
Abstract
Among the various semiconductor photocatalysts reported so far, TiO2 is still the most promising material for real applications because of its excellent chemical and thermal stability, non-toxicity, low cost and highly oxidising photogenerated holes. This review summarises the recent progress (mainly over the last five years) in photocatalytic oxidation of non-biodegradable organic pollutants (chlorophenols) and reduction of toxic heavy metal ions in aqueous solution. The review details the recently developed strategies for improving the performance of TiO2-based photocatalysts, with particular respect to the visible light activity, charge separation efficiency, stability, separability and adsorption capacity for the remediation of the aforementioned categories of water contaminants, as these factors heavily affect the practical application of this technology. Next, the underlying semiconductor photocatalytic mechanisms have been thoroughly addressed experimentally and theoretically, together with the proposed defect engineering to improve the photocatalytic performance. Finally, the prospect of TiO2 photocatalysis was discussed.
Introduction
Access to potable water is one of the biggest challenges globally, especially in developing countries. Several persistent organic pollutants (pharmaceuticals, pesticides, personal care products, endocrine disrupters) are frequently detected in wastewater effluents [1–3]. Chlorophenols (CPs) and their metabolites are common and recalcitrant environmental pollutants, believed to have high bioaccumulation capability and carcinogenic effect. Figure 1 shows four major CPs (2-CP, 2,4-DCP, 2,4,6-TCP and PCP) that have been classified as first-degree toxic pollutants by the US Environmental Protection Agency (EPA 2003) [4,5]. Chemical structure of major common chlorinated phenols.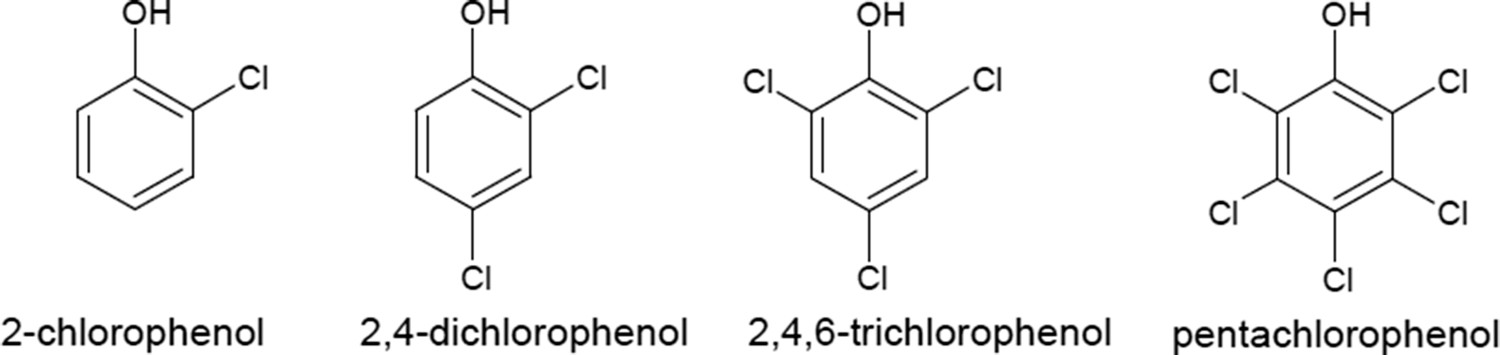
Adverse effects of phenolic compounds on the human nervous system have been reported and linked to several health disorders, e.g. kidney failure, liver and lung damage [4–6]. CPs find extensive application in the chemical, forestry and wood-working industries. They are used as herbicides, insecticides, fungicides, wood preservatives and chemical intermediates [4,7–10]. Generally, these organic pollutants are released into the environment because of several man-made activities including water disinfection, waste incineration, uncontrolled use of pesticides and herbicides and as byproducts in the bleaching of paper pulp with chlorine [11].
It is interesting to note that 2-CP, 4-CP, 2,4-DCP and 2,4,6-TCP are the most significant CPs formed as byproducts of water chlorination [12,13]. Effluents from a pulp and paper mill were analysed before treatment and subsequent discharge into the Lake Baikal in Russia, 2,4-DCP concentration of (630 ± 50) µg L−1, 2,4,5-TCP concentration of (2660 ± 210) µg L−1 and 2,4,6-TCP concentration of (320 ± 30) µg L−1 were reported [14]. After treatment, the individual chlorinated phenols were present in trace quantities, except for 2,4,5-TCP with the concentration of (560 ± 50) µg L−1 [14]. Environmental pollution agencies and health organisations recommended a maximum allowable concentration for chlorinated phenols as: 0.1 µg L−1 in drinking water and 200 µg L−1 in wastewater [12].
Conventional approaches for the removal of chlorinated phenols [15].
The current trend in wastewater treatment has moved from simple phase transfer to the destruction of pollutants such as by advanced oxidation processes (AOPs). These techniques involve reactive free radical species for non-selective mineralisation of organic compounds to harmless end products. The AOPs are generally based on the generation of the hydroxyl radicals (.OH), which have a great oxidation power, thus can almost oxidise all organic compounds to carbon dioxide and water [15]. There are several methods for generating hydroxyl radicals, e.g. Fenton-based processes, UV-based processes, Ozone-based processes and Photocatalytic processes [16,18–20].
In spite of the advantages of AOPs, there are several limitations in their use: (a) costs may be higher than competing technologies because of energy requirements, (b) harmful intermediates may be formed, (c) pre-treatment of the wastewater may be required to minimise cleaning and maintenance of UV reactor and quartz sleeves, (d) handling and storage of ozone and hydrogen peroxide require special safety precautions and (e) major challenges for the photocatalytic process are catalyst deactivation, slow kinetics, low photo efficiency and unpredictable mechanism [21]. However, AOPs are still more effective than the other techniques for wastewater treatment containing toxic and persistent pollutants [15].
Recently, application of titanium dioxide photocatalytic oxidation technology in environmental remediation has gained considerable attention as a cheap and clean alternative, though there are still various challenges to be resolved for its commercialisation. A high number of organic compounds (dyes, drug residues, pesticides and herbicides) have been eliminated from aquatic environment via TiO2 photocatalysis [22–25]. Complementary to the reviews published before, this review only concentrates on the progress of TiO2 photocatalysis over the past five years, thus providing a comprehensive view of the very recent progress in this field.
Basic principles of TiO2 photocatalysis and applications
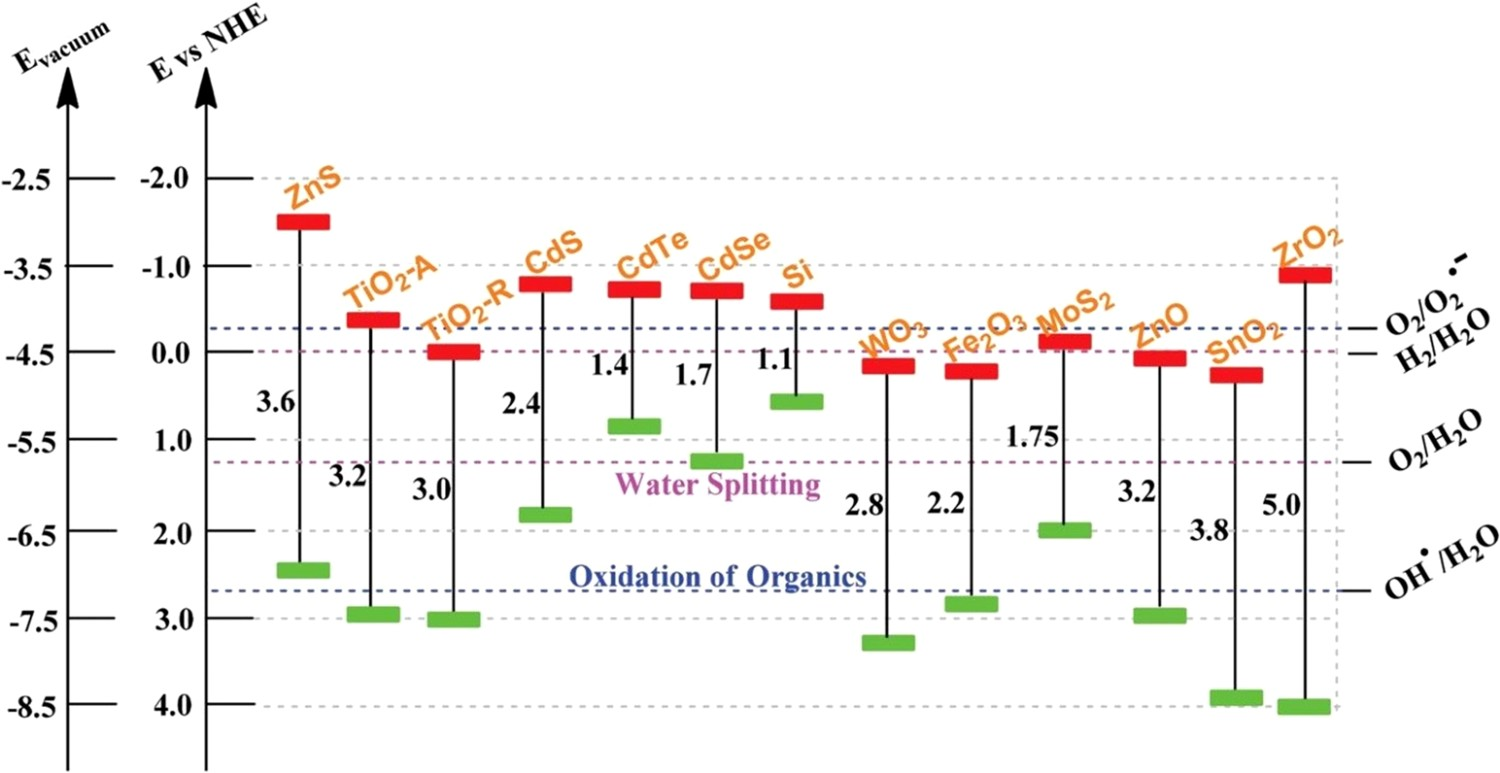
This review addressed the background in photocatalysis, and the removal of wastewater contaminants, via irradiation of semiconductors (e.g. nano-TiO2) with a light of energy higher than its bandgap energy (Figure 2). This is the first step in photocatalysis. Subsequently, the excitation of photogenerated electrons into the conduction band takes place, leaving behind energetic photogenerated holes in the valence band. Oxidation reaction is initiated in the valence band, while reduction reaction is initiated in the conduction band of the semiconductor. This requires the semiconductor to have proper band alignment required for organic pollutant decomposition (Figure 3), e.g. the conduction band minimum (CBM) should be higher (more negative in potential) than the redox potential for oxygen reduction to superoxide radicals, and its valence band maximum (VBM) should be lower (more positive in potential) than the redox potential for water or hydroxyl ion oxidation to hydroxyl radicals [13,27,28]. Diagram illustrating the principle of semiconductor photocatalysis for wastewater treatment. Diagram illustrating the bandgap energy, VB and CB positions of reported efficient semiconductors. Adapted from Wu et al. [26]. Copyright 2015, Royal Society of Chemistry. Reprinted with permission.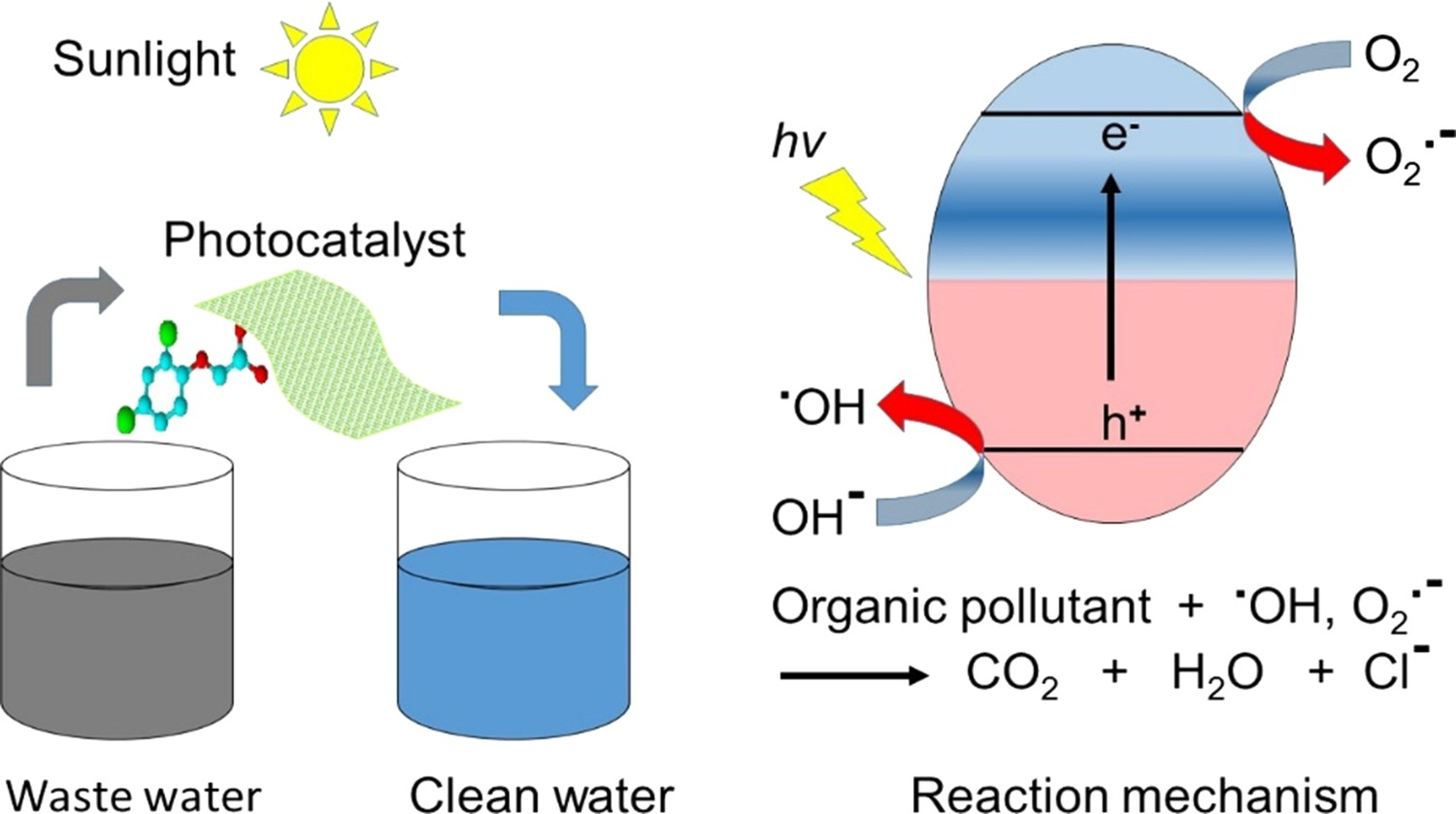
TiO2 as a photocatalyst
One of the most important aspects of environmental photocatalysis is the availability of an efficient photocatalyst, which should have several respects. For example, it is relatively inexpensive, physically and chemically stable, and non-toxic. Also, the band alignment in such a photocatalyst (Figure 3) makes the photogenerated holes to be highly oxidising. In addition, photogenerated electrons have reduction potential enough to produce superoxide radical from oxygen [26,29,30]. TiO2 is the typical material meeting these criteria. Zhang et al. [31] and Hirakawa and Nosaka [32], based on experimental evidence, proved that the conduction band electrons in TiO2 have the potential to generate superoxide radicals (O2
Challenges with TiO2 application in water treatment
The most studied phases of TiO2 are anatase and rutile. The anatase phase is known to display higher photocatalytic activity than the rutile phase. The low activity observed in rutile (E
g = 3.0 eV, λ = 413 nm), despite its lower bandgap energy than anatase (E
g = 3.2 eV, λ = 388 nm), is attributed to the rapid rate of electron–hole recombination. Also, the CBM of anatase is about 0.2 eV higher than that of rutile (Figure 3). This implies that the conduction band electrons in anatase are more reductive than those in rutile [33,34]. However, the inefficient utilisation of visible light and high recombination rate of photoelectrons and holes, that drastically reduces quantum efficiency, are the major drawbacks of photocatalytic application [28,35]. The challenges with TiO2 as a potential photocatalyst for the removal of organic pollutants in wastewater are summarised in Figure 4. Limitations in TiO2-based photocatalytic processes for the degradation of organic pollutants. Adapted from Dong et al. [34]. Copyright 2015, Elsevier. Reprinted with permission.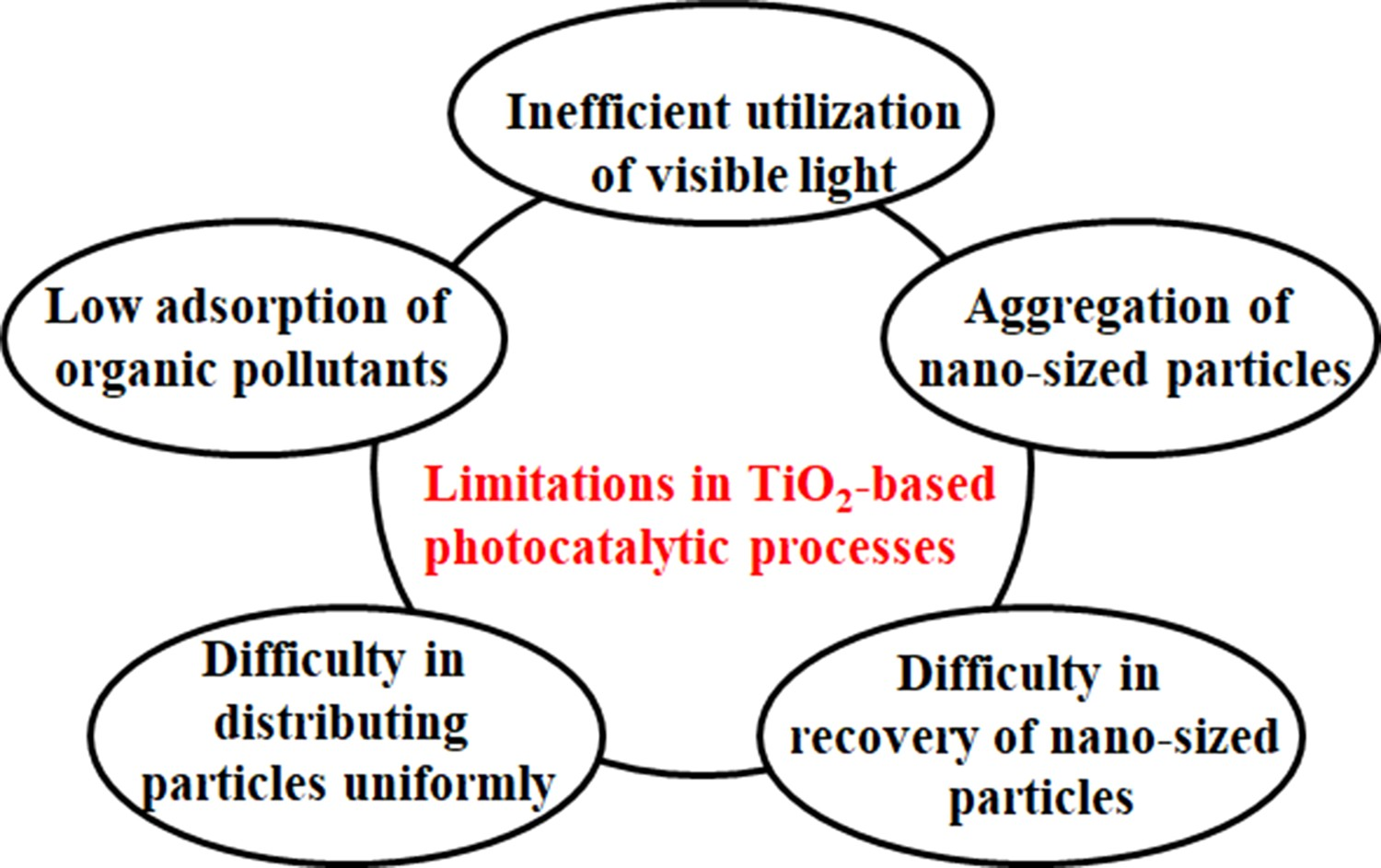
Visible light activity
The ultraviolet (UV) region only accounts for <5% of the entire solar spectrum, which TiO2 is responsive to. The strategies used in extending the optical absorption of TiO2 into the visible region include, metal-doping, non-metal doping, dyes sensitisation and direct reduction of TiO2 (e.g. hydrogenation, chemical reduction and electrochemical reduction). Three of the methods will be addressed here.
Dispersibility
TiO2 particles suffer from severe aggregation during photocatalytic application, which will reduce their active sites and light-harvesting capability. Since photocatalytic degradation occurs at the surface of TiO2, mass transfer limitations need to be reduced for its effective application in water treatment [61,62]. Two possible approaches employed are:
Separability
Recovery of fine particles of TiO2 from solution is a huge challenge in suspension systems. Thus, the recovery and reusability of TiO2 is required before the technology can proceed beyond lab scale to industrial application. Two potential approaches applied are:
Adsorption capacity
The photocatalytic activity of TiO2 is also dependent on its adsorption capacity for the pollutants in aqueous solution. Several approaches have been used to overcome the low adsorption capacity of TiO2. Modification of TiO2 with some chelating ligands (arginine and salicylic acid) and carbon nanomaterials (graphene, carbon nanotubes and activated carbon) have been reported to enhance the adsorption of organic water contaminants [77–79]. With respect to doping with carbon-based nanoparticles, the higher adsorption capability and better photocatalytic activity were reported to be related to their high surface area, high conductivity and high visible light absorption intensity [54,55].
Photocatalytic degradation of chlorophenols
Various studies show that the rate of degradation of CPs is dependent on, solution pH [80–84] and molecular structure – especially the number of substituted Cl groups and their positions on the benzene ring relative to the hydroxyl group [85–87]. It is well known that the initial degradation rate of most organic pollutants with TiO2 and other photocatalysts follows the pseudo-first-order reaction model [85,88–91]. Table 2 summarises the recent findings (mainly over the past five years) on the application of TiO2-based photocatalysts for the photocatalytic degradation of CPs in aqueous solution.
Progress on TiO2 photocatalysis for the degradation of chlorophenols over the past five years.
CTAB, cetyl trimethylammonium bromide; NTs, nanotubes; RGO, reduced graphene oxide; Pc, phthalocyanine; GO, graphene oxide; POM, poly-oxo-tungstate.
Sinirtas et al. [94] prepared a binary oxide catalyst (V2O5/TiO2) using solid-state dispersion method and evaluated the photocatalytic activity for 2,4-dichlorophenol degradation under UV–visible light irradiation. Photocatalytic degradation efficiencies of 55%, 60%, 70% and 85% were achieved in 30 min with TiO2 (synthesised), commercial TiO2 (P25), V2O5 and V2O5/TIO2, respectively. The effect of surfactant additives (CTAB, HTAB and PVA) on the photocatalytic activity of the binary oxide catalyst was also investigated. Under similar operating conditions, the (V2O5/TiO2/CTAB) sample displayed the highest activity (complete degradation in 30 min), while the samples prepared with HTAB and PVA recorded degradation efficiencies of ∼92% and ∼55%, respectively. The enhanced photocatalytic performance of V2O5/TiO2/CTAB was attributed to an appropriate pore distribution and high separation efficiency of photoinduced charge carriers. However, the photochemical stability of V2O5/TiO2/CTAB was not reported.
Photocatalytic reduction of heavy metals
Hexavalent chromium
The discharge of hexavalent chromium (Cr(VI)) from industrial processes, such as electroplating, leather tanning, metal fabrication, paint and pigment production into the aquatic environment has raised serious concerns over the potential water pollution issues [111,112]. In this regard, the reduction of toxic and carcinogenic Cr(VI) to the less harmful Cr(III) is imperative to mitigate their adverse effects towards biological and environmental systems. Over the years, photocatalytic reduction of Cr(VI) by TiO2 photocatalysts has been regarded as one of the more promising treatment alternatives owing to its direct one-step photocatalytic reduction of Cr(VI) under UV light irradiation [113–115]. Table 3 summarises the recent findings (mainly over the past five years) on the application of TiO2-based photocatalysts for the photocatalytic reduction of aqueous-based Cr(VI).
Recent studies on TiO2-based photocatalysts for photocatalytic reduction of Cr (VI).
TH, thiourea; PAN, polyacrylonitrile; CNT, carbon nanotubes; SA, sodium ascorbate; PVA, polyvinyl alcohol; CD, carbon dots.
In order to develop highly efficient TiO2-based photocatalysts with extended absorption into the visible light spectrum, the introduction of dopants into morphological-modified TiO2-based photocatalysts has been proven as an effective strategy. The introduction of dopants could hinder the rapid recombination rate of electron–hole pairs and narrowing the wide bandgap of TiO2 photocatalysts and hence, improving its visible light photocatalytic activity. Lu et al. [135] optimised the amount of vanadium (V) dopant on TiO2 nanosheets since this sheet-like structure possesses higher exposed surface areas for Cr(VI) adsorption. It is noteworthy that the introduction of V dopant resulted in the reduction of TiO2 bandgap leading to a high visible light photoactivity, and the co-existence of RhB and Cr(VI) greatly improved photocatalytic degradation kinetics as Cr(VI) is an efficient scavenger of photogenerated electrons. Besides, the V-doped TiO2 nanosheets could effectively trap photogenerated electrons and introduce a higher density defect (Ti3+), which are essential to enhance the effective separation of photogenerated charge carriers and high quantum efficiency. They extended their works in another study by synthesising TiO2 nanosheets loaded with Mn x O y and CoO x nanoparticles for simultaneous photocatalytic reduction of Cr(VI) and decontamination of organic compounds [136,137]. Similarly, the introduction of Mn x O y and CoO x nanoparticles resulted in a considerable enhancement of visible-light photoactivity, formation of active electron centres and the generation of Ti3+ density defects.
Recently, Xue et al. [138] reported the rational design of an interfacial heterojunction photocatalyst between 2D BiOBr nanoplates and 1D rutile TiO2 nanorods. Figure 5(a) shows the synthesis route of the heterojunction photocatalyst in which the 1D TiO2 nanorods are first fabricated using the molten salt flux method followed by the bonding between positively charged alkoxide complexes, Bi-(OCH2CH2OH)2+ and negatively charged TiO2 nanorods via solvothermal reaction. This eventually leads to the nucleation of BiOBr nanoplates, which are later being vertically embedded with TiO2 nanorods to synthesise the heterojunction photocatalyst (i.e. Figure 5(b)). The HRTEM image (Figure 5(c)) showed the interplanar spacings of 0.230 and 0.248 nm are well corresponded to the {200} and {101} facets of the rutile phase of TiO2, respectively. While the measured 0.277 and 0.353 nm fringes of BiOBr nanoplates can be assigned to the {110} and {101} planes of tetragonal BiOBr, respectively. Additionally, the rectangular spotty electron diffraction pattern can be clearly observed in Figure 5(d), indicating the single-crystalline rutile phase TiO2 in this heterojunction photocatalyst. More importantly, the photocatalytic results (Figure 5(e)) indicated that the TB-2 (i.e. the sample with a molar ratio of Ti to Bi, n(Ti/Bi) = 2:1) exhibited the highest photoreduction rate of Cr(VI) with an apparent rate constant of 0.025 min−1 under visible light illumination. This is ascribed to the formation of an interfacial junction between BiOBr and TiO2, which provides directional transfer nanochannels for the migration of photoinduced charge carriers and thus, enhancing quantum efficiency and reducing the charge recombination as evidenced by the PL spectra in Figure 5(f) leading to highphotoactivity as per Figure 5(g). Interestingly, similar theory on enhanced physisorption and charge transfer of PANI/MnO2/TiO2 nanocomposite shows the importance of TiO2 heterojunctions to improve the visible light-driven photoreduction of Cr(IV) [139]. (a) Schematic illustration for the synthesis route of BiOBr nanoplates/TiO2 nanorods heterojunctions; (b) TEM image; (c) HRTEM image; (d) SAED image of an individual BiOBr/TiO2 sample; (e) reaction kinetics for photocatalytic reduction of aqueous Cr(VI) over different samples under visible light illumination; (f) PL spectra with an excitation wavelength of 325 nm and (g) transient photocurrent responses under visible light irradiation for all samples. Adapted from Xue et al. [138]. Copyright 2017, American Chemical Society. Reprinted with permission.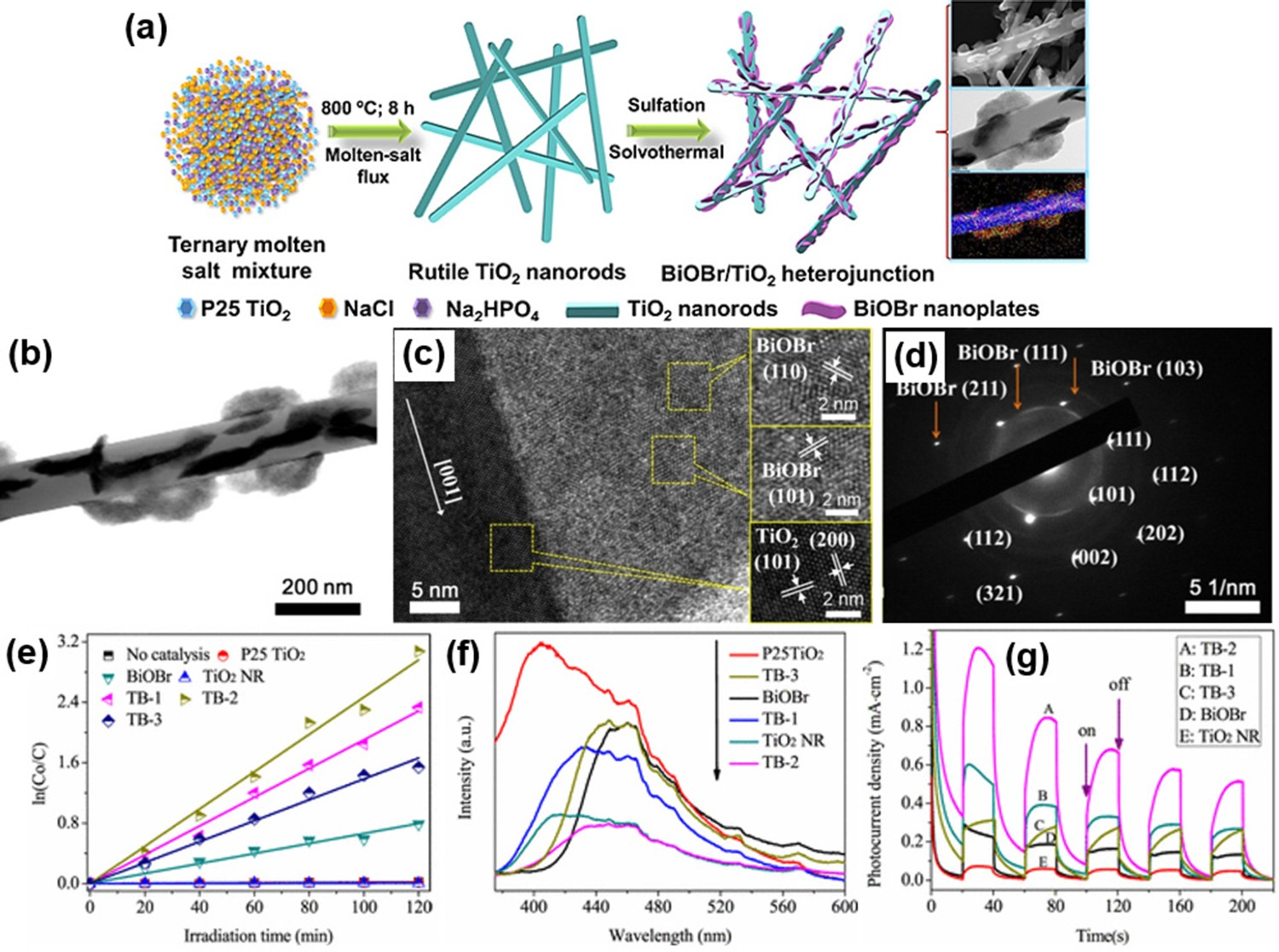
Pentavalent arsenic
Owing to their carcinogenicity and mutagenicity nature, arsenic (As) compounds contamination in natural water can present a major threat to the environmental system and public health even in a very low concentration [140–142]. Previous studies are mainly focusing on the oxidation of acutely toxic arsenite (As(III)) to arsenate (As(V)); however, the high adsorbability of As(V) on soil could adversely influence bacteria and microscopic fungi [143,144]. Particularly, this section presents the recent findings on the photocatalytic reduction of aqueous As(V) by using TiO2-based photocatalysts in detail. Previously, Levy et al. [145] have applied pristine TiO2 photocatalyst to investigate its photoreduction performance over As(V) under UV light in deoxygenated aqueous suspension. They found that the direct reduction of As(V) to As(IV) by accepting photogenerated electrons at conduction band was not thermodynamically possible. Nevertheless, the addition of sacrificial electron donors (i.e. alcohols or carboxylic acids) in the photocatalytic medium could lead to the production of strongly reductive radicals and thus, promoting the indirect reductive mechanism of As(V).
Similarly, the deposition of transition and precious metals onto TiO2-based photocatalysts has been widely attempted in order to extend their light absorption into the visible light spectrum as well as serving as active centres to inhibit the recombination of electron–hole pairs [34,146]. For instance, Andjelkovic et al. [147] prepared a Fe-doped TiO2 photocatalyst via microwave-assisted hydrothermal method in order to provide large capacity for the adsorption and removal of As(V). They found that the adsorption efficiency of As(V) was proportional to the amount of Fe(III) dopants because it tends to provide a higher specific surface area which directly responsible for faster As(V) adsorption rate. Isotherm studies showed that the Langmuir model provides the best fitting for the adsorption data of As(V) better and the Fe-doped TiO2 photocatalyst attained the maximum adsorption capacity of 17.35 mg g−1 As(V). In another study, Liu et al. [148] deposited Fe onto morphological-modified titanate nanotubes (TNTs) for sequential As(III) oxidation and As(V) adsorption. The resultant Fe-TNTs exhibited a good adsorption performance for As(V) at acidic medium (i.e. adsorption capacity of ∼49.1 mg g−1 at pH 3; removal efficiency of 98.2%) due to the opposite surface charges between Fe-TNTs and As(V) anions. It was found that there are two different forms of deposited Fe that could improve the photocatalytic activity through: (1) Fe3+ ions that locate in the interlayers to act as temporary electron- or hole-trapping sites, and (2) attached α-Fe2O3 that facilitates the charge separation from TNTs to reduce recombination of electron–hole pairs.
A more recent study by Gao et al. investigated the performance of a 3D cake-like COOH functionalised anatase (TiO2@C) (Figure 6(a,b)) for simultaneous Cr(VI) reduction and As(III) oxidisation followed by the adsorption of As(V) [149]. The simultaneous conversion of Cr(VI) and As(III) was significantly enhanced from 14.5% to 61.8% and 38.5% to 92%, respectively, in the exposure system of TiO2@C/As(III)/Cr(VI). It is worth mentioning that As(V), which was oxidised from As (III), has also been effectively removed under acidic condition, as shown in Figure 6(c). Moreover, they also reported that the removal rate of As(V) was essentially relied upon the pH, TiO2@C dosage (Figure 6(d)) and initial concentration of Cr(VI)/As(III) solution (Figure 6(e)). Before the reaction, Figure 6(f) shows the full XPS spectrum of TiO2@C containing carbon groups, oxygen and titanium elements. The characteristic peaks of As(III) (45.1 eV) and As(V) (46.1 eV) were detected on TiO2@C with the proportion of 48.5% and 51.5%, respectively, after exposure to Cr(VI)/As(III) solution (Figure 6(g)) [150]. As a result, the removal performance of As(III)/Cr(VI) using TiO2@C was improved through co-adsorption of As(III) and Cr(VI) ions, accelerated electrons transfer from As(III) to Cr(VI) initiated by carboxyl groups on TiO2, and larger binding sites provided by TiO2 surface. (a) TEM and (b) HRTEM images of TiO2@C; (c) As(V) production in various solution (As(III) concentration: 6.5 mg L−1; Cr(VI) concentration: 5.5 mg L−1; TiO2@C concentration: 0.3 g L−1; pH: 3.0; time: 90 min); (d) effect of TiO2@C dosage on removal of total As (As(III) concentration: 5 mg L−1; Cr(VI) concentration: 5 mg L−1; TiO2@C concentration: 0.1, 0.2, 0.3, 0.5, 0.7 g L−1; time: 90 min; T: 20 ± 2°C pH: 3.0); (e) effect of Cr(VI) initial concentration on adsorption of As(V) (As(III) concentration: 6.5 mg L−1; Cr(VI) concentration: 0–7 mg L−1; TiO2@C concentration: 0.3 g L−1; time: 90 min; T: 20 ± 2°C pH: 3.0); XPS spectra of (f) pure TiO2@C full spectrum before reaction; and (g) As 3d spectra of TiO2@C after exposed in As(III)/Cr(VI) solution. Adapted from Gao et al. [149]. Copyright 2018, Elsevier. Reprinted with permission.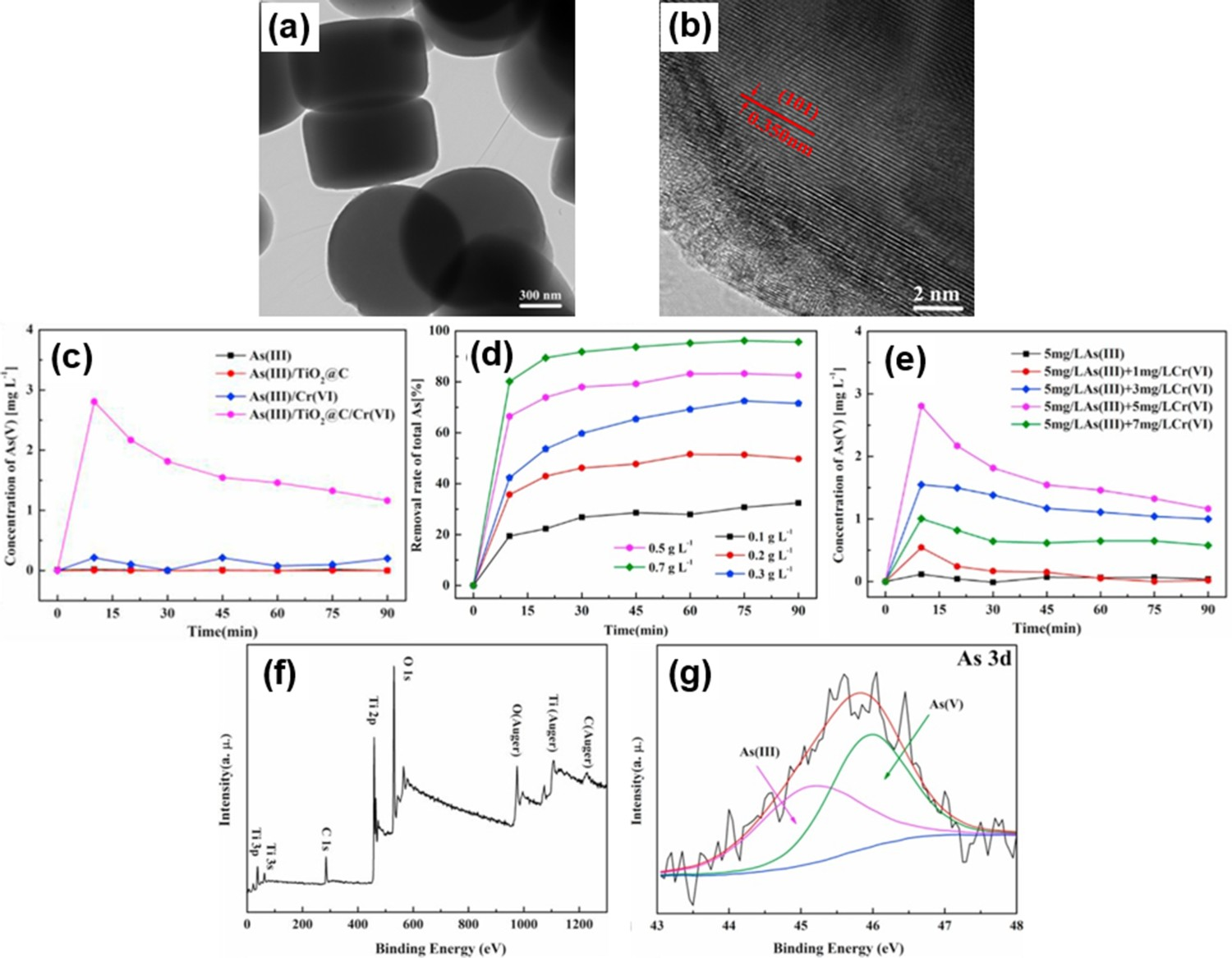
Outlook
After decade’s development on different photocatalysts, TiO2 is still widely recognized as the best semiconductor material for real application in photocatalytic water purification technology due to its low toxicity, high stability and availability. However, TiO2 is confronted with some limiting factors such as poor visible light absorption, low photonic efficiency and low adsorption capacity. In this review, we have highlighted various strategies for TiO2 modification (such as ion doping, cocatalyst decoration and heterojunction formation) not only to improve its light absorption but also to enhance charge separation. Furthermore, this review summarises the recent application of TiO2-based photocatalysts for the catalytic oxidation of CPs and reduction of heavy metal ions such as chromium and arsenic in aqueous solution, which are two typical while the most challenging topics.
The co-doping of TiO2 with a metal and non-metal is receiving considerable attention and proved to be more effective than single ion doping towards efficient photocatalytic degradation of organic contaminants in water. This improvement was attributed to an extension in the visible light absorption capacity. Apart from the suppression of photogenerated electron–hole recombination rate, the bandgap energy is also largely decreased due to the introduction of impurity energy levels above the valence band and below the conduction band. The solution pH is also an important operating variable in photocatalytic degradation of organic contaminants. In most TiO2-based studies, it was shown that acidic pH range (4–6) is the optimum for the efficient degradation of organic substrates in water. This was attributed to the positively charged surface of TiO2, favouring increased adsorption capacity for organic compounds. It is also worth noting that the fabrication of different bandgap alignments (i.e. Type I, II and III) and Z-scheme heterojunctions was proved to efficiently enhance charge separation of photogenerated electron–hole pairs and reduce the charge carriers recombination rate, beneficial to photocatalytic water treatment.
Reduction of a reactive, high oxidation state of metal ion to a lower oxidation state and less toxic species by TiO2-based photocatalysts has been regarded as an effective approach for heavy metal removal from aqueous environment. Upon UV light irradiation, the anodic and cathodic redox reactions are initiated, whereby the water oxidation by photoholes is involved and thus transfer of photoexcited electrons from the conduction band of TiO2 photocatalysts to the adsorbed heavy metal ions takes place. Acceptance of electrons into the metal atom (reduction) could suppress its reactivity and thus, the toxicity effect. Additionally, it is also showed that the equilibrium adsorption capacity of TiO2 photocatalysts is higher at lower pH medium, signifying a higher density of adsorption sites for heavy metals. In order to extend the functionality and applicability of TiO2 photocatalysts in visible light range, several material modification strategies such as morphological alteration, dopants introduction and heterojunction are proved to be effective to separate the photogenerated charge carriers and therefore, leveraging the photon responsiveness of pristine TiO2 photocatalysts.
For both photocatalytic organic contaminants decomposition and heavy metal ions reduction, cocatalysts play a proved key role, which not only help charge separation but also lower the reaction barrier. The so far best cocatalyst is still noble metal, e.g. Pt. To develop a low-cost cocatalsyt is also important for TiO2 photocatalyst application. Based on the above discussion, we proposed that the future commercial and large-scale application of photocatalysis in water treatment is feasible if the following could be solved: The preparation of low cost, reproducible and environmental-friendly cocatalyst/TiO2-based photocatalysts that can be photo-activated by solar light (not artificial UV light). The efficient recovery and reuse of the photocatalysts during water purification via immobilisation on flexible and highly stable support materials. The design of photocatalyst materials capable of selective photocatalytic oxidation of organic pollutants or reduction of heavy metal ions in water. The design of efficient photocatalytic reactor with satisfactory efficiency in acidic, neutral and basic media, reflecting the real environment of the wastewater.
The fundamental understanding underlying the photochemical process is still far behind discovery of the photocatalysts.
The photocatalytic mechanism of a single photocatalyst is relatively simple and to some extent straightforward. The photochemical reaction involves the excitation of electrons to the conduction band of a photocatalyst while leaving positive holes at the valence band upon light illumination whereby the photon energy (hv) is greater than or equal to the bandgap energy of photocatalyst. Studies on charge carrier dynamics have been successfully investigated over the years using time-resolved analytical techniques (time-resolved absorption spectroscopies) to observe the generation, trapping, and transfer of electrons and holes during photocatalytic processes [151,152]. The generation of hydroxyl radicals and superoxide radicals in aqueous systems have been widely established in the literature through spin trapping ESR experiments and fluorescence spectroscopy. The next step is still unclear. The discovery of hydrogen peroxide in oxygen-free systems justifies the generation of hydroxyl radicals by valence band holes, though hydrogen peroxide can also be generated through oxygen reduction. It is still difficult to distinguish the hydroxyl radicals generated from both different pathways, that is, hole oxidation of adsorbed water/hydroxyl ions or through oxygen reduction. Different hydroxyl radical production pathway will provide a different route for improvement of the photocatalytic activity. More importantly, although the time-resolved IR has been successfully employed to other fields, e.g. homogeneous photocatalysis, it has not been used in water treatment. By using this cutting edge technology, the detailed reaction pathway and the rate-determining step could be elucidated, which can also indicate the factor to mitigate the toxic byproducts production during photocatalytic contaminants’ treatment and heavy metal cation reduction. In addition, the development of highly efficient photocatalysts is growing intensively, the photocatalytic mechanism within the complex photocatalyst systems and compositing structures is also important while more challenging. These time-resolved spectroscopies should be applied to these complicate systems to understand the reaction pathway. Overall, a practical photocatalytic system for water treatment still requires substantial efforts.
Footnotes
Acknowledgements
A.S. and J.T. thank the Leverhulme Trust (RPG-2017-122), EPSRC (EP/S018204/2) and Royal Society-Newton Advanced Fellowship grant (NAF\R1\191163). All authors are thankful to the other Royal Society-Newton Advanced Fellowship grant (NA150418).
Disclosure statement
No potential conflict of interest was reported by the authors.