
Abstract
The purpose of this study was to assess humoral antibody responses as a function of disease progression (DP) in a well-defined HIV+ cohort. We quantified antibodies to HIV-1 gp120, Gag, and CD4 receptor by enzyme-linked immunosorbent assay in sera from a cohort of 97 HIV+ subjects at defined stages of DP. We also measured antibody-dependent cellular cytotoxicity (ADCC) as a function of the clinical status of the patients. We purified antibodies to CD4 and gp120 and assessed them for specificity, ability to block gp120 binding to target cells, ability to block virus infection, and ability to facilitate ADCC. All of the HIV+ patient samples were positive for antibodies to HIV gp120 and p24 and 80% showed evidence of hypergammaglobulinemia. Approximately 10% of cohort members were positive for antibodies to CD4, but we noted no significant correlation relevant to DP. There were statistically significant differences between the groups concerning the level of humoral response to gp120 and Gag. However, we observed no distinction in ability of anti-gp120 antibodies purified from each group to neutralize infection. In addition, there was a statistically significant difference in ADCC, with elite controllers exhibiting significantly lower levels of ADCC than the other five groups. We detected IgA anti-gp120 antibodies, but did not correlate their presence with either DP or ADCC levels. The results are consistent with the interpretation that the humoral antibody response to the antigens assessed here represents a signature of the level of viremia but does not correlate with clinical status of HIV infection.
Introduction
H
Thus, it is important to understand what constitutes an efficacious immune response and what arms of the immune system are responsible for preventing infection. From the standpoint of humoral immunity, it is generally assumed that the primary target for virus neutralizing antibody in an HIV-infected host is against the major surface glycoprotein, gp120 (Env), which is responsible for receptor binding and subsequent infection of the target cell. Antienvelope protein antibodies are prevalent in both gammaretrovirus and lentivirus infections and specific neutralization sites have been mapped on HIV 1 –5 and FIV Env. 6,7 Neutralization sites are generally in and around the V3 loop where interactions with both primary and secondary receptors occur. 1,8,9 Previous studies show that passive administration of certain neutralizing human anti-Env antibodies can afford sterilizing immunity to monkeys infected with HIV/SIV hybrid (SHIV) virus 10,11 and against HIV infection of hu/SCID mice. 5,12 Thus, it is logical that Env has been the focus for the development of efficacious vaccine responses. However, both neutralizing and non-neutralizing antibodies to Env are present in natural infections by all three lentiviruses, HIV, SIV, and FIV, with questionable correlation to disease outcome. Env is the most variable protein in the virus and thus is particularly responsive to immunological pressure, generally without penalty to virus replication. Attempts to use purified gp120 or equivalent glycoproteins from other lentiviruses as a recombinant vaccine have universally met with failure. In fact, there are several reports consistent with enhanced infection of animals 13,14 and arguably, humans 9,15,16 vaccinated with recombinant envelope proteins. The envelope glycoprotein gp120 and the Gag antigens of the virus core elicit the strongest humoral antibody responses. Antibodies are sometimes also generated to self-antigens, particularly the viral receptor proteins including the primary binding receptor, CD4 8,17 in human/HIV infections 18 –21 and to the FIV primary binding receptor, CD134 in FIV-infected cats. 22 Certain antibodies against the viral envelope and anti-receptor antibodies can interfere with virus infection by directly blocking receptor binding. Certainly, it would seem to be a selective advantage for the host as a defense against infection and generating such responses has been a primary goal in vaccine development. 1,4,23,24 Results with antiviral drug therapies show a clear correlation between limiting the viral load and better clinical outcome for the patient, 25 so any immune response that can lower the viral load should provide an advantage to the host. However, it is not clear to what extent such humoral responses influence the outcome of the infection and at best, may simply serve as surrogates for the infection and viremic state.
In this study, we examined the humoral antibody responses to gp120, full-length Gag antigen, and CD4 in a clinically defined cohort of 97 HIV+ patients 26,27 to look for correlates between the humoral responses and clinical progression. In addition, we assessed neutralization (anti-gp120 antibodies) and blocking (anti-CD4 antibodies) of infection along with antibody-dependent cellular cytotoxicity (ADCC) elicited by immunoaffinity purified anti-gp120 antibodies from each disease progression (DP) group. The results demonstrate the presence of neutralizing anti-gp120 antibodies in all six clinically defined groups. Likewise, we noted strong immune responses to Gag antigens in all groups independent of clinical DP of the HIV+ groups although differences in the levels of antibody response to gp120 and Gag were statistically significant between certain groups. Antibodies from patients in all clinical stages of DP elicited ADCC, but interestingly, we observed lower responses in elite controllers (EC) than in the other five groups. The findings suggest that virus-dictated humoral antibody responses and ADCC have at best, a limited role in protecting HIV+ individuals from DP.
Materials and Methods
Study population and clinical samples
This study obtained sera and plasma samples from subjects enrolled in the U.S. Military HIV Natural History Study (NHS). The Uniformed Services University Infectious Disease IRB and the participating site IRBs approved the study protocol. All subjects provided written informed consent. The NHS is an open enrollment, prospective, observational cohort study of HIV-infected military active duty and beneficiaries that has enrolled over 6,000 subjects since 1986. 26,27 Briefly, the cohort is ethnically balanced and geographically diverse, with open access to healthcare and medications. Subjects participating in the NHS visit a military treatment facility approximately every 6 months for study visits and collection of data and specimens (including plasma, sera, and peripheral blood mononuclear cells). The cohort is 91.6% male, therefore results may not be generalizable to HIV+ women.
Subjects for this analysis were selected from the NHS based on specimen availability, more recent study visits, and to provide an approximately equal representation across the following six mutually exclusive groupings, based on previously described stages of DP 27,28 (groups 1–4 are in the absence of antiretroviral therapy (ART), 1 and 4 for ≥1 year before categorization, and 2 and 3 ART naïve): (1) EC (12 patients) having at least three undetectable viral loads over a 12-month period; (2) long-term nonprogressors for at least 10 years (LTNP-10; 14 patients); (3) Long-term nonprogressors for at least 7 years (LTNP-7; 18 patients); (4) viremic controllers (VC; 20 patients), consistently maintaining virus loads <2,000 copies over a 12 month period; (5) rapid progressors (RP;13 patients) showing a seroconversion window <1 year with documented negative and positive serology and either (a) ≥2 CD4+ T cell counts below 350 cells/μl within 3 years of seroconversion and no subsequent rise of CD4+ T cell counts above 350/μl in the absence of ART or (b) ART-start within 3 years of seroconversion and CD4+ T cell count within 1 month of ART-start <350/μl; and finally, (6) typical progressors (TP; 20 patients) representing a cross section of HIV+ patients not in any of the other groups. Serum samples were obtained from each patient at time points just before start of ART.
Enzyme-linked immunosorbent assay
Human anti-human antibodies were tested for specificity after purification by testing against antigens human 4D-CD4 (NIH AIDS Reagent Program) and SF2 gp120 (Chiron Corp., Emeryville, CA), which were plated in 96-well Immulon 2HB plate (Thermo Scientific, Rockford, IL) overnight at 200 ng/well in 0.1 M sodium carbonate, pH9.6. Plates were washed four times with 1 × phosphate-buffered saline (PBS) before 100 μl of human anti-human antibody, diluted to 3 ng/μl in B3T Buffer (8.8 g/liter NaCl, 7.87 g/liter Tris-HCl, 334.7 mg/liter EDTA, 20 g BSA Fraction V, 33.3 ml/liter fetal calf serum, 666 μl/liter Tween-20, and 0.02% Thimerosal, pH 7.4) was added to each well and incubated at room temperature for 45 min. Wells were washed as before, then 100 μl B3T containing 1:2,000 goat anti-human IgG Fc-HRP (MP Biomedicals, Aurora, OH), or 1:4,000 goat anti-human IgA-HRP (Southern Biotech, Birmingham, AL) was added and incubated at room temperature for 45 min. Wells were washed as before, then 100 μl of enzyme-linked immunosorbent assay (ELISA) developing solution: 2 mg OPD (Sigma, St Louis, MO) per 5 ml Substrate Buffer (10.24 g/liter citric acid, 14.2 g/liter dibasic sodium phosphate, pH 5.0) + 42 μl 30% H2O2 were added to each well, and incubated for 5 min at room temperature before stopping the reaction with 50 μl 2 M sulfuric acid. A Victor 3 1420 multi-label plate reader (Perkin Elmer, Waltham, MA) read the plate at 493 nm.
Flow cytometry
Purified human anti-human gp120 and human anti-human CD4 antibodies were tested for their abilities to bind MT-2 cells (human T-Cell: CD4+, CXCR4+, CCR5+) or CEM (human T-lymphoblast: CD4+, CXCR4+, CCR5−) on their own and whether they interfere with gp120 binding cells. Two lakhs cells/sample were counted out and resuspended in 50 μl/sample of FACS Buffer (EBSS, 2% FBS, 0.2% sodium azide). Cells were incubated with either 500 ng SF2 gp120 (Chiron Corp.) or 1 μg of purified human-derived anti-human antibody and incubated for 30 min at room temp. The samples were washed by adding 200 μl FACS Buffer, centrifuging and removing the supernatant, and repeated once.
One microgram of purified human anti-human antibody was added to cells+gp120 sample, and 500 ng SF2 gp120 was added to cells+huαhu antibody; all samples were then incubated for 30 min at room temperature, before being washed twice. For samples detecting gp120 binding: human anti-gp120-Biotin was diluted 1:2,000 in FACS Buffer of which 100 μl was added to appropriate samples; the samples were then incubated for 30 min before being washed twice. Extravidin-RPE (Sigma) was diluted 1:20 in FACS Buffer, with 40 μl added to samples; samples were incubated for 30 min in darkness before being washed twice and resuspended in 200 μl of FACS Buffer. For samples detecting human anti-human antibody binding: 100 μl of 1:1,000 mouse anti-human IgG1 (hinge)-RPE (Southern Biotech) was added to samples and incubated for 30 min in darkness, before being washed twice and being resuspended in 200 μl FACS Buffer. The samples were run on a FACSCanto (BD Biosciences, San Jose, CA), and data were analyzed with FlowJo (Treestar, Ashland, OR).
ADCC assays
This protocol was adapted from Gomez-Roman et al. 29 CEM-NKr (natural killer resistant) cells, obtained from the AIDS Research and Reference Reagent Program (NIAID, NIH), were used as the Target cells, and were stained first with CFSE (5-(and 6)-carboxyfluorescein diacetate, succinimidyl ester; Molecular Probes, Eugene, OR) and then with PKH-26 (Sigma) according to each of the manufacturer's instructions. Target cells were then washed and resuspended in 300 μl of EBSS 2% FBS. Target cells were incubated with or without 15 μg of SF2 gp120 (HIV-1 subtype gp120 variant; Chiron Corp.) per 1 × 106 stained Target cells for 1 h at room temperature. Samples were washed twice with cold EBSS 2% FBS before being resuspended in EBSS 2% FBS at 1 × 105 cells/ml, of which 50 μl was aliquoted per well in a microtiter plate (5,000 cells/well).
The Human HIV+ Sera was diluted 1:40,000 in EBSS 2% FBS, and 100 μl was added to the wells with the Target cells; these samples were incubated together for 15 min at room temperature. Human monocyte cells, THP-1, were used as effector cells, they were washed with EBSS 2% FBS and resuspended at 5 × 103 cells/ml; 50 μl of effector cells were added to every well in the microtiter plate with Target cells. The plate was then briefly centrifuged for 3 min at 400 × g before being incubated at 37°C for 4 h. After the incubation period, the plate was washed once with 1 × PBS, and samples were fixed with 3.7% paraformaldehyde/PBS (v/v) and stored overnight at +4°C. Flow cytometry data were acquired by the CellQuest Software, and 30,000 nongated events where acquired within 24 h of the ADCC assay using a LSRII (BD Biosciences, San Jose, CA). Data were analyzed with Flowjo (Treestar).
Immunoaffinity chromatography
Antibodies to gp120 or to CD4 were purified by immunoaffinity chromatography (IAC) using JR-CSF gp120 or four-domain CD4 bound to an agarose-based matrix (ALD, Sterogene, Carlsbad, CA). Proteins were coupled using the manufacturer's recommended protocol at 4 mg protein per ml beads. Extensively washing the coupled beads with PBS removed any unbound protein. We decomplemented either individual sera from select patients or pooled sera from each clinical group at 56°C for 30 min and passed them over columns of the conjugated beads; where we recovered both anti-GP120 and anti-CD4 antibodies from the same patient, the sera was first depleted of anti-gp120 antibody, then anti-CD4 antibody was purified on CD4-sepharose beads. The beads were then washed with PBS and eluted with low pH elution buffer (IgG Elution Buffer; Thermo Scientific, Waltham, MA).
Follow-up elutions were also performed using a more stringent citrate-based elution buffer (100 mM citric acid, 150 mM NaCl, pH 2.1), which resulted in release of tightly bound antibodies not released by the standard glycine-based low pH elution buffer. Eluted antibodies were then concentrated and washed with PBS using centrifuge concentrators, aliquoted, and stored at −20°C for subsequent assays.
Neutralization assay
We produced viruses by transfection of 293T cells using GeneJuice (Novagen). Laboratory-adapted provirus R9 (NL4.3 derivative) was kindly provided by D. Trono. We obtained CD4+ TZM-bl cells through the AIDS Research and Reference Program and 293T cells from ATCC. All viruses were harvested 48 h post-transfection, filtered (0.2-mM filter, Pall Acrodisc), and normalized by p24 ELISA (Alliance HIV-1 p24 Antigen ELISA Kit; Perkin Elmer)
TZM-bl cells were seeded in 48-well plates at 4.5 × 104 cells per well, 24 h before infection in 200 μl of high glucose DMEM (Gibco) 10% FBS. Neutralizing antibodies were incubated with 10 ng HIV-1 (p24 equivalents) in cell media for 30 min at 37°C before being added to cells. We washed the cells with PBS and lysed them 72 h postinfection with Galacton-Star lysis buffer. We transferred 20 μl of cell lysate to a 96-well plate for detecting β-galactosidase activity. One hundred microliters of reaction buffer [Galacton-Star substrate (Applied Biosystems, Bedford, MA) diluted 1:50] was added to 20 μl of lysate, and the light emission was measured in relative fluorescence units over 1 s in a microplate luminometer after 30 min incubation.
Statistical methods
Data from ELISA results were organized and compiled in Microsoft Excel; we calculated all statistics using Microsoft Excel's Data Analysis tool. The Cohort groups were compared one-on-one using an unpaired T test with unequal variances. To reduce the chances of obtaining type 1 errors, a Bonferroni Correction was applied by multiplying the p values generated for each comparison group by 15 (the total number of comparisons made between groups). We consider all p values <.05 statistically significant (bolded in Table 1), while p values ≥.05 were not significant. We also used boxplots to illustrate ELISA results (Figs. 2 and 8a), in which quartiles represent the data sets. The line at the bottom of each box indicates the first quartile, the line in the middle of each box indicates the second quartile, and the line at the top of each box indicates the third quartile, with the whiskers indicating the minimum and maximum of all the data within the group.
Calculated p value tables comparing the different disease status groups of HIV+ sera ELISA reactivities to gp120 (A), Gag (B), CD4 (C), and IgG concentration (D). p Values <.05 are statistically significant (bold).
EC, elite controllers; ELISA, enzyme-linked immunosorbent assay; RP, rapid progressors; TP, typical progressors; VC, viremic controller.
Results
Serum samples from 97 subjects were assayed and results grouped by DP category. An informative comparison of the different groups (excluding VC) is shown in Figure 1, which represents, on average, when each patient group was placed on ART, which was a major factor in defining the six groups. The present studies employed sera or plasma from each patient immediately before being placed on drug treatment.
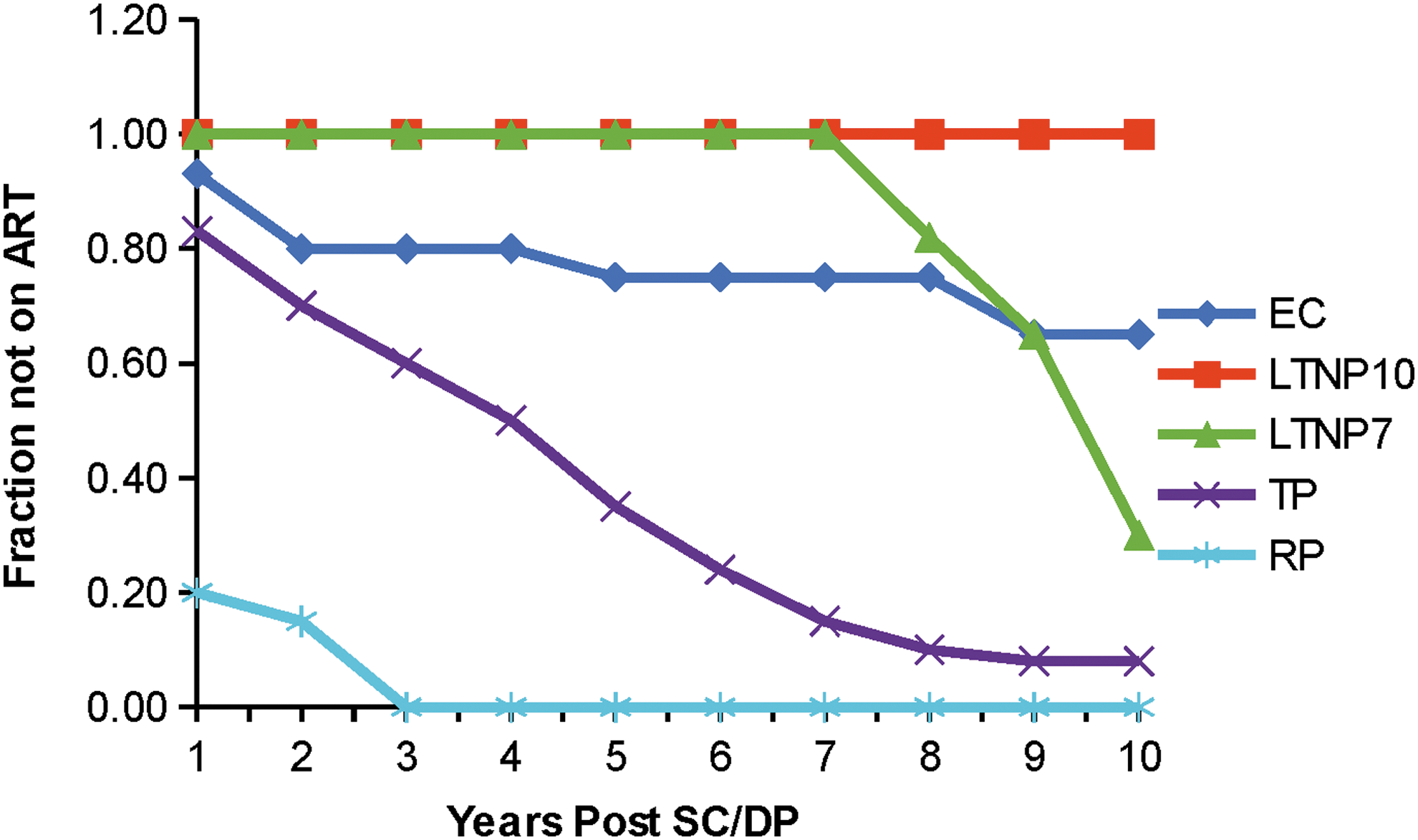
Anti-retroviral therapy (ART) history of HIV+ patients divided into groups based on stage of DP. Note that up to 8 years post sero conversion (SC) neither LTNP7 nor LTNP10 were placed on HAART, whereas all rapid progressors were on HAART by year 3. DP, disease progression.
Figure 2 shows the results of ELISA analyses of the patient sera against SF2 gp120; full-length Gag antigen (NL4-3) and 4-domain CD4 (Fig. 2A–C). In addition, we assessed the total immunoglobulin per ml serum or plasma for each patient as a measure of the degree of hypergammaglobulinemia for each patient (Fig. 2D). The majority of patient sera from the various groups exhibited high levels of antibody to both Env and Gag antigens independent of DP. Analysis of variance (ANOVA) indicated significant differences between certain groups in the level of anti-gp120 and in anti-Gag reactivities, but little difference in either anti-CD4 antibody levels or in total immunoglobulin. T tests indicate the significant differences between the groups (Table 1A–C). Although over 80% of patient sera were considered hypergammaglobulinemic (values over 10 mg/ml total Ig), there was no apparent bias toward any one group (Table 1D).
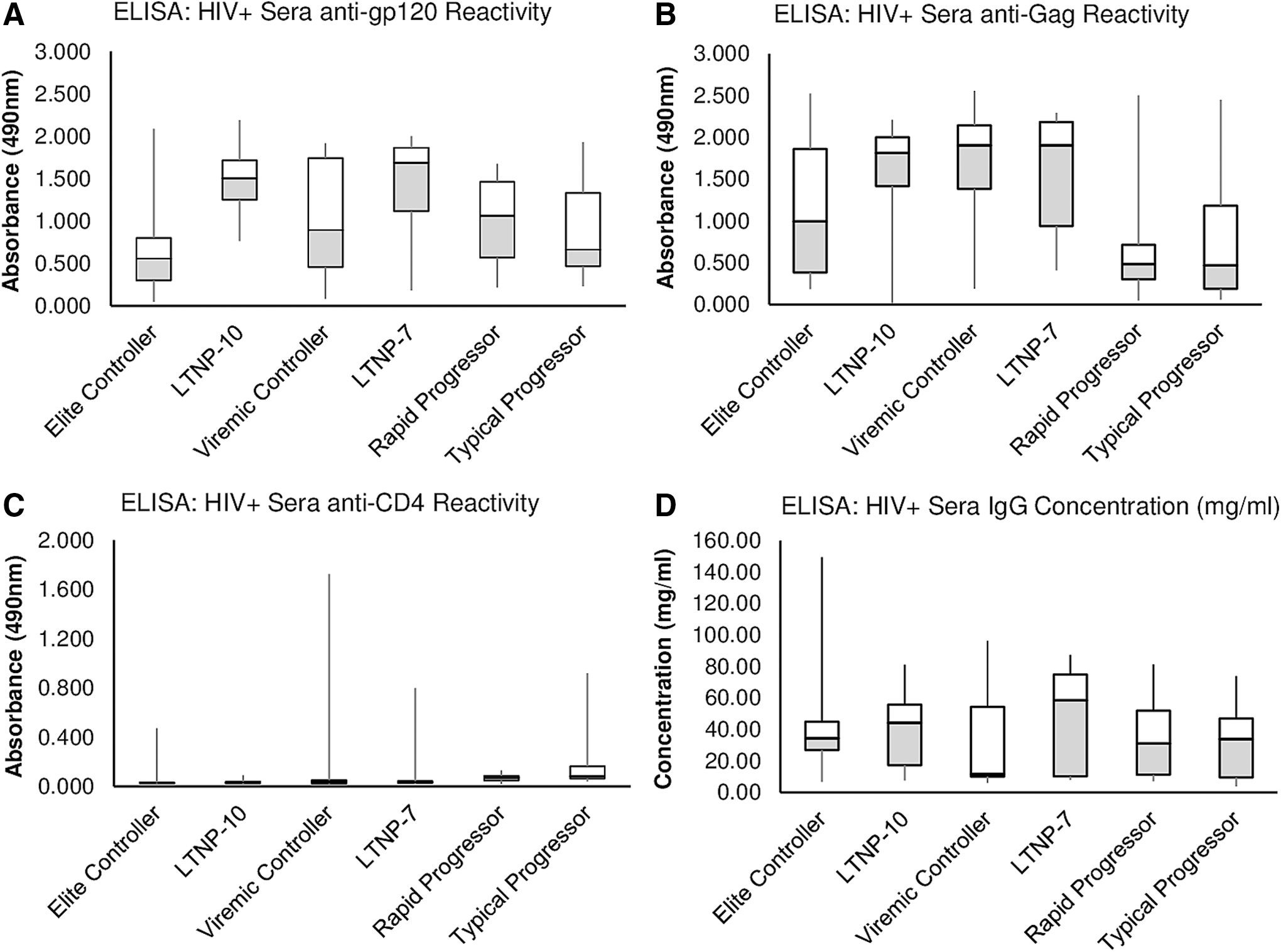
Box plots showing reactivity of HIV+ sera individuals, grouped by stage of DP, in ELISA to gp120
The most consistently high levels of anti-gp120 were noted in the two long-term nonprogressor groups (mean ELISA values of 1.519 and 1.467 for LTNP10 and LTNP7, resp) followed by VC (1.009), RP (0.983), and TP (0.908); the lowest mean value for anti-gp120 was in the EC group (0.664). The rank order for antibody levels to Gag changed somewhat from levels of anti-gp120 antibody; LTNP10 and LTNP7 also exhibited the highest mean values at 1.516 and 1.644, respectively, matched by the VC group at 1.632. The next was EC at 1.161 followed by RP and TP groups at 0.607 and 0.784, resp. Differences for both the gp120 and Gag antibody levels between the LTNP groups and the RP and TP groups were significant (p = .05–.001; Table 1); mean values for both anti-gp120 and anti-Gag in the latter two groups were less than a third the level of the LTNP and VC groups for anti-Gag reactivity. Interestingly, the latter groups did not reach statistical significance for anti-gp120 reactivity compared to RP or TP groups.
We hypothesized at the beginning of this study that there might be a correlation between clinical state in HIV infection and antibodies to the primary binding receptor for HIV, CD4. However, the number of patients with significant levels of anti-CD4 antibodies was low and we noted no significant correlations with clinical status (Table 1). There were, however, a few individuals, particularly in the VC group, that had high levels of anti-CD4 antibodies (Fig. 2). We purified these antibodies by IAC using immobilized 4-domain CD4 and also purified anti-gp120 antibodies from the same patients for comparison, using SF2 gp120 immobilized on beads. Results showed that both antibodies to CD4 and to gp120 demonstrated the expected specificity; that is, IAC-purified anti-gp120 did not recognize CD4 and anti-CD4 antibodies did not recognize gp120 by ELISA (not shown). FACS analyses using these antibodies (Fig. 3) demonstrated that antibodies to CD4 only bound to the surface of cells after gp120 was bound, consistent with recognition of a cryptic epitope on CD4 only exposed by binding of gp120 (or virus) on the cell surface (Fig. 3). Furthermore, the antibodies to CD4 did not cause substantial release of prebound gp120 from the cell surface of the target cell although gp120 binding was somewhat reduced when anti-CD4 and gp120 were added together (Fig. 4).
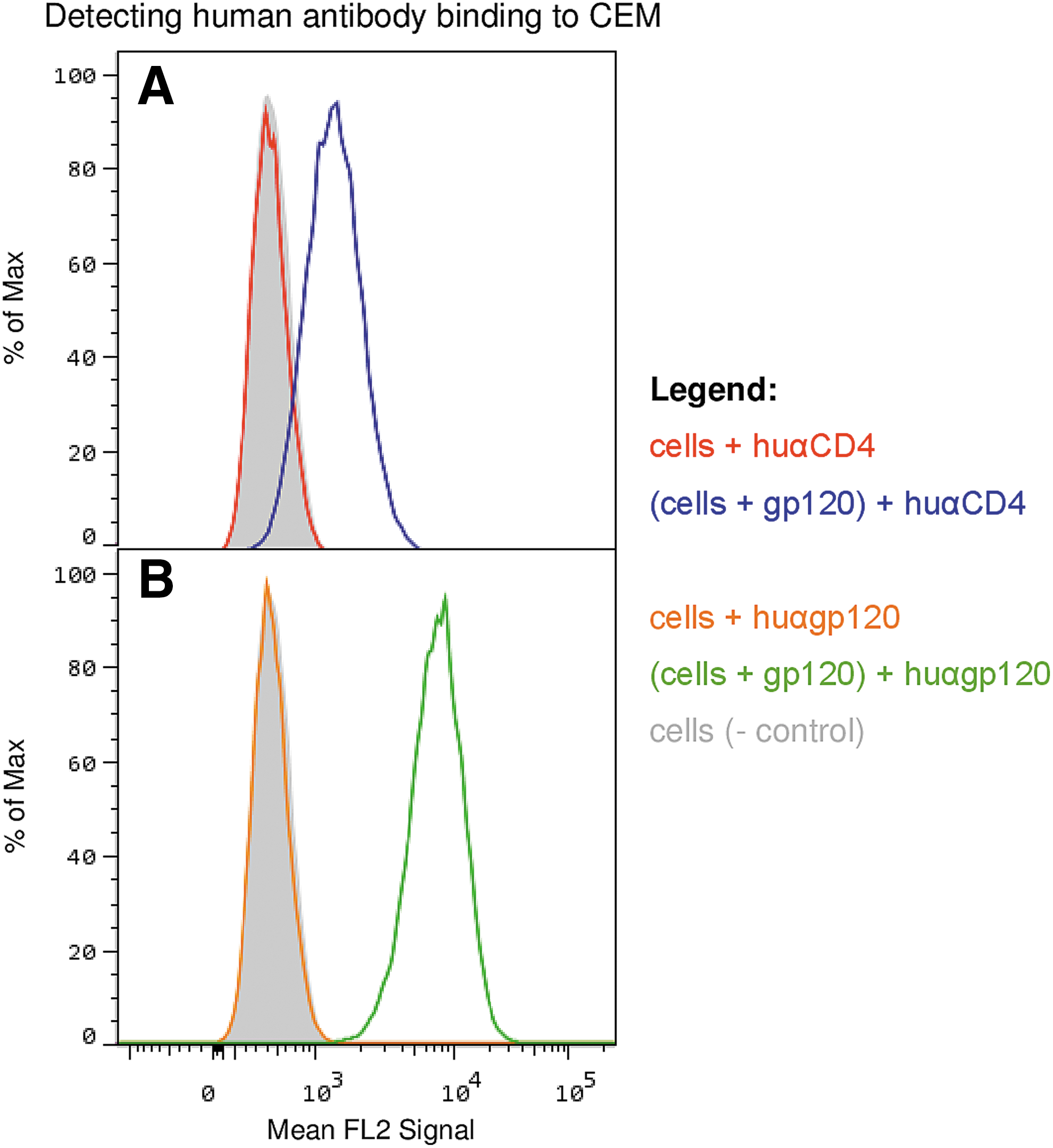
FACS results, IAC purified human anti-CD4 and human anti-gp120 where tested for their ability to bind to CEM cells (CD4+, CXCR4+, CCR5−) with or without gp120. Human anti-CD4 was not able to bind cells by itself (red line), and was only able to bind to cells when gp120 was prebound to CD4 (blue line); ELISA data (not shown) indicate the anti-CD4 detects CD4 and does not detect gp120. Human anti-gp120 does not bind to CEM (orange line), it only binds to cells when gp120 is prebound to CEM cells (green line). IAC, immunoaffinity chromatography.
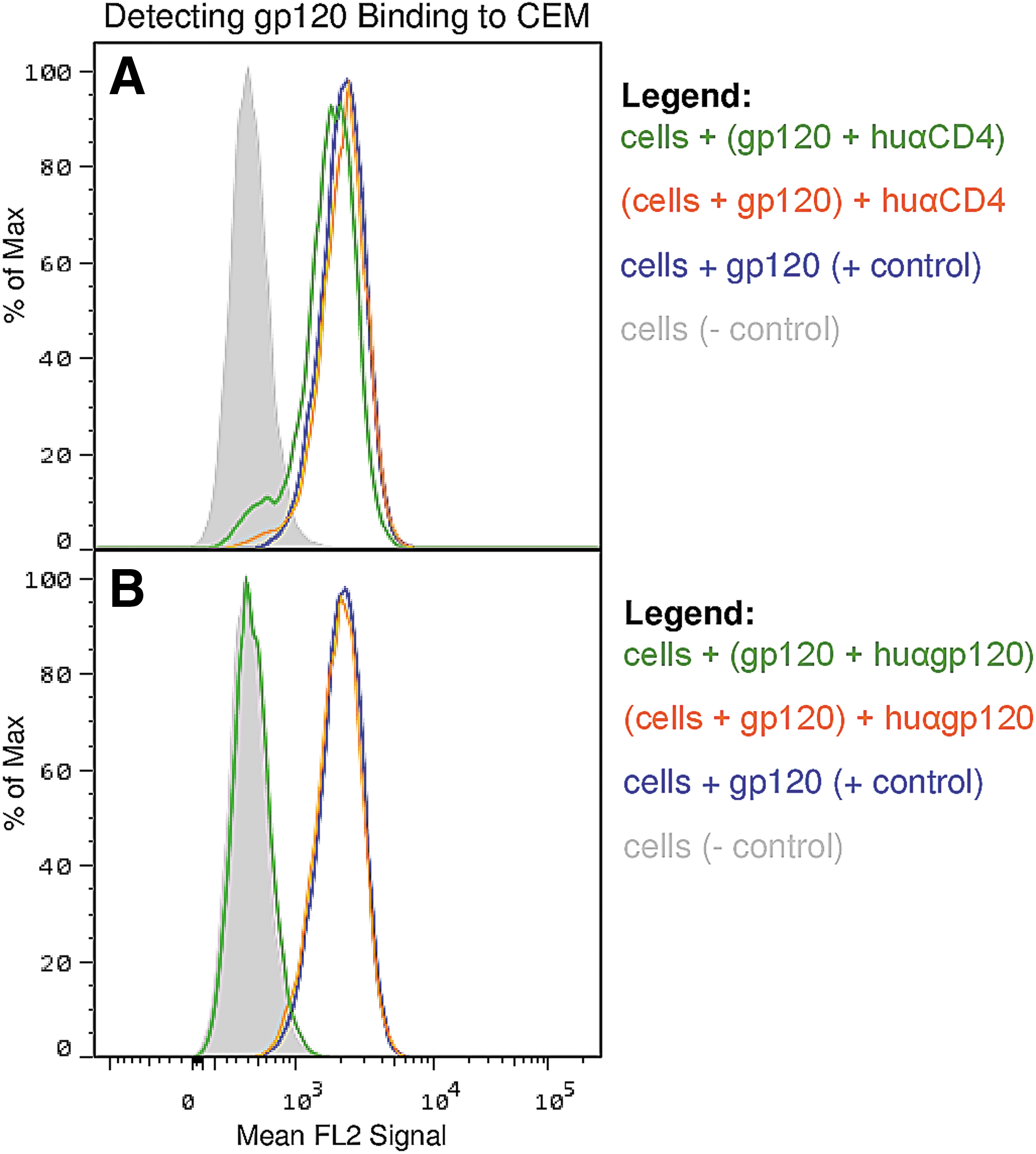
FACS results to determine the effect the human antibodies, anti-CD4
Neutralization/blocking assays were also performed and both anti-CD4 and anti-gp120 antibodies were able to interfere with virus infection (Fig. 5). However, the relative titers for neutralization were low and we noted no significant discrimination in level of neutralization for any of the groups. Furthermore, IAC-purified anti-gp120 from pooled sera of each group showed no qualitative differences in neutralization between the groups (Fig. 6).
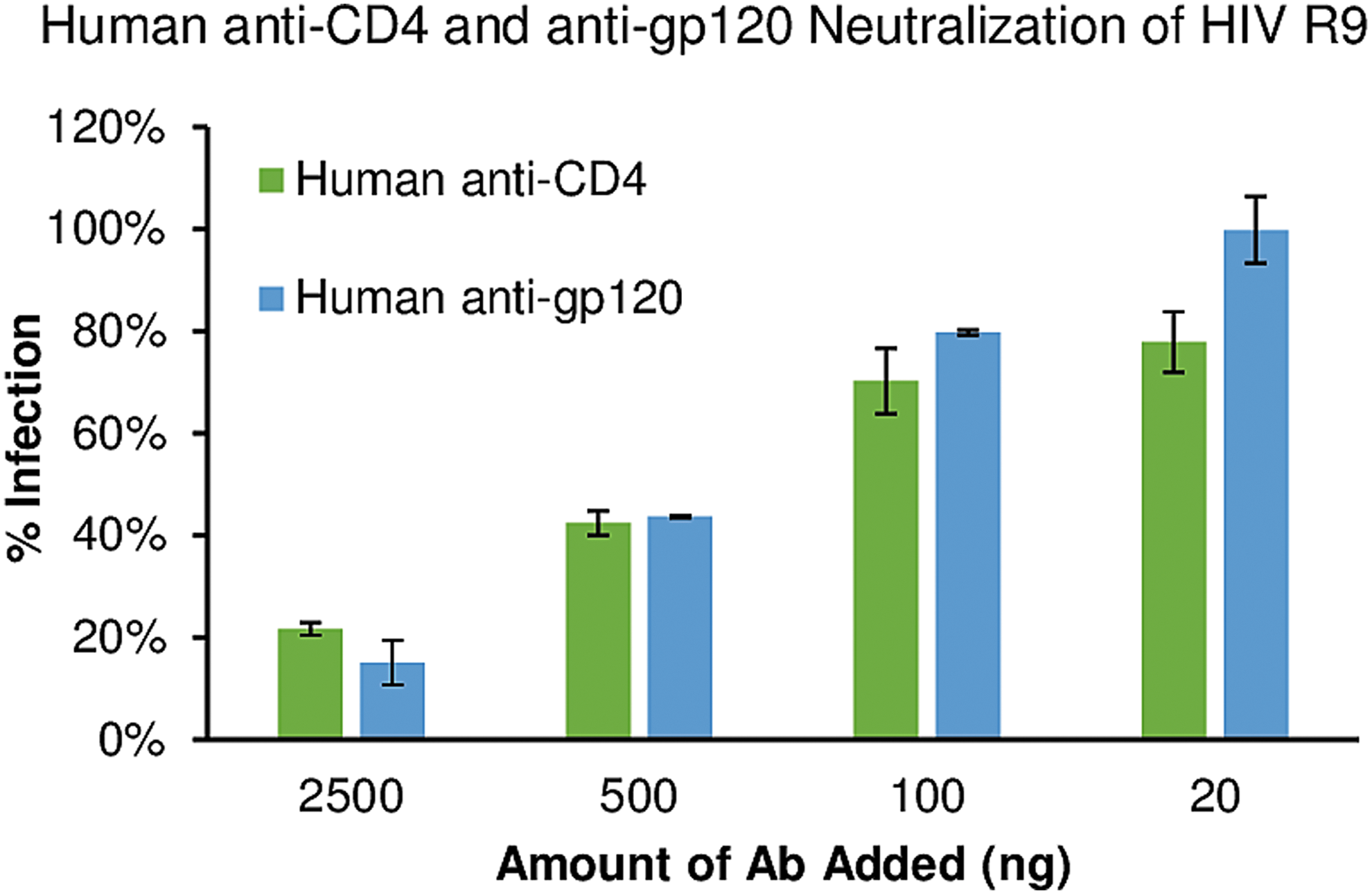
Neutralization of HIV R9 infection on TZM-bl. Various concentrations of purified VC-18 anti-gp120 (blue bars) were incubated with HIV R9 for 30 min before exposure to TZM-bl cells. Alternatively, VC-18 human anti-CD4 (green bars) was incubated with TZM-bl for 30 min before HIV R9 addition. Twenty nanograms of antibody had little effect and HIV R9 was able to infect cells at similar levels as if no antibody was added. Increasing the amount of antibody, increased the blocking effect on HIV R9 entry, with 2,500 ng inhibiting HIV R9 by 80%–85%. VC, viremic controller.
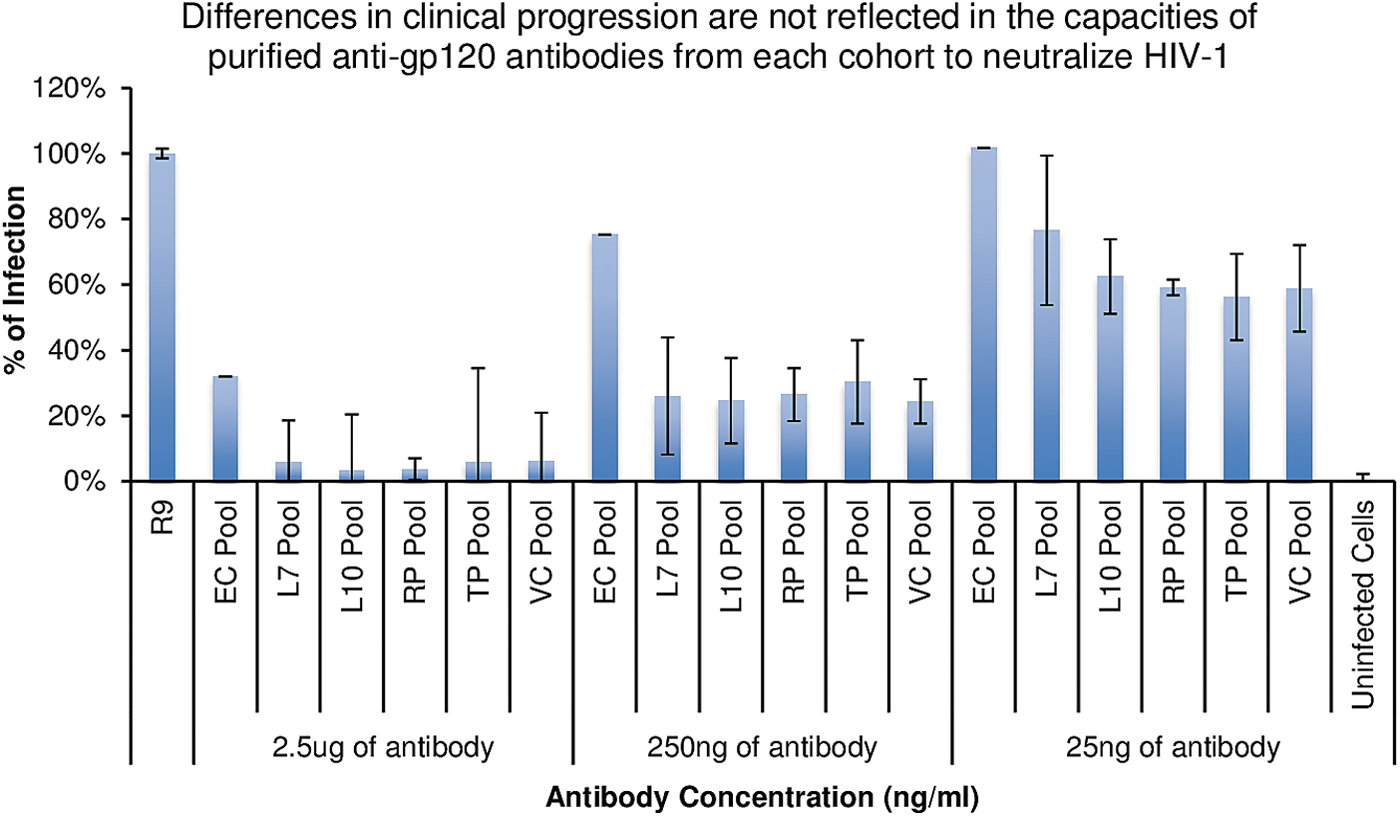
Neutralization of HIV R9 infection on TZM-bl by group pooled purified anti-gp120 antibodies. Anti-gp120 antibodies purified from the pooled groups were incubated with HIV R9 for 30 min before cell exposure. No significant difference in infection inhibition was noted between groups at the concentrations tested.
We also performed gp120 ADCC assays using all of the patient sera and compared to the relative levels of anti-gp120 antibody for each group (Fig. 7). Virtually all patient sera exhibited gp120-dependent ADCC and could not be discriminated from one another until highly diluted (1:20,000). At higher dilution, we noted a statistically significant difference in the mean levels of ADCC exhibited by the EC group compared to the other five groups, at dilutions 1:20,000 and above (Table 2).
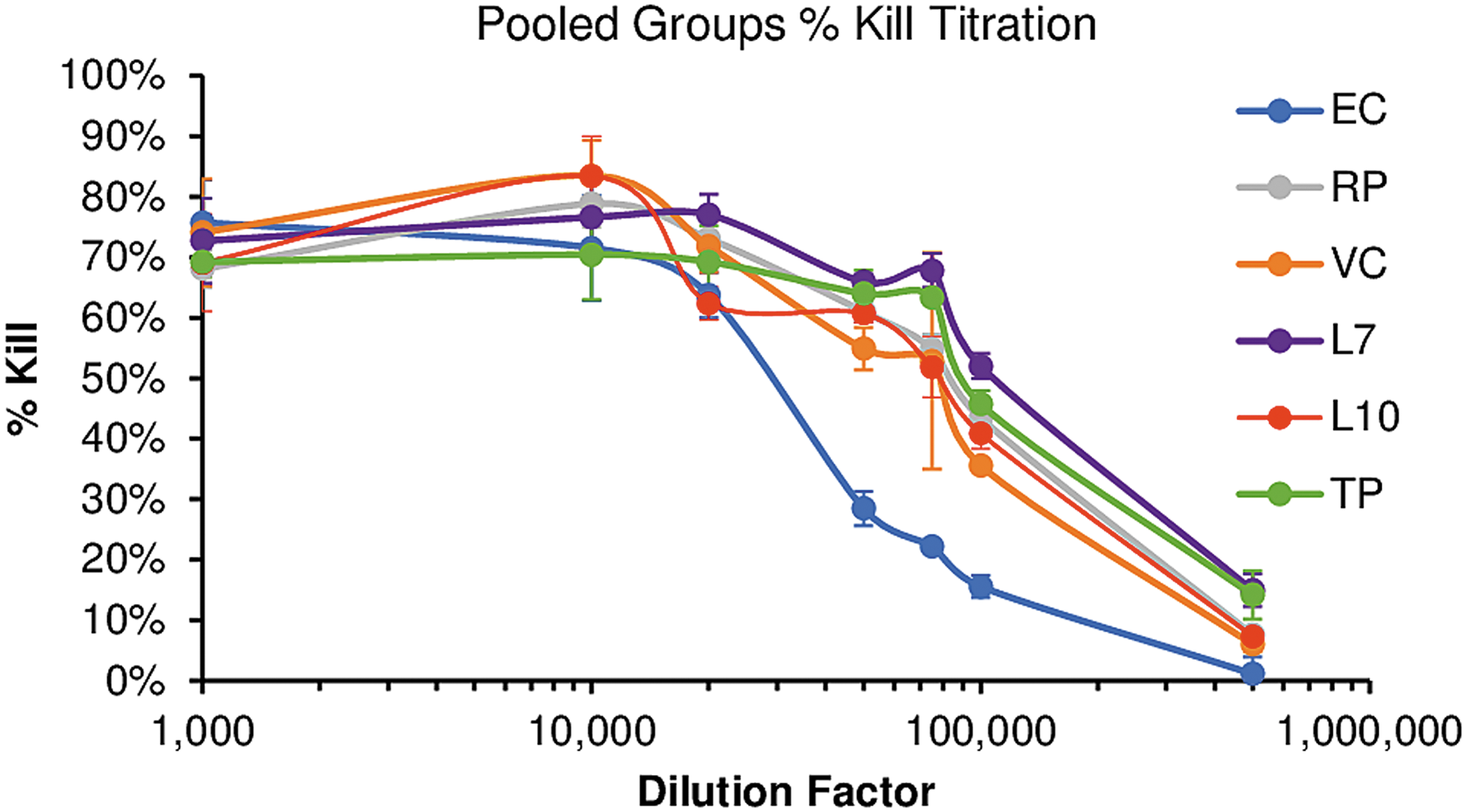
Within each HIV+ sera patient group, the individual samples were pooled together and serially diluted to 1:1k, 1:10k, 1:20k, 1:50k, 1:75k, 1:100k, and 1:500k and tested for their ability to induce ADCC killing of CEM-NKr cells coated with the Clade B HIV trimer C6a. ADCC, antibody-dependent cellular cytotoxicity.
T test of RFADCC data of HIV+ sera diluted 1:40k, p values were calculated between groups, Bonferroni adjusted, and values above ≥.05 indicate no significance. P Values <.05 are statistically significant (bold).
ADCC, antibody-dependent cellular cytotoxicity.
To determine whether there was a distinction in the level and/or nature of IgA anti-gp120 antibodies as a function of DP we assayed the individual sera by ELISA, which revealed IgA responses in a subset of patients, but we noted no significant trend in any particular group (Fig. 8a). Assessment of IgA anti-gp120 responses in pooled sera from each group yielded similar results (Fig. 8b), as did assessment of IgA antibodies in IAC-purified anti-gp120 preparations (Fig. 8c).
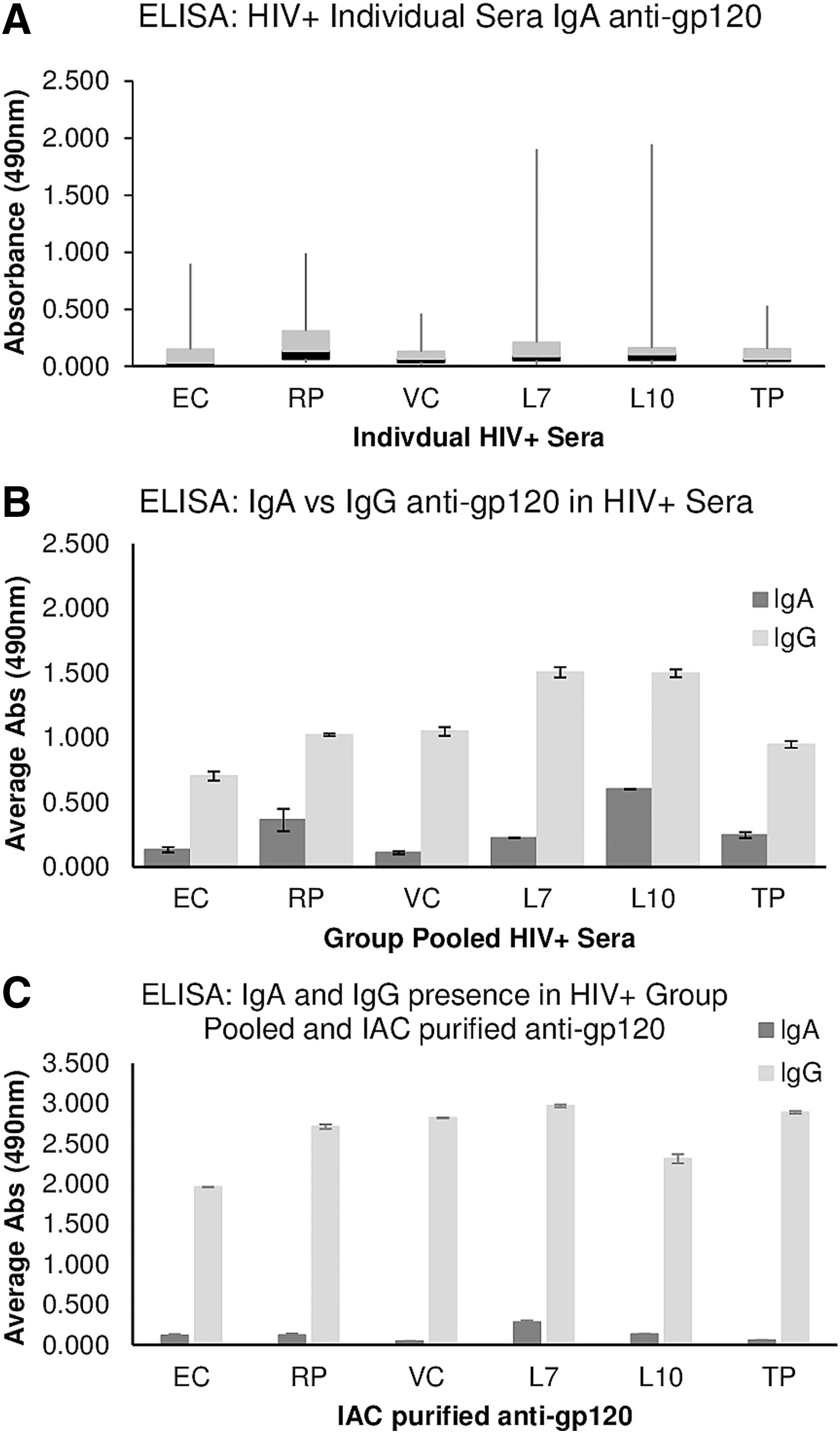
ELISA results, testing for IgA or IgG antibody specificity anti-gp120 reactivity in the individual patient HIV+ Sera
Discussion
A robust humoral antibody response occurs in most, if not all, viral infections and is thought to be a major player in clearance of the infection when it occurs. 1,4,5,30 –32 The anticipatory activation of such a response is the common goal of vaccine design. In the case of HIV-1, despite the genetic plasticity of its most immunologically exposed antigen gp120, numerous studies have shown that neutralizing antibodies are generated in the course of HIV infection. Further, a small subset of individuals produces high affinity, broadly neutralizing antibodies. 2,24 Underscoring their therapeutic potential, passive immunization with high affinity neutralizing antibody has achieved sterile immunity in animal models. 10 –12,33
Taken together these studies have strongly encouraged the hope that we can design immunogens to induce similar protective responses against HIV-1 via vaccination. To that end, interactions between these broadly neutralizing antibodies and the major protein target, gp120, have been under intense scrutiny. Unfortunately to date an efficacious vaccine has yet to emerge. One factor frustrating efforts to develop an anti-HIV-1 vaccine is that the clinical course of HIV infection is relatively protracted compared to other lethal viral infections, and patients' immune responses cycle through many evolutions of antibody against gp120. Mimicking the iterative cycles of the HIV immune response with a few antigens that can generate a response matching a desired broadly neutralizing antibody has been challenging. Perhaps, a reexamination of precisely what kind of immune response should be used as a model for a successful end point vaccine design is in order.
Numerous studies have attempted to assess the occurrence of the humoral antibody responses to HIV proteins, primarily to gp120 and gp41, as a function of the rate and severity of infection in cohorts of HIV+ individuals, with mixed results. 9,16,24,30,31 Maturation of the antibody response and development of broadly neutralizing antibodies is protracted and thus may not afford the timely protection required to prevent progression of HIV disease. Similarly, while previous studies show that ADCC develops as a consequence of infection, the relevance to the rate of DP is still unclear. 31,34 –44 In this study, we attempted to determine whether the level and/or quality of the humoral response could be correlated with the clinically defined rate of DP in a cohort of 97 patients representing six progression groups based on CD4 counts, levels of viremia and time before initiation of ART.
Results show that all of the patient pools had antibodies to both Env and Gag and neutralizing antibodies to gp120. Differences in the levels of both anti-Gag and anti-gp120 antibodies consistent with improved prognosis were small, but reached significance in LTNP groups compared to RP and TP groups. However, overall there was no distinction in the efficacy or level of neutralizing antibody per mg of anti-gp120 recovered by IAC between any of the various groups.
Another major objective of the study was to monitor levels of anti-CD4 antibodies in the patients of this cohort for relationships to patient health status. We previously observed that in FIV+ cats, antibodies to the primary receptor for FIV, CD134, occur frequently and appear to correlate with improved health status 22 and hypothesized that a similar scenario might occur in HIV-1-infected humans. However, only a few individuals developed substantial levels of anti-receptor antibodies. Although IAC-purified anti-CD4 antibodies from individuals showing the highest anti-CD4 titers could block infection the frequency of anti-CD4 antibodies was low and did not correlate with slower DP. To explain this difference, we hypothesize that the cat anti-CD134 antibodies caused release of Env bound to the surface of the cell, 22 consistent with an induced conformational change that lowers the ability of the viral glycoprotein to bind receptor. In contrast, Figure 4's results indicate that while anti-CD4 antibody binding is close to the CD4 binding site for gp120, it does not cause a conformational change, and gp120 remains bound on cell surface CD4.
ADCC activity was also prevalent in all six progression groups and when examined as pooled serum populations or as purified anti-gp120 antibody, only the EC group varied significantly. In fact, the level of ADCC in the EC group was substantially lower than the other five groups, inconsistent with a role in protection from DP in this particular EC group. Note the level of ADCC in all six groups was substantial (Fig. 7), and it was only at a very large (>10,000) dilution that we were able to discriminate between the groups.
It has been suggested that in a vaccine setting, IgA antibodies against gp120 might negatively impact ADCC elicited by IgG anti-gp120 antibodies in the immunized subjects, 45 with some argument as to whether such antibodies help or hurt during infection. 46,47 In the Thai trial, they recovered high affinity IgA anti-gp120 antibodies from vaccinees and the investigators were able to demonstrate the predicted competition with IgG. 45,48 However, these antibodies were generated in response to vaccination and it is unclear whether such responses are relevant in a natural infection. We examined the present cohort for IgA anti-gp120 antibodies and they were, indeed, present in both total serum and in IAC-purified anti-gp120 preparations.
Based on the assumption that the IgA antibodies might influence the level of ADCC, one would predict more abundant IgA anti-gp120 in the EC group. However, there was no correlation between the level of IgA anti-gp120 and the progression of the disease (Fig. 8). Although IgA interference with ADCC may factor into a vaccine setting, we find no evidence that it occurs during a natural infection. Our findings are consistent with those of Lambotte et al. 31 who compared HIV controllers against viremic expressors for such attributes as presence of nAbs and autoantibodies, competitive inhibition binding of antibodies, neutralization ability, and ADCC. They found that there was no difference in the levels of nAbs from viremic expressors and HIV controllers, there were very low levels of autoantibodies present (with no correlation to HIV status), and no specific anti-HIV antibody correlate of immunity in HIV controllers versus viremic expressors. While they had only a fraction of the number of patient sera, and only a two classification system of patient segregation (they defined controllers as patients with less than 400 copies/ml of HIV RNA), their results corroborate our conclusions.
In summary, the findings do not support a significant role of the humoral antibody response in influencing the rate of DP in this HIV+ cohort, but rather represent a signature of the infection and at best, reflect the relative viral load during infection.
Footnotes
Acknowledgments
The authors wish to acknowledge the assistance of Dr. Sonia Jain, under the auspices of the Bioinformatics and Modeling Core of the University of California San Diego Center for AIDS Research (P30 AI036214) for review of statistical analyses performed in this study. Support for this work (IDCRP-000-32) was provided in part by the Infectious Disease Clinical Research Program (IDCRP), a Department of Defense program executed through the Uniformed Services University of the Health Sciences. This project was funded in whole, or in part, with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), under Inter-Agency Agreement Y1-AI-5072 (G.V.Q., B.A.) and by R21 AI100764 (J.H.E.).
Disclaimer
The views expressed are those of the authors and do not necessarily reflect the official views of the Uniformed Services University of the Health Sciences, the National Institutes of Health, or the Department of Defense.
Author Disclosure Statement
No competing financial interests exist.