
Abstract
Epigenetics is an emerging field of research and clinical trials in cancer therapy that also has applications for pulmonary arterial hypertension (PAH), as there is evidence that epigenetic control of gene expression plays a significant role in PAH. The three types of epigenetic modification include DNA methylation, histone modification, and RNA interference. All three have been shown to be involved in the development of PAH. Currently, the enzymes that perform these modifications are the primary targets of neoplastic therapy. These targets are starting to be explored for therapies in PAH, mostly in animal models. In this review we summarize the basics of each type of epigenetic modification and the known sites and molecules involved in PAH, as well as current targets and prospects for clinical trials.
Mammalian genetic information is encoded by DNA and packaged in nucleosomes with histones to make chromatin. Changes in these complexes add another layer of genetic information to control DNA expression. Epigenetics is the modification of gene expression and cellular phenotype caused by external factors on gene transcription. This includes any process that alters gene expression without altering the DNA itself. Such transcriptional factors can be heritable or de novo and can be altered by the environment and exposures. 1 Epigenetic modification has been implicated in many disease processes, including many malignancies.2–4 Epigenetic modification has been linked to pulmonary arterial hypertension (PAH), involving each of the mechanisms discussed below. 5 What has been less well studied is the potential impact of epigenetic data on designing and understanding clinical trials in this orphan disease.
MECHANISMS OF EPIGENETIC MODIFICATIONS
Figure 1 is a schematic illustration of the three mechanisms described in this section.
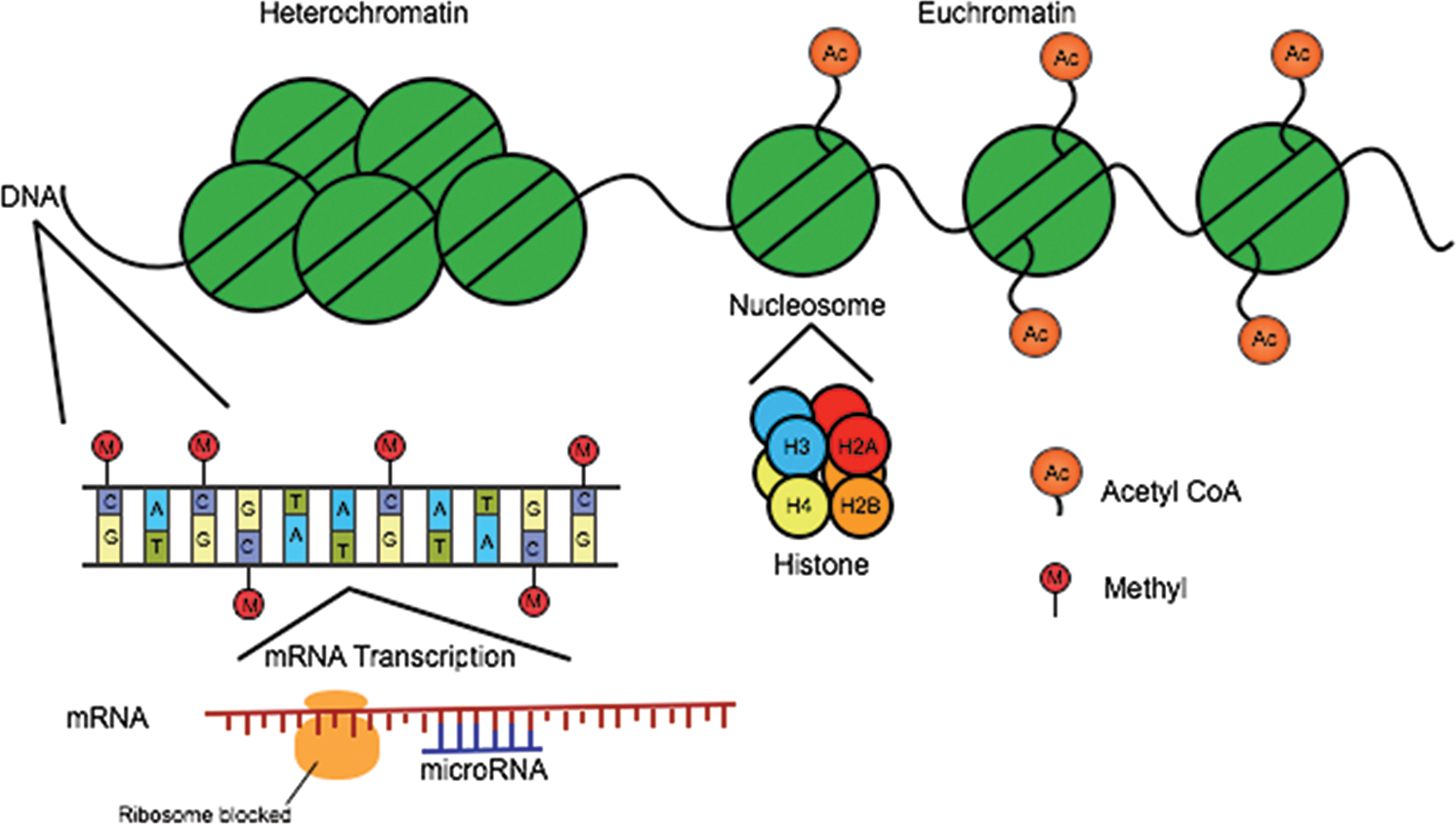
Mechanisms of epigenetic modification: schematic of epigenetic mechanisms. Histone modification, DNA methylation, and microRNA-mediated gene silencing constitute three mechanisms of epigenetic regulation. Acetyl CoA: acetyl coenzyme A; mRNA: messenger RNA.
DNA methylation
A well-characterized form of epigenetic modification is DNA methylation, which involves the transfer of a methyl group onto cytosine (C) residues in dinucleotide CpG (cytosine-guanine) sequences of DNA. DNA methyltransferases (DNMTs) moderate the process, which can both repress and activate DNA transcription. 6 There are multiple active mammalian DNMTs with different, although not exclusive, functions. Generally, DNMT3a and DNMT3b perform de novo methylation, while DNMT1 is responsible for maintenance of methylation.7,8 There is an initial wave of demethylation that occurs with embryogenesis, followed by de novo methylation that occurs in two parts, first with embryo implantation and then with primordial germ cell differentiation, which requires DNMT3L.9–11 DNMT3L and DNMT3a form a tetramer with the two 3a sites able to interact with DNA major grooves and facilitate methylation. 12 This structure is important to the possible targeting of DNMT3 for methyltransferase inhibition. In addition to the primary activity of DNMT, there is evidence that transcription-specific gene loci may recruit the methylation enzymes and may be required for imprinting. 13
Dinucleotide CpG islands at the sites of promoters are protected from methylation by multiple mechanisms, including transcription factor binding and histone-3 lysine-4 (H3K4) methyltransferases. 14 CpG islands often span promoter regions, and unmethylated islands are transcribed when histones are in an acetylated, euchromatin state. Hypermethylation of these islands can silence tumor suppressor genes, but hypomethylation can unsuppress oncogenes, leading to overexpression; both pathways have been demonstrated in malignancies. 15
Histone modification
Histones are the building blocks of the nucleosome and as such alter the exposure of the DNA to transcription machinery. In addition to DNA methylation, there are histone modifications that alter the structure of chromatin and thus DNA transcription. Many mechanisms of histone modification exist, the most common being acetylation by histone acetyltransferase with an acetyl CoA (coenzyme A) cofactor and methylation by histone methyltransferase requiring an S-adenosylmethionine. 16 Acetylation occurs at lysine residues on histones H3 and H4 and correlates with increased transcription activity. Bromodomains (BRDs) are proteins that have been shown to interact with acetylated lysine on the histone in different transcription regulators.17,18 Given their function, BRDs have become viable targets for possible epigenetic therapies.
Histone methylation is another a route of epigenetic transcriptional modification. DNMT3L has been shown to interact the amino terminal of histone, guiding the interplay between DNA and histone methylation. The interaction between H3 and DNMT3L is inhibited by histone H3K4 methylation. 19 Thus, it is thought that DNMT3L acts as a regulator for DNMT3a and that when methylation is detected on histone H3K4, DNA imprinting is inhibited. 12 Chromodomains are protein modules that appear to interact with methylated histones to control chromatin structure and function.20,21 Heterochromatin Protein 1 (HP1) and Polycomb were the first chromodomains identified in Drosophila, and three groups of mammalian chromodomains have been studied in the HP1 family: HP1α, HP1β, and HP1γ.21–23 HP1α and HP1β are located around the centromere, with HP1α having a role in mitosis via interaction with proteins that regulate progression through the mitotic phases. Alternatively, HP1γ is primarily found in the nucleoplasm. Other mammalian chromodomains also exist, all with protein interactions that influence heterochromatin formation, gene transcription, and replication.
Histone modification is not permanent. Acetylation and methylation allow for fluid alterations of the chromatin structure, with subsequent changes in the genes transcribed. The “histone code” has proven to be a very complex system that is not fully understood.
RNA interference
MicroRNAs (miRNAs) modify gene expression posttranscriptionally by base pairing with target messenger RNA (mRNA) to silence the mRNA by blocking translation or marking it for cleavage. 24 These miRNAs bind at this “seed” region as a guide for the miRNA-induced silencing complex, or miRISC. 25 The miRNAs themselves can be regulated by DNA methylation and histone modification but also have a role in expression of enzymes involved in epigenetic regulation. Thus, there appear to be regulatory pathways in which miRNAs modify both pre- and posttranscriptional gene expression.
EPIGENETICS OF PAH
DNA methylation
Superoxide dismutase 2 (SOD2) is an enzyme that is involved in H2O2 regulation, interacts with hypoxia-inducible factor 1-alpha (HIF1α), and also has a role as a tumor suppressor gene. 26 The SOD2 gene is silenced by hypermethylation, causing increasing H2O2 production and uncontrolled cell proliferation in multiple malignancies, including pancreatic cancer and myeloma. 27 By the same token, pulmonary artery smooth muscle cells (PASMCs) are under the influence of SOD2-mediated proliferation and apoptosis, seen first in fawn-hooded rats (FHRs) but also in patients with idiopathic PAH. 28 In human PAH, SOD2 deficiency has been noted in pulmonary arteries and plexiform lesions. 28 Archer and colleagues 29 demonstrated that the expression of SOD2 was reduced in a rat model of PAH and that hypermethylation of SOD2 in FHRs created a proliferative, antiapoptotic population of PASMCs. The addition of 5-azacytidine, a methyltransferase inhibitor, reversed the hypermethylation and increased SOD2 expression. Subsequent SOD2 augmentation in the FHR model decreased mean pulmonary arterial pressure and thickness of the arterial media. 29 These findings highlight a clear epigenetic pathway of PASMC proliferation through SOD2 and HIF1α that is important in the pathophysiology of PAH.
The methyltransferase inhibitor 5-azacytidine has been studied and approved for use in myelodysplastic syndrome, but it has not been studied as part of a clinical trials in humans in PAH. 30 Currently, no other DNA methylation pathway has been well characterized as contributing to the pathogenesis of PAH.
Histone modification
Histone acetylation is also involved in PAH by control of histone deacetylase (HDAC) gene expression via histone modification. HDACs are enzymes that remove the acetyl groups on lysine tails of histones, thereby modifying chromatin interaction with the transcriptional apparatus. In general, deacetylation causes more condensed chromatin and decreased gene transcription. These enzymes also regulate gene expression by interaction with transcription factors and deacetylation of proteins other than histones.31,32 Many of the HDACs are part of a complex of proteins that target specific genomic regions. 33 Thus far there are 18 mammalian HDACs (including the sirtuins) that have been identified, and they are divided into classes based on structure, function, and location in the cell (Table 1).33,34 HDAC1 and HDAC5 concentrations are higher in the lungs of PAH patients and FHR PAH models. 35 In the FHR model, the histone deacetylase inhibitors (HDIs) valproic acid and suberoylamilide hydroxamic acid induced an antiproliferative effect on the PASMCs, with a decrease in mean pulmonary arterial pressure and right ventricular (RV) hypertrophy. 35 The addition of the HDIs also appeared to inhibit PASMC proliferation driven by platelet-derived growth factor. 35 This demonstrates a role of histone deacetylation in PASMC hyperproliferation and that blocking deacetylation has apoptotic effects.
Zinc-dependent histone deacetylase classes

In the interplay between the types of epigenetic modification, histone acetylation has effects on the expression of miRNAs. Myocyte enhancer factor 2 (MEF2) is a family of transcription factors that appear to be affected by histone modification. MEF2 is important in cardiomyocyte cellular differentiation and also mediates cardiac hypertrophy as a stress response.36,37 Expression of MEF2 is suppressed by HDAC4 (as well as by sirtuin 1). 38 MEF2 activity is decreased in pulmonary arterial endothelial cells in PAH with increased HDAC4 and HDAC5 levels. 39 HDI exposure causes significant increases in miR-124 in pulmonary arterial fibroblasts. The activity of miR-124 is further discussed below. 40
miRNAs
Many miRNAs have been implicated in the pathology of PAH, as outlined in Table 2. The role of miRNA-124 in PAH appears to be via fibroblast regulation. Wang and colleagues 40 demonstrated that miRNA-124 inhibits proliferation and inflammation but is decreased in patients with PAH, compared to controls. Interestingly, HDIs have been shown to restore miRNA-124 levels. In addition, miRNA-424 and miRNA-503 have a role in the vascular homeostasis modified by transcription factor family MEF2. 41 These two miRNAs have been shown to affect cellular proliferation by action on fibroblast growth factors via the apelin signaling axis.41,61,62 Fibroblast growth factor 2 is elevated in PAH patients. 63 Downregulation of miRNA-424 and miRNA-503 results in increased fibroblast growth factor signaling and increased proliferation of pulmonary artery endothelial cells and PASMCs. 41
MicroRNAs identified as involved in PAH

Note: BMPR2: bone morphogenetic protein receptor type 2; BRD4: bromodomain 4; eNOS: endothelial nitric oxide synthetase; FGF2: fibroblast growth factor 2; HIF-1: hypoxia-inducible factor 1; miRNA: microRNA; PA: pulmonary arterial; PAEC: pulmonary arterial endothelial cell; PAH: pulmonary arterial hypertension; PASMC: pulmonary arterial smooth muscle cell; PPARγ: peroxisome proliferator-activated receptor gamma.
MicroRNA-204 is also downregulated in PAH. It also promotes oncogene expression, including Bcl-2 and survivin (Fig. 2).42,43 Bcl-2 is an antiapoptotic factor that is significantly elevated in both FHR and human PAH lung. 35 Through nuclear factor of activated T cells, the Bcl-2 oncogene appears to drive proliferation of PASMCs. 64 The suspected target of miR-204 is BRD4, which has binding sites in the nuclear factor of activated T cells promoter. 65 By binding this BRD, miRNA-204 may block nuclear factor of activated T cells–driven proliferation. Consequently, miRNA-124 and miRNA-204 may be excellent targets for therapies.
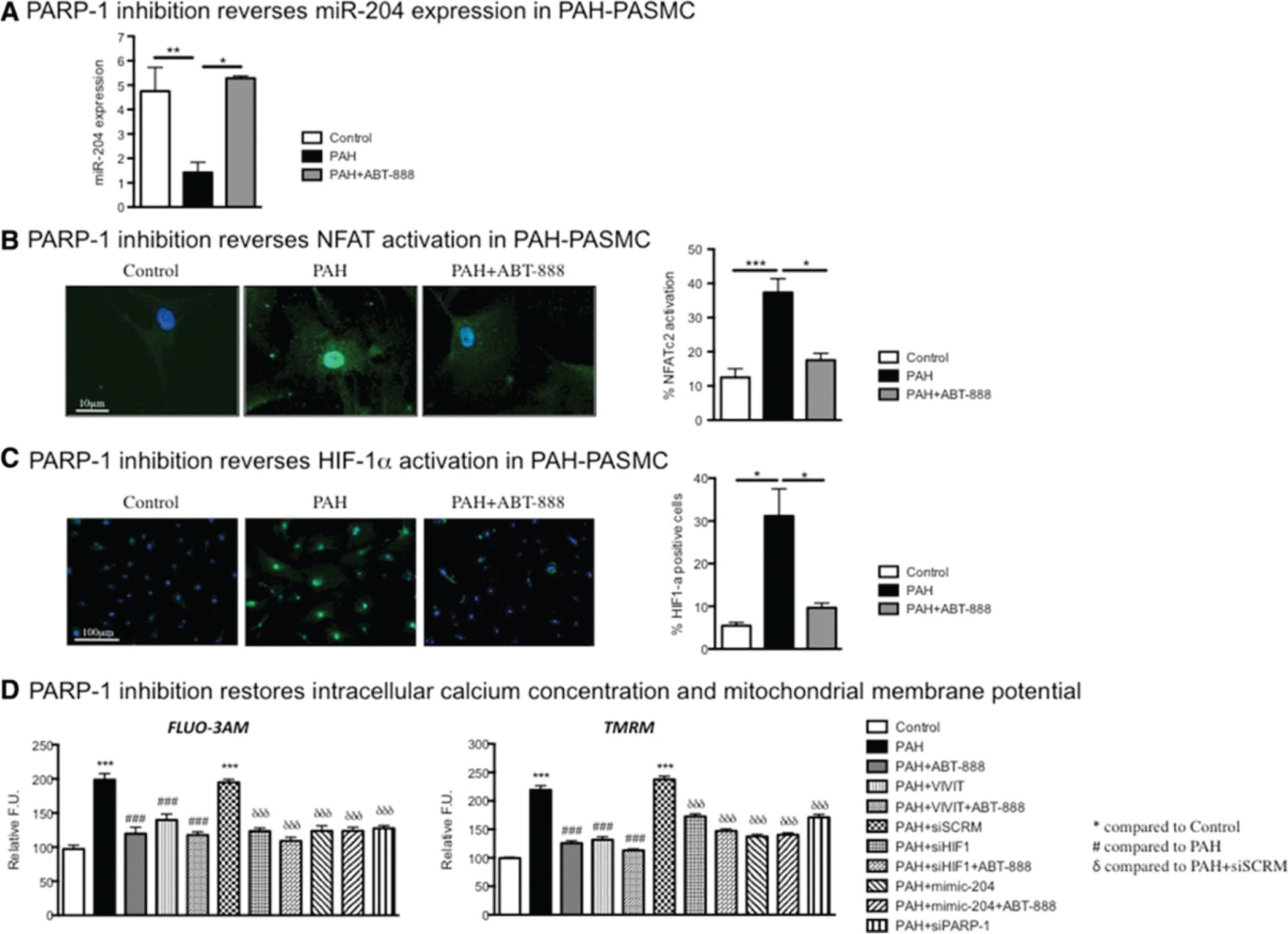
ABT-888 (a PARP-1 inhibitor) in PAH-PASMC restores the miRNA-204/NFAT/HIF-1α axis. A, miRNA-204 levels were measured in control cells (n = 5) and PAH cells (n = 4) with or without ABT-888 (10 mmol/L for 48 hours). PARP-1 inhibition increases miRNA-204 levels, as compared to those seen in control cells. B, PARP-1 inhibition decreases NFATc2 activation, measured by nuclear translocation assay (n = 4–5). C, The same phenotype is also observed for HIF-1α, as its activation is increased in PAH PASMC and decreased upon PARP-1 inhibition (n = 4). D, PARP-1 effects are mediated by miR-204/NFAT/HIF-1α, as adding ABT-888 to another treatment (miR-204 mimic, VIVIT, siHIF-1α) does not change Ca2+ or ΔΨm (for all the experiments: n = 50 cells/experiment in 3 or 4 experiments). *P < 0.05; **P < 0.01; ***P < 0.001; triple symbols in D also indicate P < 0.001; see key for detials. FU: fluorescent units; HIF-1α: hypoxia-inducible factor 1-alpha; miR: microRNA; NFAT: nuclear factor of activated T cell; PAH: pulmonary arterial hypertension; PARP-1: poly (ADP-ribose) polymerase 1; PASMC: pulmonary arterial smooth muscle cells; si: small interfering RNA. From Meloche et al.; 42 used with permission.
EPIGENETICS IN THE RV
RV failure is one final common pathway for patients with PAH; however, there appears to be a breadth of variability in each patient's ability to adapt to increased RV afterload. 66 The process of RV failure includes ischemia and metabolic transitions to less efficient glycolysis.67,68 MicroRNA factors have been implicated in epigenetic changes in RV failure.
MicroRNA-126 activates mitogen-activated protein kinase and phosphatidylinositol 3 kinase, promoting angiogenesis by vascular endothelial growth factor signaling.69,70 Bonnet and colleagues 28 showed that downregulation of miRNA-126 inhibited mitogen-activated protein kinase to decrease vascular endothelial growth factor–mediated angiogenesis in RV failure. This inhibition pushes the compensated right ventricle (also RV) into decompensated RV failure driven by a decrease in RV capillary density and resulting ischemia. No difference in left ventricular capillary density is observed. 44 In the monocrotaline rat model, increased expression of miRNA-126 correlated with improvement in echocardiographic measures of RV function. 44
In models of pulmonary banding, four miRNAs were increased in the RV that were not found in the left ventricle: specifically, miRNA-34a, miRNA-28, miRNA-148a, and miRNA-93. 71 Interestingly, miRNA-34a plays a role in apoptosis and cellular aging as proap-optotic. 72 In addition, miRNA-145/143, miRNA-204, and miRNA-17 appear to play an important role in RV function, and many more likely play a role in RV homeostasis.43,45,46
EPIGENETICS IN CLINICAL TRIALS OF PAH
Hydralazine
Hydralazine is a commonly used antihypertensive but has also been studied in the setting of epigenetic functions. The discovery of hydralazine's epigenetic effects was based on studying a side effect of hydralazine: drug-induced lupus. Treatment of cells with hydralazine results in promoter demethylation and the expression of tumor suppressor genes. On the basis of protein models, hydralazine appears to bind at the active site of DNMT; however, the interaction has not been confirmed in vivo. 73 Interestingly, in attempting to capitalize on valproic acid's HDI actions, a study of combined hydralazine–valproic acid therapy on cell lines showed inhibition of growth, although hydralazine alone had little effect. 73 To temper the enthusiasm for a role of hydralazine in PAH, there are several historical studies that demonstrate deleterious effects of hydralazine in PAH.74–76
Procainamide
Another cardiac medication, procainamide, has been investigated for its epigenetic effects. Procainamide has been shown to inhibit DNMT1 but not the DNMT3 subsets, which has potential therapeutic application for gene expression regulation in treatment of malignancy. 77 DNMT1 has been linked to epigenetic changes in colorectal cancer, and therefore procainamide could be useful in cancer prevention. 77 Currently, there are no published data about procainamide in PAH.
DNA methylation has become an important topic of research in malignancy, and the epigenetic applications of hydralazine and procainamide could be investigated in PAH. But this would require a considerable shift in data collection and clinical trial design in PAH trials.
HDIs
Therapeutic targets are difficult to isolate with techniques for epigenetic modification. Multiple spectra of targets exist, including histone deacetylation and miRNA modification. HDIs are a class of medications that can target and inhibit HDACs by zinc chelation (excluding class III), rendering the HDAC inactive (Table 3). 80 “Pan-HDIs,” or nonspecific HDIs, are associated with toxicities including thrombocytopenia, nausea, and fatigue; thus, the goal to create targeted HDIs with improved safety profiles is important. 81 HDI cardiac toxicities, specifically QT prolongation, are also a concern but thus far appear to be relatively rare in clinical trials. 82
Histone deacetylase inhibitors and targets

Note: HDAC: histone deacetylase; SAHA: suberoylamilide hydroxamic acid.
Epigenetics has been established as having a significant role in the stress response of the myocardium. Concerns about using HDIs include modification of the adaptive response and the inability of the RV to respond to stress. Mouse models have shown that the adult heart is very sensitive to alterations in HDAC expression. Class II HDACs appear to control cardiac growth and remodeling, along with class I. These two classes appear to work as antagonists: class II promotes growth while class I represses growth. 83 The opposite effects of these different classes only enhance the argument for more specific HDIs to avoid unwanted effects.
Trichostatin A (TSA) is a pan-HDI that has been studied in cell culture and effectively arrests the cell cycle in multiple cancer cell lines.84,85 TSA has been shown to increase acetylation of H3, modifying chromatin structure. 85 However, TSA is associated with negative RV effects in the rat model, including worsening RV dysfunction, fibrosis, and myocardial apoptosis. 86 Bogaard and colleagues 87 hypothesized that RV dysfunction was due to decreased capillary density and resultant ischemia, as HDIs have been shown to decrease angiogenesis via the vascular endothelial growth factor pathway. TSA reduces angiogenic factor expression in vitro, including vascular endothelial growth factor, which appears to be mediated by HDAC6 (Fig. 3). 86
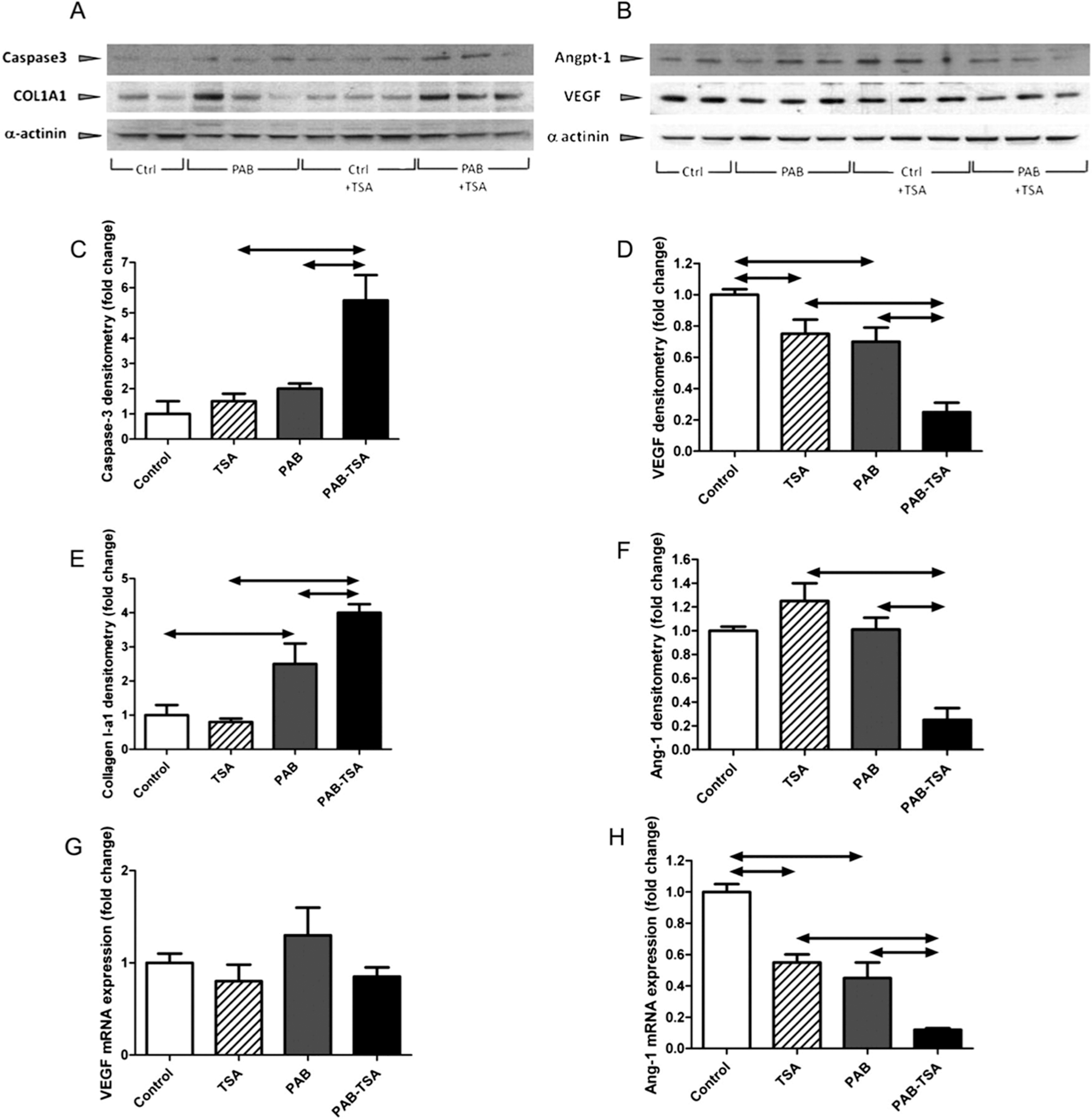
Western blots of right ventricular (RV) whole-cell lysates (A, B; note different order of experimental conditions compared with densitometries) show an increased protein expression of cleaved caspase-3 (densitometry in C) and collagen-I A1 (ColIA1; densitometry in E) and a decreased protein expression of vascular endothelial growth factor (VEGF; densitometry in D) and angiopoietin 1 (Angpt-1 [in B]/Ang-1; densitometry in F) in trichostatin A (TSA)–treated rats with pulmonary artery banding (PAB). Quantitative polymerase chain reaction does not show changes in VEGF gene expression in any of the experimental conditions (G), whereas Ang-1 messenger RNA (mRNA) expression is reduced by PAB and TSA, alone or in combination (H). 86
Valproic acid has more specific class I HDI activity and is already in common clinical use for seizure disorders. Valproic acid has been shown to decrease mean pulmonary artery pressure and RV hypertrophy in rat models of PAH but had negative effects on the RV similar to those of TSA.35,86 Valproic acid also has many other molecularly active sites, so it is difficult to attribute all of these findings to HDAC inhibition.
Suberoylamilide hydroxamic acid is a pan-HDI that had similar effects on pulmonary pressures in rat models, decreased pulmonary arterial muscularization and RV hypertrophy, although its effect on RV function has not been reported. 35 Suberoylamilide hydroxamic acid is approved for use in T cell lymphoma and is generally well tolerated.
The MEF2 pathway described above has an important regulatory role in fibroblast growth factor production, which appears to be pathologic in PAH. The class IIa HDACs bind MEF2 and suppress expression of downstream proliferative genes.80,88 Kim and colleagues 39 studied MC1568, a selective class IIa HDI that targets HDAC4 and HDAC5, in pulmonary arterial endothelial cells. In MEF2-impaired pulmonary arterial endothelial cells there is nuclear accumulation of these class IIa HDACs. 39 Previously, class IIa HDIs have been shown to reverse experimental PAH in rodent models without the negative effects of nonspecific HDIs described above. The specific class I HDIs MGCD0103 and MS-275 have positive effects on hypoxia-induced pulmonary hypertension, with decreased smooth muscle proliferation and medial thickening and without negative RV effects. 78 These findings also support targeted HDAC inhibition as a more desirable therapeutic option, especially because of the absence of the negative effects on the RV seen in pan-HDIs. 78 Clinical trials of HDIs have focused on use in malignancy; as of yet, there are no studies in human subjects with PAH.
BRD inhibitors
The bromodomain and extra terminal domain family, also called BET, has been studied in a range of cancers. These proteins bind to the histone tail and regulate transcription. BRD4 inhibition is currently the best-characterized BET therapy to be studied in oncologic processes. BRD4 is involved in mitosis and promotes gene transcription by recruiting the complex for postmitotic transcription. 89 The main targets of BRD4 are c-Myc, Bcl-2, S-phase kinase-associated protein 2, p21, and p27, all molecules implicated in PAH. 90 Meloche and colleagues 64 studied BET and BRD4 inhibition, using JQ1 and siBRD4, respectively, in human PAH PASMCs and the Sugen/hypoxia rat model and found a baseline 7-fold increase in BRD4 protein in human PAH PASMCs versus controls. In addition, treatment with an miRNA-204 mimic downregulated BRD4 expression in the rats with Sugen/hypoxia PAH. 64 BRD4 inhibition decreased levels of the oncogenes nuclear factor of activated T cells c2, Bcl-2, and survivin. Interestingly, p21 increased, suggesting an arrest of proliferation. These findings establish the role of BRD4 in oncogene expression and cellular proliferation. In addition, BRD4 inhibition led to decreased PASMC proliferation as well as reversal of Sugen/hypoxia-induced PAH in the rat model, with a decrease in mean pulmonary arterial pressure by 34%–42%. 64 BET inhibitors are currently being studied in malignancy, including lymphoma, leukemias, and solid tumors.91–93
miRNA-based therapies
In vivo miRNAs have been targeted, mostly in rodent models. Suppression of miRNA-145/143 activity in smooth muscle and endothelial cells could have potential therapeutic benefit. Mice without miRNA-145/143 are protected from hypoxia-induced pulmonary hypertension. 45 There are multiple miRNAs that are attractive targets; still, however, little is known about effects in other organs systems.
One use of RNA sequencing is to determine gene expression. Hemnes and colleagues 94 used microarrays of lymphocytes to identify genetic characteristics of vasodilator-responsive PAH. This study found differences in gene expression, via RNA microarray, between vasodilator-responsive and vasodilator-nonresponsive patients, allowing the creation of an algorithm able to identify vasodilator-responsive patients by gene expression patterns. 94 If genotype/phenotype insight such as this could be obtained from participants within PAH clinical trials, targeted therapies could be developed that capitalize on this individual variability.95,96
Modification of miRNA pathways is accomplished primarily by antagomir molecules that are targeted at a specific miRNA. 97 The method of miRNA replacement, in the setting of underproduction of an miRNA, can be accomplished by the introduction of a “mimic.” 98 The limitations to using these therapies are determining the specific target miRNAs in PAH, understanding the function of the miRNA in the disease, targeting the lung vascular tissue, and avoiding off-target effects.
METHODS TO EXPAND THE IMPACT OF EPIGENETICS IN PAH CLINICAL TRIALS
Patient selection in clinical trials
Currently, there exists a considerable effort, led by the White House, to emphasize personalized medicine and identify patient phenotypes that may be advantageous in guiding therapy. 99 Identification of different PAH patient subsets for tailored therapy is essential to advancing in treatment of this heterogeneous syndrome. Currently, patients are divided on the basis of clinical characteristics and hemodynamics; however, advances in understanding of the genetic, epigenetic, and molecular basis of disease may allow better differentiation between patient phenotypes. The current World Health Organization groupings are inadequate to characterize the breadth of the disease.96,100 Clinical trials in PAH have not focused on phenotypic expression in PAH but instead have used the World Health Organization histopathological classification to define patient populations and clinical trials. Thus, a relatively heterogeneous patient population is studied and may account for the variability seen in randomized controlled trials.
Ironically, PAH embraced personalized medicine long before it entered popular parlance, when Rich and colleagues 101 identified the vasodilator-responsive PAH phenotype in the 1980s. However, it took several decades for this clinical phenotype to be translated into identifiable gene expression patterns. 94
Alternative study designs in PAH
Previously, we have discussed study designs that are adaptable to current standards of care in the modern PAH population. 96 Multiple possible study designs that would be advantageous in PAH are outlined in Table 4. 96 The identification of specific genetically different phenotypes is also possible through genome-wide association studies (GWASs) and phenome-wide association studies (PheWASs).
Potential study designs in PAH

The Electronic Medical Records and Genomics, or eMERGE, network is an ongoing collection of electronic health records (EHRs) linked to genetic sequencing. As healthcare facilities are being compelled to adopt EHRs, the accessibility of large pools of data for relatively rare diseases will improve. As a result, the use of databased genetic information, in association with clinical phenotypes, can help drive research in PAH. This link between genotypic and phenotypic information via EHRs can enable GWASs. GWASs have been used to identify genetic variations associated with specific disease states. 102 In a similar vein, EHR-linked epigenetic or mRNA data could be coupled to phenotypes in various disease states, including PAH. Pharmacogenetic variations among patients could be utilized to determine responsiveness to a therapy or risk for adverse reactions to medication with less-invasive testing. EHR-coupled biobanks can facilitate the generation of large data banks for relatively rare diseases in a cost-effective and timely manner. 103 Another method would be PheWAS, which can identify pleiotropic genotypes.104,105 PheWAS is a viable method to identify novel associations between genes and phenotypes; this method, while in infancy, has great potential. 105
CONCLUSION
Epigenetic targets have great potential for therapeutics, prognostics, and pharmacodynamics in PAH. Currently, epigenetics-targeted therapies are being used in multiple forms of malignancy. Animal studies of PAH have shown potential for the use of HDIs as therapeutic targets; however, clinical trials in humans for PAH are not yet ongoing. The potential for EHR genetic, epigenetic, and gene expression repositories are interesting for advances in PAH diagnosis and treatment.