
Abstract
Recent research has identified specific molecular mechanisms that might account for impaired learning in particular intellectual disability syndromes. These and other findings raise the possibility that targeted drug treatments might be developed to enhance learning in subjects with intellectual disability. This review considers strategies for developing treatments, and identifies critical issues that will need to be considered in such programmes.
Intellectual disability (ID) is characterized by limitations in intellectual functioning and adaptive behaviour [1]. Psychometric testing in subjects with ID identifies slower rates of learning across multiple cognitive domains compared with subjects with normal intellectual functioning [2, 3]. This can be illustrated as differences in learning trajectories (rate of learning over time) between individuals with ID and those with normal intelligence (Figure 1). Over time, these differences lead to progressively greater disparities in measures of cognitive development between ID and non-ID populations. Efforts to improve delayed learning trajectories in ID have focused on improved socialization [4, 5], and enhanced educational and training input, especially in childhood and adolescence [6–8]. Despite these interventions, however, it has not been possible to increase learning trajectories in subjects with ID into a normal range.
Examples of learning trajectory (rate of learning as a function of age) for (A) an individual with normal intelligence; and (B) an individual with intellectual disability. Learning enhancement would be expected to produce an upwards shift in this curve (C).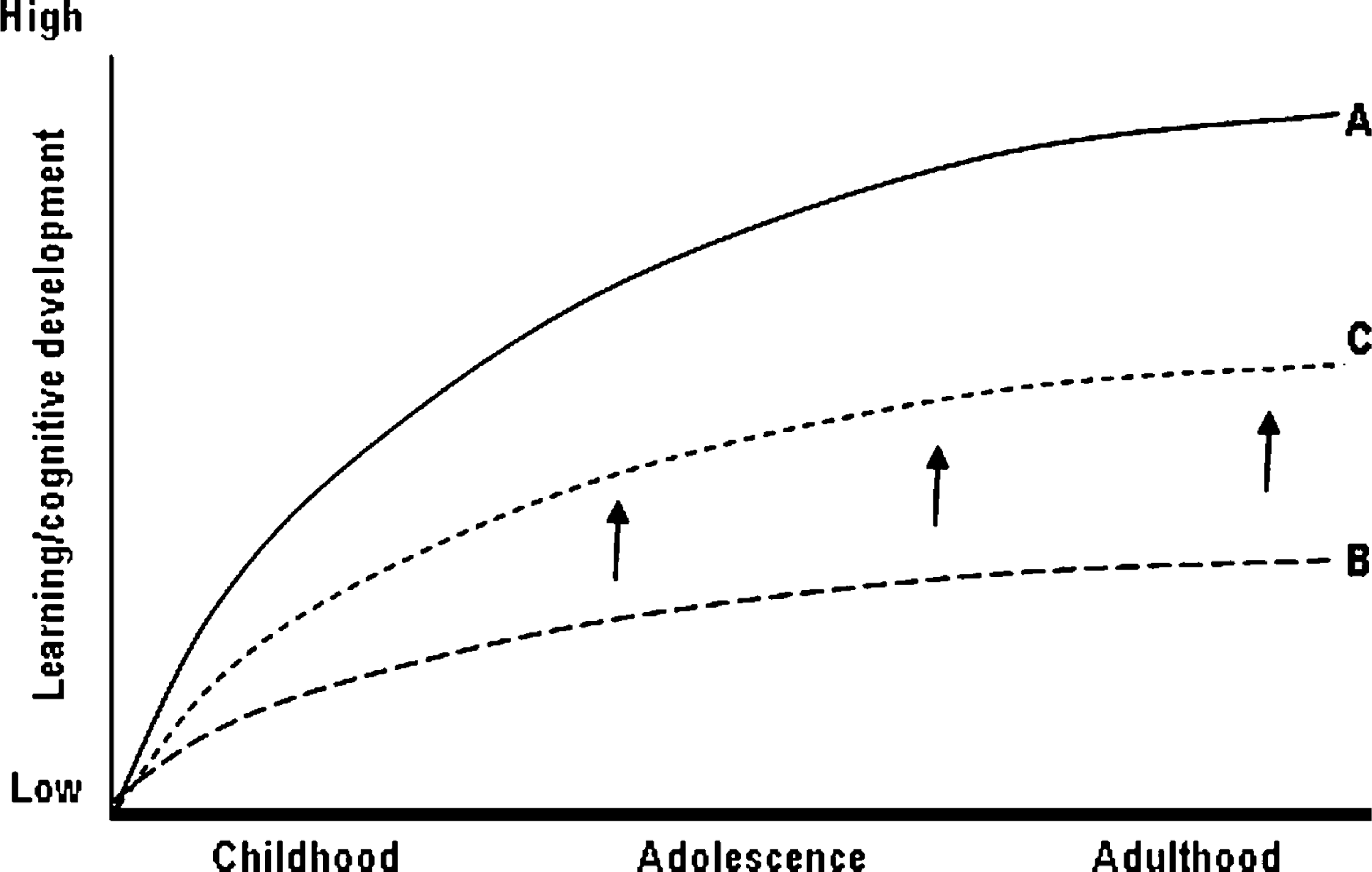
This article provides a high-level overview of several pharmacological approaches that could improve learning in subjects with ID. Such approaches are well-established for certain dementias and are under development for other disorders (e.g. schizophrenia), based on targeting identified or hypothesized pathological mechanisms. In the last decade, advances have been made in identifying genes associated with cognitive performance [9], and >300 gene mutations have been identified as being associated with cognitive dysfunction [10, 11]. It has been more challenging, however, to develop treatments based on these discoveries. Recent studies have identified specific molecular mechanisms that might affect learning in certain ID syndromes, and offer the potential for targeted treatments to enhance learning. There are also other approaches that may be of value, based on normalizing changes in brain biochemistry, or through influencing neuroregulatory processes, but these are less well developed.
Improved learning via targeting specific molecular mechanisms
There are two recent examples of this approach, which is based on identifying pathological mechanisms that may affect learning in certain syndromes.
In Down syndrome (DS), impaired learning may be caused by overexpression of a specific type of inhibitory potassium channel (G-protein coupled inwardly rectifying potassium channel or GIRK2 [12]), which is located on chromosome 21. This channel is involved in maintaining neuronal membrane excitability. Overexpression of this channel could produce excessive neuronal inhibition and thus negatively affect learning and memory. This channel is regulated by several neurotransmitters and hormones through G protein-coupled receptors (α2-adrenergic, γ-aminobutyric acid (GABA)-B, serotonin-1A, dopamine-D2,3,4, µ-opioid, metabotropic glutamate receptors (mGluRs), and others). A mouse model of DS has been developed, and has shown excessive sensitivity to the effects of the GABA-B agonist baclofen, consistent with increased GIRK2 activity [12]. Based on this finding, we predict that GABA-B antagonists [13] might be expected to reverse this excessive inhibition, and have positive effects on learning and memory in DS. Given that other receptors are associated with GIRK2, potentially other agents might also modulate learning.
Fernandez et al. have subsequently tested the effects of several GABA-A agonists and showed that these enhanced learning in the same mouse model [14]. A criticism of the latter study is that GABA-A does not appear to be associated with GIRK2, and thus the learning enhancements reported may be due to non-specific effects on neuronal excitability. A second concern is that GABA-A agonists have narrow therapeutic indices, with the potential to produce seizures. Even at lower doses, these agents are likely to be anxiogenic [15].
Fragile X syndrome (FXS) results from shutting off transcription of fragile-X mental retardation (FMR) protein. This is an RNA-binding protein that modulates processes such as synaptic plasticity and dendrite maturation, processes centrally involved in learning and memory. FMR protein inhibits group 1 mGLuRs [16, 17], and thus neurodevelopmental changes in FXS may reflect increased activation of processes controlled by this group of receptors, and inhibition should be therapeutic. This has been studied in two ways.
Symptoms and other phenotypic changes in a mouse FXS model can be reversed using mGluR5 antagonists [18], or by cross-breeding FXS mice with those lacking copies of mGluR5 genes [19]. A pilot study in human subjects with FXS using the mGlu5 antagonist fenobam has now been published [20], using prepulse inhibition (PPI) as a biomarker. Abnormal PPI is a feature of animal models of FXS and human subjects with FXS [21, 22]. The prediction is that changes in PPI in human subjects with FXS would be a biomarker (positive indicator) of improvement in other aspects of FXS, including learning. This study gave single doses of fenobam 50–150 mg to 12 adult subjects with FXS, using an auditory PPI model [20]. PPI improvement of >20% was noted in six of 12 treated FXS subjects, but only in two of 13 untreated FXS subjects, and none of the 16 controls. Improved behaviour was noted in nine of 12 FXS subjects receiving fenobam [20]. Although PPI and behavioural end-points are not markers of improved cognitive performance, these positive human findings indicate that some of the features of FXS can be modulated. Improvements in learning and memory should be included in subsequent trials, but changes may need to occur over a different (longer) timeframe than the aforementioned behavioural assessments.
Other approaches
Reported changes in excitatory/inhibitory neurotransmitter levels in DS brain tissue
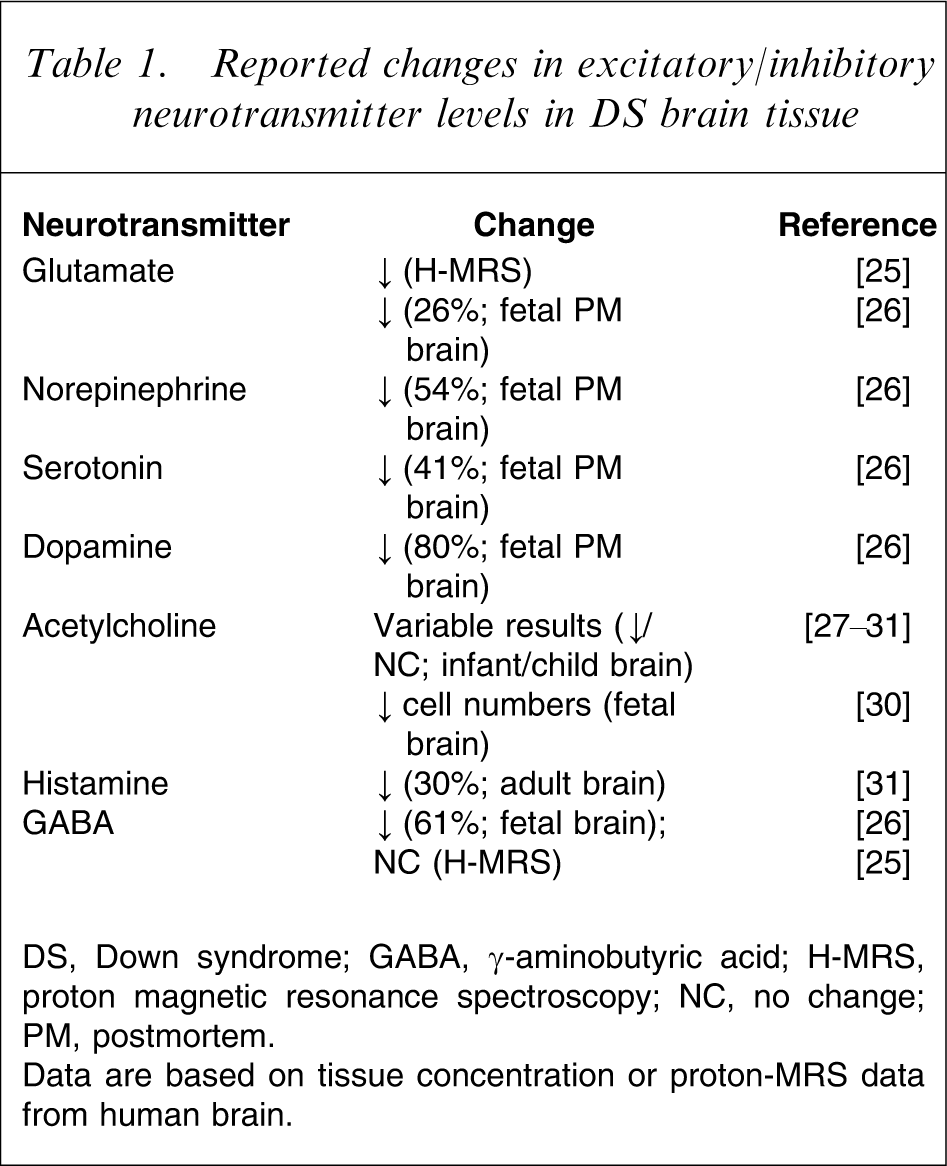
DS, Down syndrome; GABA, γ-aminobutyric acid; H-MRS, proton magnetic resonance spectroscopy; NC, no change; PM, postmortem.
Data are based on tissue concentration or proton-MRS data from human brain.
An alternative rationale for considering interventions that affect central neurotransmitters is based on the neuropharmacology of cognition and arousal. In general, agents that enhance activity of excitatory transmitters or reduce inhibitory neurotransmission might be expected to facilitate cognition and/or arousal [33]. For example, in DS, levels of excitatory or modulatory neurotransmitters are generally decreased (Table 1), and theoretically drugs that increase these might be expected to enhance learning. Positive findings have been reported for cholinesterase inhibitors in children and young adults with DS [34–36], but a more recent series of larger studies was terminated due to insufficient evidence of efficacy [37]. Studies to assess the cognitive effects of methylphenidate (a monoamine-releasing agent) in subjects with comorbid ID and attention-deficit–hyperactivity disorder (ADHD) report improved performance on a number of tasks in children with mild ID and ADHD [38, 39], but it is unclear whether benefits are observed in subjects with more severe ID [38].
A third approach is based on the hypothesis that learning and memory deficits are due to shared pathological mechanisms associated with dysfunctional neuronal development. One candidate is the Rho GTPase family, which function as molecular switches [40, 41]. When bound to guanosine triphosphate (GTP), these proteins facilitate downstream signalling of multiple pathways, regulating neuronal activities such as dendrite growth and morphology, organization of the neuronal cytoskeleton, and synaptic connectivity. Several genes associated with X-linked, autosomal and non-syndromic ID are involved in regulation of Rho GTPase signalling, and genetic abnormalities affecting this pathway may reduce neuronal ability to respond to environmental stimuli via, for example, axon growth, synapse formation and/or neuronal plasticity, and thus affect ability to learn. Although the concept of agents with positive cognitive effects in a broad range of ID disorders is appealing, at present it is still unclear which member or members of this family of enzymes would be important to regulate, and what type of regulation would be required. Furthermore, a key assumption of this approach (that mechanisms underlying cognitive delay are shared across disorders) still requires confirmation.
Discussion
There is now a considerable literature on genes associated with cognition [9], and on gene mutations associated with cognitive impairment [10, 11]. It has been challenging, however, to use this information to develop treatments that might improve cognition. Some approaches (e.g. gene therapy, in a mouse model of Rett's syndrome [42]) are interesting from a proof-of-concept perspective, but cannot be realistically considered for clinical use. Turning genetic leads into druggable targets is complicated by genetic heterogeneity, especially in non-syndromic ID [11], and developing potential treatments based on these targets has been difficult. Impaired learning and cognition have been thoroughly studied as pharmacological targets in disorders such as dementia and schizophrenia [42–44], therefore it is surprising that it has not been more extensively considered as a target in ID. The concept of developing specific pharmacological treatments for impaired learning is mentioned only once in the US National Institutes of Health's otherwise extremely detailed research plan for DS [45].
There are now plausible specific molecular targets associated with learning impairment in DS and FXS, which provide a scientific rationale for proof-of-concept trials to improve learning. It is much less clear if it will be possible to identify agents with broad efficacy across a range of ID syndromes, or in non-syndromic ID, and extensive additional work is still required to see if there are shared basic mechanisms associated with impaired learning, along with continued development and validation of animal models.
Key issues in implementing a research programme to assess effects of drugs on learning performance in ID

DS, Down syndrome; ID, intellectual disability.
Two additional factors need to be considered in relation to end-points and specific populations tested. First, ID of different aetiologies have cognitive profiles that differ in terms of patterns of strengths and weaknesses [48–50]. It is important to take into account established cognitive profiles of ID groups because intervention outcomes may have a differential effect depending on whether a specific task targets a cognitive strength or weakness [51]. Second, ID of different aetiologies may have different developmental trajectories in terms of cognitive functioning. For example, persons with DS over the age of 35 may develop cognitive decline and are at risk of developing early dementia [3, 48, 52]. Outcomes may, therefore, not be due to intervention but a by-product of differential developmental trajectories.
Further, motivational factors need to be considered [53]. Many children or adults with ID may find cognitive testing difficult and unrewarding [54]. This may not only affect performance on individual tests but also affect the number of subjects remaining on treatment [55]. Cognitive tasks that are engaging and within the range of the child's ability may assist with motivation, as well as intermittent reinforcement of effort throughout testing [52, 56]. Caregivers and subjects need to be provided with a clear rationale of the potential benefits of the study to encourage them to engage and remain in the study.
Additional issues that need to be considered in the development programme include the acceptability of the programme to the subjects and caregivers involved. Specifically, the costs and benefits of such treatments would need to documented and considered. For example, do the drugs show benefits for cognitive function but adversely affect behaviour, mood or tolerability [57, 58]? Programmes developed, therefore, need to include measures to ascertain unwanted or adverse side-effects of the medication (e.g. measures of behaviour, emotion, physiological measures).
Regarding specific drugs, appropriate work must be performed to characterize dose, duration of dosing, safety, and therapeutic index. Because much of the learning trajectory occurs in childhood and adolescence, it will be important to determine how early drugs should be started and for how long they should be used. It is possible that some of this work could be guided through studies in animal models. Extant studies already indicate age-related or dose-related adverse side-effects [57–59]. If multiple mechanisms to enhance cognition are identified, should drugs be used as monotherapy or combinations?
Addressing these points is critical if safe and effective treatments are to be developed to enhance cognition in subjects with ID. It is therefore of extreme concern that at least one group has started to advertise a ‘treatment protocol’ based on an undiscriminating review of some of the aforementioned findings, without any consideration of dose, combination, duration or safety issues (Changing Minds Foundation; http://www.changingmindsfoundation.org/home.htm).
In conclusion, recent studies have identified molecular targets in DS and FXS that are plausibly associated with impaired learning, and offer the potential for developing drug treatments to enhance learning in subjects with ID. If treatments are to be developed in an ethical and scientifically rigorous manner, there are substantial trial design and methodological issues to be considered.