
Abstract
Chronic thromboembolic pulmonary hypertension (CTEPH) is characterized by fibrotic obstruction of the proximal pulmonary arteries, and it is believed to result from incomplete thrombus resolution after acute pulmonary embolism. While treatment for this condition with surgery and medical therapy has improved outcomes, our understanding of the molecular mechanisms underlying CTEPH is incomplete. Numerous risk factors have been associated with the development of CTEPH, including but not limited to acquired thrombophilias and chronic inflammatory states. A minority of patients with CTEPH have an abnormal fibrin structure that may delay thrombus resolution. Recently, examination of resected scar material in patients with CTEPH has suggested that deficient angiogenesis may play a role in thrombus nonresolution, and there is increasing interest in factors that drive intravascular scar formation. An additional challenge in CTEPH research is understanding the etiology and implications of the small-vessel disease present in many patients. Future work will likely be directed at understanding the pathways important to disease pathogenesis through further examinations of resected tissue material, continued work on animal models, and genomic approaches to identify alterations in gene expression or gene variants that may distinguish CTEPH from other forms of pulmonary hypertension.
Chronic thromboembolic pulmonary hypertension (CTEPH) is thought to be a late complication of acute pulmonary embolism (PE), occurring in 0.1%–9.1% of patients after an acute PE.1–13 Supporting this concept is the finding in a large, prospective international registry that nearly 80% of patients diagnosed with CTEPH have a history of PE, 14 with the remainder thought to have had clinically silent embolism. The disease is characterized by pulmonary artery obstruction with intraluminal scar tissue, which is thought to arise from non-resolving pulmonary emboli. A diagnosis of CTEPH is made by the presence of an elevated mean pulmonary artery pressure ≥ 25 mmHg and the presence of at least one segmental perfusion defect despite 3 months of anticoagulation therapy. 1 CTEPH is distinct among the types of pulmonary hypertension in that many cases are amenable to pulmonary endarterectomy (PEA), a procedure where the chronic intravascular scar tissue is surgically dissected out to restore vessel patency and thereby reduce pulmonary pressures and improve right ventricular (RV) function. 15 Moreover, recent studies have demonstrated the benefits of medical therapy 16 and pulmonary angioplasty 17 as alternatives to PEA in those patients with contraindications to surgery or patients with residual pulmonary hypertension after PEA.
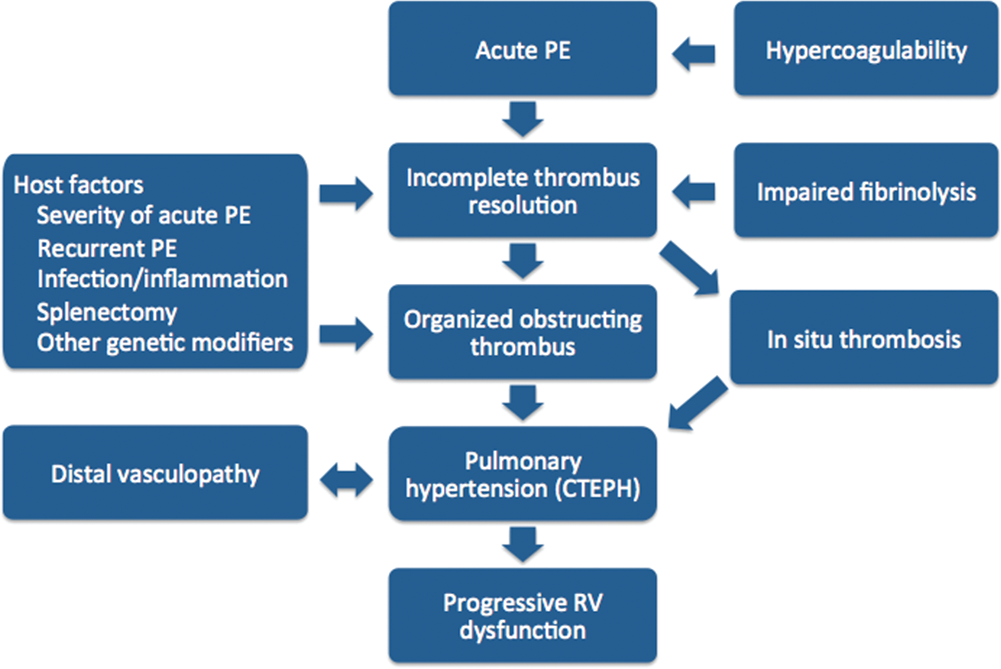
Current investigations in the pathogenesis of CTEPH suggest that it is unlikely that a single factor leads to the development of chronic pulmonary hypertension after acute PE but rather that a host of clinical factors and alterations in the postthrombotic response lead to the formation of chronic intravascular scar and ultimately pulmonary hypertension and RV dysfunction (Fig. 1).18,19 This review begins by discussing the significance of residual perfusion defects and echocardiographic findings after acute PE and then summarizes the current state of knowledge of the pathology of CTEPH and the known risk factors for CTEPH and discusses the current understanding about pathways that may be important in the development of CTEPH.
RESIDUAL PERFUSION DEFECTS AND ECHOCARDIOGRAPHIC FINDINGS AFTER ACUTE PE
Why some patients develop CTEPH after acute PE and others do not is incompletely understood. One systematic review found that more than half of patients have residual perfusion defects 6 months after an acute PE. 20 A more recent prospective study of 167 patients with acute PE, using computed tomography at the time of diagnosis and after 6 months of anticoagulation therapy, found that 85% of patients achieved complete resolution and 15% had residual perfusion defects, suggesting that complete resolution may be more common than previously believed. 21 Despite the relatively high prevalence of perfusion defects, only 1%–4% of patients have pulmonary hypertension 6 months to 2 years after PE. 12 Thus, not all patients with residual perfusion defects have CTEPH. Asymptomatic patients having perfusion defects and normal hemodynamics may simply have resolving PE, whereas symptomatic patients with perfusion defects and normal hemodynamics are described as having chronic thromboembolic disease.1,22
The presence of pulmonary hypertension on echocardiogram at the time of an acute PE diagnosis is associated with an increased risk of persistent pulmonary hypertension at 1 year. In a study of 78 patients with acute PE on whom serial echocardiography was performed for 12 months, an age of >70 years and an estimated pulmonary artery systolic pressure of >50 mmHg on echocardiogram were independently associated with an increased risk of persistent pulmonary hypertension or RV dysfunction. Despite this finding, of the 36 patients with significant RV dysfunction at the time of diagnosis, only 3 had persistent RV dysfunction at 1 year. While this study informed only on RV function and not on clot burden in the pulmonary arteries, these results suggest that the majority of patients who initially have RV dysfunction with acute PE will not develop pulmonary hypertension. However, those patients with the most severe RV dysfunction and elevations in pulmonary pressures are at increased risk of pulmonary hypertension at 1 year. 13
THE PATHOLOGY OF CTEPH CONTRASTS WITH THAT OF ACUTE PE
Resected pulmonary artery tissue in patients with CTEPH differs from pulmonary thrombectomy specimens in acute PE (Fig. 2). Microscopically, acute-PE specimens contain predominantly fibrin, inflammatory cells, and red blood cells. In contrast, resected chronic thromboemboli are considerably more complex (Fig. 3). The Jamieson classification system used to classify resected pulmonary artery tissue is shown in Table 1, along with the frequency of each lesion seen during PEA.23,24 In a series of 54 patients who underwent PEA, all but 2 had bilateral distribution of disease. The size of the lesions varied considerably, with proximal specimens as large as 13 cm and more distal specimens measuring less than 1 cm. In almost all cases, organized or organizing thrombi were seen in conjunction with fresh red thrombi. The intima was comprised of myofibroblast cells as well as collagen and extracellular matrix containing collagen (100%), elastin (67%), hemosiderin (56%), atherosclerosis (32%), or calcification (15%). Evidence of acute or chronic inflammation was also observed in more than half of the specimens. All specimens contained partial-thickness portions of media, and only one specimen contained a small portion of adventitia. 25 A second series of 200 consecutive PEAs revealed similar findings in terms of the distribution of lesions and noted a predominance of organizing or organized thrombi, compared to fresh fibrinous clot (99.2% vs. 0.8%). In addition, the authors noted predominantly mild to moderate inflammation in 76.2% of specimens, with increased cellularity within thrombi around newly formed recanalizing vessels. 24

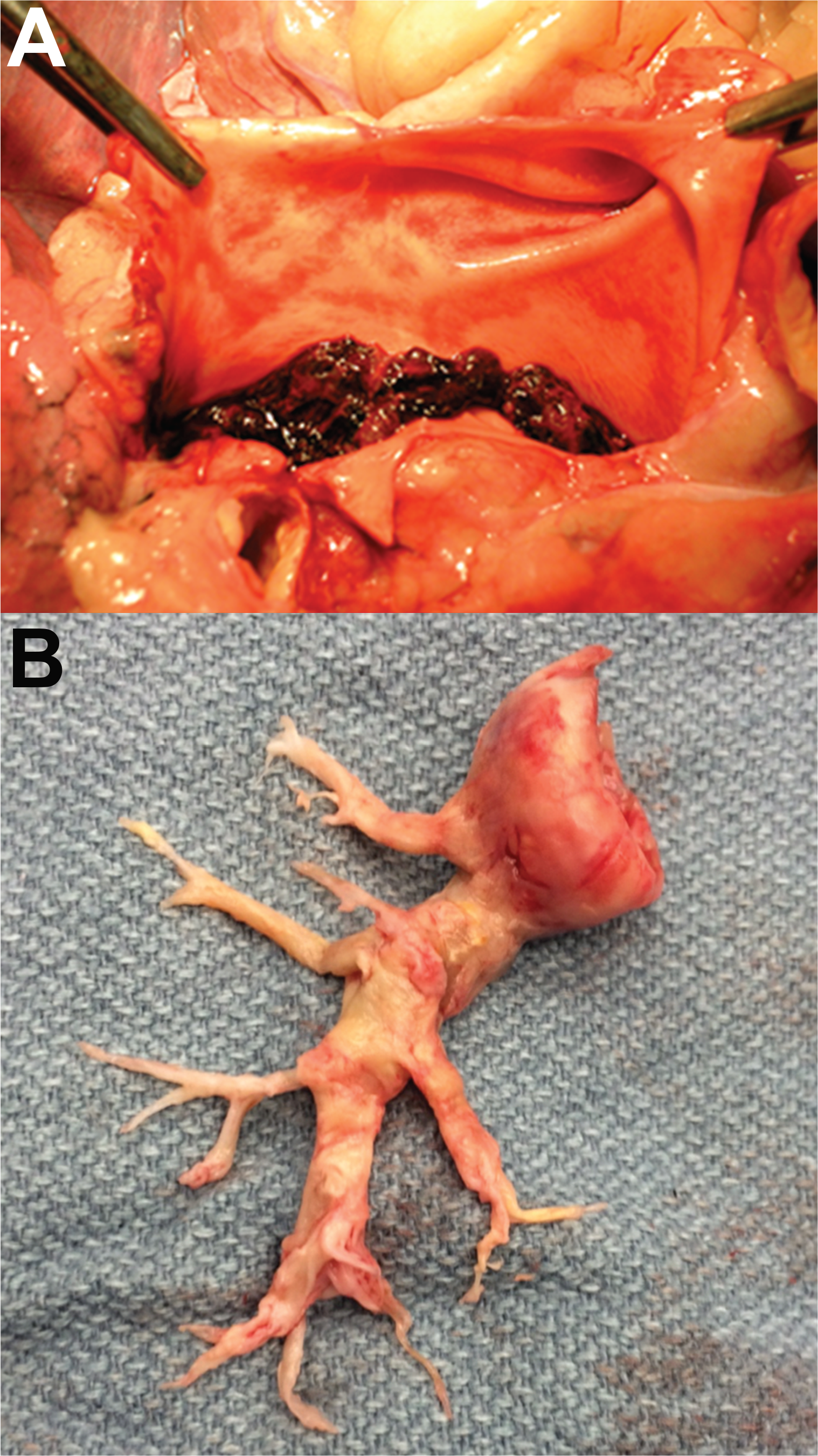
Vascular obstructive lesions in acute pulmonary embolism (A) and chronic thromboembolic pulmonary hypertension (CTEPH; B). The CTEPH patient underwent bilateral pulmonary endarterectomy; however, only the right-side specimen is shown. Images courtesy of Dr. Joyce Johnson (A) and Tarek Absi (B).
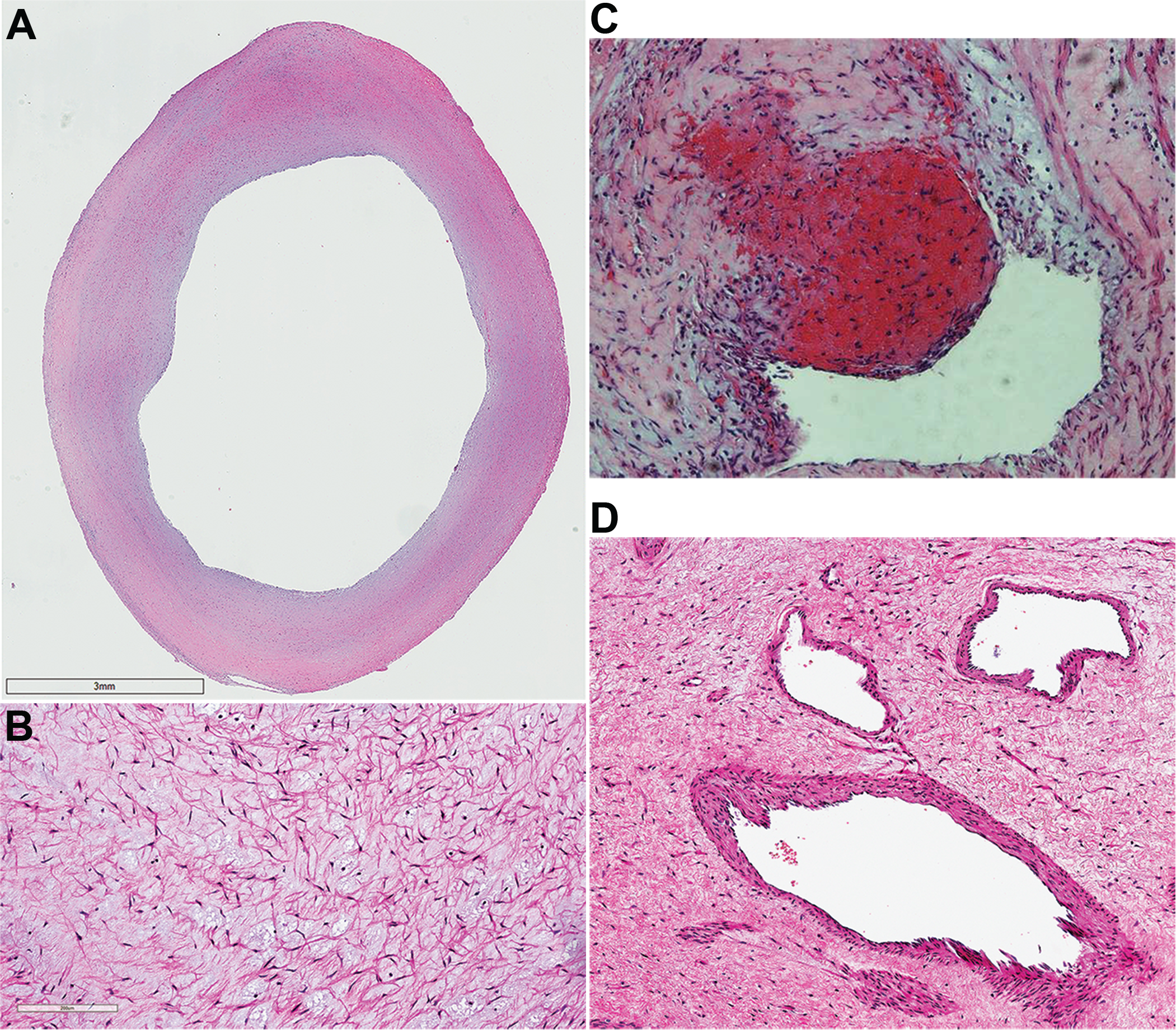
Pulmonary endarterectomy specimens stained with hematoxylin and eosin. A, Neointimal thickening, 18x; B, region of fibrotic chronic thromboembolic pulmonary hypertension clot containing extracellular matrix and spindle-shaped cells, 290x; C, fresh red clot within an area of organized clot, 100x; D, obstructing lesion with areas of recanalization, 170x.
A more recent series of 52 patients with CTEPH who underwent PEA revealed 4 distinct types of resected lesions. Neointima was characterized by the presence of α-smooth muscle actin–positive cells and was seen in 94% of patients. Thrombotic lesions, seen in 75% of patients, contained either fresh thrombi or varying levels of organization. Recanalized lesions displayed areas of angiogenesis within a thrombotic lesion and were seen in 63% of patients. Finally, atherosclerotic lesions containing macrophage-derived foam cells with lipid droplets were seen in 31% of patients. 26
ASSOCIATIONS AND RISK FACTORS FOR DEVELOPMENT OF CTEPH
A summary of conditions associated with CTEPH is provided in Table 2. Multiple studies have evaluated the incidence of CTEPH after acute PE, and many have attempted to find associations and risk factors for developing CTEPH. In one study, factors that increased the risk of CTEPH included a previous PE, younger age at diagnosis, larger perfusion defects at the time of the initial PE, and idio-pathic PE. 12 A second study confirmed idiopathic PE as a risk factor for the development of CTEPH. 5 In both studies, CTEPH was diagnosed within 2 years of acute PE, and fewer data were available examining the development of CTEPH farther out from the acute thromboembolic event.
Associations and risk factors for chronic thromboembolic pulmonary hypertension
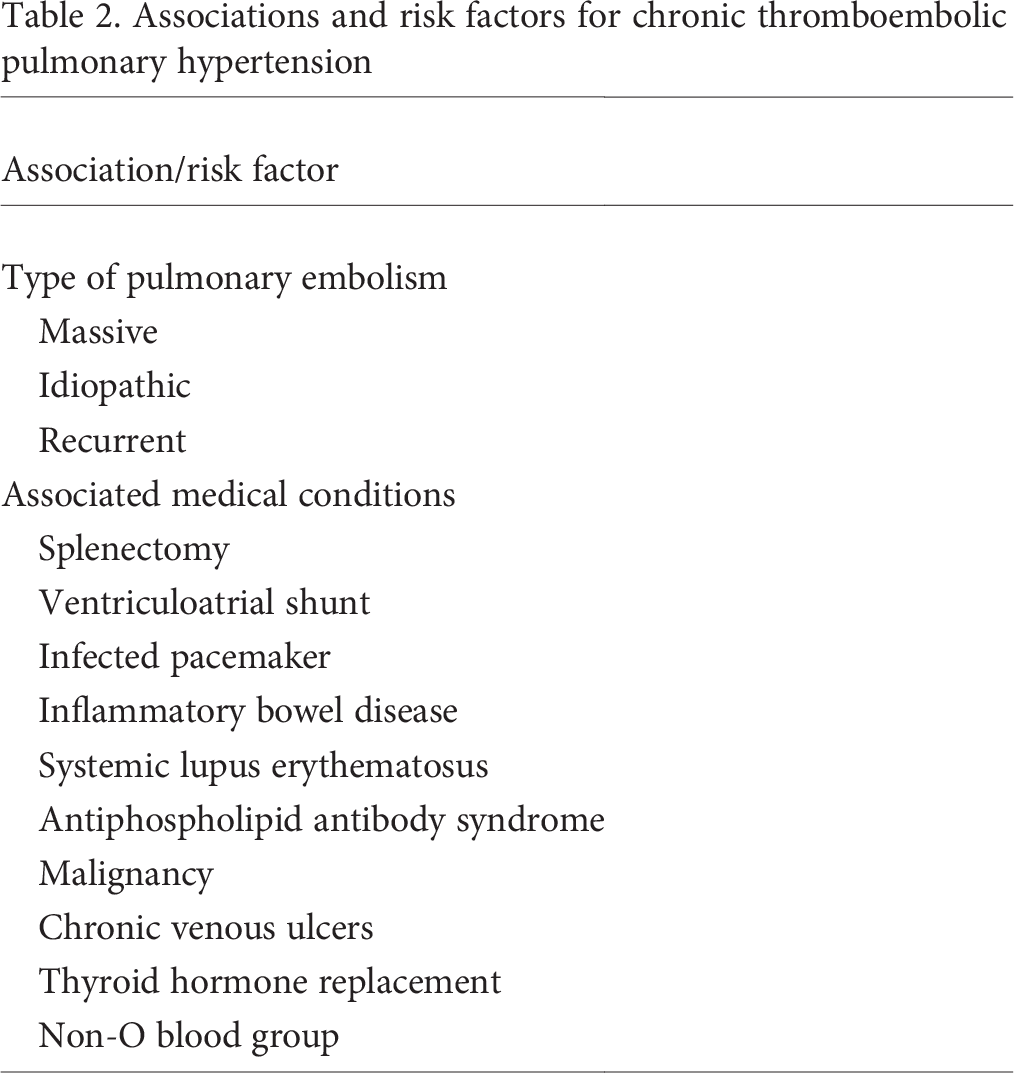
Several medical conditions have been shown to occur more frequently in patients with CTEPH than in other types of pulmonary hypertension. The association between prior splenectomy and CTEPH has been well described, and it has been postulated that abnormal erythrocytes unfiltered in the asplenic patient or reactive thrombocytosis may predispose these individuals to the development of chronic obstruction. 27 Ventriculoatrial (VA) shunts and infected pacemakers have also been shown to be present more frequently in patients with CTEPH than in patients with other etiologies of pulmonary hypertension. While the precise mechanism linking VA shunts and CTEPH is unclear, data suggest that staphylococcal infection may play an important role. In one report, 6 of 7 VA shunts in patients with CTEPH had evidence of staphylococcal infection, and staphylococcal infection delayed thrombus resolution and upregulated transforming growth factor beta in a murine model of venous thrombosis. 28
The presence of a non-O blood group has been associated with an increased incidence of venous thromboembolism (VTE) and CTEPH, and proposed mechanisms explaining this finding include increased levels of von Willebrand factor, factor VIII, p-selectin, and tumor necrosis factor α in individuals with non-O blood groups, all of which have been associated with VTE. 29 Finally, chronic inflammatory disease (including inflammatory bowel disease, osteomyelitis, and the antiphospholipid antibody syndrome [APS]), chronic venous ulcers, infected chronic indwelling venous catheters, thyroid hormone replacement, and malignancy have also been associated with an increased risk of CTEPH.27,30–32
INHERITED THROMBOPHILIAS IN CTEPH
Inherited hypercolaguable states, including the presence of mutations in factor V Leiden, the prothrombin gene, proteins C and S, and antithrombin, are known risk factors for the development of VTE. 33 The role these mutations play in the development of CTEPH is less clear. In one study, patients with CTEPH (n = 46) and idiopathic pulmonary arterial hypertension (IPAH; n = 64) were evaluated for the presence of inherited hypercolaguable states, and the frequency of these mutations was no greater than that seen in a healthy control population. 34 A more recent study looked at the same mutations in patients with CTEPH (n = 45) and pulmonary hypertension from other causes (n = 200) and found no differences in the frequencies of mutations, except for a higher incidence of the factor V Leiden mutation among the subset of Caucasian patients with CTEPH (29% vs. 7.8%, P = 0.001). 35 These findings highlight the lack of a strong causal link between inherited thrombophilia and the development of CTEPH.
ACQUIRED THROMBOPHILIAS IN CTEPH
APS is also associated with an increased tendency toward VTE, and anticardiolipin antibodies in particular have been shown to be associated with an increased risk of recurrent VTE. 33 In one study of 116 patients with CTEPH and 83 patients with IPAH, Wolf et al. 34 found that antiphospholipid antibodies or lupus anticoagulant were found in 10% of patients with IPAH and 20% of patients with CTEPH, which is higher than previously reported in patients with acute PE. Moreover, patients with CTEPH had higher titers of antiphospholipid antibodies than patients with IPAH, suggesting that these prothrombotic antibodies may be most significant in patients with CTEPH. 34 In another study looking only for the presence of lupus anticoagulant in 216 patients with CTEPH, 23 (10.6%) had a positive antibody. 36 Finally, a smaller study by Wong et al. 35 found no increased incidence of antiphospholipid antibodies in 40 patients with CTEPH compared to 174 patients with pulmonary hypertension from other causes. These findings suggest that antiphospholipid antibodies or lupus anticoagulant may contribute to the development of CTEPH in some individuals by increasing the thrombotic tendency.
FACTOR VIII LEVELS ARE ELEVATED IN CTEPH
Factor VIII is a hemostatic protein that aids in factor IXa–mediated activation of factor X. A high level of factor VIII in the blood is a known risk factor for initial and recurrent VTE. 37 Bonderman et al. 38 showed that plasma factor VIII was elevated in patients with CTEPH (n = 122), compared to patients with nonthromboembolic pulmonary hypertension (n = 88) and healthy controls (n = 82), and that these elevated levels persisted 1 year after PEA. Factor VIII levels higher than 230 IU/dL are strongly associated with recurrent VTE, and these levels were found in 41% of patients with CTEPH. 38 These findings suggest yet another potential mechanism for the development of CTEPH; however, significant factor VIII elevations are not found in all patients, and it is unclear whether factor VIII elevations are causal or are sequelae of the disease.
FIBRINOLYTIC IMPAIRMENT IN CTEPH
Fibrinolysis describes the initial phase of clot breakdown, and a fibrinolytic defect has been proposed to play a pathophysiologic role in the development of CTEPH. Fibrinolysis is a complicated process involving lysis of cross-linked fibrinogen through interactions between plasminogen, tissue plasminogen activator (t-PA), plasminogen activator inhibitor (PAI-1), and other regulatory proteins as well as interactions with the endothelium at the site of vessel injury. Reduced plasma fibrinolytic potential has been previously described as a risk factor for VTE, 39 and some data suggest that this is true in CTEPH. In dogs with experimentally induced acute PE, the administration of the antifibrinolytic tranexamic acid inhibited resolution of emboli and led to the development of persistent elevations in pulmonary artery pressure, and at autopsy these dogs demonstrated organized fibrotic thrombi within the pulmonary arteries. 40 In humans with CTEPH, it is possible that these fibrinolytic enzymes are deficient or dysfunctional or, alternatively, that the thromboemboli could be resistant to fibrinolysis. 19
Olman et al. 41 investigated plasma levels of t-PA and PAI-1 in 32 patients with CTEPH, compared to those in age-matched controls, and found that basal levels of t-PA and PAI-1 were elevated in plasma taken from CTEPH patients compared to that from controls; however, no difference in enzymatic activity was detected between the groups. Venous-occlusion experiments demonstrated that patients with CTEPH, compared to controls, had a greater rise in t-PA antigen and PAI-1 activity, suggesting that an impaired response of t-PA to venous occlusion is not critical to the pathogenesis of CTEPH. 41 Because endothelial cells (ECs) produce both of these key regulatory proteins, a follow-up study was performed investigating the production of t-PA and PAI-1 by cultured ECs harvested from patients with CTEPH, compared to that by ECs taken from organ donors. No difference was seen in basal t-PA levels or PAI-1 activity in CTEPH patients compared to the control group, and this lack of difference persisted after ECs were exposed to thrombin. 42 Taken together, these studies suggest that hypofibrinolysis due to abnormal activity or function of t-PA or PAI-1 may not explain the development of CTEPH in most cases. Nevertheless, in vitro assays of fibrinolysis are limited because they cannot replicate the in vivo environment to account for such factors as blood flow, platelet interactions with the fibrinolytic system, or the contribution ECs make to thrombus resolution.
Aside from its role in the relatively rapid phase of fibrinolysis, PAI-1 is secreted from multiple tissues, including ECs, adipose tissue, and hepatocytes, and it is believed to play an important role in tissue fibrosis.43,44 A study investigating the local expression of PAI-1 antigen and PAI-1 messenger RNA using immunohistochemistry and in situ hybridization in specimens harvested from patients undergoing PEA found that both PAI-1 antigen and PAI-1 messenger RNA were highly expressed in smooth muscle cells and ECs within thrombi, compared to noninvolved areas of patients' pulmonary arteries. 45 Since PAI-1 is the primary inhibitor of fibrinolysis, PAI-1 overexpression may stabilize vascular thrombi and promote excessive accumulation of collagen and other extracellular matrix proteins, leading to scar formation. 43
CTEPH THROMBOEMBOLI MAY BE RESISTANT TO FIBRINOLYSIS
Another possible mechanism for hypofibrinolysis is the formation of clot that is resistant to fibrinolysis. Given that fibrin is critical to the structure of acute thromboembolic material, defects in fibrin may be present in patients with CTEPH. Morris et al.19,46 showed that purified fibrin from patients with CTEPH was resistant to plasmin-mediated lysis when compared to fibrin from healthy control subjects. The beta chain of fibrin—which has been implicated in various physiological events, including heparin binding, cell signaling, and angiogenesis—was particularly resistant to fibrinolysis, leading the authors to suggest that the incompletely lysed clot material could stimulate vascular remodeling into organized scar tissue.19,46 In 33 patients with CTEPH, fibrin was analyzed by liquid chromatography–mass spectrometry to identify structural variants. Targeted genomic DNA sequencing identified 5 fibrinogen variants (dysfibrinogenemias) in 5 patients. Morris et al. 47 speculated that these dysfibrinogenemias might impair fibrinolysis and thereby contribute to the development of CTEPH. In a follow-up study, fibrin taken from those patients with dysfibrinogenemias was shown to have abnormal structure, and induced clots were shown to have lower turbidity, less porosity, and slower lysis rates when compared to clots of control fibrin. 48 This work suggests that some, but not all, patients who develop CTEPH may have abnormal fibrin, which forms an abnormal fibrin matrix and is resistant to plasmin-mediated clot lysis. Miniati et al. 49 followed up this work by examining the rate of plasmin-mediated cleavage of the beta chain of fibrin in CTEPH (n = 17) compared to that in healthy controls (n = 26), patients with pulmonary hypertension other than CTEPH (n = 14), and patients with a history of acute PE but no pulmonary hypertension (n = 39). Compared to controls, plasmin-mediated lysis of the beta chain of fibrin was impaired in each group, but most significantly in patients with CTEPH and pulmonary hypertension other than thromboembolic. 49 This work serves as validation of the work by Morris et al.19,46–48 but goes farther by suggesting that fibrin resistance to lysis may not be unique to CTEPH. Further study is needed to better understand not only the role abnormal fibrin and impaired fibrinolysis play in the development of CTEPH but also the role these factors may play in the thrombotic arteriopathy seen in PAH.
ANGIOGENESIS AND THROMBUS RESOLUTION
Thrombus resolution involves much more than the fibrinolytic system. The fibrin matrix is a scaffold for cellular infiltration, and the acute thrombus additionally consists of platelets and leukocytes encompassing a red cell mass. 50 Thrombus resolution has classically been described as a process of thrombus organization followed by recanalization. 51 ECs lining the vessel, as well as leukocytes and platelets within the fresh thrombus, are thought to secrete proteolytic enzymes, chemotactic factors, and cytokines, leading to fibrinolysis and neovascularization. Monocytes are recruited into the thrombus and express proangiogenic factors, including vascular endothelial growth factor, basic fibroblast growth factor, and interleukin 8 (IL-8).52–54
Lang et al. 55 investigated the role coagulation inhibitors play in the maintenance of vessel patency within chronic intravascular occlusions in CTEPH. Immunohistochemical studies revealed intense immunoreactivity of protease nexin 2/amyloid beta precursor protein, which is a potent inhibitor of coagulation factors, in the layer of ECs lining “neovessels” inside obstructed pulmonary arteries. 55 These data suggest that local factors, including coagulation factor inhibitors, may play a role in reestablishing and maintaining vessel patency.
Deficient angiogenesis has been proposed to play an important role in the process of organization and recanalization of pulmonary thromboemboli. Alias et al. 56 demonstrated that thrombus resolution was delayed in a mouse model with EC-specific conditional deletion of vascular endothelial growth factor 2/kinase insert domain receptor, and further work showed that several vascular-associated genes were underexpressed in resected CTEPH material. In another histologic study of 52 patients with CTEPH who underwent PEA, evidence of angiogenesis was observed in all types of resected tissue, with increasing levels of angiogenic activity seen in highly recanalized lesions compared to thrombotic occlusions or neointimal lesions. Furthermore, the degree of neovessel formation correlated positively with survival and negatively with persistent pulmonary hypertension after PEA. 26 Further study is needed to delineate the role impaired angiogenesis plays in thrombus resolution in patients with CTEPH.
INFLAMMATION AND THROMBUS RESOLUTION
Under normal conditions, ECs promote a vasodilatory and local fibrinolytic state characterized by suppressed coagulation, platelet activation, and leukocyte activation. 57 The normal physiological response to thrombus formation involves reorganization of the thrombus through fibrinolysis and angiogenesis, a process that is coordinated by leukocyte migration into the affected area. 58 While this process is incompletely understood, platelet EC adhesion molecule 1 (PECAM-1) is thought to play an important role in leukocyte migration into thrombi. Recently, Lang and colleagues 58 showed, in a mouse stagnant-flow venous-thrombosis model, that PECAM-1-deficient mice exhibited larger, more persistent thrombi and that these thrombi showed decreased macrophage invasion, decreased vessel formation, and increased fibrosis.
While transient inflammation is likely important to normal thrombus resolution, excessive inflammation may play a role in thrombus nonresolution. The clinical association between more frequent CTEPH occurrence and patients with VA shunts, infected pacemakers, and/or chronic osteomyelitis may reflect this tendency of infection-induced inflammation to stabilize thromboemboli. Bacterial infection has been shown to upregulate the profibrotic cytokine transforming growth factor beta as well as connective tissue growth factor, which regulates collagen synthesis and maintains fibrosis. 28 Furthermore, noninfectious inflammatory states, such as APS or malignancy, may also promote stabilization of thromboemboli and thereby increase the risk for the development of CTEPH.
Histologic studies of resected PEA material indicate that lymphocytes, macrophages, and neutrophils are present with chronic thrombi, 26 and while inflammatory cells are part of normal thrombus resolution, it is possible that these cells could also play a pathologic role in CTEPH. Inflammatory markers, including IL-6, monocyte chemoattractant protein 1 (MCP-1), interferon γ–induced protein 10 (IP-10), macrophage inflammatory protein (MIP)1α, and chemokine ligand 5 (RANTES), have been shown to be upregulated in resected PEA supernatant compared to lung tissue from healthy controls. 59 In addition, multiple studies have demonstrated that elevated circulating mediators of inflammation, including IL-6, IL-8, IL-10, IP-10, monokine induced by interferon γ, MIP1α, MCP-1, and matrix metallopeptidase 9, are present in CTEPH plasma compared to plasma from healthy controls.26,59Of particular interest is IP-10, which is known to induce fibroblast proliferation and thus may play an important role in thrombus nonresolution and the development of chronic intravascular scar tissue. 59
PARALLELS WITH PULMONARY ARTERY INTIMAL SARCOMA
Pulmonary artery sarcoma is an extremely rare neoplasm affecting the pulmonary arteries, characterized by tumor cells that undergo endothelial, fibroblastic, or myofibroblastic differentiation. In a series of 200 consecutive PEAs performed at University of California, San Diego, 2 cases of pulmonary artery sarcoma were diagnosed, 24 highlighting that this neoplasm can mimic CTEPH. Recently, Tatsumi and colleagues 60 isolated cells from endarterectomized tissue from patients with CTEPH and identified one cell type as sarcomalike cells (SCLs). These SCLs were characterized as hyperproliferative, anchorage independent, invasive, and serum independent, thus reflecting several cancer-defining traits. Mice injected intravenously with these SCLs developed tumors that grew along the intimal surface of pulmonary vessels. 60 While these SCLs were isolated from only a single patient with CTEPH, this work raises interesting questions about whether some cells within CTEPH tissue possess neo plastic features that may drive hyperproliferation and excessive scar tissue formation.
SMALL-VESSEL DISEASE IN CTEPH
Persistent postoperative pulmonary hypertension is the most significant predictor of mortality after PEA. 15 Two broad processes could explain this observation. First, obstructions of subsegmental pulmonary arteries may be present that are not amenable to endarterectomy. In the postoperative classification of CTEPH, these are classified as type III lesions, and they have been reported in up to 12% of patients who undergo surgery. 15 Whether these lesions are within the spectrum of CTEPH or whether they arise from local thrombosis is unclear. A second cause of persistent postoperative pulmonary hypertension is a secondary arteriopathy affecting distal muscular pulmonary arteries and arterioles in patients with chronic large-vessel obstruction. Histologically, affected regions are characterized by intimal proliferation and increased media thickness; in some cases, the histopathological findings are indistinguishable from those of PAH. 61 Moser et al. 62 showed, in a series of 15 patients undergoing PEA and simultaneous lung biopsy, that numerous lesions were observed in the small pulmonary arteries and arterioles, including eccentric intimal fibrosis (67%) as well as concentric laminar intimal fibroelastosis and internal fibromuscular proliferation (combined 80%). Furthermore, plexiform-like lesions were observed in 67% of patients. 62 These changes were present distally to both occluded and unoccluded pulmonary segments, making it difficult to explain these changes with a “high-flow” injury paradigm alone. A more recent series examining lung tissue from 17 patients with CTEPH undergoing lung transplant (8 deemed inoperable because of primarily distal disease, 9 with persistent pulmonary hypertension after PEA) found that all examined lungs exhibited distal arterial remodeling similar to that reported by Moser et al. 62 but without evidence of plexiform lesions. Furthermore, lung tissue was notable for disease involvement in postcapillary venules with venous fibrosis and remodeling, in addition to changes resembling pulmonary capillary hemangiomatosis. 63 Currently, the mechanism for these changes is unknown, but it may relate to a combination of factors including shear stress, inflammation, and the release of cytokines, and further work is needed to understand the pathogenesis of distal disease and to assess the magnitude of distal disease before surgery.
THE ROLES OF PLATELETS AND IN SITU THROMBOSIS IN CTEPH
Platelets play a critical role in coagulation and hemostasis, but the role they play in the development of CTEPH is unclear. Several platelet-activating states, such as splenectomy and thyroid hormone replacement, have been identified as risk factors for CTEPH. In a recent study comparing markers of platelet activation, including p-selectin and glycoprotein IIb/IIIa, in patients with CTEPH or PAH and control subjects, platelets from CTEPH patients were activated compared to those from controls, and patients with CTEPH were hypersensitive to thrombin stimulation. Whether the platelet activation seen in CTEPH is a secondary phenomenon caused by pulmonary hypertension is unclear, given that platelets are also activated in patients with PAH. 64
The extent to which in situ thrombosis contributes to the development and progression of CTEPH is another area needing further study. In a large prospective registry of 679 patients with a diagnosis of CTEPH, 75% of patients had a confirmed previous PE and 56% had a confirmed previous deep venous thrombosis. 65 It has been suggested that patients without a documented VTE before diagnosis of CTEPH may undergo clinically silent PE. Others have suggested a role for primary pulmonary artery thrombosis as a cause of chronic large-vessel obstruction and pulmonary hypertension, citing the difficulty of inducing CTEPH in animal models with recurrent pulmonary emboli and the lack of association between traditional PE risk factors and CTEPH. The authors propose that various insults to the pulmonary endothelium, including trauma, inflammation, or atherosclerosis, may lead to the development of CTEPH. 66 Support for this theory includes descriptions of localized pulmonary emboli after chest wall trauma or focal inflammation, where direct injury or inflammation to pulmonary vasculature leads to a prothrombotic environment and local thrombus formation.67,68 Given the strong association between CTEPH and prior VTE, it is difficult to envision in situ thrombosis as the primary mechanism for developing CTEPH in most patients. Nevertheless, it may play a causal role in some cases and exacerbate chronic occlusions in others. In patients with CTEPH, the abnormal pulmonary artery endothelium and turbulent blood flow may lead to in situ thrombosis and further vascular obstruction. Such a phenomenon could explain the progressive nature of disease seen in many patients with CTEPH and also account for the frequent finding of fresh red clot intermixed with more chronic-appearing white clot at the time of PEA.
PROGRESS IN THE DEVELOPMENT OF ANIMAL MODELS OF CTEPH
One of the challenges in understanding the molecular mechanisms of CTEPH has been the development of a suitable animal model to study the disease. A CTEPH animal model would include the ability to induce clot persistence and organization in the proximal pulmonary arteries, leading to the development of pulmonary hypertension, distal pulmonary vasculopathy, and RV remodeling. As mentioned above, thrombin-induced PE in dogs followed by tranexamic acid administration led to clot persistence, organization, and pulmonary hypertension in dogs. 40 In another canine study, induced inferior vena cava thrombi that were embolized to the lung underwent rapid dissolution. 69 Taken together, these studies suggest that repeated emboli alone do not account for the development of chronic pulmonary artery obstructions and that other factors, including impaired fibrinolysis or chronic inflammation, may play an important role.
Embolization procedures have also been performed in dogs, with coils and tissue adhesive to create pulmonary artery obstruction; however, these conditions provide little insight into the pathogenesis of CTEPH and have not been shown to result in RV remodeling or distal vasculopathy in unobstructed segments. 70 A promising piglet model of CTEPH was recently reported that involved left pulmonary artery ligation followed by 5 weekly catheter embolizations of tissue adhesive to the right lower lobe pulmonary artery. The left pulmonary artery ligation aimed to overwhelm any pulmonary reserve, and the weekly injections gradually increased the pulmonary artery pressure to allow RV adaptation. At 5 weeks, the authors reported clot persistence and organization, increased pulmonary vascular resistance, increased media thickness, endothelin 1 overexpression in distal pulmonary arteries, and RV remodeling. 71 While these models may help further our understanding the downstream consequences of chronic pulmonary artery occlusions, they do not model impaired thrombus resolution seen in human disease and therefore have limited ability to provide insight into the molecular etiology of CTEPH.
SUMMARY
Research over the past decade has improved our understanding of chronic thromboembolic disease; however, the molecular mechanisms underlying this disease remain incompletely understood. The fundamental question of why a small proportion of patients with PE develop CTEPH remains unanswered. Inherited and acquired thrombophilias play a role in some cases, but these conditions are not present in all patients with CTEPH. Abnormal fibrin that is resistant to lysis may lead chronic intravascular scar formation in some cases. Additional areas of interest include the role of chronic inflammation and impaired angiogenesis in thrombus resolution after acute PE. The development of newer animal models that more closely mimic disease in humans may help further our understanding of this devastating disease.
In conclusion, it is unlikely that a single factor explains CTEPH in the majority of affected patients; rather, different mechanisms, such as a prothrombotic tendency, impaired fibrinolysis, and chronic inflammation, combine with other clinical factors to alter the prothrombotic response toward thrombus nonresolution and eventual scar tissue formation. Novel approaches, such as gene microarray, RNA sequencing, and broad-based proteomic approaches, which have previously provided clues to the mechanisms underlying PAH,72–74 may supplement current work and provide insight into the differing molecular signatures of patients with CTEPH and patients with acute PE or other types of pulmonary hypertension. Continuing current lines of research, combined with novel approaches adapted from other fields, may enhance our mechanistic understanding of CTEPH and provide insights applicable to the care of patients suffering from chronic thromboembolic disease.