
Abstract
Combination antiretroviral therapy fails in complete suppression of HIV-1 due to drug resistance and persistent latency. Novel therapeutic intervention requires knowledge of intracellular pathways responsible for viral replication, specifically those untargeted by antiretroviral drugs. An understudied phenomenon is the nucleolar localization of Rev phosphoprotein, which completes nucleocytoplasmic transport of unspliced/partially spliced HIV mRNA through multimerization with intronic cis-acting targets—the Rev-response element (RRE). Rev contains a nucleolar localization signal (NoLS) comprising the COOH terminus of the arginine-rich motif for accumulation within nucleoli—speculated as the interaction ground for Rev with cellular proteins mediating mRNA-independent nuclear export and splicing. Functionality of Rev nucleolar access during HIV-1 production and infection was investigated in the context of deletion and single-point mutations within Rev-NoLS. Mutations induced upon Rev-NoLS are hypothesized to inactivate the HIV-1 infectious cycle. HIV-1HXB2 replication ceased with Rev mutations lacking nucleolar access due to loss or replacement of multiple arginine residues. Rev mutations missing single arginine residues remained strictly nucleolar in pattern and participated in proviral production, however, with reduced efficiency. Viral RNA packaging also decreased in efficiency after expression of nucleolar-localizing mutations. These results were observed during propagation of variant HIV-1NL4-3 containing nucleolar-localizing mutations within the viral backbone (M4, M5, and M6). Lentiviral particles produced with Rev single-point mutations were transducible at extremely low frequency. Similarly, HIV-1NL4-3 Rev-NoLS variants lost infectivity, unlike virulent WT (wild type) HIV-1NL4-3. HIV-1NL4-3 variants were capable of CD4+ host entry and reverse transcription as WT HIV-1NL4-3, but lacked ability to complete a full infectious cycle. We currently reveal that viral integration is deregulated in the presence of Rev-NoLS mutations.
Introduction
A
The “A” conformation involves Rev binding sites IIB and IA that compose the opposite legs of the “A” shape. 8 Arginine-rich motif (ARM) of Rev monomer binds IIB with high affinity, 9 –11 exposing a dimerization interface for a second Rev monomer to bind. 12 Rev monomers achieve multimerization throughout the RRE, 13,14 mainly within the nucleolus. 15 Nuclear export of Rev-RRE RNP (ribonucleoprotein) complex occurs through the Rev nuclear export signal (NES) interaction with chromosomal region maintenance 1 (CRM1) 16,17 and GTP-bound Ran, 18 leading to accumulation of intron-containing unspliced (9 kb full-length genome containing gag/pol) and partially spliced (4 kb fragment containing vif, vpr, vpu, and env) HIV transcripts in cytoplasm for viral protein expression and viral particle assembly. 19,20
HIV-1 transcripts were previously identified in the nucleolus of infected peripheral blood mononuclear cells (PBMC) during electron microscopy and in situ hybridization with probes complementary to gag, env, and nef. 21 A nucleolar therapeutic model was hence developed against HIV-1 production, utilizing a hammerhead ribozyme placed within U16 small nucleolar RNA platform (U16Rz) and targeted against conserved motifs within HIV-1 5′LTR. U16Rz was expressed in HeLa T4, where nucleolar specificity was observed. Upon HIV challenge, nucleolar U16Rz strongly suppressed HIV-1 production, 22 selectively cleaving unspliced and partially spliced transcripts that traversed through the nucleolus to the sequestered ribozyme. Similarly, modified U16Rz targeting both U5 and gag (RzC36) significantly reduced viral production in CEM during HIV challenge. 23
The potent catalytic activity of nucleolar-specific U16Rz against HIV RRE-containing transcripts suggests an HIV-1 nucleolar pathway facilitated through Rev nucleocytoplasmic activity.
The dynamic and multifunctional nucleolar proteome enables ribosome subunit biosynthesis, cell cycle control, apoptosis, DNA replication/repair, RNP biogenesis, and stress response within subnucleolar compartments—fibrillar centers, dense fibrillar components (DFC), and outermost granular components (GC). 24,25
Viral pathogenesis induces cellular stress, leading to transformation in nucleolar proteomic morphology as nucleolar-accessible viral proteins recruit replication factors. 26,27 Similar to HIV-1 Rev, the following single-strand RNA viruses express nucleolar RNA binding proteins of diverse functionality: severe acute respiratory coronavirus nucleocapsid (N) harbors an NES predicted in nucleocytoplasmic shuttling activity 28 ; dengue virus core protein facilitates encapsidation and potentially modulates transcription 29 ; and the Semliki Forest virus nonstructural protein (nsP2) regulates viral mRNA synthesis. 30
Although nucleolar trafficking is prevalent in many viral infectious models, the purpose of this localization pattern is unknown. In the case of HIV-1, the nucleolus may serve as the interaction ground for Rev with cellular proteins that facilitate rapid mRNA nuclear export and protect HIV mRNA from spliceosomal complexes. 31
Rev subnucleolar localization within DFC and GC 32 occurs through a nucleolar localization signal (NoLS) 45 WRERQRQ 51 , directly downstream of the nuclear localization sequence (NLS) 34 TRQARRNRRRR 44 within Rev ARM. 33,34 Rev nucleolar localization was originally demonstrated to occur through amino acids 40–45 (NRRRRW) within the Rev NLS. 35 Studies by Cochrane et al. later identified amino acid residues within 35–50 of the Rev ARM as vital for nucleolar accumulation. 34 Using site-directed mutagenesis to alter amino acids surrounding the Rev NLS and indirect immunofluorescence, Rev localization of mutations lacking amino acids 48–51 (RQRQ) maintained nucleolar accumulation in the absence of HIV production.
However, when deletions were expanded to residues WRE, resulting in complete elimination of WRERQRQ (amino acids 45–51), nuclear accumulation was observed. 34 Although a potential Rev-NoLS was identified at amino acids 45–51 downstream of the NLS, functionality of Rev nucleolar mutations was not investigated in the context of HIV-1 production and infection.
To characterize Rev nucleolar function, HIV-1 production and infection were examined in the presence of Rev nucleolar mutations. We currently reveal that Rev nucleolar access is necessary for the completion of the HIV-1 infectious cycle. Nucleolar activity allows efficient HIV-1 mRNA splicing and nucleocytoplasmic transport. In the background of Rev nucleolar mutations, the frequency of viral integration increases dramatically in comparison to WT (wild type) HIV-1. Extreme integration frequencies result in cell death and the arrest of the HIV-1 infectious cycle. We further demonstrate the loss of Rev interaction with B23 as a result of nucleolar mutations, and discuss the involvement of B23 in other viral infectious models requiring nucleolar access for infectivity.
Materials and Methods
Cell culture
HeLa cells containing stably integrated copies of a Rev-deficient HIV-1HXB2 molecular clone (HLfB) were obtained from the NIH AIDS Research and Reference Reagent Program (#1300). HLfB, HeLa (#CCL-2; American Type Culture Collection), CD4+ HeLa (T4), human embryonic kidney 293T (HEK293T), and human fibrosarcoma (HT1080) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM
Site-directed mutagenesis of Rev-NoLS
CMV-driven Rev-EGFP fusion within pcDNA platform 36 was a template for QuickChange II site-directed mutagenesis (Agilent Technologies). Primer sequences were designed according to mutations of interest within Rev-NoLS (Table 1). Primer sequences in Table 1 were also used to generate 3′Flag-tagged Rev mutations within pcDNA3.1. Single-point mutations and deletions were confirmed through DNA sequencing.
WT, wild type.
Site-directed mutagenesis of pNL4-3 Rev-NoLS
The Rev-NoLS fragment (∼1,200 bp) was excised from pNL4-3 (NIH AIDS Research and Reference Reagent Program #114) using NheI and BamHI restriction digests and ligated into pcDNA3.1. The restriction fragment was a template for site-directed mutagenesis using primer sets (Table 2) designed to establish mutations of Rev-NoLS M4, M5, M6, and M8 specifically. Mutant-positive fragment was excised and reintroduced into the pNL4-3 backbone.
DNA sequencing was used to confirm the presence of each Rev-NoLS mutation and deletion. WT pNL4-3 contains reintroduced, nonmutated Rev-NoLS restriction fragment, ligated alongside single-point mutant fragments into pNL4-3. Bacterial colony PCR screens using forward primers specific to each Rev-NoLS mutation (Table 3) in combination with Env reverse primer 5′-CGAATAGCTCTATAAGCTGCTTGTAA-3′ were used to amplify the following mutant clones: pNL4-3 M4; pNL4-3 M5; and pNL4-3 M6. DNA sequencing was used to confirm the presence of mutation cDNA fragments within pNL4-3.
Nucleotides bolded in black represent each single-point mutation present within the mutated clone. Nucleotides bolded in gray represent the region located directly downstream of the M8 RQ deletion. List of primer sets used in mutant pNL4-3 bacterial colony PCR screen.
PCR, polymerase chain reaction.
Restriction digest sets—BbvCI with AhdI and DraII with PstI—were used to analyze WT, pNL4-3 M4, and pNL4-3 M8 for chromosomal rearrangement using original pNL4-3 plasmid as a control. Restriction digest sets—BbvCI with AhdI and DraIII with PstI—were used next to analyze pNL4-3 M5 and pNL4-3 M6. All pNL4-3 mutations chosen to propagate viral particles were subjected to a final analysis for chromosomal rearrangement with three restriction digest sets—PstI and DraIII, BbvCI and AhdI, and NdeI and AhdI (Supplementary Fig. S3A–C). Mutant plasmids matching in digestion fragment pattern with WT pNL4-3 were used to propagate HIV-1NL4-3 particles.
Confocal microscopy
HLfB and HeLa (1 × 105 cells) were cultured onto 0.01% poly-
Cells were washed in triplicate with 1× phosphate-buffered saline (PBS) and subjected to 1-h incubation in Alexa Fluor 555 goat-anti-mouse IgG1 diluted to 1:100. Cells were rinsed and stained with 4′,6-diamino-2-phenylindole (DAPI, 1 μg/ml; Sigma-Aldrich) for 5 min. Glass cover slips were mounted onto cells using Prolong Gold anti-fade reagent (Invitrogen) and stored at 4°C in darkness. Images were taken with a Zeiss Upright LSM510 2-Photon Microscope at 63 × magnification in oil immersion and 1.7× zoom.
HIV-1HXB2 production
HLfB cells (1 × 106 cells) were grown in six-well tissue culture plates to 50% confluency and transfected with 5 μg of vector plasmid containing Rev-EGFP or Rev-3′Flag (WT and mutations) using the CaCl2 method. Supplemented DMEM (1 ml) was replaced 5 h later. Viral-containing supernatant was collected 48 h post-transfection and centrifuged at 1,000 rpm for 5 min to remove cell debris. Viral supernatant was quantified for p24 capsid protein using HIV-1 p24 ELISA (PerkinElmer Life Sciences, Inc.).
HIV-1HXB2 RNA packaging
Viral particles generated in the presence of Rev-NoLS mutations were analyzed for packaged RNA using Quantigene 2.0 bDNA detection (Affymetrix–Panomics Solutions), using probe sets specific to highly conserved regions within HIV-1 vif (#SF-10183-01; Affymetrix, Inc.).
HIV-1NL4-3 infection
HIV-1NL4-3 cDNA harboring Rev-NoLS mutations was transfected into HEK293T (50% confluent) with CaCl2 (1 μg/well) within 12-well culture plates. Viral supernatant was collected 48 h later, cleared of cell debris, and quantified for p24 production. Viral supernatant normalized to 2 ng p24 was used to infect JLTRG-R5 precultured at 1 × 106 cells per well. HIV-1-exposed cells were washed thrice in 2× HBSS 24 h after infection and resupplemented with RPMI media. To assess infectivity of mutant HIV-1NL4-3 variants, cells were collected every 5 days, stained with LIVE/DEAD aqua dead cell stain (Life Technologies), fixed in 3.7% paraformaldehyde, and measured for aqua fluorescence (405 nm excitation) with CyAn ADP 9 Color FACS analysis. Live cells were analyzed for HIV-1 infection through EGFP emission (488 nm excitation).
PCR detection of total HIV and 2-LTR circle DNA
Supernatant of HIV-1NL4-3—WT and Rev-NoLS mutations—was normalized to 20 ng capsid p24 for infection of JLTRG-R5 populated at 1 × 106 cells per well of 12-well culture plates. Infected cells were washed through suspension in 2× HBSS, thrice, 24 h after viral exposure and replenished with RPMI media. Cells were harvested every 5 days for genomic DNA extraction using QIAamp DNA Mini kit (Qiagen). Amplification of total HIV-1 and 2-LTR circle DNA was achieved using 1 μg genomic DNA subjected to PCR primer sets (0.5 μM in final volume) and annealing conditions described. 37
Positive control fragments were derived through PCR amplification of genomic DNA from HIV-infected CEM (total HIV DNA) and infected CEM treated with integrase inhibitor 118-D-24 (2-LTR circle) (NIH AIDS Research and Reference Reagent Program #9957, 20 μm in final volume). Primer sets specific to β-globin gene—Forward 5′-CCC TTG GAC CCA GAG GTT CT-3′ and Reverse 5′-CGA GCA CTT TCT TGC CAT GA-3′—were used as amplification and gel loading controls under identical annealing conditions alongside total HIV-1 and 2-LTR circle primer sets.
Lentiviral transduction
Using CaCl2, the following HIV-1-based self-inactivating (SIN) vector and lentiviral packaging plasmids were co-transfected into HEK293T at 50% confluency: 15 μg pHIV7/C-GFP 38 ; 15 μg pCHGP-2; 5 μg pCMV-G; and 5 μg pCMV-Rev—WT or NoLS-mutation. Lentiviral supernatant was collected and concentrated as previously mentioned. 39 Five hundred picograms p24 supernatant was used to transduce, through polybrene transduction (4 μl/ml), HT1080 cells grown to 1 × 105 cells per well in 12-well plates. Transduced cells were collected 48 h post-transduction, fixed in 3.7% paraformaldehyde, and EGFP reporter quantified using CyAn ADP 9 Color FACS analysis.
Northern blot analysis of HIV-1 mRNA splicing
Total RNA from HEK293T transfected with 5 μg HIV-1 pNL4-3 (50% confluent in 10 cm culture plates), WT and Rev-NoLS variants, was collected 48 h later using RNA STAT-60 (Tel-Test, Inc.) according to the manufacturer's instructions. Forty micrograms total RNA was electrophoresed on 1% agarose/formaldehyde, ethidium bromide-stained gels, and transferred onto Hybond –N nucleic acid membrane (GE Healthcare).
RNA membrane was ultraviolet-crosslinked and blocked in 1× PerfectHyb Plus hybridization buffer (Sigma) for 1 h at 70°C. HIV RNA was detected using PCR-amplified nef fragment originating from nucleotide positions 8475 to 890040 of pNL4-3. Amplification was accomplished with the following primer sets: forward primer (anneals upstream of nef and introduces the unique restriction site BamHI
Before overnight hybridization, nef probes were radiolabeled with [α-
32
P] dCTP using the Rediprime II DNA labeling system (GE Healthcare). Blots were rinsed afterward in wash buffer containing 2× saline sodium citrate (SSC) and 0.1% sodium dodecyl sulfate (SDS) for 10 min, thrice, and finally in wash buffer containing 1× SSC and 0.1% SDS for 10 min. Radiolabeled RNA membrane was exposed to BioMax MS film (Kodak) overnight and developed for detectable RNA fragments. Relative density of each spliced fragment was analyzed using ImageJ software (
EGFP-Vpr infectious transfer for host cell entry
HIV-1NL4-3 cDNA (1 μg WT or Rev-NoLS mutation) were co-expressed with 1 μg pEGFP-Vpr (NIH AIDS Research and Reference Reagent Program #11386) in HEK293T (50% confluent) within six-well culture plates. CaCl2 transfection media were replaced with 1 ml supplemented DMEM 5 h later. Viral supernatant was collected 48 h afterward and quantified for p24 levels. One microgram p24 viral supernatant was used to infect 70% confluent CEM and HeLa T4 seeded in 12-well culture plates for 1.5 h. Cells were cleared of virus with three rinses of 1× PBS, centrifugation at 1,000 rpm, and preserved in 3.7% paraformaldehyde. EGFP positivity was quantified using FACS.
RT assay
Viral supernatant containing WT or mutant HIV-1NL4-3 was harvested 48 h after production in HEK293T cells. Supernatant was lysed to release intraviral RT. RT activity was measured as described in the Reverse Transcriptase Assay, colorimetric kit (Roche).
Cre-loxP lentiviral dual reporter
HEK293T cells were grown in 10 cm tissue culture plates to 50% confluency. SIN lentiviral backbone containing LoxP sites flanking CFP reporter with RFP reporter directly downstream (pBB-XCXR) was co-transfected (15 μg) with packaging plasmids described. 39 Transfection media were replaced with 6 ml supplemented DMEM and 60 μl (0.6 M) butyric acid 5 h post-transfection. Lentiviral supernatant was collected 48 h later and titrated through polybrene transduction (4 μl/ml) of HT1080 (1 × 105 cells/well in 12-well plates).
A portion of transduced HT1080 was collected 5 days later, fixed in 4% paraformaldehyde, and CFP transduction multiplicity of infection of one viral copy per cell established through CyAn ADP 9 Color FACS. This HT1080-XCXR population was propagated and sorted based on CFP positivity for stable BB-XCXR reporter cells using BD AriaII SORP (COH Analytical Cytometry Facility).
Cre-loxP recombination reporter cells (XCXR) were transduced with 10 μg Cre-Vpr lentivirus (CVITG–Cre-Vpr-IRES-Tat-GFP backbone) produced in HEK293T with WT or Rev-NoLS mutations. CFP transition to RFP from lentiviral transduction, in addition to GFP fluorescence from integration, was assessed with BD Fortessa Cytometer (COH Analytical Cytometry Facility) after preservation in 3.7% paraformaldehyde 7 days post-transduction. Integrase inhibitor-118-D-24 (20 μm in final volume) and RT inhibitor—AZT (NIH AIDS Research and Reference Reagent Program #3485, 10 μm in final volume)—served as negative controls for CVITG reverse transcription and integration. HT1080 (−) and XCXR (−) reporter cells were used as negative controls for RFP, GFP, and CFP expression.
Quantification of integrated proviral DNA by Alu-gag PCR
Integrated viral DNA quantification was performed using Alu-gag PCR as described previously. 41 –45 Total genomic DNA was isolated from uninfected PBMC and HIV-infected cells using QIAamp DNA mini kit (Qiagen). The amount of isolated human genomes in the infected samples was determined by β-globin PCR using iTaq universal probe supermix (Bio-Rad). The PCR reactions were performed on CFX96 Real-Time PCR Detection System (Bio-Rad) for 40 cycles at following conditions: 95°C for 15 s and 60°C for 30 s. The primer sequences are as follows: β-globin FP 5′-CCC TTG GAC CCA GAG GTT CT-3′ and β-globin RP 5′-CGA GCA CTT TCT TGC CAT GA-3′. The genomic concentrations of samples were calculated by using a standard curve obtained from known dilution of PBMC DNA.
The genomic DNA samples were then diluted to 2 μg/ml. Quantification of integrated DNA was performed by two-step Alu-gag nested PCR. The first step reaction was performed using OneTaq 2x Master Mix (New England Biolabs) on C1000 Thermal Cycler (Bio-Rad) at following conditions: 95°C for 30 s, 40 cycles of 95°C for 15 s, 50°C for 15 s, and 68°C for 3.5 min. The primer sequences are follows: Alu FP -GCC TCC CAA AGT GCT GGG ATT ACA G-3′ and gag RP 5′-GTT CCT GCT ATG TCA CTT CC-3′.
The product from the first step reaction was then subjected to the second step quantitative PCR (qPCR) to quantify integrated viral DNA. The qPCR reaction was performed on CFX96 Real-Time PCR Detection System (Bio-Rad) using iTaq universal probe supermix (Bio-Rad) for 40 cycles at following conditions: 95°C for 15 s and 60°C for 30 s. The primer sequences are follows: RU5 FP 5′-TTA AGC CTC AAT AAA GCT TGC C-3′ and RU5 RP 5′-GTT CGG GCG CCA CTG CTA GA-3′. Copy number of integrated viral DNA was calculated using a standard curve obtained from integration standard. 41 –45
Immunoprecipitation
HLfB was grown to 50% confluency in six-well culture plates and CaCl2 transfected with 5 μg Rev-3′Flag cDNA per well (WT and Rev-NoLS mutations). Total protein lysates were collected 48 h later with lysis buffer (50 mM Tris-HCl pH 8.0, 137 mM NaCl, and 1% Triton-X 100) treated with protease inhibitor cocktail (Roche). One milligram per milliliter protein lysate was subjected to IP with 25 μl prerinsed anti-Flag M2 affinity gel (Sigma-Aldrich) in lysis buffer for 2 h, rotating at 4°C. Flag beads were rinsed afterward 3× in lysis buffer and boiled at 95°C for 10 min in 50 μl 2× sample loading buffer (without β-mercaptoethanol). Eluates were separated from anti-flag gel and analyzed through Western immunoblotting.
Western immunoblot
Protein eluates were subjected to an SDS-polyacrylamide gel electrophoresis (SDS-PAGE) 15% acrylamide gel run, transferred onto PVDF transfer membrane (GE Healthcare), and incubated in blocking solution (5% milk with 1× TBS and 0.1% Tween—1× TBST) for 1 h at room temperature. Membranes were submerged in primary antibodies diluted with blocking solution—B23 mouse monoclonal IgG1 (1:500, #sc-47725; Santa Cruz Biotechnologies)—and anti-Flag M2 mouse monoclonal IgG1 (1:1,000, #F3165; Sigma-Aldrich) overnight at 4°C and rinsed 3× in 1× TBST.
Membranes were incubated in secondary goat-anti-mouse IgG1-horseradish peroxidase (1:5,000; Santa Cruz Biotechnology) diluted in blocking solution for 1 h at room temperature, rinsed 3× with 1× TBST, and protein bands developed in enhanced chemiluminescence substrate (Pierce) for 5 min. Protein chemiluminescence was detected upon exposure to BioMax MS film (Kodak) at varying time points, and relative density of protein signals was analyzed using ImageJ.
Results
Rev-NoLS requires two arginine residues for strict nucleolar access during HIV-1 production
Rev mutations lacking four amino acid residues from the NLS C-terminal end ( 48 RQRQ 51 ) were previously demonstrated to maintain nucleolar localization pattern in COS-7 cell lines; deletion mutations that extended three residues upstream ( 45 WRERQRQ 51 ) exhibited nucleolar mislocalization. 34 We replicated these nucleolar mutations and examined localization pattern in the presence of HIV production. Rev-NoLS mutations containing amino acid deletions or single-point substitutions of each arginine residue within NoLS— 45 WRERQRQ 51 —were generated and fused to enhanced green fluorescent protein (EGFP). Rev-EGFP fusion variants were examined for intracellular localization pattern within Rev-deficient HIV-1HXB2 during proviral production (HLfB, Fig. 1).
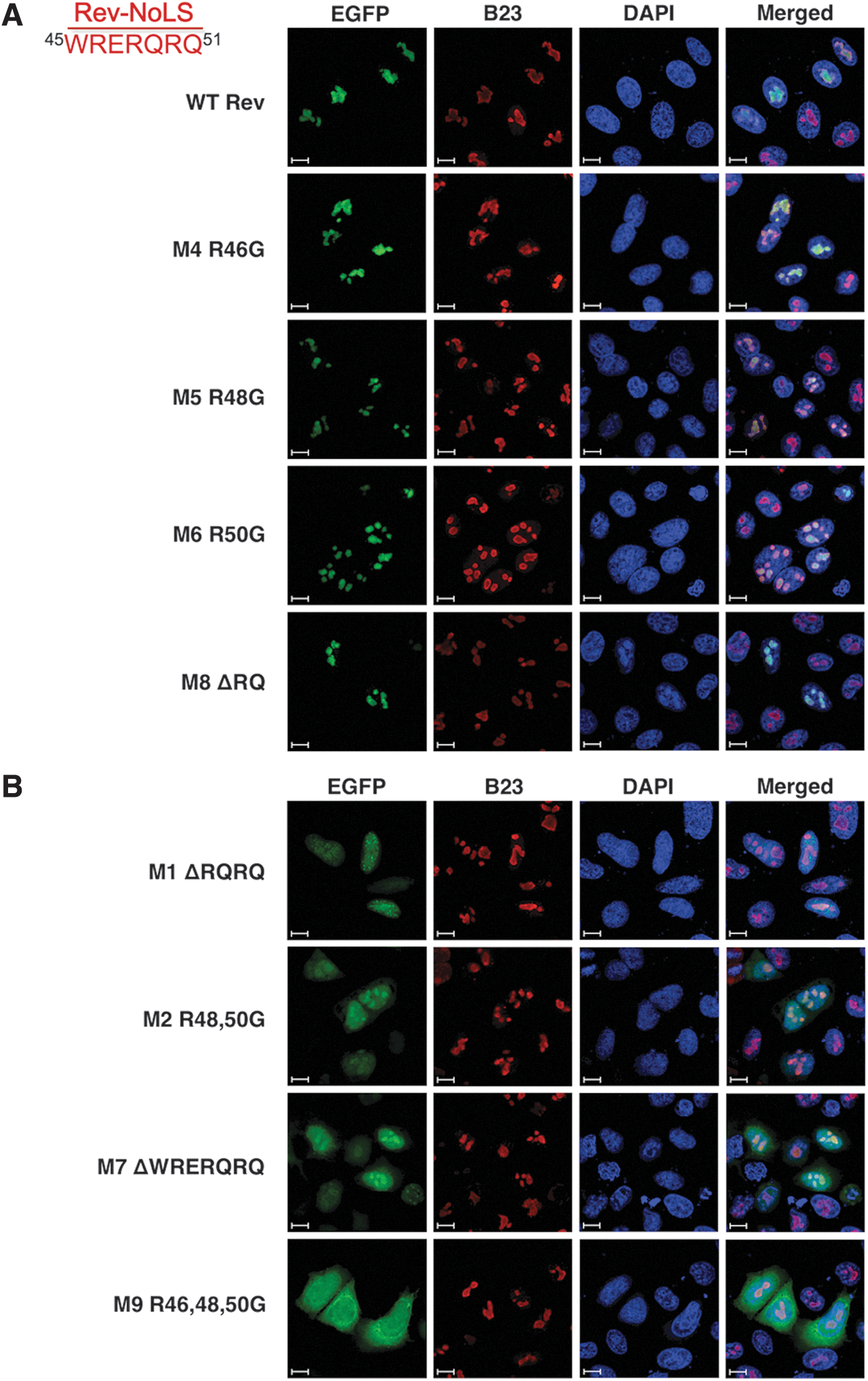
Localization of Rev-NoLS mutations in the presence of HIV-1HXB2 production. HLfB cells stably expressing Rev-deficient HIV-1HXB2 provirus were transfected with Rev-EGFP mutations (green), fixed, immunostained with anti-B23 nucleolar control (red), and treated with DAPI nuclear stain (blue). Rev-EGFP fusion mutations were examined for intracellular localization pattern and captured at 63× magnification, oil immersion 1.7× zoom, and represented against 10 μm scale bars.
Localization pattern of Rev-EGFP fusion variants was also examined in the absence of viral production (HeLa, Supplementary Fig. S1; Supplementary Data are available online at
In the absence of HIV-1, the nucleolar patterns of M4, M5, M6, and M8 were similar, however, included subnuclear distribution outside the nucleolus (Supplementary Fig. S1A). Rev-NoLS maintaining one arginine residue after deletion (M1-ΔRQRQ) and glycine substitution (M2-R48 and 50G) allowed nucleolar entry and caused nuclear dispersion of Rev (Fig. 1B). Similarly, M1 and M2 localized within nuclei and nucleoli in the absence of HIV-1 (Supplementary Fig. S1B). Rev-NoLS missing all arginine residues through deletion (M7-ΔWRERQRQ) caused nuclear dispersion during HIV-1HXB2 production (Fig. 1B) and intracellular distribution without HIV-1 (Supplementary Fig. S1B). Glycine substitution of all arginine residues (M9-R46, 48, and 50G) resulted in nucleocytoplasmic distribution of Rev with HIV-1 (Fig. 1B) and nuclear localization without HIV-1 (Supplementary Fig. S1B).
Functionality of Rev-NoLS mutations in HIV-1HXB2 production and RNA packaging
Functionality of Rev-NoLS mutations in the rescue of Rev deficiency during HIV-1HXB2 production was investigated in HLfB, which upon expression of WT Rev generates viral particles. 46 Each Rev-EGFP NoLS variant was introduced into HLfB cells and viral particles collected 48 h afterward. Viral output of HIV-1 p24 capsid protein was quantified with enzyme-linked immunosorbent assay (ELISA) antigen capture assays (Fig. 2A).
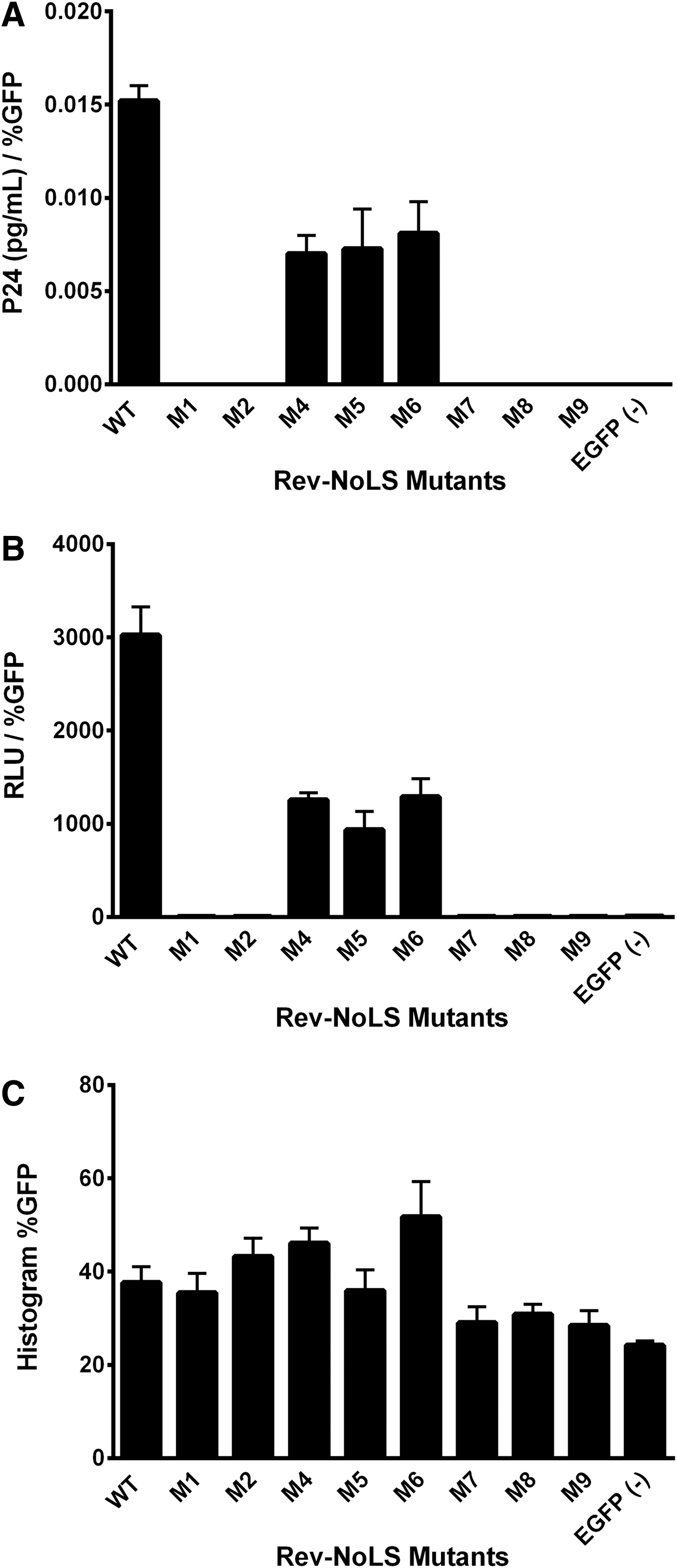
Functionality of Rev-NoLS mutations in HIV-1HXB2 proliferation and RNA packaging. Rev-EGFP mutations were expressed within HLfB cells stably expressing Rev-deficient HIV-1HXB2 provirus. Viral supernatant was collected 48 h later and subjected to ELISA p24 antigen capture assays for the quantification of viral production. Viral supernatant was also quantified for the presence of HIV-1HXB2 RNA using colorimetric bDNA assays incorporating probes specific to vif. In this system, WT Rev rescues Rev deficiency and leads to HIV-1HXB2 proviral production
Rev-NoLS mutations exhibiting non-nucleolar localization pattern in the nucleus and cytoplasm (M7 and M9) did not rescue viral production. Simultaneous nuclear and nucleolar access were also not sufficient for the rescue of viral production (M1 and M2).
Single-point mutations retaining strict nucleolar access (M4, M5, and M6) rescued viral production, although with reduced efficiency compared to WT. Rev-NoLS M8, which contains a deletion of two amino acid residues—arginine and glutamine (ΔRQ)—was strictly nucleolar, yet nonfunctional. p24 (pg/ml) was normalized to EGFP transfection efficiency (Fig. 2C).
Functionality of Rev-NoLS mutations was examined without EGFP fusion. Rev-NoLS mutations containing a 3′Flag tag were expressed in HLfB and viral supernatant collected 48 h later. Viral productivity through HIV-1 p24 capsid protein was quantified with ELISA antigen capture assays.
Similar results were observed without the EGFP tag (Supplementary Fig. S2A). EGFP fusion and the addition of the 3′Flag tag to Rev-NoLS mutations did not alter the function of Rev.
Rev mutations were next tested for ability to package viral RNA using branched DNA (bDNA) colorimetric assays and probes specific to HIV-1 vif. Each Rev-EGFP NoLS variant was introduced into HLfB cells and viral particles collected 48 h afterward. Intraviral RNA was detected in particles produced with nucleolar-localizing Rev mutations (M4, M5, and M6), however, with less efficiency than WT Rev (Fig. 2B). Nucleolar-specific M8, in addition to all other nuclear and cytoplasmic mutations, did not participate in RNA packaging during HIV-1HXB2 production. Intraviral RNA, represented as chemiluminescent relative light units, was normalized to EGFP transfection efficiency (Fig. 2C).
Rev-NoLS mutations without EGFP fusion were examined for ability to package intraviral RNA. Rev-NoLS mutations containing a 3′Flag tag were expressed in HLfB and viral supernatant collected 48 h later. Intravirally packaged RNA was measured using bDNA colorimetric assays with probes specific to vif. Similar to Rev-EGFP, only nucleolar-localizing single-point mutations (Rev-NoLS M4, M5, and M6), except Rev-NoLS M8, were able to package intraviral RNA (Supplementary Fig. S2B). Rescue in RNA packaging was not as efficient as WT Rev-3′Flag. Rev mutations shown to achieve simultaneous nuclear/nucleolar localization (Rev-NoLS M1 and M2) or cytoplasmic localization (Rev-NoLS M7 and M9) lost functionality in intraviral RNA packaging. EGFP fusion and 3′Flag tag addition did not alter the ability of Rev-NoLS mutations to package intraviral RNA.
HIV-1NL4-3 harboring Rev nucleolar single-point mutations is unable to complete a full infectious cycle
Mutations corresponding to Rev-NoLS M4, M5, M6, and M8—all nucleolar in pattern—were induced within HIV-1NL4-3 cDNA (Supplementary Fig. S3) and used to propagate virus from HEK293T. Viral supernatant was collected 48 h later and quantified for p24 capsid through p24 ELISA antigen capture assays. Single-point mutations within HIV-1NL4-3 backbone (M4, M5, and M6) did not prevent viral production and the release of p24 (Fig. 3A). In comparison to WT HIV-1NL4-3, viral production of Rev-NoLS mutants decreased. Supernatant (20 ng p24) was subjected to bDNA assays using vif probe specific to packaged genomic RNA. Single-point mutations within HIV-1NL4-3 backbone did not prevent genomic RNA packaging within viral particles (Fig. 3B). In comparison to WT HIV-1NL4-3, M8NL4-3 was unable to produce virus or package genomic RNA as efficiently as WT HIV-1NL4-3.
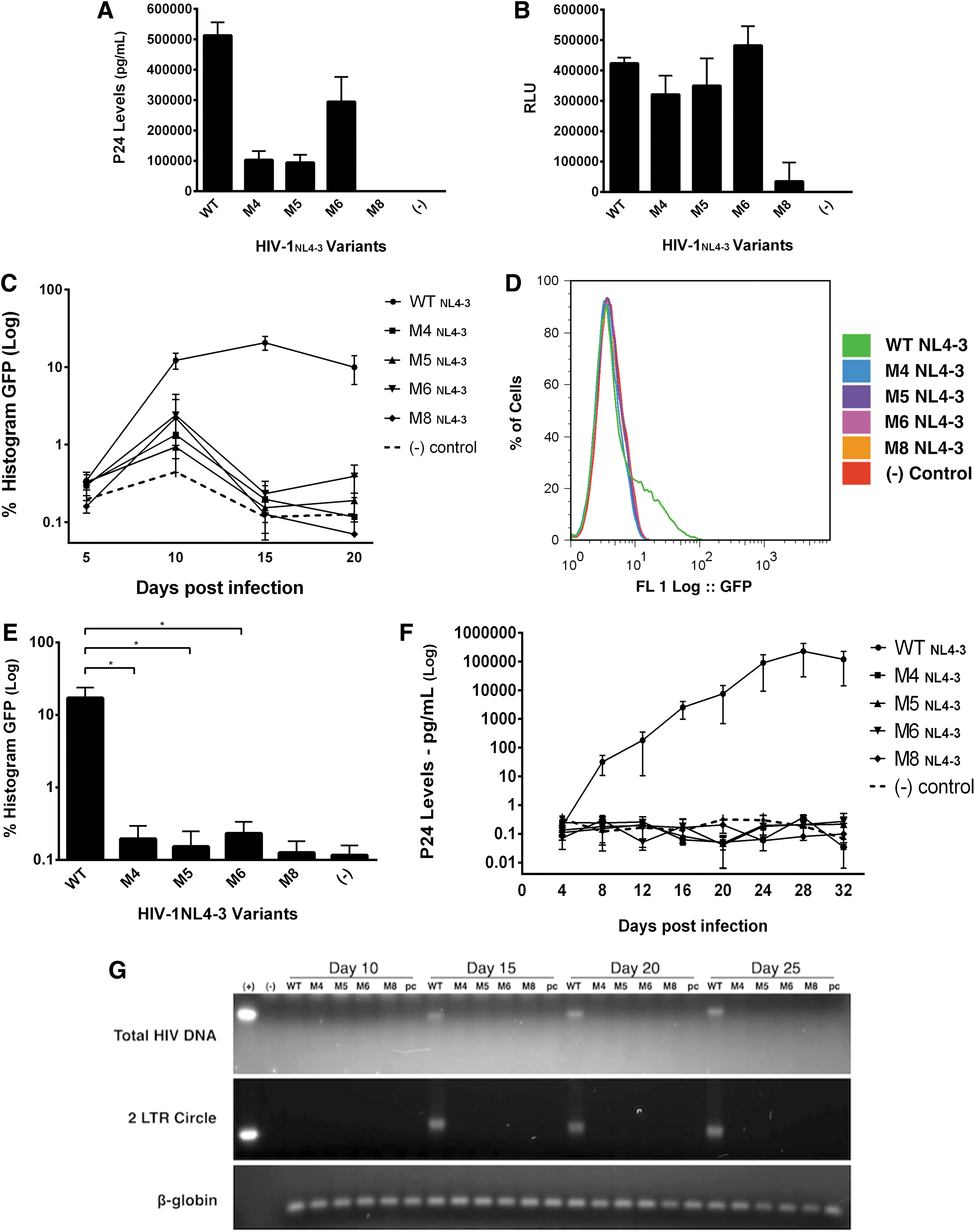
HIV-1NL4-3 harboring Rev nucleolar single-point mutations are unable to complete a full infectious cycle. Mutations corresponding to Rev-NoLS M4, M5, M6, and M8—all nucleolar in pattern—were induced within HIV-1NL4-3 cDNA and used to propagate virus. Virus was collected 48 h later and quantified using p24 ELISA antigen capture assays. Single-point mutations within HIV-1NL4-3 backbone (M4, M5, and M6) allowed viral production to occur, however, not as efficiently as WT HIV-1NL4-3
HIV-1NL4-3 particles harboring nucleolar mutations within the viral backbone were examined for infectivity of CD4+ JLTRG-R5. JLTRG-R5 stably expresses LTR-EGFP, inducible upon Tat production after HIV-1 infection. 47,48 Viral particles containing each of the single-point mutations—nucleolar-localizing Rev-NoLS M4, M5, and M6—were used to infect JLTRG-R5. HIV-1NL4-3 backbone containing the nucleolar-localizing M8 deletion did not yield viral particles and was therefore used in this study as a negative control along with the pcDNA negative control. JLTRG-R5 was subjected to infection with 2 ng p24 viral supernatant containing each HIV-1NL4-3 variant and washed 24 h later with 2× HBSS (Hank's balanced salt solution). Infected cells were resupplemented in RPMI media. JLTRG-R5 was collected afterward at 5-day intervals postinfection.
HIV-infected cells were measured for EGFP positivity (inducible upon Tat expression after integration) using fluorescent-activated cell sorting (FACS) analysis. Unlike infectious WT HIV-1NL4-3, variants of HIV-1NL4-3 harboring single-point mutations (M4, M5, and M6) lacked the ability to replicate and produce abundant Tat for the internal activation of LTR-EGFP. Low frequency in LTR-EGFP activation after replication of HIV-1NL4-3 variants, unlike WT HIV-1NL4-3, was evident up to 20 days postinfection (Fig. 3C). During the replication peak at day 15, HIV-1NL4-3 variants exhibited significant defects in replication compared to WT HIV-1NL4-3, which was able to replicate and infect 20% of the entire cell population (Fig. 3D). HIV-1NL4-3 variants infected host cells at extremely low frequency at the 15-day time point (Fig. 3E).
Quantification of infected JLTRG-R5 is as follows for WT versus HIV-1NL4-3 variants: M4 p value = .01, M5 p value = .01, and M6 p value = .01. Each column represents a sample size of three individual infectious processes during unpaired t-tests.
Ability of HIV-1NL4-3 variants to gain function into the HIV-1 infectious cycle was monitored through p24 viral output. Viral supernatant from JLTRG-R5 was collected at 4-day intervals until 32 days postinfection. Unlike infectious WT HIV-1NL4-3 exhibiting active viremia, HIV-1NL4-3 variants lacked the ability to fully regain functionality and infectivity up to 32 days postinfection (Fig. 3F).
This was also evident upon examination of genomic DNA harvested from HIV-infected JLTRG-R5 (WT HIV-1NL4-3 vs. single-point variants). Genomic DNA was collected every 5 days postinfection for 25 days and subjected to polymerase chain reaction (PCR) amplification for detection of total HIV DNA and preintegrated 2-LTR circles. Total HIV DNA was undetectable from JLTRG-R5 infected with HIV-1NL4-3 variants (M4, M5, M6, and M8), unlike detectable total HIV DNA from WT HIV-1NL4-3 starting at day 15 and beyond (Fig. 3G). WT HIV DNA was undetectable up to 10 days postinfection. Similarly, preintegrated 2-LTR circles were not detected in DNA genomic samples from the HIV-1NL4-3 variants; 2-LTR circles accumulated in the presence of WT HIV-1NL4-3 infection beginning at day 15. Gel loading controls were obtained through amplification of β-globin DNA.
Lentivirus generated with nucleolar-localizing Rev mutations is transducible at extremely low frequency
Lentiviral backbones lacking the following genes: gag/pol, vif, vpu, env, and nef were developed previously for safe delivery of therapeutic genes and small interfering RNAs inhibiting HIV-1 infection. 38,39,49 Lentiviral HIV7/CMV-GFP vector containing a GFP reporter for quantification of transduction events (Fig. 4A) was co-expressed with packaging vectors, including Rev-NoLS mutations (M4, M5, and M6), in HEK293T. Viral supernatant was collected 48 h afterward and normalized to 500 pg p24 before the transduction of HT1080 (Fig. 4B).
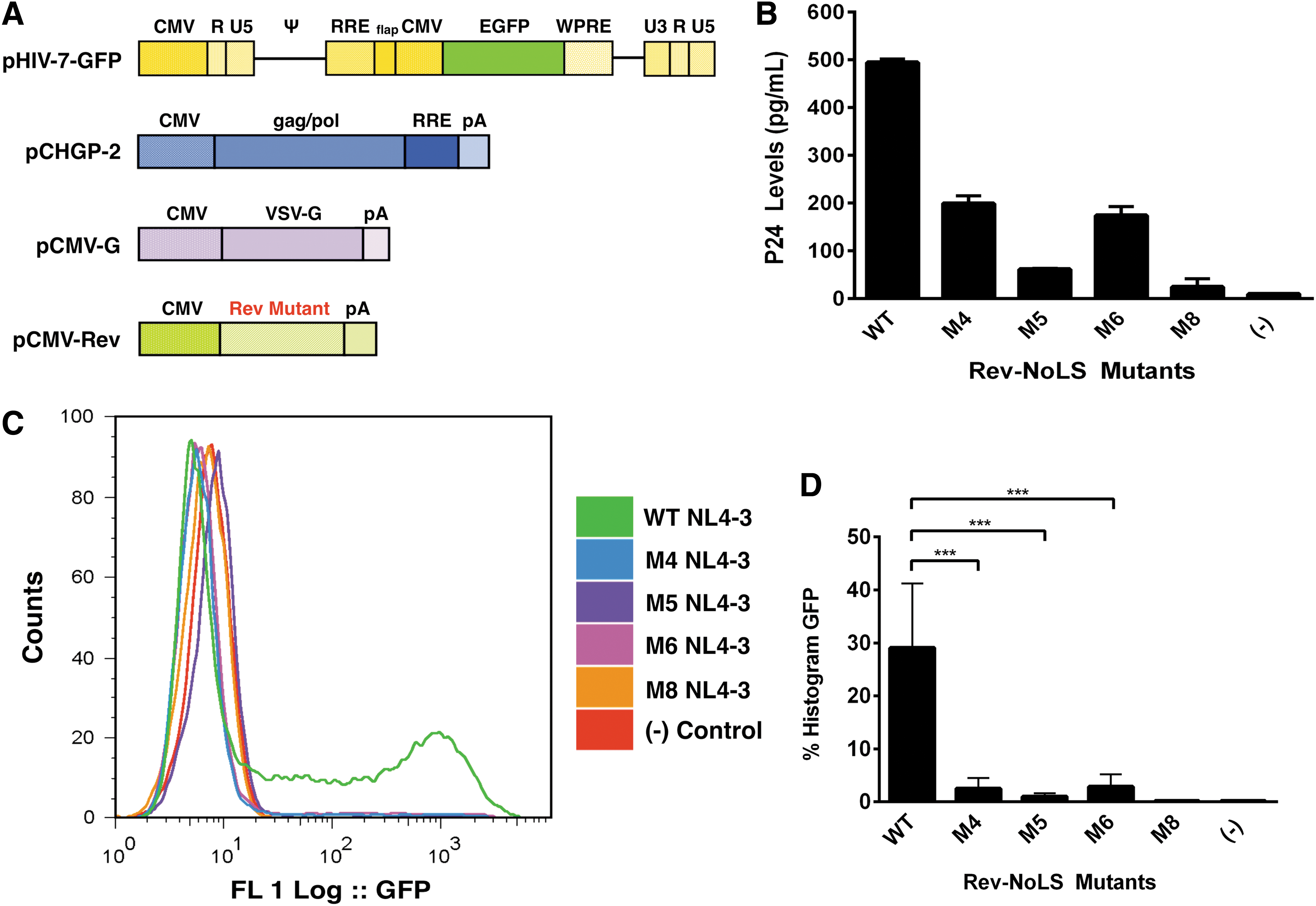
Transducibility of HIV/7-C-GFP lentivirus packaged with nucleolar-localizing Rev-NoLS mutations. Lentiviral HIV7/CMV-GFP vector was co-expressed with packaging vectors, including α-flag-tagged Rev-NoLS mutations (M4, M5, and M6), in HEK293T
Rev M8, previously shown to lack function in viral production and viral RNA packaging, was analyzed as a negative control. Viral particles were introduced into HT1080 cells, and transduced cells quantified 48 h later using EGFP FACS analysis. Lentivirus packaged with Rev-NoLS mutations exhibited significantly defective transduction below 6% in comparison to lentivirus packaged with WT Rev, in which 40% of HT1080 were transduced (Fig. 4C, D). Each column represents a sample size of six separate transduction events during unpaired t-tests (Fig. 4D).
Rev single-point nucleolar mutations lead to irregularities in HIV-1NL4-3 mRNA splicing
Nucleolar mutations of interest resulting in proviral production (Rev-NoLS M4, M5, and M6) were examined further to identify the step of the HIV-1 infectious cycle interrupted in the presence of Rev-NoLS mutations. The HIV-1 mRNA splicing machinery was initially examined, as HIV-1 mRNA splicing is dependent on the host mRNA-splicing machinery to produce three mRNA species for expression of proteins required in viral assembly and release—unspliced 9 kb mRNA, partially spliced 4 kb mRNA, and fully spliced 2 kb mRNA. Upon infection and viral integration into host genomes, fully spliced viral mRNA leads to expression of Tat, Rev, and Nef. Rev in turn achieves nucleocytoplasmic transport of RRE-containing RNAs through affinity with CRM1, involved in an mRNA-independent nuclear export pathway. 50
Rev transports partially spliced viral mRNA to the cytoplasm for expression of Vif, Vpr, and Vpu accessory proteins. Rev additionally stabilizes and transports unspliced viral mRNA to encode Gag and Gag-/Pol-derived proteins; unspliced HIV mRNA serves as viral genomic RNA to be packaged within viral particles for release and re-infection of other hosts. It is currently unknown if HIV-1 mRNA stabilization for this process involves a nucleolar pathway mediated by the nucleocytoplasmic activity of Rev. Rev-NoLS single-point mutations and deletions corresponding to nucleolar-localizing M4, M5, M6, and M8 were induced within the HIV-1NL4-3 backbone. HIV-1 mRNA splicing was observed as a result of nucleolar mutations to further understand the role of the nucleolus on viral mRNA splicing.
Single-point mutations incorporating glycine in replacement of arginine residues were induced within the cDNA of HIV-1NL4-3 Rev-NoLS motif (M4, M5, and M6). Deletion of the last two amino acids of Rev-NoLS (M8) was also generated. Expression plasmid containing HIV-1NL4-3 Rev-NoLS mutations was transfected into HEK293T and total RNA collected 48 h later. Total RNA (40 μg) was subjected to Northern blot analysis for the detection of HIV splice variants (unspliced 9 kb, partially spliced 4 kb, and unspliced 2 kb) using radiolabeled probe specific to nef (Fig. 5A). Ribosomal RNA loading controls (60S and 40S) were captured before blot transfer and used as standards in densitometry of three independent experimental blots.
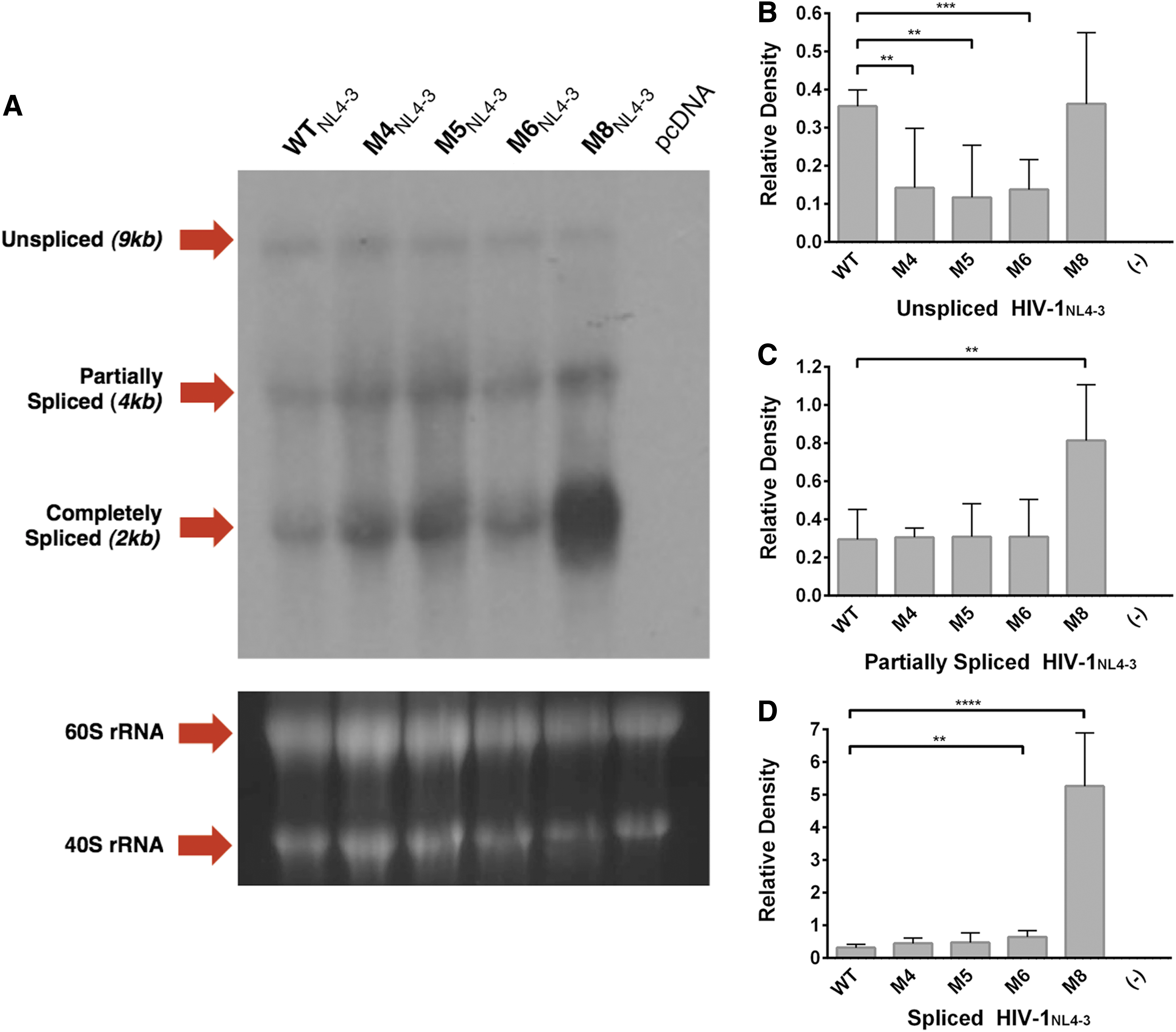
Northern blot analysis of HIV-1NL4-3 in the presence of Rev-NoLS mutations. pNL3-4 plasmid was transfected into HEK293T culture and total RNA collected 48 h later. Total RNA (40 μg) collected during HIV-1NL4-3 propagation in HEK293T was subjected to Northern blot analyses using radiolabeled Nef probe. The known spliced variants of HIV-1 mRNA in lane 1-WT (9 kb unspliced, 4 kb partially spliced, and 2 kb spliced) were compared to HIV-1NL4-3 variants harboring Rev-NoLS single-point mutations (lane 2: M4NL4-3, lane 3: M5NL4-3, lane 4: M6NL4-3, lane 5: M8NL4-3, and lane 6: pcDNA)
Unpaired t-tests (N = 6) were used in the statistical analyses of WT mRNA versus Rev-NoLS mutant mRNA. Relative density of unspliced Rev-NoLS variant expression (M4NL4-3, M5NL4-3, and M6NL4-3) reduced significantly in comparison to unspliced WT mRNA; unspliced M8NL4-3 mRNA was similar in expression to unspliced WT mRNA (Fig. 5B). Partially spliced HIV-1NL4-3 mRNA of WT and Rev-NoLS variants (M4NL4-3, M5NL4-3, M6NL4-3) was similar in relative density, unlike abundant M8NL4-3 mRNA (Fig. 5C). Fully spliced mRNA of M6NL4-3 and M8NL4-3 was significantly abundant compared to fully spliced WT mRNA (Fig. 5D). M4NL4-3 and M5NL4-3 relative densities were comparable to fully spliced WT mRNA ratio.
Rev-NoLS HIV-1NL4-3 single-point mutant particles are capable of host cell entry
Despite nucleolar mutations, HIV-1NL4-3 particles resembled WT in the ability to replicate and package intraviral RNA. We next tested the ability of mutant viral particles to achieve host cell entry, quantified through intraviral EGFP-Vpr infectious transfer. The direct cellular uptake of HIV-1 virions to the CD4 surface receptor has been observed with fluorescence microscopy by co-packaging EGFP-Vpr fusion protein to create GFP+ HIV particles. 51 EGFP-Vpr was later utilized to monitor virion uptake into CD4+ and CD4− HeLa cells during HIV-1 Nef regulation of viral entry into the host cytoplasm. 52
Thus, we implemented this technique to quantify the cellular uptake of GFP+ HIV-1 particles co-packaged with our Rev-NoLS variants. HIV-1NL4-3 cDNA—WT or Rev-NoLS variants—was co-packaged with EGFP-vpr vector to propagate fluorescent viral particles. CEM and CD4+ HeLa T4 cells were subjected to infection with 1 μg p24 supernatant for 1.5 h, preserved, and quantified for EGFP positivity using FACS analysis. Despite the nucleolar single-point mutations, viral particles are as capable of host cell entry as WT HIV-1NL4-3 in CEM (Supplementary Fig. S4A) and HeLa T4 cells (Supplementary Fig. S4B). Quantification of host cells with EGFP-Vpr-M8 NL4-3 contained fluorescent background comparable to pcDNA (−) control.
Rev-NoLS HIV-1NL4-3 single-point mutant particles are able to achieve reverse transcription
Mutations corresponding to Rev-NoLS M4, M5, M6, and M8—all nucleolar in pattern—were generated within HIV-1NL4-3 cDNA and used to propagate virus in HEK293T culture. Single-point HIV-1NL4-3 particles were normalized to 20 ng of p24 supernatant and examined intravirally for functional reverse transcriptase (RT) using a colorimetric RT kit. HIV-1NL4-3 variants were able to produce and package functional RT (M4, M5, and M6), unlike M8 and pcDNA negative controls absent in RT activity (Supplementary Fig. S5). Colorimetric values represent DNA concentration (ng) after release of RT from viral particles and reverse transcription thereafter.
Cre-loxP dual fluorescent reporters indicate lentiviral entry, reverse transcription, and integration in the background of Rev-NoLS mutations
As another test for viral entry, reverse transcription, and integration of virus packaged with the Rev-NoLS variants, we designed a new system based on the ability to detect viral entry using virus carrying EGFP-Vpr fusion protein. We created a reporter cell line of HT1080 cells integrated with a LoxP-mCFP-LoxP-TagRFP (HT1080-XCXR) gene cassette, which would undergo an mCFP (cyan) to TagRFP (red) conversion upon the delivery or expression of Cre protein (Fig. 6A). In this new system, we wanted to test for viral entry, reverse transcription, and transgene integration into infected cells. Thus, we created a Cre-Vpr fusion protein that was expressed from a Tat-responsive lentiviral vector (HIV LTR–Cre-Vpr fusion–IRES–Tat/GFP fusion or CVITG) (Fig. 6B).
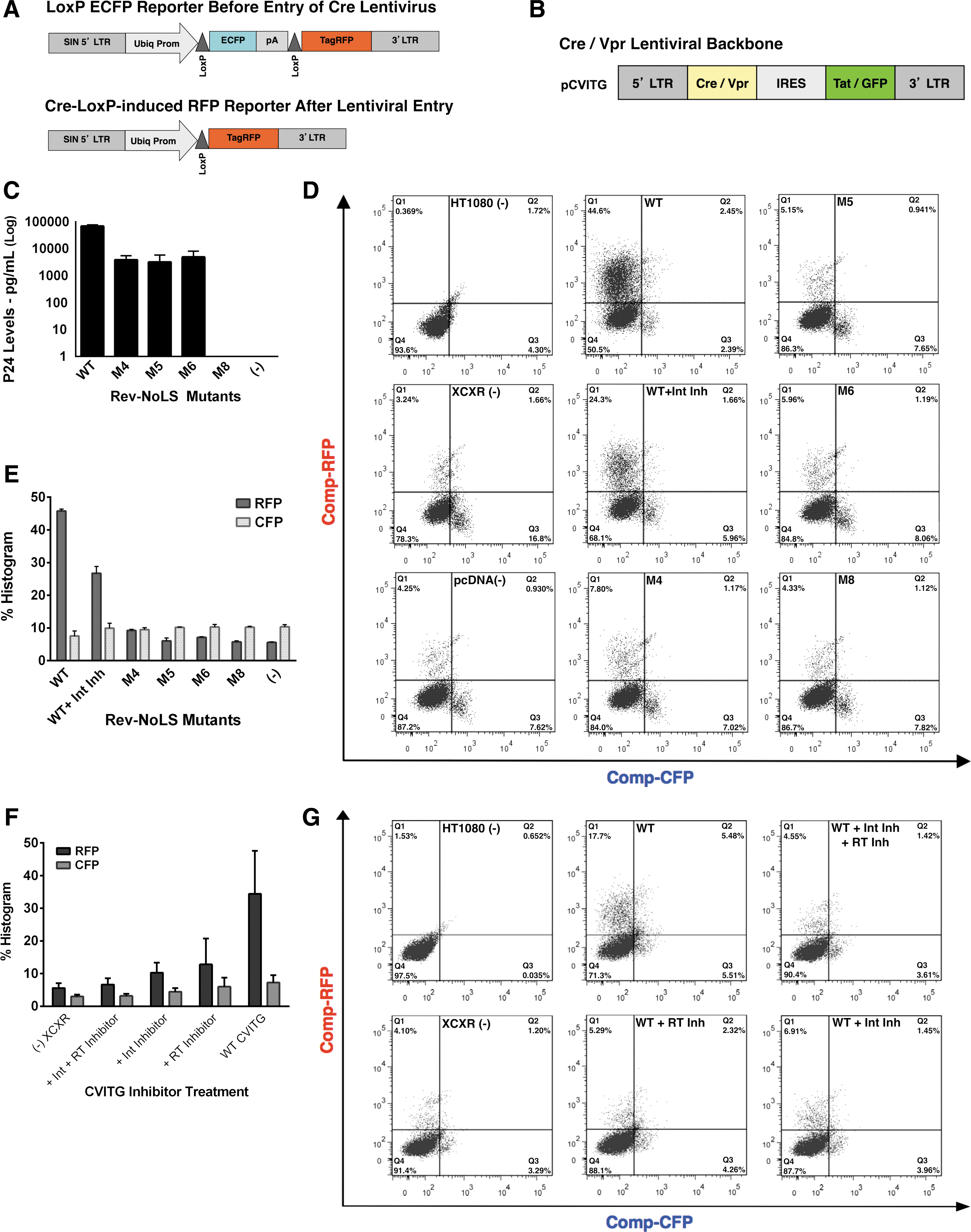
Cre-loxP dual fluorescent reporters indicate lentiviral entry, reverse transcription, and integration in the background of Rev-NoLS mutations. An HT1080 reporter cell line was generated to test for viral entry, reverse transcription, and transgene integration. HT1080 reporters contain a LoxP-mCFP-LoxP-TagRFP (XCXR) gene cassette, which undergoes conversion from mCFP (cyan) to TagRFP (red) upon the viral delivery of Cre protein
Therefore, entry of the CVITG virus carrying Cre-Vpr fusion protein would induce a conversion from mCFP expression to TagRFP expression, while successful integration of the CVITG virus would also lead to GFP production. CVITG lentiviral vector was co-expressed with packaging vectors, including α-flag-tagged Rev-NoLS mutations (M4, M5, and M6) in HEK293T.
Viral supernatant was collected 48 h later and quantified using p24 ELISA antigen capture assays. Rev-NoLS mutations function in CVITG lentiviral production, yet not as efficiently as WT Rev (Fig. 6C). Cre-loxP recombination reporter cells (HT1080-XCXR) were transduced with 10 μg CVITG Cre-Vpr lentivirus produced using WT or Rev-NoLS mutations (M4, M5, and M6). Cyan fluorescent protein (CFP) transition to red fluorescent protein (RFP) from lentiviral transduction, in addition to GFP fluorescence from integration, was assessed using FACS analysis.
From these experiments, we observed that CVITG virus co-packaged with M4, M5, and M6 Rev variants was able to induce TagRFP expression at extremely low frequency—an indicator of lentiviral entry (Fig. 6D, E). CVITG virus co-packaged with M4, M5, and M6 Rev variants was also able to induce Tat/GFP expression at extremely low frequency—an indicator of lentiviral reverse transcription and integration (Supplementary Fig. S6A, B) in a small fraction of cells.
Lentivirus co-packaged with Rev variant M8 did not induce viral entry, reverse transcription, or integration similar to pcDNA negative control. Control experiments using WT Rev with RT inhibitor (azidothymidine [AZT]) and/or integrase (IN) inhibitor (118-D-24) demonstrated the functionality of the system to detect either viral entry (RFP+ and GFP− cells, Fig. 6F, G) or expression of Cre and integration (RFP+ and GFP+ cells, Supplementary Fig. S6C, D).
Rev nucleolar mutations lead to the deregulation of HIV-1NL4-3 integration
Supernatant of HIV-1NL4-3—WT and Rev-NoLS variants M4, M5, and M6—was normalized to 20 ng capsid p24 for infection of JLTRG-R5 populated at 1 × 106 cells per well of 12-well tissue culture plates. Cells were harvested for genomic DNA starting 6, 12, 24, and 48 h postinfection. The genomic DNA samples were diluted to 2 μg/ml and quantified for integrated viral DNA using a two-step Alu-gag PCR described previously. 41 –45 Copy number of integrated viral DNA was calculated using a standard curve obtained from integration standard consisting of JLTRG-R5 infected with WT HIV-1NL4-3. Viral integration of HIV-1NL4-3 variants (M4 and M5) occurred at extremely higher frequency than WT HIV-1NL4-3 beginning 6 h after infection (Fig. 7). Variant M6 exhibited similar integration copies to that of WT.
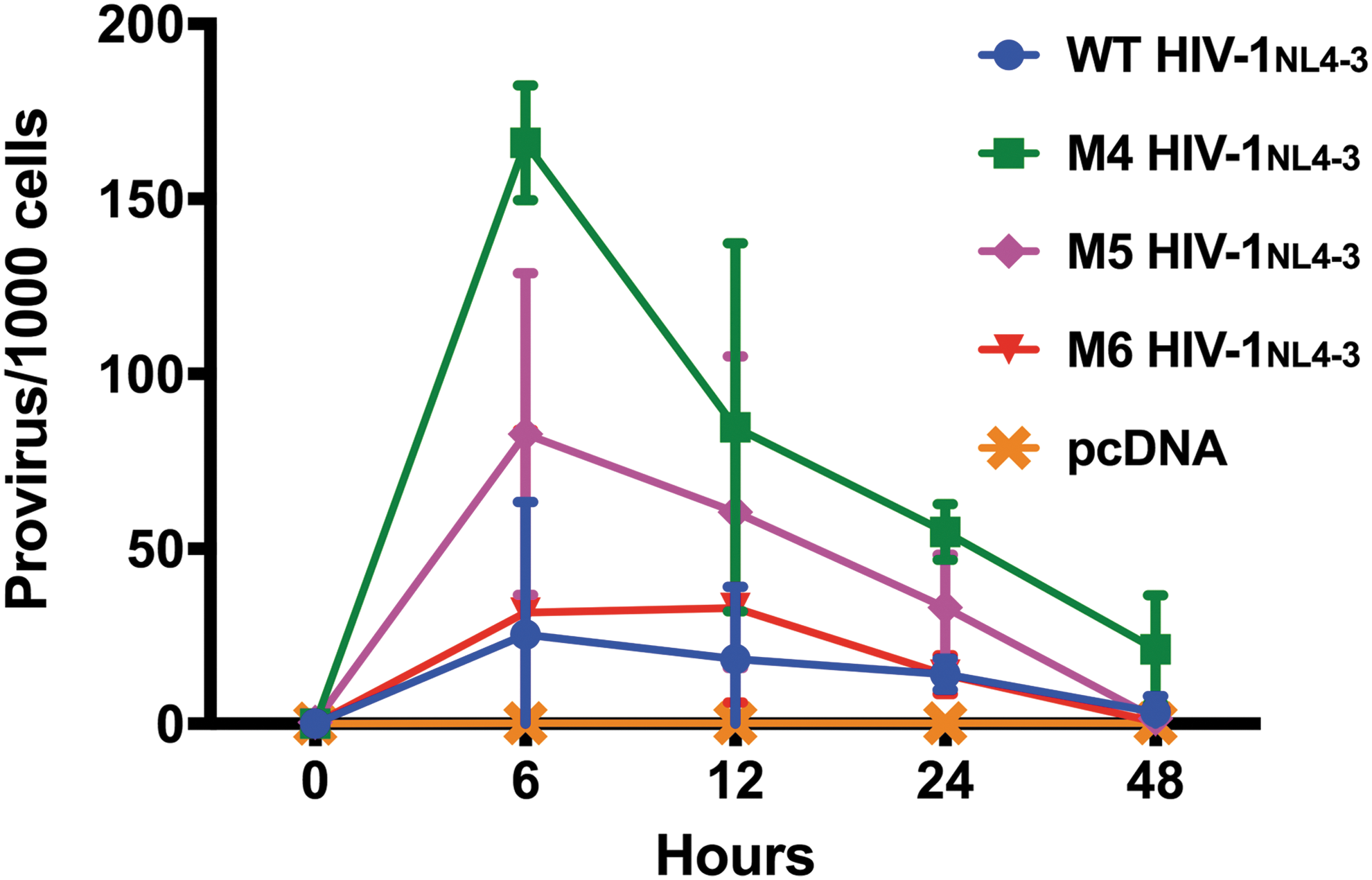
Rev nucleolar mutations lead to the deregulation of HIV-1NL4-3 integration. Viral supernatant (20 ng p24 capsid) collected during the propagation of WT HIV-1NL4-3 and HIV-1NL4-3 variants M4, M5, and M6 was used to infect JLTRG-R5. Genomic DNA was harvested from JLTRG-R5 hosts starting 6, 12, 24, and 48 h postinfection. Genomic DNA was diluted to 2 μg/ml and quantified for integrated viral DNA using a two-step Alu-gag PCR method. Viral integration of HIV-1NL4-3 variants (M4 and M5) occurred at extremely higher frequency than WT HIV-1NL4-3 beginning 6 h after infection. Variant M6 exhibited similar integration copies to that of WT. Integrated copies began to decrease 12 h postinfection and continued to decrease gradually until the 48-h time point. Supernatant collected from pCDNA during viral propagation was used as a negative control for viral integration.
Integrated copies were detected at lower frequency starting 12 h postinfection and continued to decrease gradually until the 48-h time point after viral infection. Supernatant of pCDNA was used as a negative control for integration in these studies.
Rev-NoLS single-point mutations lose interaction with B23 during viral production
Nucleolar B23 has high affinity to Rev-NoLS and functions in nucleolar localization of Rev 53 and nucleocytoplasmic transport of Rev-bound HIV mRNAs. 54 The affinity of B23 with Rev containing single-point arginine substitutions and deletion was assessed through immunoprecipitation (IP) of Rev-3′Flag (WT and mutations) and Western immunoblotting. HLFB were transfected with Rev-NoLS mutations (M4, M5, M6, and M8). Protein extract for IP reaction (1 mg/ml) was collected from HLFB after 48 h of HIV-1HXB2 production. IP eluates were subjected to Western immunoblotting using monoclonal antibodies specific to α-Flag-Rev (Fig. 8A–C) and B23 (Fig. 8B).
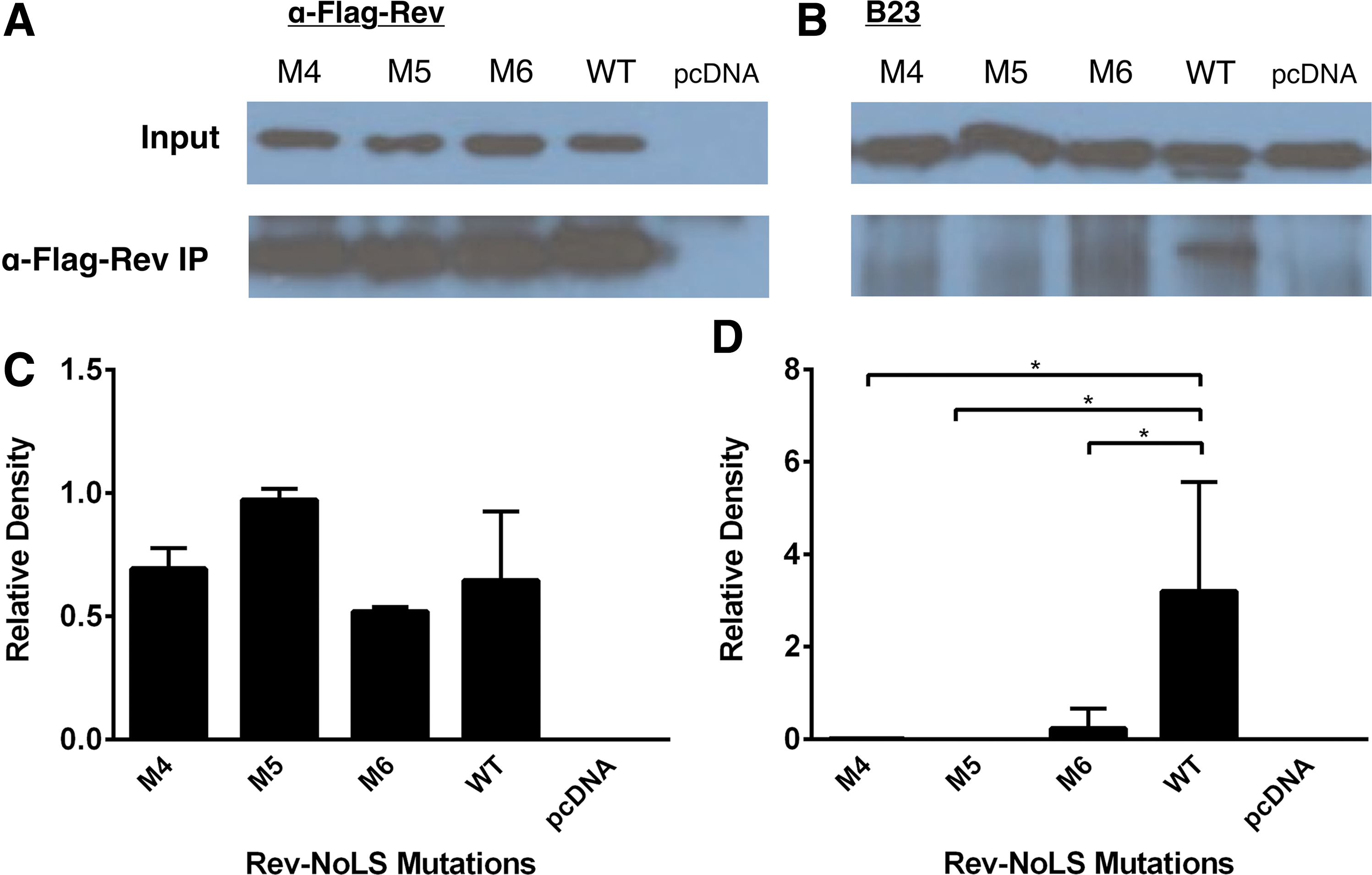
Affinity of nucleolar Rev-NoLS mutations to B23 during viral production. HLfB culture was transfected with Rev-NoLS mutations and protein lysate was collected 48 h after viral proliferation. Lysates were subjected to α-Flag-Rev IP and immunodetection against B23, and assessed through Western immunoblotting.
In the background of Rev single-point mutations (Rev-NoLS M4, M5, and M6), B23 binding affinity was significantly reduced (Fig. 8D). B23 maintained affinity with WT Rev during HIV production. Each column represents a sample size of three IP experiments, compared during unpaired t-tests.
Discussion
Rev-NoLS in viral production and infection
Deletion of amino acids 48–51 (RQRQ) from the C-terminal end of the Rev-NoLS maintained nucleolar accumulation in COS-7 culture. Deletion of the entire Rev-NoLS (amino acids 45–51, WRERQRQ) caused the intracellular dispersion of Rev. 34 In our current model, removal of amino acids 48–51 (M1-RQRQ) from Rev-EGFP resulted in the nucleolar/nuclear accumulation of Rev during HIV-1 production in HeLa culture expressing Rev-deficient proviral HIV-1HXB2. Similarly, nucleolar/nuclear accumulation was observed upon removal of the entire NoLS (M7-WRERQRQ).
Differences in subcellular localization in the previous study in comparison to this observation are attributed to the presence of an HIV-1 production model. In the absence of viral production in HeLa culture, removal of amino acids 48–51 (Rev-NoLS M1-RQRQ in our study) from Rev resulted in nucleolar accumulation, previously observed in COS-7 cells. 34 Removal of amino acids 45–51 (M7-WRERQRQ in our study) caused nuclear dispersion, also previously observed in COS-7.
With and without HIV-1 production, Rev mutations containing two arginine residues within the NoLS motif are exclusively nucleolar in pattern (Rev-NoLS M4, M5, M6, and M8) as WT Rev originally containing three arginine residues within the NoLS motif. We currently demonstrate the dependence of Rev nucleolar accumulation on the presence of at least two arginine residues within Rev-NoLS.
Nucleolar localization of HIV-1 Rev and HTLV-1 Rex was previously demonstrated unnecessary for accumulation of cytoplasmic unspliced RNA when tethered to an RNA operator through MS2 fusion in the absence of viral response elements 55 ; nucleolar trafficking was therefore believed to occur through the high affinity of viral proteins with nucleolar constituents that were described as unessential for Rev activity.
We investigated the significance of Rev nucleolar trafficking and localization in HIV production models through the rescue of Rev-deficient HIV-1HXB2 with Rev-NoLS mutations. In contrast to the previous finding, we revealed that Rev mutations lacking nucleolar entry are nonfunctional in HIV proliferation and viral RNA packaging (M9). Partial nuclear/nucleolar localization did not rescue viral productivity (M1, M2, and M7).
Mutations M1, M2, and M7 lack two arginine residues within the Rev-NoLS seuqence, and M9 lacks three arginine residues; this would lead to conformational changes within the α-helical structure of Rev-NoLS and interrupt Rev/RRE binding activity.
The nonfunctional activity of M1, M2, M7, and M9 to rescue viral production could be attributed to the inability of Rev mutations to bind to the RRE of HIV-1 mRNAs. Strictly nucleolar Rev mutations functioned in viral production (M4, M5, and M6), however, with reduced efficiency compared to WT Rev. Nucleolar-localizing M8 contains two missing amino acids from NoLS (Arg 50 and Gln 51 ) and resulted in defective viral production. This insufficiency in the rescue of viral production and viral RNA packaging, despite nucleolar accessibility of M8, provides evidence that HIV-1 infection is also dependent on efficient splicing and transport of HIV-1 mRNA—both processes that take place within the nucleolus.
Substitution of arginine residues at the carboxyl terminus of the Rev NLS to alanine (Arg 46 , Arg 48 , and Arg 50 ) was previously demonstrated to reduce Rev/RRE binding threefold. 4 Our observations yielded similar results upon substitution of these arginine residues to glycine. p24 capsid expression originates from unspliced HIV-1 mRNA transported to the cytosol through Rev/RRE interactions. We observed a decrease in p24 expression in our Rev-deficient HIV-1HXB2 model upon viral production with the expression of nucleolar-localizing single-point mutations (Rev-NoLS M4, M5, and M6). In comparison to WT Rev, single-point mutations led to a three-fold decrease in p24 protein as well as decreases in packaged genomic RNA.
Mutation of arginine residues to a similar basic amino acid—lysine—did not affect Rev/RRE binding. 4 Arg 46 , Arg 48 , and Arg 50 at the carboxyl terminus of the Rev ARM are moderately important for phosphate and van der Waals interactions between Rev and RRE of the HIV-1 mRNA. 5 These interactions stabilize the α-helical orientation of Rev ARM within the RRE RNA groove. The inability of nucleolar-localizing Rev-NoLS M8 to rescue Rev deficiency during viral production is postulated to result from two missing amino acids from the NoLS motif (Arg 50 and Gln 51 ).
This would lead to the destabilization of the Rev ARM and the RRE. Rev-NoLS M8 deletions would additionally lead to a loss in nucleolar-specific protein interactions with Rev-NoLS. Inability to rescue viral production and genomic RNA packaging despite nucleolar access reveals that Rev nucleolar localization is insufficient for the progression of the HIV-1 infectious cycle. HIV-1 infection may be able to manipulate host cellular pathways at the subnuclear level, and nucleolar access is too simple an explanation for Rev function. Misplaced protein interactions from nucleolar-localizing Rev-NoLS mutations (M4, M5, M6, and M8) in comparison to WT Rev could identify nucleolar pathways required for HIV-1 production.
Viral particles generated in the background of Rev-NoLS single-point mutations are capable of transduction and infection at extremely low frequency. VSVG-pseudotyped lentivirus generated with Rev-NoLS mutations replicated, yet lacked transducibility.
Similarly, mutations introduced into the HIV-1NL4-3 cDNA did not hinder viral production, yet resulted in the inability of HIV-1 variants to infect host cells. We quantified the potency of Rev-NoLS single-point mutations on viral infection through viral output 32 days postinfection. Mutant virus lacked the ability to regain function and participate in the viral infectious cycle. Unlike Rev-NoLS, single-point mutation R55Q acquired within the HIV-1 Tat basic domain— 49 RKKRRQRRRAHQ 60 required for TAR RNA binding, 56,57 NLS, 58 and NoLS 59 —delayed HIV-1NL4-3 production 12 days in comparison to WT Tat. 60 Other single-point mutations R52Q, R55Q, and R57Q did not significantly affect Tat function in the transactivation of viral genes.
Rev-NoLS in HIV-1 mRNA splicing
HIV-1 mRNA splicing involves the interplay of core splicing signals, exonic/intronic splicing enhancers and silencers, and the secondary structure of the pre-mRNA. 61 Core splicing signals are composed of the following three sites commonly found in pre-mRNA: 5′-splice site (5′ss); 3′-splice site (3′ss); and branch point sequence (BPS). 5′ss recruits the splicing factor U1 snRNP, and the 3′ss allows interaction with another splicing factor—U2AF. BPS binds to the SF1/mBBP branch point binding protein, later displaced by U2 snRNP during formation of the spliceosomal complex.
5′ss and 3′ss are categorized in pairs throughout mammalian pre-mRNA and are classified as strong or weak splice sites based on affinity with corresponding splicing factors (5′ss with U1 snRNA, or 3′ss with U2AF and SF1/mBBP of the BPS). Exon definition and bridging between 5′ss and 3′ss lead to formation of an early, irreversible splicing complex (E complex), resulting in splicing reactions. 62,63 Core splicing signals are surrounded by cis-elements that either enhance (exonic splicing enhancers [ESEs]) or repress (exonic splicing silencers [ESSs]) exon definition during alternative splicing reactions. 64,65
ESEs are sequence elements within exons that interact with serine-arginine-rich splicing factors (SR proteins). ESSs interact with the heterogeneous ribonuclear splicing inhibitors (hnRNP) to repress exon definition. Intronic splicing silencers (ISSs) and intronic splicing enhancers are sequences within introns that facilitate exon definition. The pre-mRNA secondary structure is able to modulate alternative splicing reactions through exposure of core splicing signals and regulatory cis-elements. 66 Accumulation of partially spliced and unspliced viral mRNA is dependent on the presence of the RRE, located between 5′ss D4 and 3′ss A7 (Supplementary Fig. S7). mRNA encoding Vif, Vpr, and Tat is expressed at extremely low levels, indicating the infrequent activity of 3′ss A1, A2, and A3. 67
Partially spliced 4 kb vif mRNA, a product of 5′ss D1 to 3′ss A1, composes 1% of incompletely spliced viral mRNA in infected cells. Vpr mRNA, a product of 5′ss D1 to 3′ss A2, composes 2% of total partially spliced mRNAs. Approximately half of all spliced viral mRNA is fully spliced 2 kb mRNA, indicating frequent splicing at 3′ss A7. Tat mRNA composes 9% of completely spliced mRNA (two-exon Tat) and 5% of partially spliced mRNA (one-exon Tat). Tat mRNAs are formed through splicing activity at 5′ss of D1, D2, D3, or D4 to 3′ss A3.
Transcripts leading to Vpu, Env, and Rev are abundantly expressed, indicating the frequent use of 3′ss A4c, A4a, A4b, and A5. The center of HIV-1 pre-mRNA contains 3′ss clusters leading to the accumulation of fully spliced rev mRNA (5′ss D3 or D4 to 3′ss of A4c, A4a, or A4b), fully spliced nef mRNA (5′ss D3 or D4 to 3′ss A5), and partially spliced env/vpu mRNAs (5′ss D2 or D3 to 3′ss cluster). Both populations compose 90% of completely spliced mRNA and 92% of partially spliced mRNA.
Alternatively, mRNA leading to expression of two-exon Tat, Rev, and Nef utilizes 3′ss A7. Efficient splicing activity at A7 requires ESS, ESE, and ISS elements. 68,69 Novel 5′ss D1A and 3′ss A1A identified within gag/pol mRNA are utilized at low frequency compared to other splice sites within the HIV-1 pre-mRNA. 70 The 5′ss D1A is suggested as a necessary feature to prevent the degradation of unspliced viral mRNA. U1 snRNP bound to 5′ss D1A recruits SR proteins that may stabilize the binding of Rev to the viral RNA, leading to the nuclear export of unspliced HIV-1 RNA.
Based on the observations of Rev mutations (M4, M5, and M6), the inability of lentivirus to replicate and package mRNA may result from single-point mutational defects affecting Rev/RRE binding and nucleocytoplasmic transport. Specifically, Arg 35 and Arg 39 of Rev ARM interact with nucleotides U66, G67, and G70 of RRE major groove; Asn 40 and Arg 44 interact with U45, G46, G47, and A73, opposite the major groove. 4 Arg 46 , Arg 48 , and Arg 50 at the COOH terminus of Rev ARM are suggested as being moderately important for specific binding to achieve phosphate and van der Waals contacts in an α-helical orientation within the RRE groove. 5
In our studies, substitution of Rev Arg 46 (M4), Arg 48 (M5), and Arg 50 (M6) with glycine within proviral cDNA leads to spliceosomal irregularities. The 9 kb mRNA of HIV-1NL4-3 variants significantly reduced in expression compared to WT HIV-1NL4-3; Arg 50 -Gln 51 deletion within the M8 variant exhibited abundant 4 and 2 kb mRNA than WT HIV-1NL4-3. These findings exemplify the importance of Rev-NoLS in RRE binding and HIV-1 mRNA transport.
We observed that Rev single-point mutations decrease the cytoplasmic export of the 9 kb HIV-1 mRNA, thereby leading to the decrease of p24 production, since p24 is derived from gag/pol precursors of 9 kb mRNA. 46,71 Two amino acid deletions of M8 did not significantly change the levels of the 9 kb mRNA, although disproportionately higher levels of 2 and 4 kb mRNA splice variants were observed. Impairment of Rev in facilitating nucleocytoplasmic transport of unspliced and partially spliced transcripts could result in the accumulation of fully spliced 2 kb transcripts, which encode Tat, Rev, and Nef proteins. Therefore, an increase of Tat expression would activate the Tat-positive feedback loop, leading to the enhanced transactivation of HIV gene expression and the subsequent accumulation of 2 and 4 kb mRNAs. 6,72
This is also consistent with computational models, which predict an increase of 2, 4, and 9 kb transcripts upon inhibition of Rev. 73 The proportionality of spliced/unspliced mRNA variants may be a prerequisite for viral production and infection. In addition, host cell protein interactions with Rev in the nucleolus may regulate HIV mRNA transport and splicing.
Nucleolar B23 is involved in nucleocytoplasmic shuttling of Rev-bound HIV mRNA and is well characterized in its interactions with the NoLS of host factors—p120 growth factor (amino acids 40–57) 74 and C23 pre-rRNA processor (amino acids 540–628) 75 —and retroviral proteins HTLV-1 Rex (amino acids 1–22) 76 and HIV-1 Tat (amino acids 49–57). 77 Currently, we reveal that Rev-NoLS single-point mutations abolished B23 interaction with Rev. This may lead to the disruption of nucleocytoplasmic shuttling of HIV mRNAs. We also reveal that the loss of B23 interaction with Rev does not hinder viral production, yet diminishes viral infectivity, suggesting that other nucleolar factors are involved in viral production.
Rev-NoLS in the viral entry, reverse transcription, and integration of HIV-1
Rev single-point mutations were demonstrated to allow the entry of EGFP-Vpr-labeled HIV-1NL4-3 particles into host cells. We postulated that defects in HIV-1 production in the presence of nucleolar mutations occur postentry during reverse transcription and/or integration. Rev-NoLS mutants M4NL4-3, M5NL4-3, and M6NL4-3 exhibited reduced expression of p24 capsid, RT, and packaged RNA; this would result from the reduced expression of Gag/Pol precursors as well as the Ψ packaging element, all originating from 9 kb HIV mRNA, in the presence of single-point mutations. Rev mutant M8NL4-3 produced p24 capsid, RT, and packaged RNA at extremely low quantities in comparison to HIV-1 mutants M4NL4-3, M5NL4-3, and M6NL4-3; this would result from the constant splicing of the 9 kb mRNA and accumulation of 4 and 2 kb mRNA fragments in the presence of nonfunctional Rev M8.
Nullbasic, a transdominant negative mutation of Tat, contains glycine/alanine substitutions of the Tat basic domain (amino acids 49–57). Nullbasic was demonstrated to reduce HIV-1 infection of MAGI, Jurkat, and human primary CD4+ T cells during reverse transcription. 78,79 The mechanism by which Nullbasic interrupts reverse transcription is unclear. Nullbasic Tat was further demonstrated in redistribution of nucleolar Rev to cytoplasm and downregulation of Rev-mediated mRNA transport and expression of Gag-derived proteins. 78 Nullbasic co-expression with Rev caused dispersion of nucleolar CRM1, B23, and nucleolin to the nucleus. 80
Absence of the Tat basic domain in Nullbasic, which interacts with B23, 77 would suggest alternative indirect interactions between Tat and Rev besides B23. Mutation within Rev-NoLS may in turn interrupt Tat function through this unknown pathway; identification of host factors involved in this mechanism would explain reverse transcription deficiencies in the presence of Nullbasic Tat as well as Rev-NoLS single-point mutations. Alternatively, nucleolar factors such as B23 may be required within viral particles to directly facilitate reverse transcription postentry, and reduction of cellular factors through Rev-NoLS mutations could lead to the defects and reduction of reverse transcription.
Lentivirus generated in the background of Rev-NoLS single-point mutations (M4, M5, and M6) transduced host cells, however, at extremely low frequency in comparison to WT lentivirus. Transducibility in this model is defined by the ability of lentivirus to achieve host cell entry, reverse transcription, and integration. These events are quantifiable through reporters placed within the lentiviral backbone (HIV7/C-GFP contains a GFP reporter to detect reverse transcription and integration; CVITG contains GFP for detection of reverse transcription and integration, and produces Cre to induce Cre-LoxP-dependent cleavage of stably expressed LoxP-CFP-LoxP-RFP reporters for detection of lentiviral entry in HT1080-XCXR hosts).
The inability of HIV7/C-GFP and CVITG lentivirus to fully transduce host cells in the presence of Rev-NoLS mutations reveals a deficiency during the integration step. In a separate infectious model, deficient integration was observed upon inability of HIV-1NL4-3 variants to integrate into JLTRG-R5 hosts, preventing the Tat-inducible activation of internal LTR-EGFP reporters.
A previous study demonstrated the intracellular interaction of HIV-1 integrase and Rev using bimolecular fluorescence complementation and co-IP assay systems. 81 Rev protein is initially transcribed from unintegrated viral DNA at the early stages of infection before the integration step occurs, and is believed to have inhibitory effects on the enzymatic activity of integrase. An HIV-1-infected cell typically contains a limited number of integrated viral genomes per host cell—namely one or two integration events have been observed. Several integration events are documented in other retroviruses—murine leukemia virus and Rous sarcoma virus—suggesting different regulatory mechanisms of viral integration in other retroviral models.
Two integrase-derived peptides (INr-1 and INr-2) were shown to diminish the inhibitory effects of Rev on integrase activity and stimulate multiple viral DNA integration events during kinetics studies. These findings suggest the involvement of the Rev nucleolar pathway in the regulation of lentiviral integration. Formation of the lens epithelium-derived growth factor p75 (LEDGF/p75) complex with Rev was previously observed as a crucial step in regulating viral cDNA integration. 82 Co-IP experiments revealed that an IN-LEDGF/p75 complex is formed postinfection, which is later replaced by Rev-LEDGF/p75 and Rev-IN complexes. Nucleolar single-point mutations, namely in the background of M4 and M5, may hinder the Rev-dependent regulation of integrase. This would result in the high frequencies of viral integration observed in this study.
Cellular host factors involved in the HIV-1 nucleolar pathway
Single-point mutations induced within Rev-NoLS were expected to decrease binding affinity to other cellular host factors facilitating Rev functionality in HIV mRNA binding and transport. Nucleolar B23 is involved in the nucleocytoplasmic shuttling of Rev-bound mRNA. 53,54 Rev-NoLS single-point mutations in this model abolished B23 affinity to Rev, indicating disruption in nucleocytoplasmic shuttling and HIV mRNA transport. Disruption of B23 interaction with Rev in turn does not hinder viral production, yet diminishes ability to produce infectious viral particles. We postulate that nucleolar factors, transport factors, and splicing factors involved in B23 function further participate in the HIV-1 infectious cycle.
B23-mediated transport of nucleolar viral proteins, viral assembly, encapsidation, production, and latency is documented in other viral infectious models. B23 is characterized in interactions with NoLS of cellular factors for nucleocytoplasmic transport—p120 growth factor (amino acids 40–57) 74 and C23 pre-rRNA processor (amino acids 540–628) 75 —and with retroviral proteins HTLV-1 Rex (amino acids 1–22), 76 HIV-1 Tat (amino acids 49–57), 77 and HIV-1 Rev (amino acids 37–47). 83 Juvenile Encephalitis virus (JEV) infection is transmitted through mosquito vectors, leading to acute encephalitis in humans. The JEV genome encodes a nucleolar-localizing core protein, through which amino acids Gly42 and Pro43 interact with N-terminal region of B23 during JEV infection, resulting in transportation of viral core protein/B23 into the nucleus. 84
The single-strand RNA HBV causes cirrhosis and hepatocellular carcinoma during infection. The HBV genome is composed partially of double-stranded DNA, which encodes a nucleolar core protein. HBV core protein associates with nucleolin and B23 in the nucleolus 85 ; B23 was demonstrated in HBV assembly through interaction with the core protein N-terminal domain. Specifically, B23 amino acids 259–294 bind the N-terminal domain of HBV core protein to allow viral encapsidation. 86
The negative-sense, single-strand RNA hepatitis D virus (HDV) expresses HDVAg antigen in two isoforms; the small isoform aids in RNA replication and the large isoform facilitates viral assembly. RNA replication takes place within the nucleolus and requires B23 interaction with HDVAg. 87,88 HDV infection causes an upregulation of B23, which interacts mostly with the small HDVAg isoform and less with the large HDVAg isoform. Interactions take place through the small HDVAg NLS domain, through which B23 binds and achieves nuclear accumulation. Upon deletion of HDV binding site to B23, RNA replication was impaired. HDVAg was shown to co-localize with B23 and nucleolin in the nucleolus. Nucleolin was discovered to possess transcriptional properties as a repressor, 89 revealing the nucleolus as a compartment for regulation of HDV replication.
B23 is involved in latency of the double-strand DNA Kaposi sarcoma herpesvirus (KSHV), which leads to Kaposi's sarcoma and is associated with AIDS-induced non-Hodgkin lymphoma. KSHV latent protein—v-cyclin—with host CDK6 kinase phosphorylates B23 at Thr199, facilitating B23 interaction with latency-associated nuclear antigen. 90 The latency-associated nuclear antigen acts to prevent viral lytic replication. Depletion of B23 leads to KSHV reactivation, revealing B23 as a regulator of KSHV latency. B23 function in the HIV production cycle is characterized in nucleocytoplasmic transport activity of Tat and Rev, and it is unknown if B23 can induce latency during HIV infection. B23 involvement in the production, encapsidation, and assembly of HIV is also unknown.
Conclusion
We currently demonstrate Rev nucleolar access a requirement for HIV-1 production and pathogenesis. This localization pattern allows efficient HIV-1 mRNA splicing and nucleocytoplasmic transport. We reveal an extra step during the HIV-1 infectious cycle in the nucleolus through Rev nucleocytoplasmic activity, required for production of infectious viral particles. Interference with nucleolar activity through mutation of Rev-NoLS diminishes the ability of HIV-1 to regulate viral integration within host cells, thereby leading to host cell death and termination of the viral infectious cycle.
In summary, we find that HIV-1 utilizes a nucleolar pathway to maintain an infectious cycle. Our findings suggest that this understudied pathway is a novel therapeutic target for the interference of HIV-1 infection. Nucleolar manipulation could be applicable against other viral infections and disease models requiring nucleolar access.
Footnotes
Acknowledgments
We acknowledge Dr. Barbara K. Felber and Dr. George N. Pavlakis for HLfB adherent culture and Dr. Olaf Kutsch for JLTRG-R5 suspension cells, provided by the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. We kindly thank Dr. Ulrike Jung for pNL4-3 cloning expertise, and Dr. John Zaia for support. We finally acknowledge financial sources provided by the National Institutes of Health, Grants AI042552 and AI029329.
Authors' Contributions
J.A.C.A. generated all Rev-NoLS mutations and HIV-1NL4-3 variants, created Nef probe for Northern blotting, and carried out all experiments discussed throughout the article. M.T. contributed Alu-gag qPCR experiments for detection of integrated HIV-1 DNA. J.C.B. created the dual Cre-LoxP lentiviral reporter and CVITG backbone, participated in designing the dual reporter study, and helped to draft the ![]() section of the article. D.L.O. conducted the co-localization of Rev-NoLS mutations with B23 in HeLa (absence of HIV-1 production) and generated the Flag-WT Rev fusion, later used as a template for Rev-NoLS-3′Flag mutagenesis. J.J.R. proofread and edited the final article.
section of the article. D.L.O. conducted the co-localization of Rev-NoLS mutations with B23 in HeLa (absence of HIV-1 production) and generated the Flag-WT Rev fusion, later used as a template for Rev-NoLS-3′Flag mutagenesis. J.J.R. proofread and edited the final article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.