked high-starch foods. It is formed during the heating of foods to temperatures >120°C with the amount of AA formed being a function of food composition and processing conditions.7–9 The presence of AA in foods is concerning because it has been reported to be a carcinogen in rodent studies and is classified as a probable carcinogen in humans.10–12 AA in food is rapidly absorbed in the intestine and distributed to multiple organs. AA and its major metabolite glycidamide can bind covalently to cellular nucleophiles such as -SH. Hence the depletion of intracellular reduced glutathione (GSH) could predispose tissues to toxicity. The ability to perform a rapid screen for absorption, distribution, and organ toxicity by using an in vitro system would provide a means for regulators to identify potential hazards and predict relevant human exposures.
Materials and Methods
Chemicals and reagents
The test articles AA (CAS# 79-06-1), phosphate buffered saline (PBS pH 7.4) and human serum albumin (HSA) were purchased from Sigma-Aldrich (St. Louis, MO). EpiIntestinal™ tissues and culture medium were from MatTek Corp (Ashland, MA). Human primary hepatocytes and cell culture medium were obtained from LifeNet Health LifeSciences (Virginia Beach, VA). The human renal proximal tubule cell line (HK-2) was purchased from The American Type Culture Collection (ATCC) (Manassas, VA). Renal Epithelial Cell Basal Medium was from ATCC. The HK-2 culture medium was supplemented with the recommended growth kit from ATCC.
Preparation of test compound
A stock solution [300 mg/mL prepared in the vehicle PBS (pH 7.4)] was prepared and then used to prepare final dosing solutions in PBS of 3, 5, and 10 mg/mL AA. A 100 μL aliquot was added to the apical surface of the EpiIntestinal chamber to begin the experiment. This translates to a total amount applied of 0.3, 0.5, and 1 mg AA. These doses were selected based on rat studies in which the animals received an oral dose of AA that resulted in a Cmax value of approximately 0.03 mg/mL AA. 13 Therefore, a dose 10 times greater than the Cmax was selected to simulate a low oral dose. The lower dose used in this study resulted in compartmental values similar to those reported by Doerge et al. 13
Description of the test system
The MPS plate system used in this study is based on the importance of biology and relevant biological barriers. The system uses a standard six-well culture plate, however, in order to allow for the culturing of tissues without the entire plate assembly, polystyrene cups (surface area of 8 cm2) were designed to fit inside the standard six-well compartment. The insert cup was coated to reduce non-specific adsorption and allow for optimal cell or tissue culture. Each plate was then assembled with the simulated vasculature tubing inserted as shown in a top view (Fig. 1A) and in the side view (Fig. 1B). The tubing is held in place by a grommet that seals around the tubing to prevent leaking. The tubing runs just above the cells on the bottom of the plate for the liver and kidney chambers. The intestine tissue was cultured on a Transwell, and therefore the tubing is on the basolateral side of this compartment.
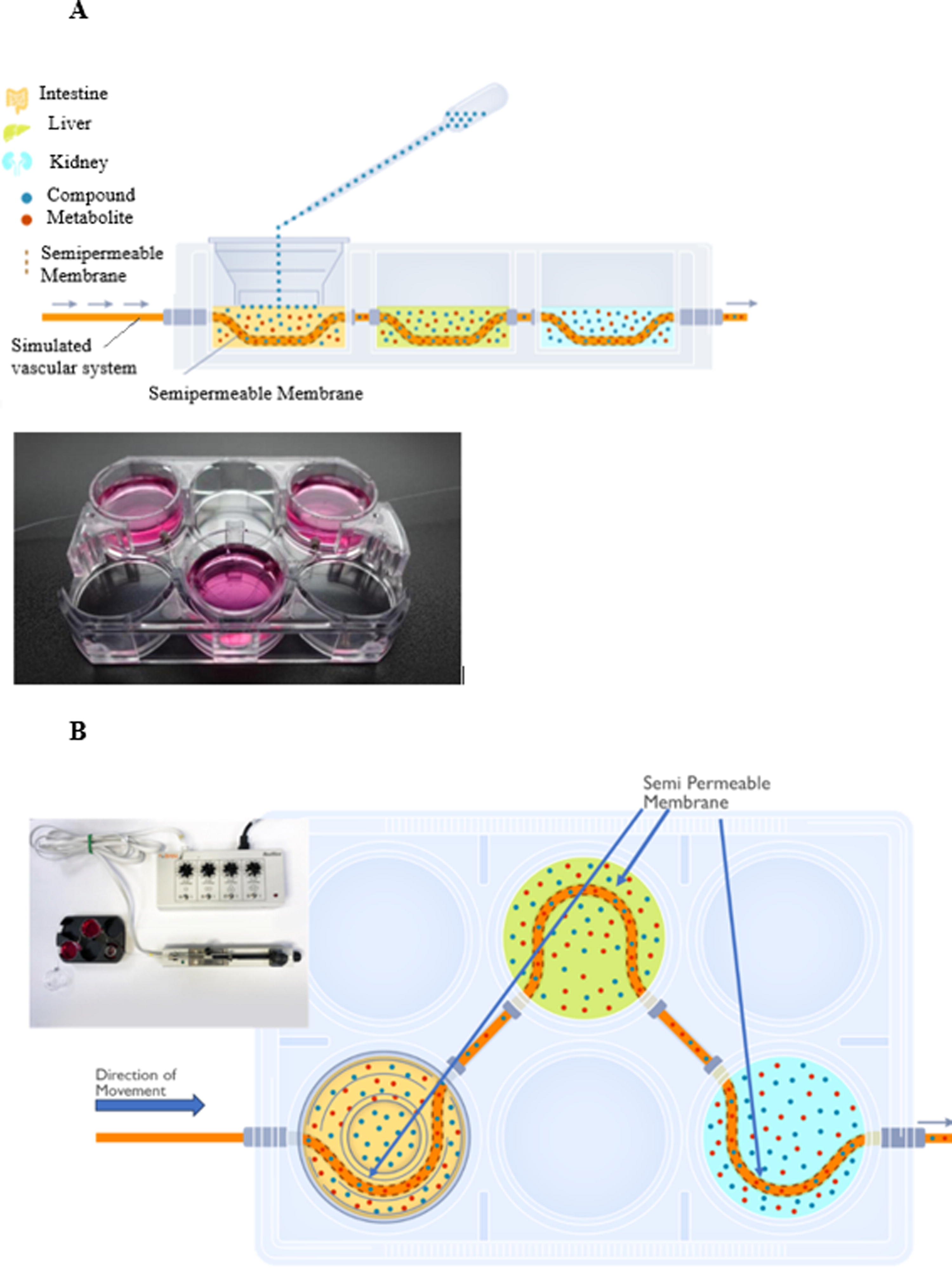
The meso-scale MPS plate system shown is formatted to fit a standard 6-well plate. Customized well inserts (cups) were designed to fit into the 6-well plate
Fluid was then pumped through the system from left to right in Figure 1A and B by a micro syringe pump purchased from BASi (Model MD-1020) at a rate of 5 μL/min. The perfusion medium was composed of PBS (pH 7.4) and 0.1% HSA. The tubing used in the system was also from BASi (DL-3). The dimensions of the tubing were as follows: flexible FEP tubing (1.2 mm OD) with a 3 cm length of dialysis membrane in the center of each organ compartment. The membrane has a molecular weight cut off (MWCO) of 30 kDa with an effective MWCO of 7–8 kDa. The membrane has an inside diameter of 240 μM. For this study the flow was straight through the system from intestine to liver to kidney, and then the perfusion fluid was collected at the designated time-intervals for analysis by LC/MS/MS.
LC/MS/MS detection of acrylamide
The test articles were analyzed using a Waters Acuity UPLC system in-line with a Waters TQ-S mass spectrometer via an electrospray interface.
Multiple reaction monitoring (MRM) is a targeted mass spectrometry (MS) technique that allows the detection and quantification of specific molecules in a complex mixture. AA at 90 ng/mL in methanol was manually optimized for sensitivity and MRM parameters using the TQ-S Intellistart fluidics. Final MRM parameters are shown in Table 1.
Final MRM Parameters For Acrylamide
AA standards were prepared as a 1 mg/mL solution in methanol, serial diluted in methanol, and spiked into intestinal, liver, kidney, and simulated blood media at a 5% (v/v) ratio of standard to media. An aliquot (50 μL) of standard in media was precipitated with 150 μL of methanol via vortex mixing, then centrifuged at 11,000 g for 5 minutes at room temperature. A 100 μL aliquot of the resultant supernatant was removed and placed in a 200 μL PCR plate with a silicone cover and held at 8°C during analysis.
AA was separated on a Waters HSS PFP 1.8 μm 2.1 × 100 cm column, using 0.1% formic acid/H2O as mobile phase A and 0.1% formic acid/acetonitrile as mobile phase B. For gradient elution, initial B was 1%, increased to 95% B at 2 minutes, 95% B at 3.8 minutes, and decreased back to 1% B at 3.9 minutes. The total run time was 4.5 minutes and 5 μL of sample was injected. Data were collected using a Waters TS-Q mass spectrometer via electrospray ionization and processed using Waters Target lynx application manager.
Tissue quality and determining tissue viability prior to administration of test material
The EpiIntestinal tissue from MatTek Corp has been well characterized in the literature and has been shown to possess polarized transporters, drug metabolizing enzymes, alcohol and alcohol dehydrogenases, and a full inflammatory response mechanism.15–17 Normal primary human hepatocytes (PHH) were purchased from LifeNet Health (Virginia Beach, VA). Each donor lot of these cells was tested for plate ability, transporter function, and cytochrome P450 (CYP) induction. In addition, a complete clinical history of the donor is provided, and the cells are genotyped so that extensive and poor metabolizers can be identified. The renal cells were normal human renal proximal tubule cells (HK-2). These cells have been used extensively to evaluate drug and chemical toxicity in the kidney and have been shown to express key renal specific markers of cytotoxicity.18,19 Each organ compartment was assessed for viability prior to beginning the study. Intestine, liver, and kidney were evaluated using the assays described below. A viability of ≥80% was considered to be acceptable. In addition, the barrier function of the intestine was verified by trans-epithelial electrical resistance (TEER) analysis and is described in greater detail in the sections below.
Integrated organ plate setup
A three-organ integrated system was set up as shown in Figure 1. The EpiIntestinal tissue was added on a Transwell® insert cultured at an air-liquid interface and was handled according to the manufacturer’s instructions. PHHs were seeded on to collagen-coated cups at a density of 1.2 × 106 cells/2.5 mL per cup or 0.15 × 106 cells/cm2 in complete growth medium. The human renal proximal tubule cells (HK-2) were seeded at a density of 1.25 × 106 cells/2.5 mL per cup or 0.156 × 106 cells/cm2 in complete culture medium. The assembled plate with cells was then allowed to equilibrate overnight at 37°C with 5% CO2. These seeding densities resulted in a starting confluency of approximately 80–90%. Each cell type was seeded in its optimum growth medium. The final volume of complete culture medium was 2.5 mL per cup.
AA was administered to the apical surface of the intestinal chamber by adding a 100 μL aliquot of the dosing solutions (3, 5, or 10 mg/mL) to the apical side (air side) of the EpiIntestinal chamber to yield a final applied mass of 0.3, 0.5, and 1.0 mg. After the addition of AA, the perfusion pump was started at a flow rate of 5 μL/min. The perfusion medium (simulated blood) was PBS plus 0.1% HSA at pH 7.4. The test articles and samples were collected at times of 0, 0.083 (5 minutes), 0.25 (15 minutes), 0.5, 1, 1.5, 2, 4, 8, 24, 48, and 72 hours. Samples in 100 μL aliquots were collected at each time point from the intestinal basolateral (lower) compartment, liver media, kidney media, and simulated blood. The collection volume for the simulated blood was cumulative for the sampling period and based on flow rate. For example, if the time between samples was 1 hour the volume of perfusate collected (5 μL/min) would be 300 μL.
After the 72-hour sampling time point the remaining media in each organ cup was collected and saved at −80°C for analysis. The remaining tissue or cells were prepared for various biochemical tests for cell health and are described below.
Verifying the intestinal barrier
To determine intestinal barrier integrity prior to compound administration, the inserts were tested using TEER analysis. Prior to beginning the experiment, the intestinal chamber was evaluated using an EVOM2 Epithelial Voltammeter equipped with an STX2 probe. Resistance values were considered acceptable if they were 400–600 ohm. This range of TEER acceptance was determined based on the manufacturer’s pre-ship tests and by confirming barrier integrity with lucifer yellow.
MTT assay
This assay was performed essentially as described by Mossman et al. 20 Briefly, the apical surface of EpiIntestinal inserts was aspirated and then assayed for MTT metabolic activity. A 1 mg/mL MTT solution was made in complete media and pre-warmed before use to 37°C. Media was aspirated from the wells and a 0.3 mL aliquot of the MTT solution was added to the intestinal tissue and incubated for 3 hours at 37°C with 5% CO2. Following the incubation time, the MTT solution forms a purple formazan crystal in metabolically active cells. To solubilize and extract this formazan product the initial solution was removed from the wells and replaced with 2 mL of isopropanol. The extraction was done by placing the cup on a shaker for 2 hours at room temperature at 500 rpm. The extracted solution was then added to a 96-well plate and the absorbance read at 570 nm and at 630 nm for background subtraction on a BioTek Synergy H1 Plate Reader.
Liver LDH assay
Lactate dehydrogenase (LDH) is a ubiquitous enzyme which is contained with cell cytosol. If LDH leaks out of cells into the culture medium it is an indication that cell membranes are losing integrity and cells are dying. Therefore, the presence of LDH was measured following the 72-hour exposure period using the CytoTox-ONETM Homogeneous Membrane Integrity Assay. Briefly, 50 μL aliquots of the culture medium were assayed according to the manufacturer’s instructions. The fluorescence signal was read on a BioTek Synergy H1 Plate Reader at an excitation wavelength of 560 nm and an emission wavelength of 590 nm.
Liver ATP assay
Liver cultures were washed with PBS, detached with TrypLETM and cell scraper, then evenly divided to be assayed for LDH, ATP, and reduced GSH.
Intracellular ATP was measured using the CellTiter Glo® Luminescent Cell Viability Assay according to the manufacturer’s instructions. Rotenone was used as a positive control for exposure. Following the exposure period, the media was removed from the liver cup, and 50 μL fresh media and 50 μL CellTiter-Glo® Reagent were added to the cells. The plates were then shaken for 2 minutes to free and preserve intracellular ATP and then allowed stand at room temperature for 6 minutes to allow the signal to stabilize. The plate was then read using a BioTek Synergy H1 Plate Reader in luminescent mode.
Liver glutathione assay
Changes in intracellular glutathione (GSH) and oxidized glutathione (GSSG) levels were determined with the GSH/GSSG-Glo Assay kit from Promega (Cat. # V6611). The assay was performed according to the manufacturer’s instructions. DL-Buthionine-(S,R)-sulfoximine was used as positive control for this assay.
Kidney β-N-acetylglucosaminidase assay
β-N-Acetylglucosaminidase (NAG, EC 3.2.1.52) is a lysosomal enzyme that is expressed in various tissues, including the kidney. NAG can cleave N-acetylglucosamine, a monosaccharide derivative of glucose. Its concentration in urine is minimal due to its inability to cross the glomerular basal membrane. An increased concentration of NAG in urine indicates renal tubular cell breakdown. Therefore, the presence of NAG in the kidney medium was used as an indicator of kidney cell toxicity. NAG activity in the kidney medium following the 72-hour exposure was measured with the kit from Abcam (Cat. # ab204705) or from Sigma (Cat. # CS0780). Cyclosporin A was used as a positive control. Each kit performed in a similar manner based on positive control data and on assay standard curves using NAG as the standard. The assay was performed according to the manufacturer’s instructions. The absorbance was then read at 400 nm on a BioTek Synergy H1 Plate Reader.
Kidney injury marker
The presence of KIM-1 in the kidney medium was assessed with the KIM-1 ELISA kit from MyBioSource (Cat. # MB264966). After the 72-hour exposure period, the medium was collected and assayed according to the manufacturer’s instructions. The optical density was read at 450 nm on a BioTek Synergy H1 Plate Reader.
Data analysis
All data were compiled and organized in Excel. The mean, standard deviation, and standard error of the mean were all determined using Excel. All results were graphed using GraphPad Prism 9. Statistical analysis was performed by ANOVA followed by Tukey’s posthoc test. *p = < 0.05.
Results
Concentration versus time kinetic profiles
A simulated oral administration was achieved by adding the test material at three concentrations to the apical side of the intestinal chamber. The kinetic curves showing absorption and distribution in the other organ compartments are shown in Figure 2A–D.
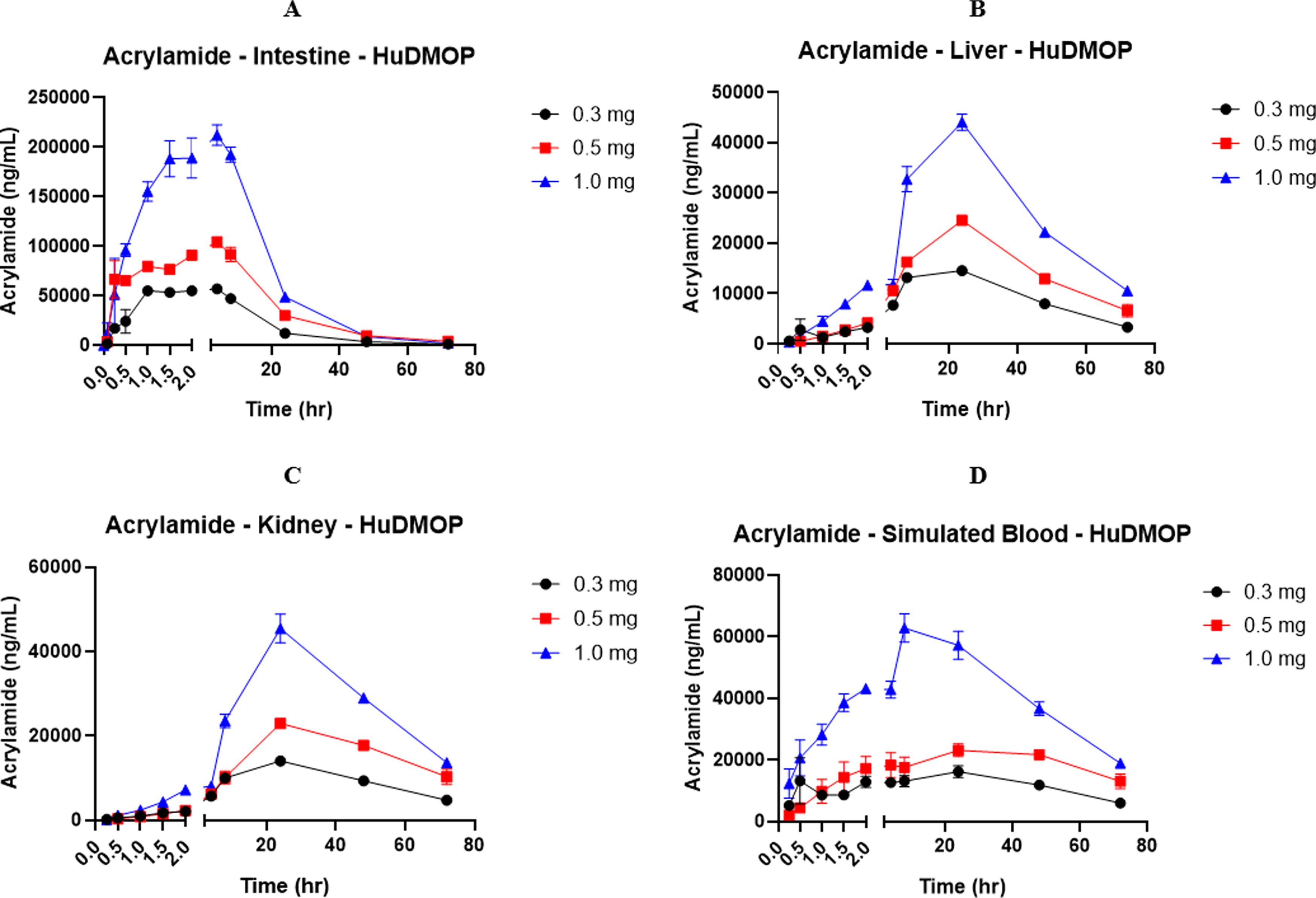
Concentration versus time curves for acrylamide (AA) over a 72-hour period. There was a clear dose-dependent profile observed in all organ compartments. A rapid absorption phase, a peak concentration, and an elimination phase were observed. This profile of AA movement was observed in all compartments. In the liver there was a lag in AA peak concentrations, but the initial appearance of AA in the liver was rapid. The concentration of AA in the liver and kidney compartments was lower due to dilution that occurs as AA moves from one compartment to the other, metabolism, and tissue distribution. Each value represents the mean ± SEM of n = 3 separate plate systems.
A rapid absorption phase occurred within the first 30 minutes of AA exposure in the intestinal basolateral (lower) chamber followed by a peak concentration absorption and an elimination phase (Fig. 2A). In the liver compartment (Fig. 2B) there was also a clear absorption and elimination phase. The lag in liver uptake was most likely due to tissue uptake of the AA, metabolism, and binding of the AA to intracellular macromolecules and nucleophiles such as reduced GSH. This pattern of uptake and elimination was also seen in the kidney (Fig. 2C) and in the simulated blood compartments (Fig. 2D). There was a clear dose response in the kinetic curves in all compartments, and if viewed in total a clear similarity to a drug plasma concentration versus time curve can be seen.
Intestinal cytotoxicity
The effects of AA on intestinal, liver, and kidney health are shown in Figures 3, 4, and 5. There was a small reduction in intestinal viability, based on LDH release into the culture medium (Fig. 3A). In comparison, the MTT assay, which focuses on mitochondrial function, indicated 30–40% loss in cell viability (Fig. 3B). It is likely that AA had direct effects on mitochondria, causing a more pronounced result with the MTT assay.
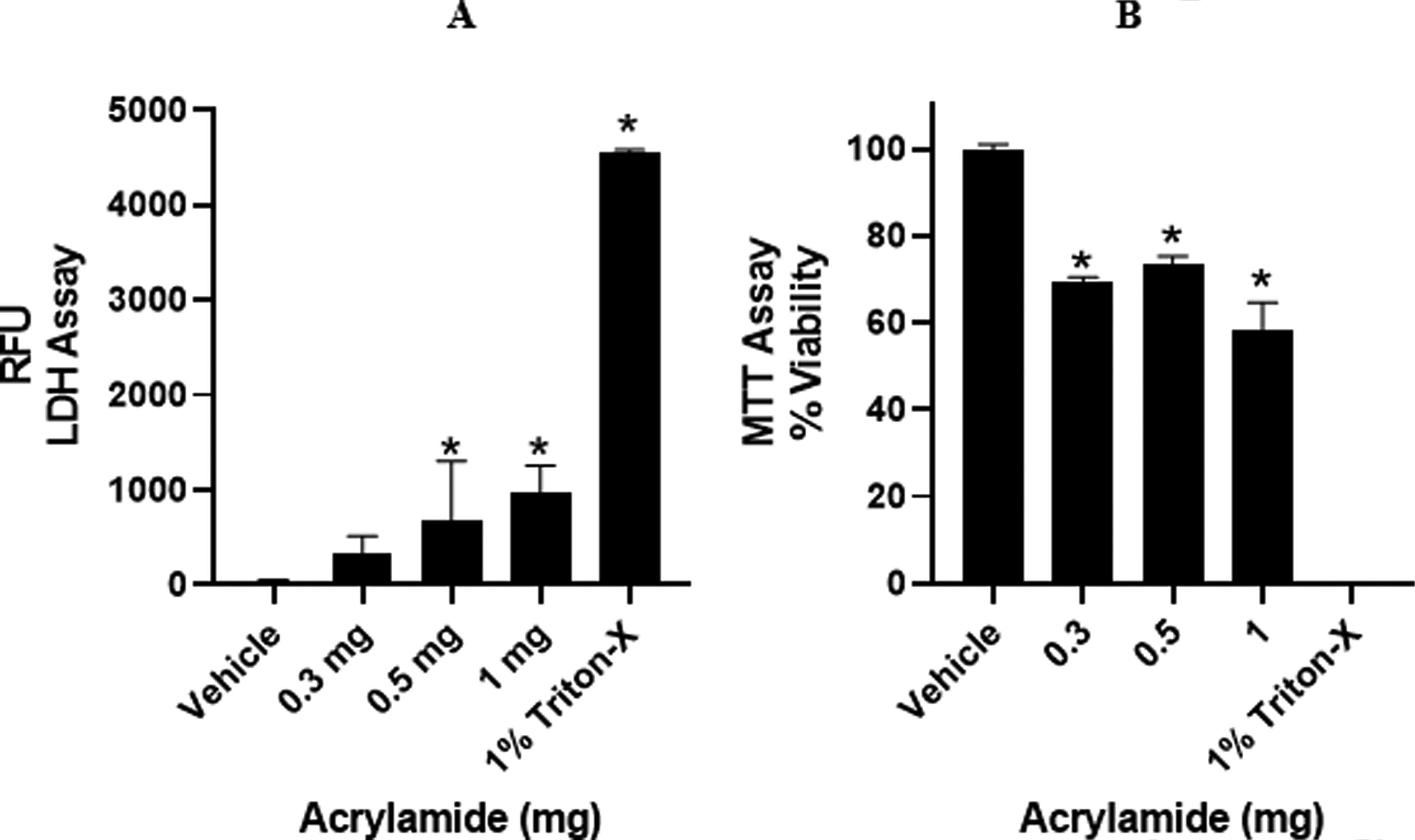
Effect of acrylamide (AA) on intestinal health following 72-hour. Cytotoxicity was assessed by Lactate Dehydrogenase (LDH) leakage and loss of mitochondrial function (MTT). An increase in LDH leakage was observed along with a 30–40% loss in mitochondrial function. Values represent the mean ± SEM of n = 3 separate circuits. Triton-X represents total tissue that is non-viable (dead). *p < 0.05 compared to vehicle (PBS + medium) group, ANOVA with Tukey post hoc test.
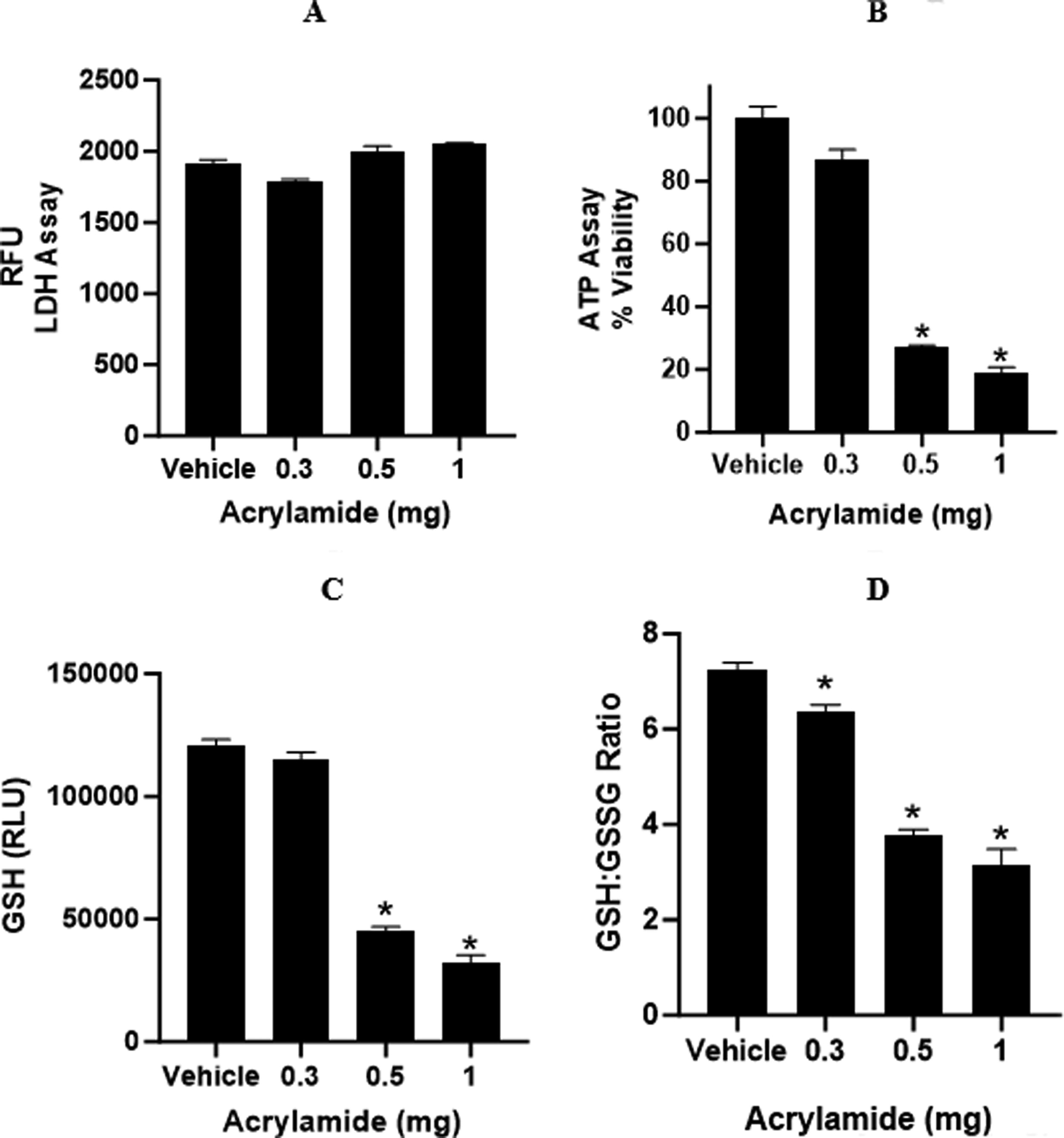
The effect of acrylamide (AA) on liver health. The effect of AA on human primary hepatocytes was determined by LDH which measures membrane integrity, intracellular ATP for mitochondrial health, and changes in glutathione (GSH) and oxidized (GSSG) ratios. All measurements were done following a 72-hour exposure period. There was no significant loss in cell viability based on LDH leakage
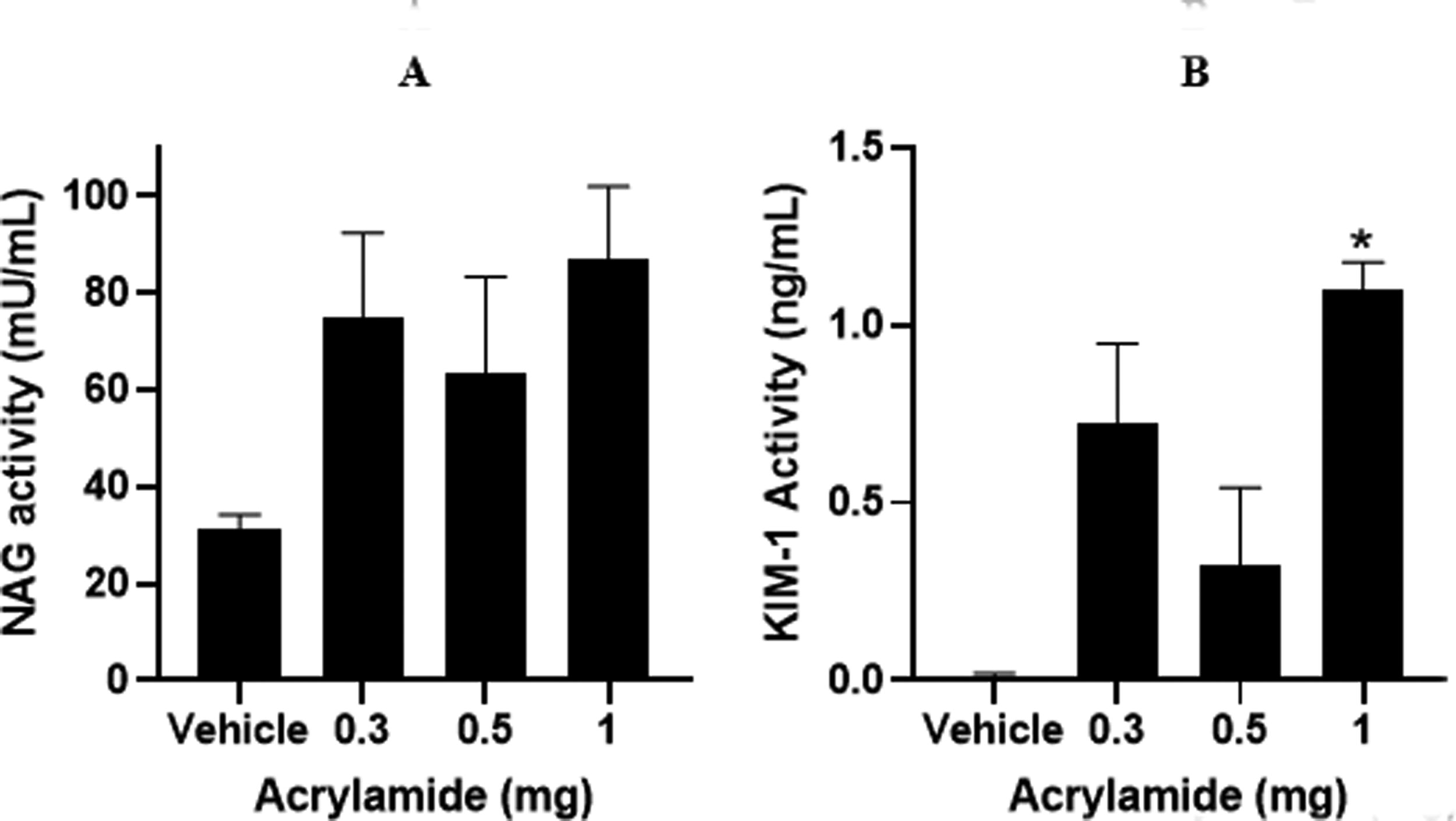
Effects of acrylamide (AA) on kidney (HK-2) cell health after 72 hr. N-acetyl-β-glucosaminidase (NAG) is a lysosomal enzyme that is present in renal proximal tubular cells and increasingly excreted as an indicator of renal tubular dysfunction. There was a clear increase in NAG activity at all doses of AA
Liver cytotoxicity
In the liver, AA exposure had no measurable effect on membrane integrity (LDH) but did cause a marked loss in cellular ATP (Fig. 4A and B). This response is seen when chemicals have an initial and potent effect on mitochondrial function. Liver GSH was also significantly reduced at the 72-hour exposure time and there was also an increase in the formation of GSSG, indicating that direct conjugation of GSH resulted in an increased oxidative environment in the cell (Fig. 4C and D).
Kidney cytotoxicity
AA also caused toxicity in the kidney (HK-2 cells) at 72-hour with a marked increase in NAG leakage into the medium and an increase in KIM-1 leakage into the medium (Figs. 5A and B).
Discussion
AA is formed during high-temperature baking of many foods, including potatoes, coffee, and biscuits. It is now apparent that dietary exposure to AA is prevalent. According to a European Food Safety Authority report released in 2015, human exposure is between 0.4 and 1.9 μg/kg/day in adults, with exposure doubling for children.21–24 These exposure scenarios mean that AA exposure to humans is a real concern. The use of an in vitro integrated organ MPS system would be a rapid and lower-cost approach that could greatly improve the hazard identification process and ultimately the risk assessment process.
AA readily binds to cellular nucleophilic sites on nucleotides, proteins, and other cellular components. AA at high exposure levels is a carcinogen and neurotoxin, along with being genotoxic and hepatotoxic.13,24,25 AA causes extensive oxidative stress due to its direct binding to reduced GSH in cells and subsequent disruption of cellular antioxidant defense mechanisms.
Regulatory agencies are faced with the challenge of identifying chemicals that can cause adverse effects in humans. When there are large groups of suspect agents, the task becomes time-intensive and costly when animals are the only choice for testing. In vitro integrated organ models with the ability to rapidly screen suspect chemicals for organ toxicity and provide reliable estimates of human exposure would greatly improve the risk assessment process. 26
In this study the primary objective was to evaluate a meso-scale human-based integrated organ MPS platform with simulated blood flow. The simple design of this model combined with the integration of complete in vivo-like biology makes this a promising device for the rapid assessment of chemical groups. To evaluate this model AA was selected as a case study. The test material was applied to the intestinal chamber of a three-organ (intestine, liver, and kidney) system. Each cell or tissue type was cultured in its optimal growth medium. In this system there is no movement of culture medium from one compartment to the other. Communication between organ compartments was via a simulated blood or vascular system. This system consists of tubing connected to a micro syringe pump (flow rate 5 μL/min) that pushes the simulated blood through the system. The portion of tubing that is inside the organ compartment consists of a semipermeable membrane (Fig. 1). This allows osmotic diffusion of the test chemicals and their metabolites into and out of the perfusion system. There is no net fluid exchange between the organ chambers because all other constituents are osmotically balanced or are too large to pass through the semipermeable membrane. This recapitulates the in vivo conditions controlled by blood flow, diffusion into and out of the blood, and delivery to organs.
Before beginning an experiment, the system was evaluated for non-specific binding of the test agent by first adding the test agent to the system without cells or tissues. For AA, the amount recovered was 87%. An in vitro maximum dose was also determined by administering a range of AA concentrations to identify the highest doses that would not cause more than 20% cell death. Rodent pharmacokinetic studies have reported doses of AA of 1.0, 10, and 100 mg/kg.27,28 Assuming an average adult rat weight of 300 g, the total amount of AA received was 0.3, 3, and 30 mg. In a pharmacokinetic study done in humans, the total administered amount of AA was reported to be 0.94 mg. 9 The doses selected for this study bracketed the rat and human values. The initial uptake of AA across the intestinal barrier was dose-dependent and rapid (Fig. 2A). There were clear absorption and elimination phases observed. This pattern was also observed for liver, kidney, and simulated blood compartments (Fig. 2B–D). It is important to note the dilution effect of multiple compartment models. This is similar to what would be expected in vivo when a chemical moves from one compartment to another and undergoes metabolism and compartmental dilution. For example, the volume of an adult human stomach is approximately 1500 mL, and therefore when a 400 mg oral dose is taken the first dilution occurs in the stomach. In rats the stomach volume is 3.4 mL.29,30 In animal and human studies, it is the fraction of the dose absorbed that drives most pharmacokinetic work, however, when using in vitro systems, it is important to account for the chamber dilution effects for organ exposure and mass balance calculation in the final parameterization of the plate system. This information will be an important component for the future development of models that enable extrapolation of the in vitro data to in vivo effects. When these in vitro kinetic profiles are taken together, they represent the uptake, dilution, distribution, metabolism, and elimination of AA. This also suggests that the data from the system described here could be used to develop models that would enable extrapolation from the in vitro situation to animal or human exposure and hazard identification. However, before this can happen, there must be an accumulation of multiple datasets consisting of drugs and chemicals with diverse physical and chemical properties.
In addition to developing in vitro chemical kinetic data, it is also important to relate the in vitro distribution of a test chemical to specific organs and identify toxicity at relevant concentrations. In this study, AA administration produced intestinal toxicity by reducing mitochondrial function by 30–40%, as measured by the MTT assay (Fig. 3B). This is consistent with recent in vivo studies demonstrating AA-induced intestinal stress, inflammation, and oxidative stress in mice. 31
It is well known that AA causes significant hepatotoxicity, and this is mediated through oxidative stress.21,32 The primary mechanism is AA-induced disruption of the GSH homeostatic system. Both AA and its primary metabolite glycidamide (GA), can bind to GSH directly or via glutathione S-transferase (GST) activity. 33 The formation rate of AA with GSH is 1.5-3 times higher than for the formation of GA-GSH adducts.34,35 AA causes damage to mitochondria, which also contributes to oxidative stress via the release of reactive oxygen species (ROS).35,36 AA and its primary metabolite (GA) can also activate NF-kB, increasing inflammatory cytokines (IL-18, IL1β, IL-6, and TNFα). AA can induce apoptosis via activation of Caspase-9 and Caspase-3. 22 In this study, AA caused a significant depletion of intracellular ATP (Fig. 4B), reduced GSH levels, and increased GSSG, indicating direct effects on mitochondrial function, direct chelation of GSH, and increased oxidative stress (Fig. 4C–D) following administration of AA. It is important to note that the loss in ATP and GSH occurred without a significant loss in membrane integrity (LDH), which is a common observation when evaluating compounds with a primary effect on mitochondrial function. 37
AA has also been shown to cause significant kidney damage with increased blood urea nitrogen (BUN) and creatinine levels in the blood of rats following oral exposure. 38 In this study, AA in the renal compartment caused an increased release of NAG and KIM-1 at relevant concentrations (Fig. 5A and B), indicating that renal cell toxicity had occurred.
Conclusions
The human-based multi-organ MPS system described in this study successfully provided kinetic data for AA following a single (simulated) oral administration. The doses applied to this novel in vitro system were consistent with doses used in rodent studies, and the toxic effects observed in all organ compartments were consistent with in vivo cytotoxic effects reported for a single oral administration of AA. Finally, the new multi-organ MPS can be described mathematically, and the movement of chemicals modeled.39,40 Future work will focus on building PBPK models that allow for the prediction of in vivo exposures and organ toxicity.
Footnotes
Acknowledgments
The authors thank Ms. Ella Betz for her expert assistance in the formatting and proofreading of this article, as well as for her assistance in preparing the graphs.
Author’s Contributions
J.M.M. designed the MPS plate system, led the study, served as project manager, and wrote the article. D.J.A. was the lead scientist in the laboratory who performed the work described. R.S., S.H., W.M., and S.F. all worked to study design, including compound selection, dose levels, and literature searches. In addition, these authors reviewed the article and provided important feedback.
Authors Disclosure Statement
All authors declare that they have no conflict of interest.
Funding Information
No funding was received for this article.