
Abstract
Abstract
Hypertension is a cardiovascular risk factor that has a profound influence on cardiovascular morbidity and mortality. While good progress has been made in terms of identifying and managing this risk factor for patient care, methods to assess the potential of chemical compounds to induce hypertension or to assess the efficacy of consumer products (e.g., e-cigarettes) targeted at reducing disease burden remain largely limited to epidemiological associations and in vivo studies. The field of toxicology has undergone a paradigm shift toward the replacement of in vivo testing in toxicological risk assessment. The adverse outcome pathway (AOP) framework could facilitate improved knowledge-based risk/benefit assessment of chemicals or consumer products, respectively, without the necessity of animal testing. Furthermore, to facilitate a more timely, cost effective, and ethical solution for risk/efficacy assessment purposes, integrated testing strategies are required, which do not heavily rely on in vivo studies. In this study, we present the supporting information on an AOP describing how vascular endothelial peptide oxidation leads to hypertension through perturbation of endothelial nitric oxide bioavailability, leading to impaired vasodilation. We also discuss the essentiality of the key events (KEs), and biological plausibility and empirical support of KE relationships, in accordance with the Organisation for Economic Cooperation and Development (OECD) handbook. This AOP could be a useful tool to serve as a foundation for a future integrated testing strategy for the regulatory assessment of the harm reduction potential of e-cigarettes relative to conventional tobacco products and other consumer products, which aim to reduce cardiovascular disease risk.
Introduction
C
Given the associated morbidity and costs to manage the condition through pharmacological interventions, recent efforts have focused on preventative initiatives to raise awareness of cardiovascular health among healthcare practitioners and the general population while various industries work upon therapeutic interventions and/or consumer products aimed at risk reduction.
Hypertension is a multifactorial disorder not only primarily associated with increasing age but also influenced by a number of other risk factors, including xenobiotic exposure. 4 Endothelial dysfunction underlies the development of hypertension and is defined as the imbalance between the production and bioavailability of endothelium-derived relaxing and contractile factors. Furthermore, endothelial dysfunction is associated with increased bioavailability of reactive oxidant species and decreased antioxidant capacity, also characterized as oxidative stress. 5 By (i) eliminating or reducing the exposure to stimulators of endothelial oxidative stress or (ii) using a consumer product that offers reduced risk compared with a currently used product, it follows that one can reduce the overall risk of developing hypertension in a population.
From a toxicological point of view, the focus of risk factor reduction resides in (i) the understanding and control of exposure to xenobiotic compounds, which have the potential to cause and/or accelerate the onset of hypertension, and (ii) efficacy assessments of consumer products, which either ameliorate the effects of (e.g., health supplements) or reduce exposure to (e.g., e-cigarettes) harmful xenobiotics. To determine which compounds could be of toxicological concern for the development and progression of hypertension, an understanding of the biology of endothelial dysfunction and hypertension is required. Furthermore, given the timescales required to develop hypertension from chronic xenobiotic exposure, toxicological studies need to focus on early events in the development process to predict the risk of onset as long-term clinical/epidemiological studies may not be practical for timely and cost-effective risk assessment.
One of the most widely reported hypertension risk factors is tobacco smoking. While smoking cessation remains the best way to reduce the harmful effects of tobacco smoking, tobacco harm reduction is being considered by some regulators (e.g., US Food and Drug Administration [FDA]) as a complementary strategy to reduce smoking-related disease burden. The US FDA has published guidance on assessing a modified risk tobacco product, either through demonstration of reduced toxicant exposure or reduction in health risks. 6 By gaining an understanding of how, and to what extent, tobacco smoke initiates biological mechanisms of hypertension, such knowledge could be utilized as a baseline for comparison purposes in toxicological assessments of the risk reduction potential of e-cigarettes.
Given the wide variety of data requirements to support such risk assessments, for example, human exposure studies, behavioral studies, efficacy studies, in vitro studies, clinical biomarker studies, pharmacokinetic studies, quality of life studies, a data integration framework is required to organize these data types into a comprehensive story that (i) characterizes the toxicological problem, (ii) demonstrates the likely outcome of an intervention, and (iii) can be utilized to monitor the performance of any intervention over time. Adverse outcome pathways (AOPs) offer scientists and regulators such a vehicle.
In this article, we briefly describe the principles of AOP development and present the supporting information, which substantiates a proposed AOP for hypertension through a mechanism of endothelial cell oxidative stress. We believe that such an AOP would be of benefit for both toxicological risk assessment purposes and for monitoring efficacy of therapeutics that aim to treat or protect against the condition.
AOP Development Process
The AOP development process has been well described by Villeneuve et al. 7 and Horvat et al. 8 Briefly, the basic principles of AOP development require the author(s) to establish a molecular initiating event (MIE), adverse outcome (AO), and a series of key events (KEs), which describe the critical intermediary steps in between the MIE and AO. Furthermore, KE relationships (KERs) are required to scientifically describe the mechanistic relationship between any two stated KEs, one of which is upstream and the other downstream. Thus, the status of the downstream KE should be able to be predicted or inferred by measurement of the upstream KE. By definition, AOPs are chemically agnostic and the AOP description should be independent from any specific chemical initiator and/or mode of action. 7 However, this is not to say that experimental data using specific chemicals for the purposes of establishing mechanism and biological response patterns should be avoided; in fact, quite the opposite, such studies are critical in determining KE essentiality. Therefore, an AOP development process starts by identifying the compounds (or compound classes) that have been clinically proven to induce the particular AO of interest.
In this specific case, given that tobacco smoking is one of the major clinically proven risk factors for the development of hypertension, 3 we have included information on compounds traditionally found in the aerosols of combusted organic matter, alongside known inducers of endothelial oxidative stress. A literature review was undertaken to map out the KEs for the purposes of weight of evidence assessment. By initially focusing on the AO and working backward through progressively lower levels of biological organization, we could be sure that the KEs remained focused toward the AO. Hence, we applied a top–down strategy for the development of this AOP.
KEs have been defined as changes in biological state, which have to be both measurable and essential for the specific AO. 8 Given the considerable complexity of the biology underlying hypertension, it is tempting to define as many inter-related pathways as possible to provide a comprehensive overview. However, an AOP does not provide a comprehensive molecular description of every aspect of the biology involved, but focuses on the critical steps in the pathway, which are essential for the progression of AOP toward the AO. Ideally, AOPs are simplified pragmatic frameworks, which are linear in nature and connect a single MIE to a single AO by means of nonbranching and directional sequences of KEs. We have tried to adhere to these principles as much as possible where the biology allowed, in our proposed AOP, for oxidative stress-induced hypertension. The following information in this article will substantiate the AOP according to the guidelines laid out in the Organisation for Economic Cooperation and Development (OECD) AOP handbook. 9 Supporting evidence for KERs is presented and evaluated according to three principles, namely biological plausibility of the KEs (most important), biological essentiality of the KEs, and quantitative evidence of the KERs (least important). We have assessed the relative strength of each of these KERs according to modified Bradford-Hill criteria, 10 as suggested by the OECD handbook.
AOP MIE, KEs, and AO
A summary schematic of the AOP is presented in Figure 1. Further information on this AOP is presented online at the OECD AOPwiki site: https://aopwiki.org/aops/149
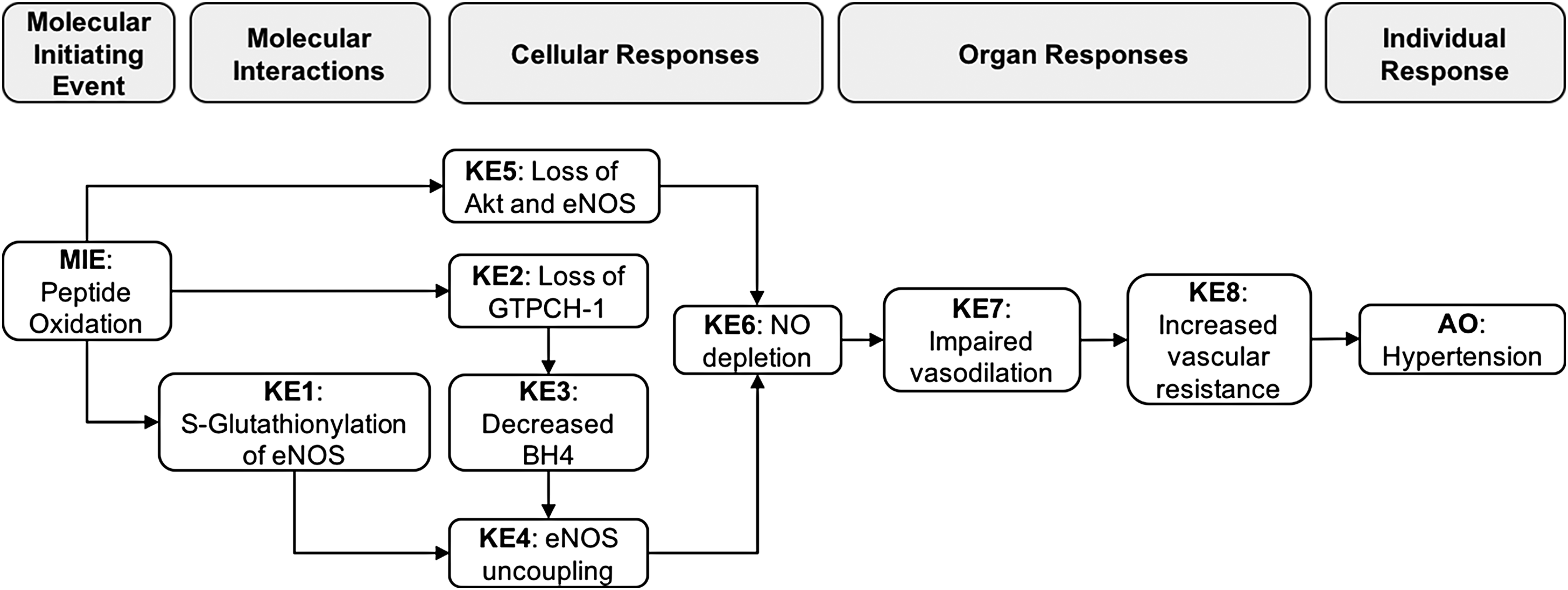
Schematic of the adverse outcome pathway, linking peptide oxidation to hypertension. MIE, molecular initiating event; KE, key event; AO, adverse outcome; eNOS, endothelial nitric oxide synthase; AKT, protein kinase B; GTPCH-1, guanosine triphosphate cyclohydrolase 1; NO, nitric oxide; BH4, tetrahydrobiopterin.
Molecular initiating event
The MIE of this AOP is peptide oxidation within endothelial cells lining the circulatory vasculature. The rationale for this is due to the broad (yet critical) effects of oxidation on the downstream target molecules in the AOP, which ultimately culminate in reduced endothelial nitric oxide (NO) bioavailability (forstermann, whitsett piclo).11,12 Peptide oxidation within endothelial cells can arise as a result of endothelial oxidative stress, which can occur following exposure to a direct oxidant(s) or through xenobiotic exposure leading to the generation of secondary oxidants, for example, reactive oxygen (ROS) nitrogen species by intracellular processes.12–14 Intracellular antioxidant peptides such as reduced glutathione (GSH) act as scavenger molecules to limit oxidative damage to critical biomolecules such as DNA, proteins, and lipids.15,16 As intracellular antioxidant protection is overwhelmed by increasing levels of oxidative species, antioxidant depletion occurs, which in turn can lead to protein/lipid oxidation.15–17 A wide range of chemicals are capable of causing MIE as any chemical exposure, which causes a shift in the redox balance of vascular endothelial cells toward oxidation (oxidative stress), can theoretically lead to MIE. Given the sealed nature of the circulatory system and high reactivity of oxidative radicals, vascular oxidative stress tends to arise from secondary generation of oxidant species following the action of xenobiotics and/or dietary and lifestyle factors, as opposed to exposure to direct acting oxidants, which are more likely to react with more proximal tissues related to the route of exposure.18–20 Known secondary oxidant species include superoxide, peroxynitrite, hydroxyl radicals, lipid/protein peroxides, and carbonyls. 21 In vitro studies in endothelial cells have shown that exposure to chemicals, such as hydrogen peroxide, tertbutyl hydroperoxide, glucose, ultrafine particles (UFPs) derived from ambient air, 4-hydroxynonenal (4-HNE), and methylglyoxal, lowers the intracellular GSH content of human umbilical vein and pulmonary aortic endothelial cells.12,22–26
Measurement of GSH itself and the GSH:GSSG ratio within endothelial cells can be achieved by the use of commercially available luminescence-based assays on the cellular lysates. Measurement of specific oxidative modification of target proteins can be achieved by immunoblotting techniques.27,28
KE1: S-glutathionylation of endothelial nitric oxide synthase
Oxidation of GSH results in the formation of a disulfide-bridged glutathione dimer (GSSG). GSSG is either rapidly rereduced back to GSH by nicotinamide adenine dinucleotide phosphate (NADPH)-dependent GSSG-reductases or extruded from the cell by adenosine triphosphate-dependent translocases. However, when these mechanisms become overwhelmed by high local oxidant concentrations, GSSG can interact with protein thiol groups to form protein-GSSG adducts, a process termed S-glutathionylation. 29 Interestingly, glutathione disulfide protein formation has been suggested to occur with a certain degree of specificity to cellular proteins since protein thiol groups exhibit considerable heterogeneity in terms of their individual pKa values and their location in protein structures. 29 The oxidation of GSH to GSSG elevates levels of GSSG, which then covalently bind to critical serine residues on endothelial nitric oxide synthase (eNOS).22,27,30
Measurement of KE1 can be achieved as described above by immunoblotting techniques on endothelial cell lystates,27,28 and also through mass spectrometry analysis of eNOS protein-SG adducts. 28
KE2: loss of guanosine triphosphate cyclohydrolase-1
GTPCH-1 converts GTP to 7,8-dihydroneopterin triphosphate and is the rate-limiting enzyme responsible for the production of 5,6,7,8-tetrahydrobiopterin (BH4) in a three-step process within endothelial cells. 31 Loss of enzyme activity leads to depletion of cellular BH4 within endothelial cells.
Measurement of KE2 can be achieved in cell lysates using immunoblotting as described by Wang et al., 32 also GTPCH-1 ELISA kits are commercially available.
KE3: decreased BH4
BH4 is a critical cofactor of eNOS, which facilitates the production of NO.31,33 Oxidative stress-related BH4 depletion occurs primarily by two mechanisms: (1) by oxidation of GTPCH-1, the rate-limiting enzyme responsible for BH4 production; or (2) by oxidation of BH4 itself by other oxidant species. As already described, BH4 is synthesized from guanosine triphosphate in a three-step process, in which the first step, mediated by GTPCH-1, is the rate-limiting step. 31
Measurement of KE3 can be achieved both in cellular lysates and supernatants, also in plasma. Analysis is through high-performance liquid chromatography, and its oxidized form (BH2) is commonly measured in tandem to give a ratio of the two analytes. This method is described by Biondi et al. 34
KE4: eNOS uncoupling
eNOS is the primary enzyme responsible for the production of NO in the vascular endothelium. 35 eNOS is a modular enzyme, which contains a C-terminal reductase domain (which binds NADPH, flavin mononucleotide, and flavin adenine dinucleotide) linked to an N-terminal oxygenase domain (which binds l-arginine, molecular oxygen, and BH4). L-arginine is converted into l-citrulline, with NO produced as a by-product. 35 The net result of KEs 1, 2, and 3 is the uncoupling of the eNOS enzyme, in which eNOS retains its NADPH oxidase function and produces superoxide, but loses the ability to produce NO.22,27,30,36
Measurement of KE4 can be achieved through the use of redox-sensitive probes such as dihydroethidium and hydroethidine, which form oxidation products with the superoxide that is liberated from uncoupled eNOS. The abundance of oxidation products is measured through HPLCs, and a detailed method was described by Zhao et al. 37 Furthermore, detailed methods for EPR spin trapping of superoxide using DMPO/BMPO have been published.28,38
KE5: loss of AKT and eNOS
The predominant signaling pathway leading to activation of eNOS (by phosphorylation) is also perturbed by oxidative stress. The phosphatidylinositol 3-kinase (PI3K)-AKT pathway is responsible for the phosphorylation of human eNOS at the serine 1177 residue.39,40 There are also an increasing number of hormones or bioactive substances found to modulate eNOS function through the PI3K/AKT pathway. These include insulin, insulin-like growth factor, angiopoietin-1, estrogen, leptin, sphingosine 1-phosphate, vascular endothelial growth factor, ROS, and corticosteroids. 41 Thus, phosphorylation of human eNOS serine 1177 by AKT is a central mechanism of eNOS regulation and NO production.42–44 Oxidative damage to these enzymes by radical species results in both loss of enzyme function and decreased enzyme levels.45,46
Measurement of KE5 can be achieved in cellular lysates through immunoblotting techniques and commercially available ELISA kits. It is recommended that the phosphorylation status of enzymes is measured (also by immunoblotting/ELISA) in addition to total enzyme to provide information on the activation status.
KE6: NO depletion
NO is a critical endothelium-derived hyperpolarizing factor, responsible for relaxation of vascular smooth muscle and vasodilation. The primary regulator of endothelial vasodilator function through NO is vascular shear; the frictional force exerted on the vascular wall during the flow of blood through the vessel. Vascular shear opens calcium channels on endothelial cells and leads to the calcium-dependent activation of eNOS and thus NO production. NO then diffuses to the underlying vascular smooth muscle, where it activates soluble guanylate cyclase, causing an increase in cyclic guanosine monophosphate, potassium ion efflux, hyperpolarization, and smooth muscle relaxation. 47 Depletion of vascular NO bioavailability causes an imbalance in the maintenance of vascular tone, which shifts in favor of vasoconstriction, and hence elevates blood pressure (BP). 48
Measurement of KE6 can be achieved both directly by measurement of NO itself through EPR spectrometry 30 or indirectly using chemiluminescence methods. Kapuganti et al. describe various chemiluminescence methods and provide comments on the advantages and disadvantages of their use. 49
KE7: impaired vasodilation
Vasodilation and its various modulating stimuli are well described by Giles et al. 47 Vascular tone decreases during vasodilation, increasing the size of vessel lumen as the underlying vascular smooth muscle relaxes. Impairment of vasodilation can arise as a result of perturbation of the key vasodilatory mechanisms, which includes NO bioavailability. Impairment is characterized by narrowing of the vessel lumen and stiffening of the vessel due to increased vasoconstriction.
This KE can be quantified by measuring isometric tension using ex vivo tissues (for example, aortic rings or sections of other relevant arterial tissue) as described by Dhar et al. 23 In live animals and human subjects, flow-mediated dilatation (FMD) (usually of the branchial artery in humans and femoral artery in rodents) through B-mode ultrasound is routinely used and is well described by Raitakari and Celermajer. 50
KE8: increased vascular resistance
Vascular resistance is the resistance that must be overcome to push blood through the circulatory system and create flow. The resistance offered by the systemic circulation is known as the systemic vascular resistance (SVR) or may sometimes be called by the older term—total peripheral resistance (TPR). BP is commonly described as a function of cardiac output and vascular resistance. Although a simplification, this emphasizes that an elevation of mean BP can only come about as a result of an increase in cardiac output, an increase in vascular resistance, or a combination of both. 51
Measurement of this KE can be achieved noninvasively as described by Stefadouros et al. 52
Taxonomic applicability/species concordance and other considerations
With respect to the MIE, GSH-mediated oxidant scavenging is highly conserved in both plants and animals.53,54 Differences with respect to how GSH is generated/recycled through enzymatic pathways could differ, however. Similarly, amino acid (AA) oxidation within proteins occurs throughout the plant, animal, and bacterial kingdoms.
The biology of KEs 1–6 is likely to be conserved across mammalian species. This is specifically evident from the numerous studies utilizing bovine, rodent, and human endothelial cells in vitro, rodent in vivo models of hypertension, and human clinical studies, in which the KERs follow the same biology. (De)phosphorylation sites specifically for eNOS (de)activation are conserved between cows, mice, and humans.55–57 Therefore, oxidation of these specific AA residues is likely to show similar effects on eNOS function and NO bioavailability (although the magnitude may vary). While the overall function of AKT, eNOS, and GTPCH-1 is conserved in mammalian species, the effect of oxidation/modification of other AA residues on the function of these enzymes has not been sufficiently characterized and hence differences are possible.
The biology of KEs 7 and 8 and KERs KE7→KE8 and KE→AO is highly complex, and numerous sex, age, tissue, and species differences are evident, also between healthy and diseased animals and humans. Durand and Gutterman highlight such differences in great detail 58 and we refer the reader to this article.
With respect to gender differences, Scotland et al. 59 noted differences in acetylcholine-induced vasodilatory responses in C57/BL6 mice, in which vasodilation in male mice was abolished with eNOS blockade alone, whereas female mice required both eNOS and COX blockade to observe the same effect. Therefore, it is possible that mechanisms of vasodilatory plasticity could differ between males and females. Although some of the evidence presented in this article is derived specifically from males or females, the processes described are applicable to both genders. This is corroborated by epidemiological data, which indicate that hypertension affects women and men from age 45 to 64 years nearly equally. 3
To conclude this subsection, the support for the proposed AOP combines well-documented in vitro evidence from multiple organisms, including mouse, rat, cow, and humans. However, the current view relates these differences to changes in physiological status or to age-related comorbidities such as obesity and type II diabetes, which are also linked to increased oxidative stress. 60 While eNOS-mediated NO signaling clearly plays a key role in the development of hypertension (see Empirical Evidence Supporting KERs), other compensatory mechanisms are capable of maintaining vascular tone in the short-medium term. However, the specific mechanisms of these compensatory changes in humans over the longer term are poorly understood and hence require further study.
Empirical Evidence Supporting KERs
MIE (peptide oxidation) to KE1 (S-glutathionylation of eNOS)
Hypoxia/reoxygenation-induced oxidative stress (associated with ischemia/reperfusion injury) was shown to deplete GSH in bovine aortic endothelial cells, which led to S-glutathionylation of eNOS and eNOS uncoupling. This phenomenon was partially reversible, in bovine aortic endothelial cells and rat aortic rings, by raising intracellular GSH levels upon administration of N-acetylcysteine.23,27
Chen et al. have shown that eNOS is particularly sensitive to S-glutathionylation at cysteine residues 689 and 908 of the reductase domain, a phenomenon that is dose dependent with application of exogenous GSSG. 30 This finding was corroborated by Peng et al. using mutated eNOS constructs in Escherichia coli, demonstrating that superoxide was produced by the eNOS phosphorylation site in the reductase domain. 61 Wu et al. studied responses in human lung microvascular endothelial cells to lipopolysaccharide (LPS) in vitro. Upon LPS administration, NADPH oxidase 2 (NOX2) expression levels increased with a subsequent rise in superoxide production, which led to S-glutathionylation of eNOS. 62 Furthermore, in mice, coimmunoprecipitation studies revealed that NOX2 associated with eNOS and that S-glutathionylation in response to LPS was much more apparent in elderly animals compared with younger animals. 62 Similar observations were made by De Pascali et al. following hypoxia-induced oxidative stress in bovine aortic endothelial cells. 27 Finally, Chen et al. demonstrated that coadministration of glutaredoxin-1 and GSH reversed GSSG-mediated eNOS S-glutathionylation and restored eNOS-mediated NO production, also in bovine aortic cells. Interestingly, inhibition of eNOS function occurred when the GSH/GSSG ratio was >0.2 and function was restored at a ratio of <0.1. 63
MIE (peptide oxidation) to KE2 (loss of GTPCH-1)
Although biologically plausible, based upon widely accepted existing knowledge that oxidative modification of protein AA residues can cause protein dysfunction, evidence specifically linking the MIE to KE2 is limited. 4-HNE is a carbonyl known to induce oxidative stress. Bovine aortic endothelial cells exposed to 4-HNE had reduced levels of GTPCH-1 compared with controls. Control experiments with cycloheximide (a protein synthesis inhibitor) confirmed that 4-HNE did not act to inhibit synthesis of GTPCH-1, but rather it damaged the protein and induced proteasomal degradation (confirmed by studies with proteasomal inhibitors). 12 In murine lungs where localized vascular oxidative stress was induced by the selective depletion of endothelial superoxide dismutase (SOD), levels of active GTPCH-1 were shown to be reduced in the knockout animals compared with controls. 64 Furthermore, in atrial samples from patients undergoing cardiopulmonary bypass surgery, NOX2 expression and superoxide production were elevated following tissue reperfusion (known to induce oxidative stress), with a corresponding 50% reduction in GTPCH-1 activity and a 32% decrease in BH4. Although eNOS activity was significantly decreased by ∼60% following reperfusion, BH4 supplementation did not recover eNOS activity, whereas treatment with dithiothreitol reversed eNOS S-glutathionylation and restored eNOS activity. 36 This final example is, however, a published abstract, and no follow-up publication could be found at the time of writing to permit scrutiny of the study.
KE2 (loss of GTPCH-1) to KE3 (decreased BH4)
With respect to KE essentiality, numerous studies have demonstrated that deletion of GTPCH-1 led to the deficiency of BH4 in bovine and murine endothelial cells31,33,65 and in knockout mice. 28 Exposure of bovine or mouse aortic endothelial cells to GTPCH-1 inhibitors (DAHP or NAS) or GTPCH-1-siRNA significantly reduced BH4 and NO levels and increased superoxide levels. The increase in superoxide was abolished by the BH4 precursor sepiapterin. 32 In vivo, gene silencing of GCH1 (the gene that codes for the GTPCH-1 enzyme) and GTPCH-1 inhibition lead to elevated BP in mice and rats.32,66 Furthermore, GTPCH-1 gene transfer into diabetic rats increased BH4 bioavailability and NO production. 67
In C57/BL6 mice, gene silencing through siRNA of GTPCH-1 reduced BH4 levels and raised BP compared with control animals and, furthermore, GTPCH-1 siRNA was unable to elicit these effects in eNOS null mice. Sepiapterin supplementation, which had no effect on high BP in eNOS−/− mice, partially reversed GTPCH-1 siRNA-induced elevation of BP in wild-type mice. 32 In another study, endothelial cells from diabetic rats were shown to have decreased expression of GTPCH-1, significantly reduced BH4 levels, and impaired NO synthesis compared with cells from normal animals and diabetes-prone animals, which did not develop disease. 68 In a follow-up study by the same group, artificial restoration of BH4 with sepiapterin in diabetic rats increased ACh-induced relaxation in the GTPCH-1-inhibited animals. 67 Similarly, in diabetic wild-type mice, endothelial superoxide levels were elevated, with associated oxidation of BH4 to BH2 and impaired vasodilation. The importance of GTPCH-1 was evident when in contrast, a transgenic clone in which GTPCH-1 was overexpressed showed that superoxide levels were decreased and BH4 levels and NO-mediated vasodilation were maintained compared with wild-type animals. 69 Inhibition of GTPCH-1 with 2,4-diamine-6-hydroxypyrimidine in rats yielded a 30 mmHg rise in BP compared with control rats. 66 Overexpression studies showed that the rise in BP in mice with salt-sensitive hypertension and also in mice undergoing myocardial ischemia/reperfusion injury was ameliorated by GTPCH-1.70,71 In hypertensive rats, clofibrate treatment was associated with activation of GTPCH-1, elevated BH4, and attenuation of hypertension. Correspondingly, superoxide and lipid peroxides were decreased in the same animals. 72
In humans, BH4 supplementation was shown to reverse smoking-induced impaired vasodilation, 73 improve endothelial function, and decrease arterial stiffness in estrogen-deficient postmenopausal women 74 and in older men. 75
Furthermore, localized oxidative stress is generated during an intermediary step where uncoupled eNOS and coupled eNOS in the same locale produce superoxide and NO, respectively, which interact to form peroxynitrite and can further deplete GSH 24 and perpetuate the condition. Endothelial cells treated with peroxynitrite exhibited reduced eNOS activity and disruption of eNOS dimers. 76 In ApoE-deficient mice, peroxynitrite formation from uncoupled eNOS was shown to be primarily responsible for oxidation of BH4 to BH2. 77 Furthermore, Li et al. also state that the oxidized form (BH2) can compete with BH4 for eNOS interaction as they share the same binding domain. However, since BH2 cannot support eNOS coupling, this displacement results in eNOS uncoupling. Therefore, under oxidative stress conditions, BH4-dependent eNOS function is reliant upon recycling mechanisms and de novo synthesis of BH4 by GTPCH-1. 78
KE1 (S-glutathionylation of eNOS) and KE3 (decreased BH4) to KE4 (eNOS uncoupling)
eNOS uncoupling is a phenomenon where the reductase domain and the oxygenase domain of the enzyme are no longer held together in an active dimer. BH4 is a highly redox-sensitive eNOS cofactor, which promotes the coupling of both domains, leading to efficient production of NO.79,80 Upon BH4 depletion, electron transfer from the C-terminal reductase domain to the N-terminal oxygenase domain is blocked, resulting in production of superoxide, as opposed to NO. 77 The molecular mechanisms of eNOS uncoupling have been extensively reviewed, with reduced BH4 bioavailability and S-glutathionylation of eNOS playing a critical role in the deleterious production of superoxide by uncoupled eNOS.81–83
It has been demonstrated in vivo and in vitro that eNOS is uncoupled when BH4 bioavailability is limited. The mechanism leading to BH4 depletion is generally attributed to oxidation of BH4 by ROS and/or peroxynitrite (the product of the chemical reaction between NO and superoxide), and supplementation with BH4 has been reported to restore eNOS activity.84–87 De Pascali et al. demonstrated the onset of eNOS uncoupling in vitro, which was due to a combination of S-glutathionylation of eNOS protein and reduced BH4 bioavailability. 27 Elevation of intracellular GSH by administration of N-acetylcysteine and coadministration of exogenous BH4 reversed hypoxia/reoxygenation-induced eNOS uncoupling. These findings were supported by Crabtree et al. in murine fibroblasts expressing mutated eNOS. 88 The authors concluded that both S-glutathionylation and BH4 deficiency—although functionally related—were mechanistically distinct events. However, a study in atrial tissue from human subjects demonstrated that eNOS activity was only restored following reversal of eNOS S-glutathionylation, while BH4 supplementation had no effect 36 (abstract only).
MIE (peptide oxidation) to KE5 (loss of AKT and eNOS)
eNOS activity was reduced by several stressors (methylglyoxal, high glucose, SIN-1, hydrogen peroxide, cigarette smoke) in human and bovine endothelial cells, resulting in increased ROS and decreased eNOS activity.23,30,44,89,90 Exposure of endothelial cells to CSEaq has been shown to induce oxidative stress, inhibit AKT/eNOS phosphorylation, and decrease production of NO.45,91 Oxidative stress was shown to derive from increased intracellular ROS production following CSEaq exposure, which inhibited eNOS activity and was reversible by N-acetylcysteine, a known ROS scavenger. 91
KE4 (eNOS uncoupling) and KE5 (loss of AKT and eNOS) to KE6 (NO depletion)
Taken together, the KEs leading to eNOS uncoupling and the loss of AKT/eNOS activity collectively led to a decrease in NO bioavailability. Multiple experiments demonstrated that eNOS uncoupling results in increased superoxide formation and decreased NO production in vitro with bovine aortic endothelial cells27,30,32 and isolated mouse aortas.12,32,76,85 Various stress inducers (ischemia, peroxynitrite, SIN-1, insulin, uric acid, orotic acidura, etc.) showed that a decrease in Akt and/or eNOS activity led to decreased NO in bovine, rat, and human endothelial cells and rat aortic rings.23,85,89,92–94
While many studies support KERs KE4>KE6 and KE5>KE6, studies also support the fact that secondary oxidative stress generated by superoxide production, as a result of eNOS uncoupling and subsequent peroxynitrite formation, can also lead to loss of eNOS activity and NO depletion. Exposure of human aortic endothelial cells to UFP of <200 nm, commonly found in airborne pollution, significantly reduced NO production. UFP-mediated reduction in NO production was restored in the presence of the NADPH oxidase inhibitor apocynin (indicating the presence of uncoupled eNOS), N-acetyl cysteine, and SOD mimetics (Tempol and MnTMPyP). The depletion of NO was associated with an increased GSH/GSSG ratio and eNOS S-glutathionylation, whereas overexpression of glutaredoxin-1 (which reverses S-glutathionylation of eNOS in the presence of GSH) attenuated the UFP-mediated reduction in NO production by nearly 80%, compared with overexpression of the LacZ control gene. 22 Given that the restoration by glutaredoxin-1 was partial, it is possible that oxidative damage to AKT and eNOS itself could account for the remaining NO production. This was specifically confirmed by Zou et al. who demonstrated a reduction in AKT phosphorylation, AKT activity, AKT-dependent eNOS phosphorylation, and eNOS activity following increasing concentrations of peroxynitrite (formed from uncoupled eNOS and NO) in bovine aortic endothelial cells. 76 Similar results were reported by Das et al. in the same cells utilizing SIN-1 as a source of peroxynitrite and the spin trap DMPO to recover eNOS function. 89
KE7 (NO depletion) to KE8 (impaired vasodilation)
In vivo, eNOS knockout mice develop hypertension, with mean arterial BP values that are 20–30 mmHg greater than values observed in wild-type animals, and such models are routinely used as models for the study of hypertension. 95 Aortic rings from eNOS mutant mice do not relax in response to acetylcholine, but function normally upon application of NO donors such as sodium nitroprusside (SNP). These results confirm a role for basal eNOS activity in BP regulation and vascular tone. 96
Du et al. showed that wild-type C57BL/6J mice fed a high-fat diet developed hypertension. 42 The aortic tissue harvested from these animals showed elevated expression of NOX2 and increased superoxide generation (confirmed by SOD ablation). Application of L-NG-nitroarginine methyl ester (L-NAME; a known inhibitor of eNOS) resulted in 20% reduction in superoxide production, indicating that most of the superoxide production originated from sources other than eNOS, such as NOX2 and other isoforms, but that uncoupled eNOS also contributed to the local superoxide levels. The elevated superoxide reduced phosphorylation of AKT and eNOS and impaired endothelial-dependent relaxation to acetylcholine (eNOS specificity confirmed by L-NAME coadministration). Superoxide was further implicated as a causal factor, in that application of an SOD mimetic restored acetylcholine-mediated vasorelaxation. In contrast, NOX2 knockout animals, animals treated with apocynin (an NOX2 inhibitor and ROS scavenger), and wild-type chow-fed animals showed none of these deleterious effects. 42 Scotland et al. studied the effects of various agonists and antagonists of vasodilation in the mesenteric resistance arteries of eNOS and cyclooxygenase 2 (COX-2) double knockout C57BL/6 mice. They reported that pharmacological blockade of eNOS and COX-2 individually had no effect on vasodilation, whereas coadministration of antagonists impaired vasodilation, and female mice developed hypertension. This was in contrast to male mice, in which acetylcholine-mediated vasodilation was abolished by eNOS blockade alone, inducing hypertension. The data indicate that gender differences could play an important role in vascular aging due to the increased importance of prostaglandin (PGI2) signaling in females. 59
Studies with aortic rings from hyperuremic rats showed that insulin-induced vasodilation was impaired relative to control animals treated with allantoxanamide ( uricase inhibitor). Furthermore, the hyperuremic animals developed hypertension, whereas animals treated with allopurinol to lower serum uric acid levels did not. Complimentary studies in HUVECs treated with increasing concentrations of uric acid confirmed that the in vivo effects were mediated by a corresponding decrease in PI3K/AKT/eNOS phosphorylation and reduced bioavailability of NO. 92 Similar findings were published by the same group following induction of hypertension in rats treated with orotic acid. 93
In humans, application of L-NAME and/or NG-monomethyl-L-arginine (L-NMMA) resulted in elevated mean arterial pressure through impairment of NO-mediated vasodilation. Application of both drugs compounded the effect. 97 A study in chronic smokers showed that smoking impairs FMD compared with nonsmoking subjects, respectively (5.6% ± 3.0% vs. 8.1% ± 3.7%; p < 0.01), administration of BH4 improved FMD in both cohorts (6.6% ± 3.3% vs. 9.8% ± 3.2%; p < 0.01), and smoking cessation for 1 week also improved FMD (from 5.0 ± 2.9 to 7.8% ± 3.2%; p < 0.01). These data indicate that improvement in arterial vasodilation was partially improved by BH4 administration. 98 Another study in which current smokers switched to an e-cigarette product for 1 week showed significant reductions in FMD impairment compared with baseline, which was associated with improvement in levels of 8-iso-prostagladin F2α (a biomarker of oxidative stress). 99
KE8 (impaired vasodilation) to KE9 (increased vascular resistance)
Reduction in blood vessel NO bioavailability has been reported to increase arterial BP in rat aortic rings and mesenteric arteries, mice, rabbits, and humans by a mechanism of impaired vasodilation.100–106 Where vasodilation is a key factor underlying vascular tone, impaired vasodilation leads to increased vascular resistance as the balance of vascular tone is shifted toward vasoconstriction. 5 SVR or TPR is the resistance against which the heart must pump to maintain blood tissue perfusion. Treatment with eNOS inhibitor L-NG-monomethyl arginine citrate (L-NMMA) caused an increase in SVR and a reduction in NO in human subjects,102,104,107–109 while L-NAME decreased NO-dependent relaxation and increased BP in rats. 102 In patients with malignant hypertension, treatment with the NO donor SNP, infused using a regimen in which dose was gradually increased to a maximum of 5 μg/(kg·min), was reported to decrease mean arterial pressure by ∼28% and SVR by ∼53%, indicating the potent effect of SNP (and NO) on systemic vasculature. Interestingly, the effects on cerebral vascular resistance were much less pronounced with ∼7% reduction in SV. 110 Similarly, intravenous infusion of increasing doses of SNP was also shown to decrease SVR in a dose-dependent manner in patients with other cardiovascular complications.111–113
Impaired vasodilation and increased peripheral resistance are associated with vascular remodeling, arterial stiffening, and vascular aging, all of which are key contributors to the development of hypertension.114–116
KE9 (increased vascular resistance) to AO (hypertension)
It is well established that increased vascular resistance contributes to the onset of hypertension through a mechanism of vascular remodeling and arterial stiffening over time, although the true extent is unknown.114–116
Several human studies showed a dose-dependent change in SVR and BP, following treatment with eNOS inhibitors.32,101,103,107–109 In patients with hypertension, SVR was elevated in almost 66% of cases. 117 It was reported that high normal BP (130–139/85–89 mmHg) progresses to stage 1 hypertension (>140/>90 mmHg) in 37% of individuals under the age of 64 and in 49% of individuals over the age of 65. In patients with hypertension, the 10-year risk of a cardiovascular-related event (i.e., coronary or cerebrovascular) was 10% in males and 4.4% in females. 116
Overall Assessment of the AOP
KE essentiality
Supporting evidence for KE essentiality, KER biological plausibility and empirical evidence are explained in detail in the subsections above and summarized in Tables 1–3, respectively. We rated essentiality as high for most KEs due to the existence of experimental evidence demonstrating that blocking them would prevent or attenuate the downstream KEs. KEs 7 and 9 were rated as moderate due to the influences of other major pathways/compensatory systems on downstream KEs discussed above.
AO, adverse outcome; eNOS, endothelial nitric oxide synthase; KE, key event; MIE, molecular initiating event; NO, nitric oxide; ROS, reactive oxygen.
KERs, key event relationships.
KER biological plausibility
There is extensive published literature to support the biological plausibility of the KERs in vitro, in vivo, and from human clinical studies and the overall mechanisms are well understood. In light of this, we have rated the strength of all KERs as high, with respect to plausibility.
KER empirical evidence
Direct quantitative evidence supporting the KERs is lacking in some aspects of the AOP. However, many of the cited studies do measure multiple KEs in concert and evidence is derived by association of toxic effects and reversibility by chemical interventions to recover the pathway. For example, with respect to the KER MIE > KE1 while many studies demonstrate dose-dependent glutathione oxidation in response to chemical stressors, studies which subsequently used GSSG specifically to induce eNOS S-glutathionylation are limited. The linkages are inferred by the use of chemicals, which induced S-glutathionylation (hence derived from elevated intracellular GSSG, which was not measured), and investigated the effects of GSSG-eNOS adduct removal on downstream KEs. Similarly, the KER MIE to KE2 is rated as moderate due to the lack of quantitative chemical stressor data, although some consistent data are available. Supporting evidence is derived from the use of ischemia/reperfusion injury as a stressor, which does induce vascular oxidative stress, but cannot be quantified. KERs (KE6 > KE7), (KE7 > KE8), and (KE8 > AO) are also rated as moderate due to the possibility of interaction with compensatory pathways described earlier. Furthermore, the long-term effects of such mechanisms (for example, COX-mediated prostaglandin and hydrogen peroxide compensatory vasodilation) are not well understood over years of life. Ratings and justifications are also shown in Table 3.
Conclusion
In summary, the weight of evidence for a role of each of the KEs described in this AOP and their inter-relationships in the development of hypertension over time are strong. However, as Brandes states, “to assume that endothelial vasodilator release through an increase in peripheral resistance directly translates into hypertension would be naïve”. 115 Hypertension is a very complex disorder, in which in addition to endothelial dysfunction, multiple organs/tissues (e.g., kidney, heart, brain, central/peripheral innervations, and blood vessel smooth muscle) and their associated chemical mediators also play a key role. That said, what is described here does represent the perturbation of one of the most important protective mechanisms for the maintenance of healthy blood vessel function and systemic tissue perfusion by vascular oxidative stress. Where peptide oxidation is induced by chronic chemical exposure, either intentional or otherwise, is of toxicological significance for regulatory purposes. The efficacy of health-promoting/harm-reducing interventions (e.g., health supplements), which purport to improve vascular health and alternatives to commonly used consumer products (e.g., smokers switching from combustible cigarettes to electronic cigarettes), could be assessed by using an integrated testing strategy built upon the foundations of this AOP. Finally, from the ethical perspective, the evidence presented to support this AOP demonstrates that the KEs can be measured using in vitro test systems and human clinical data in concert. While existing animal data remain an important resource to support the weight of evidence assessment of the AOP, the wealth of in vitro and human data presented in this article suggest that future testing strategies focused on nonanimal test systems are achievable.
Footnotes
Acknowledgments
The authors wish to thank Ruth Dempsey and Christopher Proctor for their helpful discussions and critical review during the preparation of this manuscript.
Author Disclosure Statement
F.J.L., L.E.H., and M.G. are full-time employees of British American Tobacco (Investments) Ltd. K.L., M.T., and J.H. are full-time employees of Phillip Morris International. V.H. was employed at Selventa at the time this project was undertaken, and Selventa received financial compensation from PMI for their services. The authors declare no further conflicts of interest with respect to the research, authorship, and/or publication of this article.