
Abstract
Pulmonary endothelial cell (EC) barrier dysfunction and recovery is critical to the pathophysiology of acute respiratory distress syndrome. Cytoskeletal and subsequent cell membrane dynamics play a key mechanistic role in determination of EC barrier integrity. Here, we characterizAQe the actin related protein 2/3 (Arp 2/3) complex, a regulator of peripheral branched actin polymerization, in human pulmonary EC barrier function through studies of transendothelial electrical resistance (TER), intercellular gap formation, peripheral cytoskeletal structures and lamellipodia. Compared to control, Arp 2/3 inhibition with the small molecule inhibitor CK-666 results in a reduction of baseline barrier function (1,241 ± 53 vs 988 ± 64 ohm; p < 0.01), S1P-induced barrier enhancement and delayed recovery of barrier function after thrombin (143 ± 14 vs 93 ± 6 min; p < 0.01). Functional changes of Arp 2/3 inhibition on barrier integrity are associated temporally with increased intercellular gap area at baseline (0.456 ± 0.02 vs 0.299 ± 0.02; p < 0.05) and thirty minutes after thrombin (0.885 ± 0.03 vs 0.754 ± 0.03; p < 0.05). Immunofluorescent microscopy reveals reduced lamellipodia formation after S1P and during thrombin recovery in Arp 2/3 inhibited cells. Individual lamellipodia demonstrate reduced depth following Arp 2/3 inhibition vs vehicle at baseline (1.83 ± 0.41 vs 2.55 ± 0.46 µm; p < 0.05) and thirty minutes after S1P treatment (1.53 ± 0.37 vs 2.09 ± 0.36 µm; p < 0.05). These results establish a critical role for Arp 2/3 activity in determination of pulmonary endothelial barrier function and recovery through formation of EC lamellipodia and closure of intercellular gaps.
Introduction
Acute respiratory distress syndrome (ARDS) remains a common and highly morbid condition in the intensive care unit. 1 Central to the pathology of ARDS and sepsis is dysfunction of the endothelial barrier resulting in leakage of protein-rich fluid from the vascular to interstitial space.2,3 Subsequent flooding of the alveolar space leads to impaired gas exchange and hypoxemia. 4 The pulmonary endothelial cell (EC) is critical to maintenance of the barrier between the vascular and alveolar spaces.5,6 While both transcellular and paracellular routes are involved in the transit of fluid and solute across the endothelial monolayer, the paracellular route predominates in pathologic vascular leak.7,8 The breakdown of cell–cell junctions and opening of gaps between individual endothelial cells is thought to be the primary driver of this process.9–11 The closure of these gaps has been implicated in the restoration of barrier integrity. 12
EC gap formation and closure is regulated by cytoskeletal dynamics and force generation which determine cell shape through membrane protrusion or retraction.7,9,10 In vitro, thrombin-induced barrier disruption is characterized by transcellular actin filaments, termed stress fibers, which generate contractile forces associated with cell rounding and reduced cell junctions.7,13,14 In contrast, barrier enhancement stimulated by the endogenous lipid sphingosine-1-phosphate (S1P) is associated with peripheral cortical actin arrangements and cell–cell and cell–matrix tethering forces.10,15–17 These peripheral actin dynamics induced by S1P include membrane projections, termed lamellipodia, which serve to decrease intercellular gaps and strengthen cellular junctions.12,18,19 The direct role of lamellipodia in the determination of barrier integrity has recently been described. 20
Formation and dynamic protrusion of lamellipodia is dependent on rapid branched actin polymerization. 21 The actin related protein complex 2/3 (Arp 2/3) is a key regulator of this process by initiating the nucleation of new actin filaments on the side of existing actin strands, the growth of which produce forces necessary for membrane protrusion. 22 Subunits 2 and 3 of the Arp 2/3 complex are homologous to actin monomers and compose the functional portion of the complex. 23 Crystalized structures of inactive Arp 2/3 reveal an end to end or “splayed” interaction between Arp2 and Arp3. The active conformation is characterized by movement of Arp2 in line with Arp3 in a “short-pitch” conformation, which closely approximates an actin dimer and initiates the polymerization of a daughter actin strand. 24 The complex is intrinsically inactive and requires the participation of nucleation promotion factors (NPFs), proteins which approximate actin monomers, actin filaments, and Arp 2/3 to begin growth of a branched daughter strand.25,26 A number of these NPFs are recruited to EC lamellipodia during barrier enhancing conditions.27,28 This includes cortactin, an important regulator of actin structure and filament branching, 29 which interacts with Arp 2/3 through its N-terminal acidic domain. 26 Furthermore, Arp 2/3 has been implicated in maintenance and repair of junctional complexes.30–32
While the known actin modulating properties and subcellular localization of Arp 2/3 have intriguing implications for EC membrane dynamics, the specific role of Arp 2/3 in determination of pulmonary endothelial barrier function remains incomplete. The small molecule CK-666 is a well described specific chemical inhibitor of Arp 2/3 which binds between Arp 2 and Arp 3 to block the complex from transitioning from the inactive to active conformation.33,34 In the current study, we employ this inhibitor in cultured human pulmonary EC to explore the role of Arp 2/3 in both maintenance and recovery of barrier function. We also examine the mechanisms of barrier determination through characterization of intercellular endothelial gaps and individual lamellipodia. The data demonstrate Arp 2/3 activity is essential for maintenance of baseline pulmonary endothelial barrier integrity and significantly contributes to recovery of barrier function after disruption. These experiments also provide strong evidence supporting EC gap closure, mediated by cytoskeletal and membrane dynamics, as the critical determinant of barrier function.
Methods
Cell culture
Human pulmonary artery EC (HPAECs) or human lung microvascular EC (HLMVECs) (Lonza, Walkersville, MD, USA) were cultured in Endothelial Basal Medium (EBM)-2 complete medium (Lonza) supplemented with 10% (v/v) fetal bovine serum (FBS) in a humidified incubator with 5% CO2 at 37°C. Passages 5–7 were used for experimentation. Prior to experimentation, media were replaced with 2% (v/v) FBS.
Transendothelial electrical resistance assay (TER/ECIS)
EC monolayer barrier function was assessed by an electrical cell-substrate impedance sensing system (ECIS) (Applied Biophysics, Troy, NY, USA) as we have described previously.35–37 In this assay, ECs are grown in wells containing gold microelectrodes on the basal surface allowing the measurement of impedance to a small AC electrical current at a frequency of 4000 Hz. Once a baseline resistance (TER) was established, the monolayer was treated with vehicle (0.3% DMSO) or CK-666 (50 µM) followed by thrombin (1 U/mL) or S1P (1 µM) and monitored continuously for up to 12 h. Values are expressed as mean resistance, measured in ohms, or normalized resistance compared to baseline values with the means of several wells pooled in each independent experiment. Baseline resistance in HLMVECs was calculated from three independent experiments with 2–4 repeats of each condition. The maximal change in HPAEC normalized resistance as well as the rate of change (Δ normalized resistance/h) following S1P was calculated from seven independent experiments with each experiment composed of 2–4 repeats of each condition. The time to recovery of baseline barrier function following thrombin (measured in minutes) was calculated from nine independent experiments with each experiment composed of 2–4 repeats of each condition.
Intercellular gap assay
An assay to quantify interendothelial cell gaps was conducted as described previously. 38 A solution of porcine skin gelatin (G 2500, Sigma) dissolved to a concentration of 10 mg/mL in bicarbonate buffer (0.1 mol/L NaHCO3, pH 8.3) and clarified by low speed centrifugation (10 K × g for 5 min at room temperature [RT]) was prepared. Next, EZ-Link NHS-LC-LC-Biotin (Thermo Scientific, Rockford, IL, USA) dissolved in DMSO (5.7 mg/mL) was added to the gelatin solution to a final concentration of 0.57 mg/mL and allowed to conjugate for 1 h at RT while stirring. Aliquots of biotinylated gelatin were then frozen at −20°C for later use. Prior to use, aliquots were thawed in a water bath at 37°C × 10 min and diluted to 0.25 mg/mL with 0.1 mol/L bicarbonate buffer. This diluted solution was filtered through a 0.22 -µm filter and used to coat the bottom of a 6-well plate (2 mL/well). Plates were then stored at 4°C overnight to allow protein adsorption. The next day, each well was washed with 1 mL sterile PBS (pH 7.4) × 2 and ECs plated in 10% FBS media in sufficient numbers to reach confluence within 24 h. On the day of experimentation, media were replaced with 2% FBS and cells were treated with S1P (1 µM) or thrombin (1 U/mL) in the presence or absence of Arp 2/3 inhibition (50 µM CK-666 × 1 h). Next, FITC conjugated avidin (Invitrogen) was added to the media to a final concentration of 12.5 µg/mL 15 min after S1P treatment or 30 min after thrombin followed by gentle rocking to mix. After 3 min, media containing unbound FITC-avidin were removed and cells washed twice with 2 mL of PBS, pH 7.4, 37°C followed by addition of 1 mL FluoroBrite™ DMEM Media (Thermo). Immunofluorescent images of intercellular gaps were then immediately obtained with the 40 × objective of a Nikon TE2000-S microscope (Nikon, Tokyo, Japan) equipped with a digital camera and imaging software (Spot RT, Diagnostic Instruments, Inc.). Two or three random fields were imaged for each condition. The Image J threshold function was used to define and quantify the area of green immunofluorescence in each image which was expressed as a fraction of the total area.
Immunofluorescent confocal microscopy
HLMVECs were grown to confluence in a 12-well plate containing a gelatin-coated 18 mm glass coverslip in each well and were fixed at baseline and time points (2–60 min) following treatment with S1P (1 µM), or thrombin (1 U/mL) in the presence or absence of Arp 2/3 inhibition (50 µM CK-666 × 1 h). At prescribed time points, the cover slips were washed in sterile PBS and then transferred to a 4% paraformaldehyde PBS solution, pH 7.4 for 20 min at RT. Next, unreacted aldehyde groups were quenched with a 50 mM NH4CL/PBS, pH 7.4 (5-min wash × 3). Membrane permeabilization and protein blocking was conducted in a solution of 2% normal goat serum/0.25% fish skin gelatin/0.01% saponin/0.1% NaN3/PBS, pH 7.4 for 30 min at RT. All cells were treated with phalloidin-Alexa Fluor 633 (Invitrogen) diluted in blocking solution in order to visualize actin. As indicated, cells were also treated with the monoclonal anti-cortactin 4F11 Alexa Fluor 555 conjugate antibody (EMD Millipore) and/or polyclonal anti-Arp 2 antibody (Cell Signaling) overnight followed by secondary anti-rabbit Alexa Fluor 488 (Invitrogen). After washing, coverslips were mounted on glass slides with Prolong Gold with DAPI (Invitrogen), dried overnight, and imaged the following day with a Zeiss 710 LSM confocal microscope with an alpha Plan-Apochromat 63X/1.46 NA oil Korr M27 objective lens. Three independent experiments were conducted with a minimum of 6–8 cell fields imaged in each condition.
Lamellipodia imaging and quantification
Subconfluent HLMVECs were seeded in 12-well plates on gelatin-coated cover slips and treated with CK-666 (50 µM × 1 h) or vehicle followed by S1P (1 µM) to induce lamellipodia. As above, cells were fixed and permeabilized at baseline and 2 min or 30 min following S1P treatment. Actin was stained with phalloidin-Alexa Fluor 633 (Invitrogen) and cells imaged by confocal microscopy. Both low (pixel size = 100 nm) and high (pixel size = 15 nm) magnification images were obtained for analysis by Image J software. The threshold function was used to better visualize and identify actin fibers. Lamellipodia were defined as peripheral flat protrusions containing thin actin fibers arranged in a bidirectional dendritic array extending from the cell edge inward 1–5 µm.39–41 A line was then traced around lamellipodia and maximal depth (membrane edge to central border) was measured at several locations. A total of 8–16 cells were analyzed with 2–3 lamellipodia quantified per cell for each condition.
Statistical analysis
Quantified values are expressed as means ± standard error. Statistical analyses were conducted with GraphPad Prism 6 software. Comparisons between groups were made using paired t-test or two-way analysis of variance (ANOVA) followed by Sidak’s correction as indicated. In all cases, significant differences were defined as P < 0.05.
Results
Arp 2/3 function regulates baseline barrier function and recovery from thrombin
The effect of Arp 2/3 inhibition on overall barrier function was measured by transendothelial electrical resistance (TER). HPAECs were treated with the Arp 2/3 inhibitor CK-666 (50 µM) and resistance measurements, a surrogate for monolayer barrier integrity, were monitored over time. A rapid and significant drop in mean baseline barrier function in the CK-666 treated cells was observed compared to vehicle (1241 ± 53 versus 988 ± 64 ohm; P < 0.01) (Fig. 1a and 1c). Baseline TER measurements were also conducted in HLMVECs. As expected, microvascular endothelial cells exhibited much higher resistance (2527 ± 54 ohm) prior to experimentation. The addition of vehicle and CK-666 (50 µM) both decreased barrier function; however, the subsequent resistance measurement in CK-666 treated cells was significantly less than vehicle (1536 ± 43 versus 1969 ± 70 ohm; P < 0.01) (Fig. 1b and 1d). Next, the impact of Arp 2/3 inhibition on the response to the barrier enhancing lipid S1P (1 µM) was assessed. The maximal absolute increase in normalized resistance was less in HPAECs treated with CK-666 versus vehicle (0.47 ± 0.04 versus 0.37 ± 0.04; P < 0.05) (Fig. 1f). Additionally, the maximal rate of increase in the normalized resistance after S1P was diminished in the Arp 2/3 inhibited cells (Fig. 1g). The impact of Arp 2/3 inhibition on TER after treatment with the barrier disruptive compound thrombin (1 U/mL) was also characterized. As seen in Fig. 1h, thrombin produced similar nadir resistance values in CK-666 and vehicle treated cells. However, Arp 2/3 inhibition resulted in significantly delayed time to recovery of pre-thrombin baseline resistance following thrombin (143 ± 14 versus 93 ± 6 min; P < 0.01) (Fig. 1i). These dynamic studies were repeated in HLMVECs, and while the baseline starting resistance values in these cells were consistently above 2000 ohm, we obtained similar trends following S1P and thrombin treatment as seen in HPAECs (data not shown).
Effect of Arp 2/3 inhibition on EC barrier function and recovery. HPAECs or HLMVECs were grown on gold electrodes to obtain measurements of TER. Depicted plots are mean normalized resistance values with standard error from 2–4 repeated measures in each condition. (a, b) Representative normalized TER vs. time plot in HPAECs or HLMVECs subjected to CK-666 (50 µM) or vehicle (0.3% DMSO). (c, d) Quantification of mean baseline resistance values (ohms) 1 h after vehicle or CK-666 in HPAEC or HLMVEC. (e) Representative TER vs. time plot of HPAECs obtained after pretreatment with CK-666 or vehicle followed by S1P (1 µM). (f) Maximal change in normalized resistance following S1P. (g) Maximal rate of change in normalized resistance per hour following S1P. (h) Representative TER vs. time plot of HPAECs obtained after pretreatment with CK-666 or vehicle followed by thrombin (1 U/mL). (i) Mean time to recovery of baseline barrier function following thrombin treatment. Quantified values were obtained by pooling 7–9 independent experiments each with 2–4 repeats of each condition (*P < 0.05 vs. vehicle, paired t-test).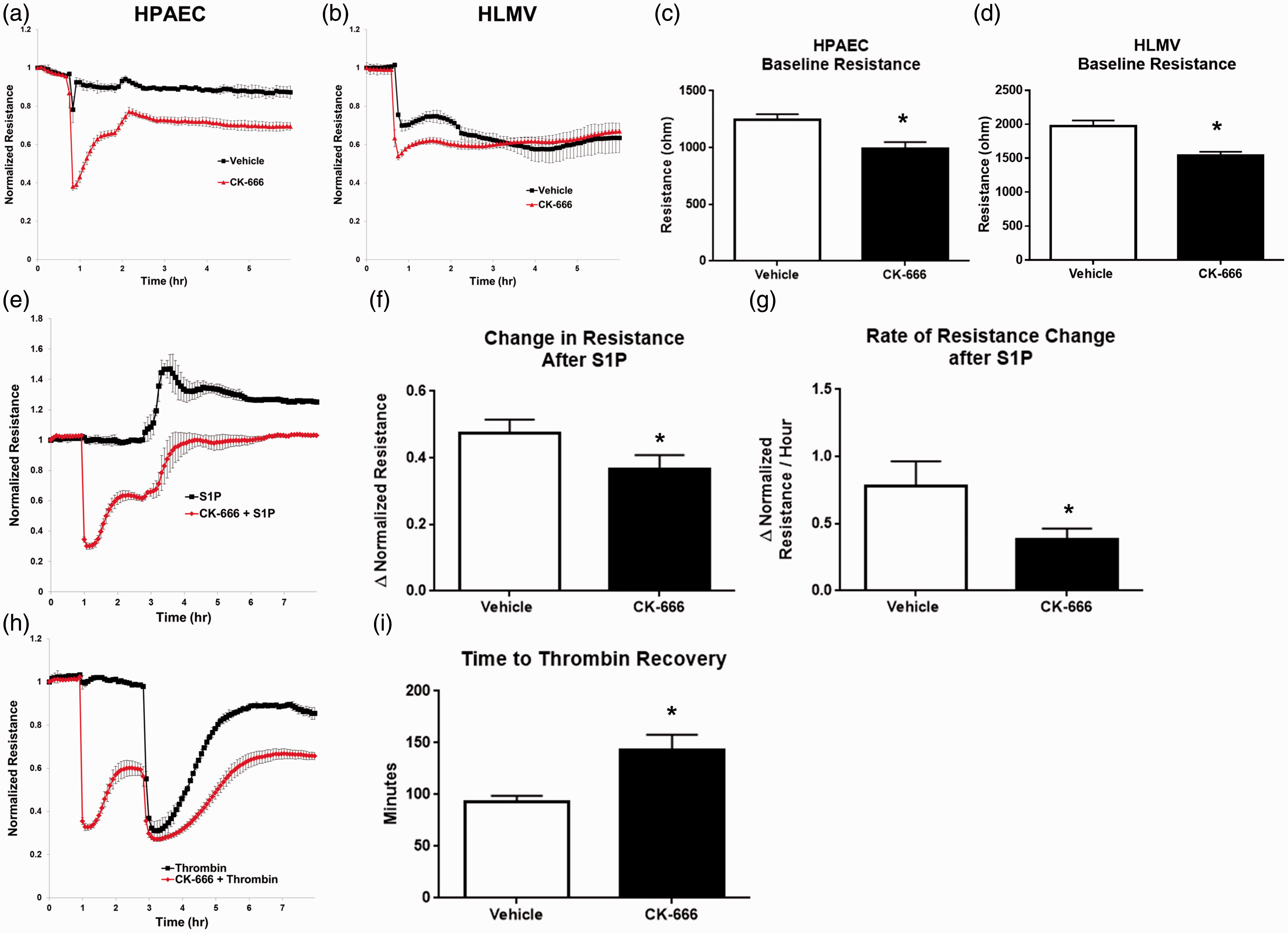
Intercellular gaps are increased in cells subjected to Arp 2/3 inhibition
The decreased barrier function seen in pulmonary EC treated with CK-666 may be the result of paracellular gap formation between cells. Localized changes in EC barrier function produced by intercellular gap formation can be assessed by measuring permeability to FITC-avidin as described recently
38
and in the “Methods” section. This assay allows visualization and quantification of the gaps between individual cells. In support of the hypothesis that paracellular gaps drive barrier dysfunction and consistent with the functional data obtained by TER, FITC-avidin permeability assays revealed a significantly increased gap area at baseline in HPAECs treated with CK-666 (50 µM × 1 h) compared to vehicle (0.456 ± 0.02 versus 0.299 ± 0.02; P < 0.05) (Fig. 2b). As expected, gap area decreased in both groups after S1P (1 µM × 15 min) compared to baseline values but did not reach statistical significance. However, the significant difference in gap area between Arp 2/3 inhibited and control cells remained (0.423 ± 0.02 versus 0.181 ± 0.02; P < 0.05). Thrombin treatment (1 U/mL × 30 min) significantly increased mean gap area in control cells by 152% and in CK-666 treated cells by 94% compared to baseline. Once again, there was a significant increase in intercellular gaps after thrombin between the CK-666 and vehicle treated cells (0.885 ± 0.03 versus 0.754 ± 0.03; P < 0.05). Localized permeability assays were repeated in HLMVECs. These microvascular cells exhibit slightly decreased gap formation when compared to ECs from large conduit vessels (Fig. 2a and 2c), consistent with increased barrier function as measured by TER. Similar to HPAECs, a significant increase in gap area was observed in HLMVECs treated with CK-666 compared to vehicle at baseline (0.437 ± 0.06 versus 0.233 ± 0.05; P < 0.05) and following thrombin treatment (1 U/mL × 30 min) (0.767 ± 0.05 versus 0.563 ± 0.04; P < 0.05) (Fig. 2D). Gap area was reduced in both vehicle and CK-666 groups following S1P (1 µM × 15 min), and there was a trend toward increased gap area in Arp 2/3 inhibited versus vehicle cells but this did not reach statistical significance.
EC gap formation is increased after Arp 2/3 inhibition. HPAECs or HLMVECs were grown to confluence on biotinylated gelatin. After 1 h pretreatment with CK-666 (50 µM) or vehicle, FITC-avidin was added to media and allowed to permeate cells to reach biotin substrate at sites of paracellular gaps. Shown are representative IF microscopy of interendothelial gaps (green fluorescence) between (a) HPAECs and (c) HLMVECs obtained at baseline and after S1P (1 µM × 15 min) or thrombin (1 U/mL × 30 min). (b, d) Quantification of gap area per imaged field in HPAECs and HLMVECs. Bar graphs represent mean values with standard error from n = 4 (HPAEC) and n = 2 (HLMVEC) independent experiments with 2–3 images quantified per condition. (α = P < 0.05 vs. baseline vehicle, *P < 0.05 between groups, by two-way ANOVA with Sidak’s multiple comparisons test).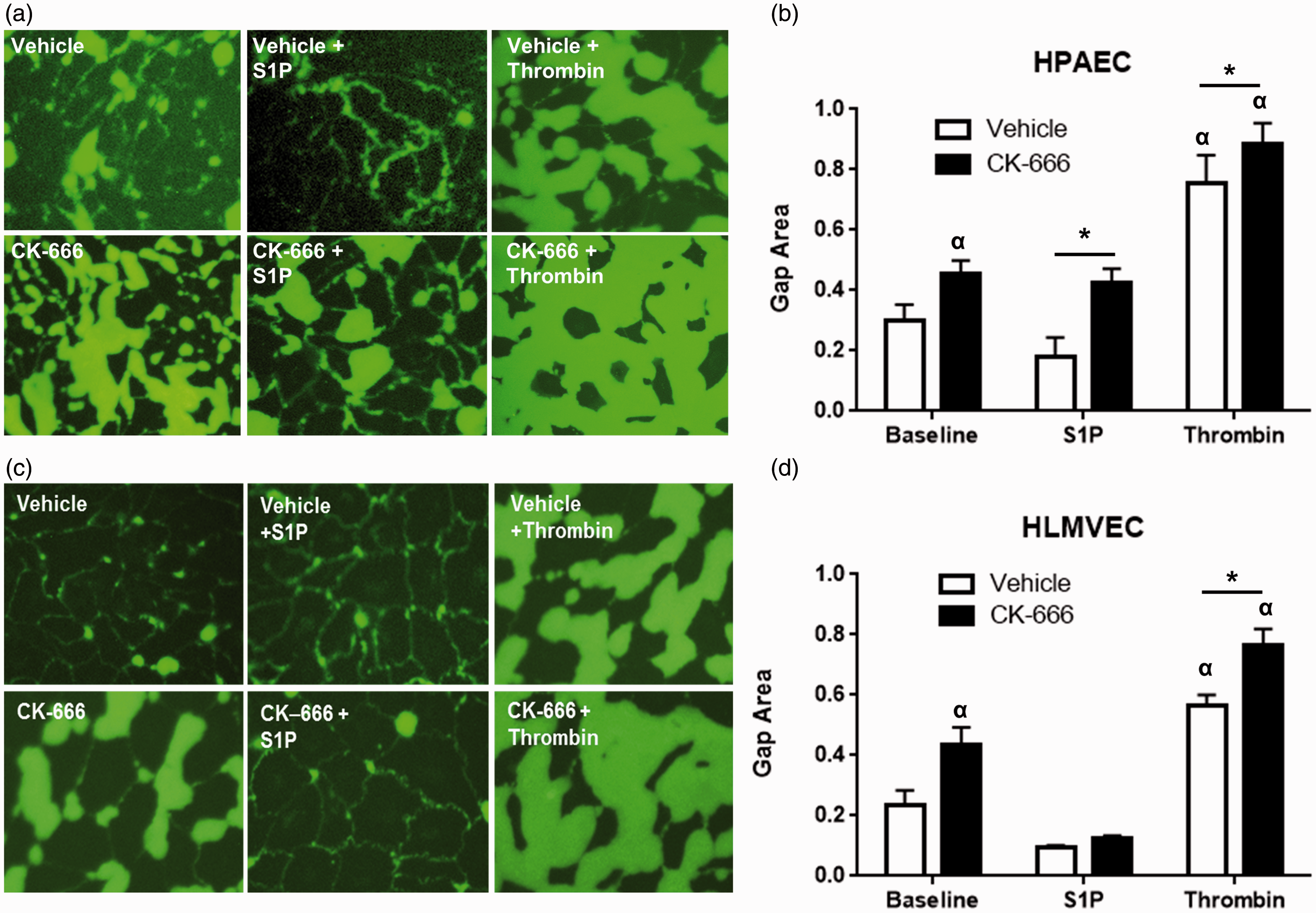
Arp 2/3 modulates actin structures and protein interaction
Cytoskeletal dynamics are critical to the regulation of endothelial cell shape and gap formation.7,9,10,20 Therefore, we explored alterations in actin structure and regulatory protein localization after Arp 2/3 inhibition. HLMVECs were used in these studies due to their higher degree of barrier integrity and presumed greater physiologic relevance in that they are located within capillary vessels, the primary site of vascular leak in ARDS. After pretreatment with CK-666 (50 µM × 1 h) or vehicle, cells were treated with barrier enhancing S1P (1 µM), then fixed and stained for proteins of interest at various time points. Confocal IF images revealed the robust development of membrane ruffles enriched with actin (red) and cortactin (green) 2–5 min following S1P which persisted (with colocalization of actin and cortactin indicated by yellow in these images), although to a lesser degree, at 15-min and 30-min time points (Fig. 3a, top row). These structures were notably absent in cells treated with CK-666 (Fig. 3a, bottom row). Images of S1P stimulated HLMVECs after co-staining actin (blue), Arp 2/3 (green), and cortactin (red) were also obtained (Fig. 3b). In these cells, colocalization of all three proteins appears white and is dramatically increased at the membrane edge 2–5 min after S1P in vehicle treated cells but substantially reduced in the Arp 2/3 inhibited cells.
S1P induced peripheral actin structures and regulatory protein interactions are altered by CK-666. HLMVECs were treated with CK-666 (50 µM × 1) or vehicle. Cells were fixed, stained, and confocal IF images were obtained at baseline and at 2, 5, 15, and 30 min after S1P (1 µM). (a) Representative images of vehicle (top row) or CK-666 (bottom row) treated cells after staining for cortactin (green), actin (red), and nuclear structure (DAPI, blue). Robust lamellipodia formation with membrane ruffling (white arrows) and associated colocalization of actin and cortactin (yellow) is observed rapidly (2–5 min) after S1P. (b) Confocal IF images of HLMVEC stained for Arp 2 (green), cortactin (red), and actin (blue). Colocalization of all three proteins appears white while cortactin-actin colocalization appears purple. Representative images of vehicle (top row) or CK-666 (bottom row) treated cells are shown. Scale bar = 10 µm.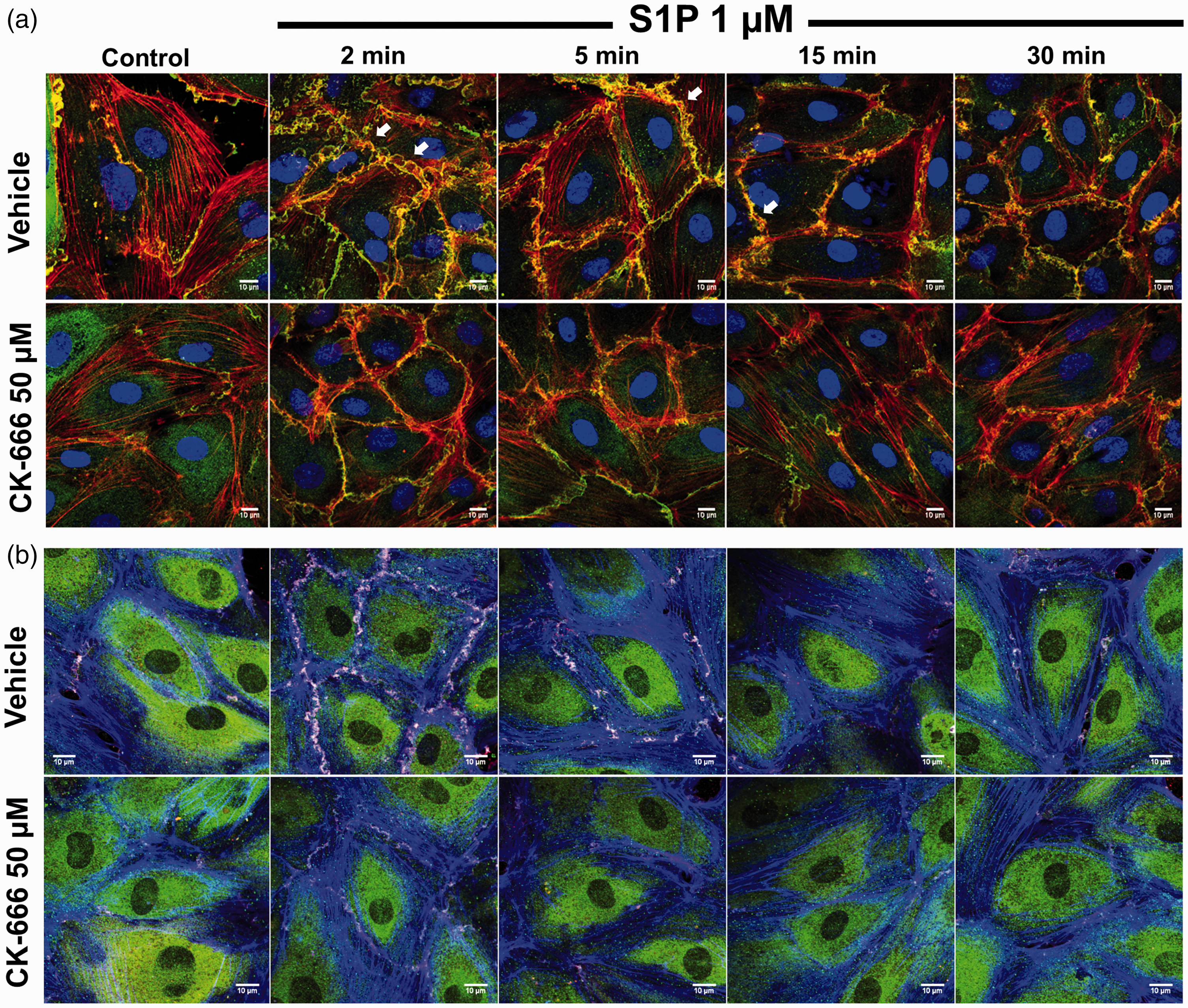
Changes in actin structure and cortactin localization under barrier disruptive conditions were determined by imaging HLMVECs after thrombin (1 U/mL) treatment. Time points of 30, 45, and 60 min after thrombin administration were chosen to assess peripheral actin and membrane structures during the time period associated with active restoration of barrier function as suggested by TER studies (Fig. 1h). In vehicle treated cells, numerous membrane lamellipodia containing cortactin (green) protruding into the gaps between ECs were observed at both 30 and 45 min (Fig. 4, top row). There were markedly fewer lamellipodia at these time points in cells treated with CK-666 (Fig. 4, bottom row). At 60 min following thrombin in vehicle treated cells, interendothelial gaps have mostly resolved and dense areas of actin-cortactin colocalization begin to appear at sites of membrane contact between two individual cells, suggestive of junctional associated intermittent lamellipodia (JAIL).32,42 In contrast, Arp 2/3 inhibited cells 60 min after thrombin exhibit persistent intercellular gaps and transcellular stress fibers.
Arp 2/3 inhibition reduces lamellipodia formation and EC contacts during thrombin recovery. Confluent HLMVECs were exposed to CK-666 (50 µM × 1 h) or vehicle and confocal IF microscopy images were obtained 30, 45, and 60 min following thrombin (1 U/mL). Cells were fixed and stained for cortactin (green), actin (red), and nuclear structure (DAPI, blue). Representative images from three independent experiments are shown (6–8 cells imaged per experiment). Cortactin-enriched lamellipodia (white arrows) are observed at 30 and 45 min in vehicle treated cells (top panel) but mostly absent in CK-666 treated cells at the same time points (bottom panel). At 60 min after thrombin, punctate colocalization of actin and cortactin are observed at sites of intercellular contact (yellow ovals) while Arp 2/3 inhibited cells demonstrate persistent stress fibers and intercellular gaps (white stars). Scale bar = 10 µm.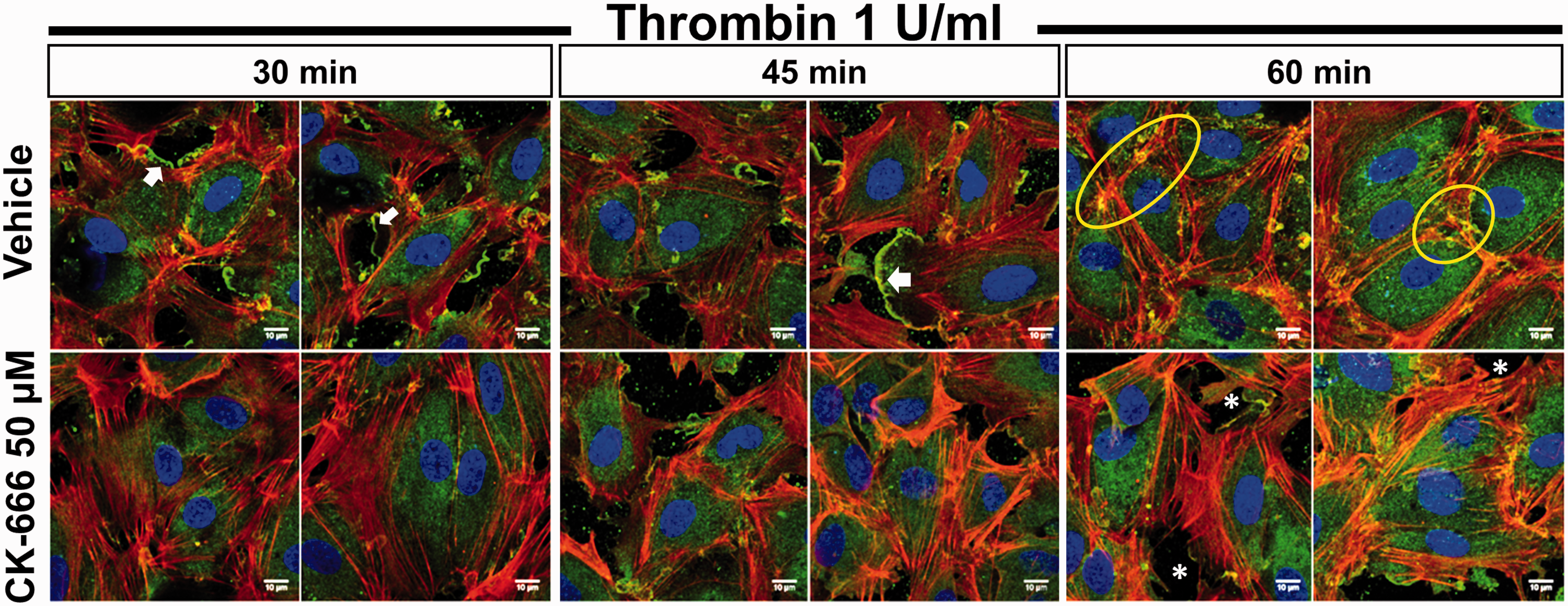
Arp 2/3 inhibition alters lamellipodia structure
The differences observed in actin structure between ECs subjected to Arp 2/3 inhibition after S1P and during recovery from thrombin could lead to alterations in membrane lamellipodia. To investigate this possibility, imaging of individual lamellipodia was pursued. High resolution confocal microscopy images were obtained from subconfluent HLMVECs at baseline and after S1P (1 µM × 2–30 min) (Fig. 5a). These images were used to identify and measure the depth of individual lamellipodia as described in the “Methods” section. Subconfluent cells spontaneously produce lamellipodia, and under basal conditions a significant decrease in lamellipodia depth was observed in cells treated with CK-666 (50 µM × 1 h) versus vehicle (1.83 ± 0.41 versus 2.55 ± 0.46 µm) (Fig. 5b). Interestingly, while the number of lamellipodia per cell was increased by S1P (data not shown), there was no significant change in lamellipodia depth which was similar in both CK-666 and vehicle treated cells. Thirty minutes following S1P, lamellipodia depth was again significantly diminished following Arp 2/3 inhibition compared to vehicle (1.53 ± 0.37 versus 2.09 ± 0.36 µm).
Arp 2/3 inhibition reduces lamellipodial depth. Subconfluent HLMVECs were exposed to CK-666 (50 µM × 1 h) or vehicle followed by S1P (1 µM). (a) Representative high resolution (pixel size 15 nm) confocal images of phalloidin stained peripheral actin in vehicle (top row) or CK-666 (bottom row) treated cells at baseline, 2 min, and 30 min following S1P. Individual lamellipodia were identified and outlined. The distance from membrane leading edge to central border was measured at several points (yellow lines). Scale bar = 1 µm. (b) Quantification of mean lamellipodia depth at baseline and following S1P. n = 8–15 cells per group with 2–3 lamellipodia measured per cell. (*P < 0.05 vs. vehicle, by two-way ANOVA with Sidak’s multiple comparisons test).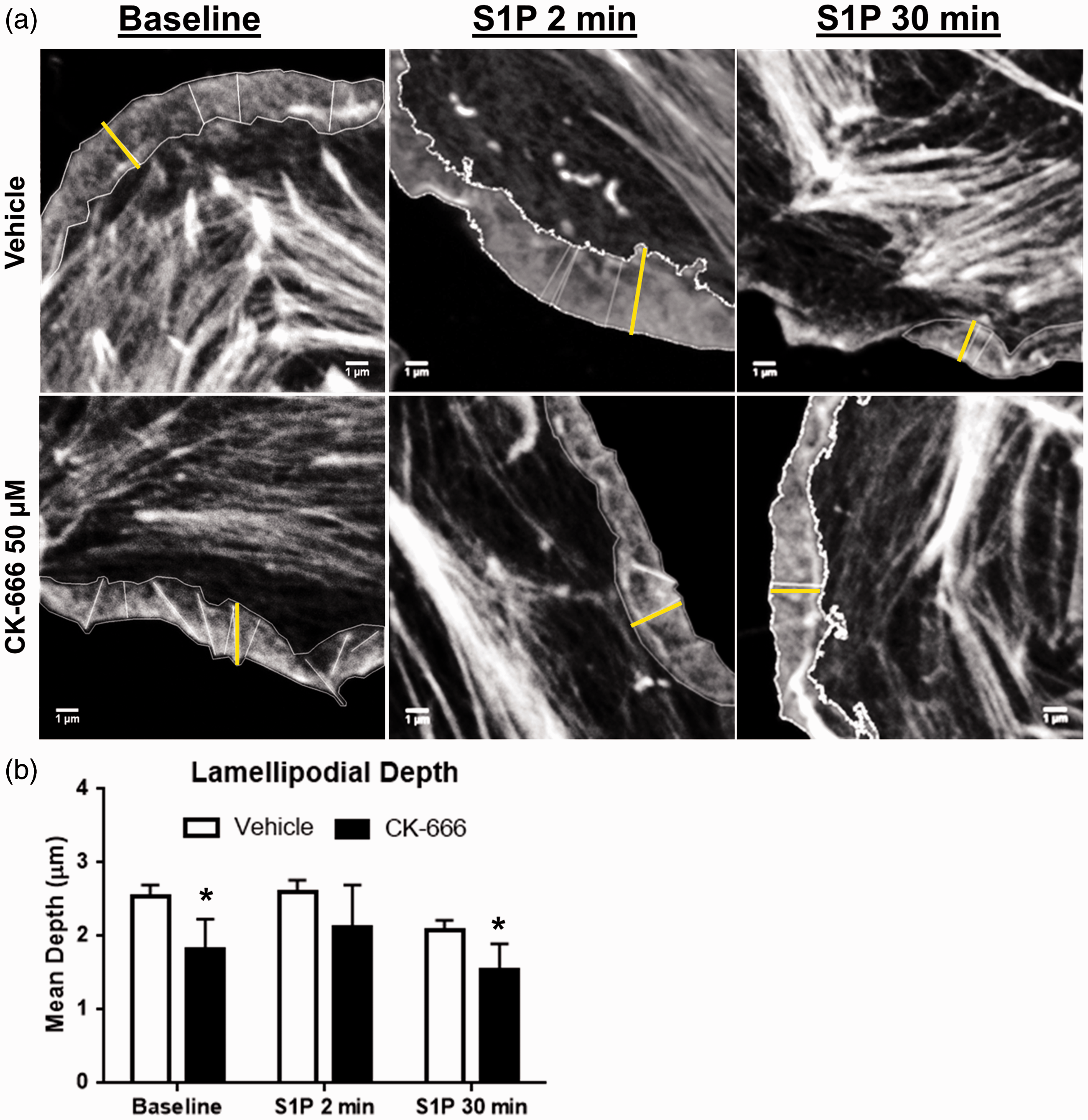
Discussion
To summarize, these results establish a critical role for Arp 2/3 function in maintenance and recovery of pulmonary endothelial barrier integrity. Furthermore, the current study provides strong evidence connecting endothelial cell gap formation and closure to barrier function. Finally, we provide mechanistic insights of Arp 2/3 function by highlighting alterations in actin-based cellular structures following inhibition of the complex.
Both epithelial and endothelial cell layers in the alveolar-capillary barrier become dysfunctional in ARDS. 43 Important differences have been described between primary insults to the epithelium versus endothelium leading to ARDS. 44 The pulmonary endothelial layer is uniquely positioned compared to endothelium in other tissues. It receives the entire right ventricular stroke volume and is continuously exposed to a variety of systemic inflammatory cytokines and cells.45,46 As such, its barrier properties also differ from the epithelial layer as well as other endothelial tissues.47,48 Therefore, the mechanisms responsible for this dynamic control of barrier function are of particular interest. The Arp 2/3 complex in determination of epithelial barrier integrity has been investigated previously.49–51 These studies offer insights into roles for Arp 2/3 in cell–cell junctional assembly and interestingly, terminal epidermal cell differentiation. 50 Previous work in a variety of EC types has implicated Arp 2/3 function in cellular processes highly relevant to vascular endothelial barrier integrity. These include EC lymphocyte transmigration, 52 the regulation of junctional complexes,30,32 and the cytoskeletal and peripheral membrane dynamics responsible for flow-induced EC alignment. 53 Directly relevant to the lung vasculature, a study conducted in HPAECs demonstrates the role of Neural Wiskott-Aldrich syndrome protein (N-WASP) function in regulation of adherens junctions, cortical actin formation, and, ultimately, barrier function. 54 Like cortactin, N-WASP induces Arp 2/3 actin polymerization as a nucleation promotion factor (NPF) via interaction with Arp 2. Our study contributes additional details to these previous findings and specifically implicates Arp 2/3 function in altered membrane shape, which may represent the final common pathway of a number of upstream signaling pathways to ultimately determine endothelial barrier function. A limitation of our study is that it only assesses the effects of loss of Arp 2/3 function. Future gain of function studies would be helpful to better characterize the role of Arp 2/3 in this process.
While the importance of cytoskeletal dynamics in gap formation leading to paracellular permeability has been known for some time,7,9,10,55 only recently have the specific cell membrane structures produced by these actin rearrangements, namely lamellipodia, been studied in the context of endothelial barrier integrity.12,20 The finding of decreased functional barrier integrity after Arp 2/3 inhibition, as measured by TER (Fig. 1), corresponding to increased EC gap formation, as measured by the intercellular gap assay (Fig. 2), is consistent with these studies. Our data also support the hypothesis of paracellular permeability as a critical cause of inflammatory vascular leak as our studies demonstrate that EC gap resolution after thrombin is associated with return to baseline functional integrity. Important differences exist between ECs from large conduit vessels and alveolar capillaries. 56 By evaluating responses in both pulmonary artery (HPAEC) and microvascular cells (HLMVEC), this study further characterized this heterogeneity with increased barrier function and decreased EC gaps in HLMVECs, which broadens the relevance of these results.
Consistent with earlier work on cell motility and membrane shape, 57 these new data demonstrate reduced lamellipodia formation following Arp 2/3 inhibition both in response to S1P (Fig. 3) and during recovery from thrombin (Fig. 4). The observation of robust lamellipodia formation 30 and 45 min following thrombin injury in vehicle treated cells (Fig. 4) demonstrate the critical role of these membrane projections, not only in barrier enhancement but also during recovery of barrier integrity after injury. Colocalization of Arp 2/3 and cortactin within lamellipodia (Fig. 3b) was also observed, consistent with prior reports of the role of this interaction in determining cellular protrusions through branched actin polymerization in other cell types.58–61 Cortactin has diverse functions related to dynamic cytoskeletal rearrangement.29,62–65 Both cortactin51,66 and Arp 2/330,31 have been implicated in the formation of cell–cell and cell–matrix junctions. Speculation based on these separate reports raises the interesting possibility that the Arp 2/3–cortactin interaction may play a more complicated signaling role in these complexes independent of its known effects on actin polymerization. This interaction as it relates to endothelial barrier integrity is an intriguing area of future research.
The observation of diminished lamellipodial depth following Arp 2/3 inhibition (Fig. 5) strongly supports the idea of Arp 2/3-driven actin polymerization providing the force to protrude the membrane leading edge in these structures. It is somewhat surprising that S1P did not significantly increase lamellipodial depth in these experiments, in vehicle or CK-666 treated cells, as would be expected due to S1Ps effects on peripheral actin dynamics and cytoskeletal regulatory proteins.10,15–17,19,67 These prior studies using IF and atomic force microscopy largely focused on cortical actin which lies at the periphery of the cell but not within individual lamellipodia and may partially explain our seemingly contradictory findings. Alternatively, it may not be the degree of protrusion of an individual lamellipodium but rather the overall number of lamellipodia per cell that is the key factor in determination of barrier integrity induced by S1P. Indeed, we did observe an increased number of lamellipodia in vehicle versus Arp 2/3 inhibited cells (Fig. 3). Finally, these studies were necessarily performed in subconfluent cells to allow better visualization of the cell edge. Subconfluent EC produce robust lamellipodia at baseline in an effort to contact neighboring cells so it is possible the addition of S1P was unable to significantly augment these structures in already stimulated cells.
Despite the high cost of ARDS as measured by healthcare utilization dollars 68 and prolonged deficits and disability among survivors, 69 there remains no targeted therapy to resolve or decrease the fundamental pathologic derangements that characterize this syndrome. Recent reports by our group and others have identified several potentially therapeutic agents which act through modulation of inflammation and/or vascular leak.37,70–74 These studies target numerous upstream regulators of pulmonary EC cytoskeletal dynamics. Modulation of these regulatory elements may ultimately produce alterations in cell membrane shape and junctional properties relevant to both endothelial barrier function20,55 and EC interaction with inflammatory cells.75–79 The current study suggests that the Arp 2/3 complex, as an end effector of peripheral actin polymerization and hence determination of membrane shape, has implications in both of these EC processes with direct relevance to ARDS pathophysiology.
Footnotes
Acknowledgments
The authors thank Lakshmi Natarajan for her expertise and technical assistance in cell culture.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This work was supported by American Lung Association Award RG-349765 (PB) and National Institutes of Health grants HL058064 (JGNG) and HL088144 (SD).