
Abstract
The current paradigm for treating major depressive disorder (MDD) is initial treatment with monotherapy. Initial antidepressant monotherapy for MDD results, however, in only 25–35% remission rates even under research or enhanced care conditions [1]. For example, the Sequenced Treatment Alternatives to Relieve Depression (STAR∗D) effectiveness trial, a National Institute of Mental Health (NIMH)-funded study that recruited a large sample from both psychiatric and primary care, resulted in a remission rate of only 33% with citalopram as the initial monotherapy based on the modified intent-to-treat sample [2].
When an initial monotherapy fails, it is typically followed by switching to another monotherapy or adding a second treatment to the first medication (augmentation or combination therapy) [3]. In STAR∗D, those who did not reach remission (score ≤5 on the 16-item Quick Inventory of Depressive Symptomatology–Self-Rated (QIDS-SR16)) with or who were intolerant to citalopram [4–7] and moved on to a second monotherapy treatment (medication switch) [8] had a remission rate of approximately 20%. Those who moved on to a second treatment in which another medication was given in addition to the citalopram had a 30% remission rate (medication augment) [9]. The overall cumulative remission rate of STAR∗D patients after two medication treatment steps (either sequential monotherapies or monotherapy followed by augmentation/combination) was slightly over 50%.
Longer term studies with one or more monotherapies have produced low remission rates. For example, the Improving Mood-Promoting Access to Collaborative Treatment (IMPACT) trial examined older outpatients with MDD, dysthymia, or both and found that 12% reached remission after 12 months in treatment [10]. The Texas Medication Algorithm Project (TMAP) examined public sector psychiatric outpatients after 12 months in treatment and found that 11% reached remission for those in algorithmic care, and 7.8% reached remission for those who received treatment as usual [11–13].
When remission is achieved in acute treatment, longer term relapse/recurrence is common [14]. For example, of the STAR∗D patients who entered follow up from each treatment step, the proportion of participants who relapsed was higher if remission was not achieved at entry into follow up, and the greater the number of treatment steps needed to achieve remission, the higher the risk of relapse. Clearly, new treatment strategies to enhance the likelihood of remission, and to sustain it in the long run, are needed [3].
Theoretically, two antidepressant medications used together at the onset of treatment could increase remission rates over those found with monotherapy by (i) treating a broader range of patients (some remitting specifically on one or the other medication); (ii) creating additive or synergistic pharmacological effects such that each agent increases the efficacy of the other; and (iii) potentially increasing patient retention by increasing efficacy. The downsides of combining two antidepressant medications are greater cost, additive side-effect burden, risk of drug–drug interactions, and reduced patient adherence when two agents must be taken [3].
For the present study we chose to test the combination of venlafaxine-XR and aripiprazole for initial MDD treatment because venlafaxine-XR is a well-tolerated monotherapy. Atypical antipsychotic agents are widely prescribed as augmenting agents for treatment-resistant psychotic MDD and there is increasing evidence from uncontrolled and controlled clinical trials [15–20]. Aripiprazole was chosen from among the available atypical antipsychotic agents due to its ease of dosing (15-30 mg day–1); its minimal effect on weight and glucose regulation [3, 21–23]; its lack of cardiovascular side-effects including QTc prolongation; and open trial evidence that indicates its safety, tolerability and potential efficacy at 15–30 mg day–1[22, 24–27]. Aripiprazole is a dopamine D2 receptor partial agonist. This property renders it a functional agonist in certain parts of the brain with low dopamine activity such as the hippocampus, and a functional antagonist in other areas such as ventral striatum/nucleus accumbens with higher dopamine levels [28]. It is also a partial agonist at serotonergic 5HT1A receptors and an antagonist at 5HT2A receptors [28, 29]. Theoretically, these effects could complement the effects of the serotonin/norepinephrine re-uptake inhibitor venlafaxine-XR.
The primary objective of the present study was therefore to evaluate and assess the tolerability, side-effect profile, and rates of remission and response with the medication combination venlafaxine-XR and aripiprazole in the treatment for chronic or recurrent MDD (CRMD).
Methods
This effectiveness trial was conducted as part of the NIMH-funded Depression Trials Network, a national infrastructure for clinical research in depression that included the National Coordinating Centre (NCC) at the University of Texas Southwestern and the Data Coordinating Centre (DCC) at the University of Pittsburgh. The study was conducted at five clinical sites (Massachusetts General Hospital, Boston MA; University of Texas Southwestern Medical Centre, Dallas TX; University of North Carolina School of Medicine, Chapel Hill NC; Laureate Psychiatric Clinic and Hospital, Tulsa, OK; VA Medical Centre Tuscaloosa, AL; Vanderbilt University, Nashville TN), which were typical research clinics in academic centres. Clinical sites were identified based on the availability of depressed outpatients, clinicians, administrative support, and representative minority populations. The protocol was approved and monitored by the Institutional Review Boards at the NCC, DCC, and each participating academic centre. An independent, external Data Safety and Monitoring Board also oversaw the study.
Participants
The study enrolled participants, between 18 and 65 years of age, who met DSM-IV criteria for chronic (index episode >2 years) or recurrent non-psychotic MDD as documented by a checklist and by the Mini-International Neuropsychiatric Interview Plus (MINI Plus) [30]. Recurrent MDD was defined as three or more lifetime MDD episodes. Enrolled participants had to have the capacity to understand the study, signed a written informed consent, and were fluent in English. They also had to have a baseline score ≥14 on the 17-item Hamilton Rating Scale for Depression (HRSD17) [31, 32]. Clinician approval was required, indicating that initial treatment with antidepressant medications in combination was clinically appropriate and safe, especially for those participants who were taking other non-excluded concomitant medications. Participants were reimbursed up to US$80 for the extra time spent filling out forms and for travel expenses.
Participants were excluded if they had a lifetime history of bipolar disorder (I, II, or not otherwise specified (NOS)), schizophrenia, schizoaffective disorder, psychosis NOS, anorexia nervosa or bulimia nervosa, a current primary obsessive–compulsive disorder, a history of clear-cut intolerability to either medication used in the combination under study or lack of response to an adequate trial of the combination in the current MDD episode (lack of response to one of the medications was acceptable). The study also excluded patients who had received and not responded to seven or more sessions of electroconvulsive therapy in the current episode and women who were sexually active and not using adequate contraception, or who were pregnant or breast-feeding. Additional exclusion criteria included any general medical condition or concomitant medication(s) that contraindicated the use of the study medication combination; the need for immediate hospitalization for substance/alcohol detoxification, or treatment or immediate hospitalization for psychiatric disorder; the need for antipsychotic medications or mood stabilizers; or the use of exclusionary medications (anticonvulsant medications, central nervous system stimulants, or antidepressant medication used for the treatment of depression or other purposes such as smoking cessation). Patients taking thyroid medication for hypothyroidism could be included if they had been stable on the medication for 3 months, as could patients who were in a modality of psychotherapy that was not targeting depressive symptoms (e.g. supportive therapy, marital therapy). Those receiving therapy that was depression specific, such as cognitive therapy or interpersonal psychotherapy of depression, were excluded. Patients were excluded if they had current alcohol dependence or abuse (last 6 months), significant liver disease for which use of the study medications was contraindicated, a known hypersensitivity to either study medication, uncontrolled narrow-angle glaucoma, or were taking monoamine oxidase inhibitors in the 14 days prior to study entry.
Intervention
Participants were started on venlafaxine-XR (37.5 mg day–1) at baseline with an increase to 75 mg day–1 on day 4, and aripiprazole (5 mg day–1) was started at week 2, with oral administration for both. They proceeded through a dosing schedule to a maximum of 300 mg day–1 venlafaxine-XR and 30 mg day–1 aripiprazole, with dose increases given per schedule if remission was not achieved at the established dose. Venlafaxine-XR was increased to 150 mg day–1 by week 1. At week 4, venlafaxine-XR could be increased to 225 mg day–1 and aripiprazole to 15 mg day–1. Aripiprazole could be increased to the final dose of 30 mg day–1 at week 6. Venlafaxine-XR could be increased to the final dose of 300 mg day–1 at week 8. Medications could be decreased or participants could exit the study if significant side-effects could not be otherwise managed.
Participants were evaluated at baseline, and at weeks 1, 2, 4, 6, 8, 10, and 12. Clinical management was guided by the 16-item Quick Inventory of Depressive Symptomatology–Clinician-rated (QIDS-C16) [4–7], which evaluates depression symptom severity, and the Frequency, Intensity, and Burden of Side-effects Rating (FIBSER) [33], which assesses overall side-effects. These two measures, and procedures using a measurement-based care approach, were successfully used in STAR∗D to implement high-quality care with vigorous but tolerable dosing in both primary care and psychiatric settings [2].
Measures
The QIDS-SR16 was measured at baseline and at each treatment visit as a primary outcome measure of remission and response rates. The QIDS-SR16 is highly reflective of results obtained with the HRSD17[7, 34], reduces patient burden, and is as reliable and valid as the clinician-rated QIDS-C16[5, 7]. Self-reports (as opposed to clinician ratings) have commonly been used in effectiveness trials (e.g. IMPACT) [10], and the PATHWAYS study [35]. The QIDS-C16 was gathered at baseline and at each treatment visit to measure depression symptom severity and to guide dosing. This instrument has been used as the principal instrument driving the critical decision points for the algorithmic pharmacotherapy of several large multi-centre trials such as Research Evaluating the Validity of Augmenting Medication with Psychotherapy (REVAMP) (1UO1MH61562) and STAR∗D (NO1MH90003).
The FIBSER self-report was given at each treatment visit. It provides three global ratings scored 0–6 (higher score indicating greater impact) on a Likert-type scale: frequency, intensity, and the overall burden or degree of interference in day-to-day activities and function due to the side-effects attributable specifically to the antidepressant treatment.
The following measures were gathered to evaluate safety issues and to gauge medication tolerability at each treatment visit: the Systematic Assessment for Treatment Emergent Events–Systematic Inquiry (SAFTEE-SI) Butler/MGH modified [36, 37], the Barnes Akathisia Rating Scale (BARS) [38], and the Abnormal Involuntary Movement Scale (AIMS) [39, 40], and measures of blood pressure, pulse and weight. The SAFTEE-SI Butler/MGH modified is an easy-to-use 55-item self-report that rates most commonly reported side-effects expected with the study medications. The clinician-rated BARS is a four-item instrument that measures motor restlessness. The AIMS is a 12-item clinician rating of abnormal involuntary movements, which includes evaluations of facial and oral movements, as well as evaluation of extremities and trunk movements. Each symptom is rated from 0 (none) to 4 (severe). The Self-Administered Comorbidity Questionnaire [41] self-report was administered to assess the presence of a range of common medical conditions (e.g. heart disease, high blood pressure, lung disease, diabetes, ulcer or stomach disease etc.), as well as their severity and whether or not the conditions limited function.
Data analysis
The two aims of the present study were to estimate the proportion of participants who went into remission and to evaluate the side-effects experienced by participants. Remission was defined as QIDS-SR16 score ≤5 at the last clinic visit. Response was defined as an improvement in QIDS-SR16 score ≥50% over baseline. Intolerable side-effects were defined as attrition due to side-effects or an indication of at least severe impairment due to side-effects as rated on the FIBSER.
A 95% confidence interval was used to estimate the variability of each proportion. Given the sample size of 50, if one assumes a remission rate of 50%, then a 95% confidence interval can be estimated with 14% precision. That is, the 95% confidence interval will include the sample remission rate ±14%. Likewise, if one assumes an intolerable side-effect rate of 15%, a 95% confidence interval will include the sample intolerable side-effect rate ±9%.
Descriptive statistics are presented as mean±SD for continuous variables and percentages for discrete variables. Kaplan–Meier methods were used to estimate the cumulative proportion with first remission, first response, and study discontinuation.
Results
Subject characteristics
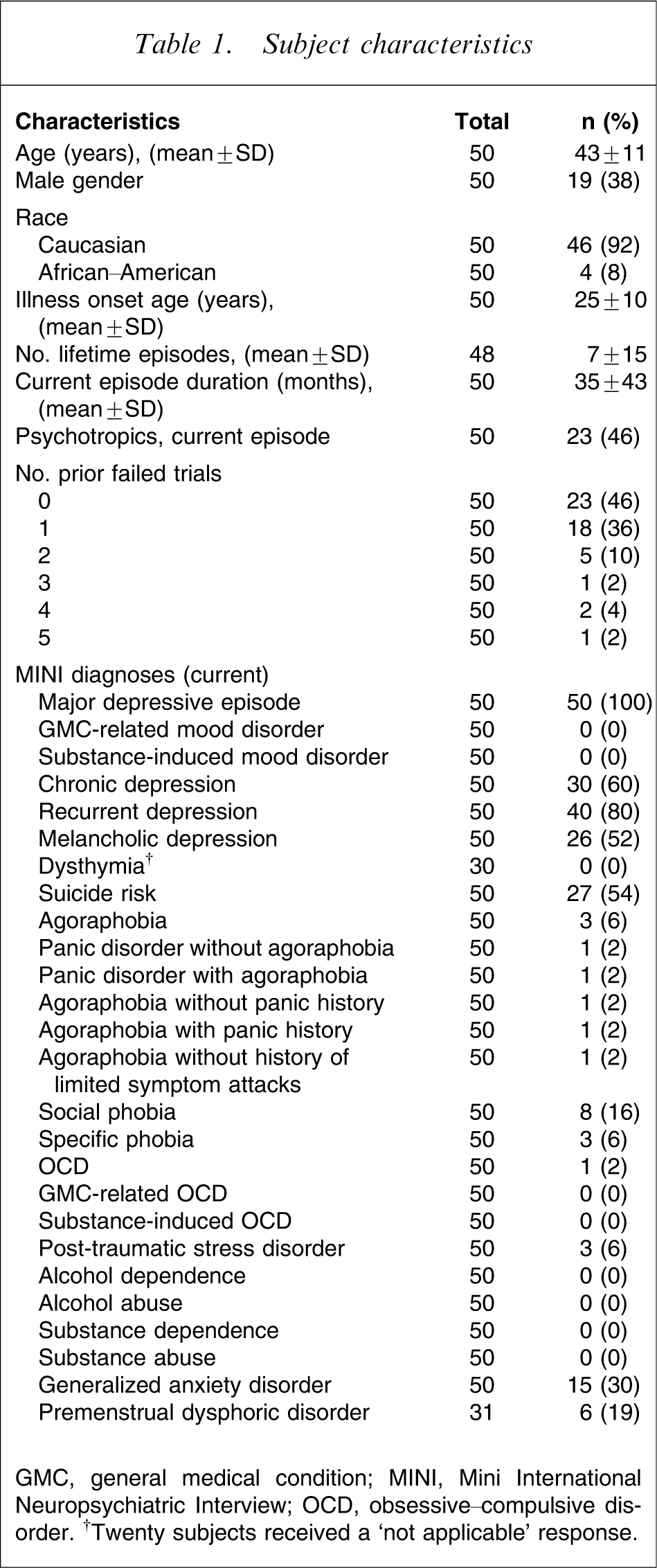
GMC, general medical condition; MINI, Mini International Neuropsychiatric Interview; OCD, obsessive–compulsive disorder. †Twenty subjects received a ‘not applicable’ response.
Dosage, discontinuation, and adverse events by week
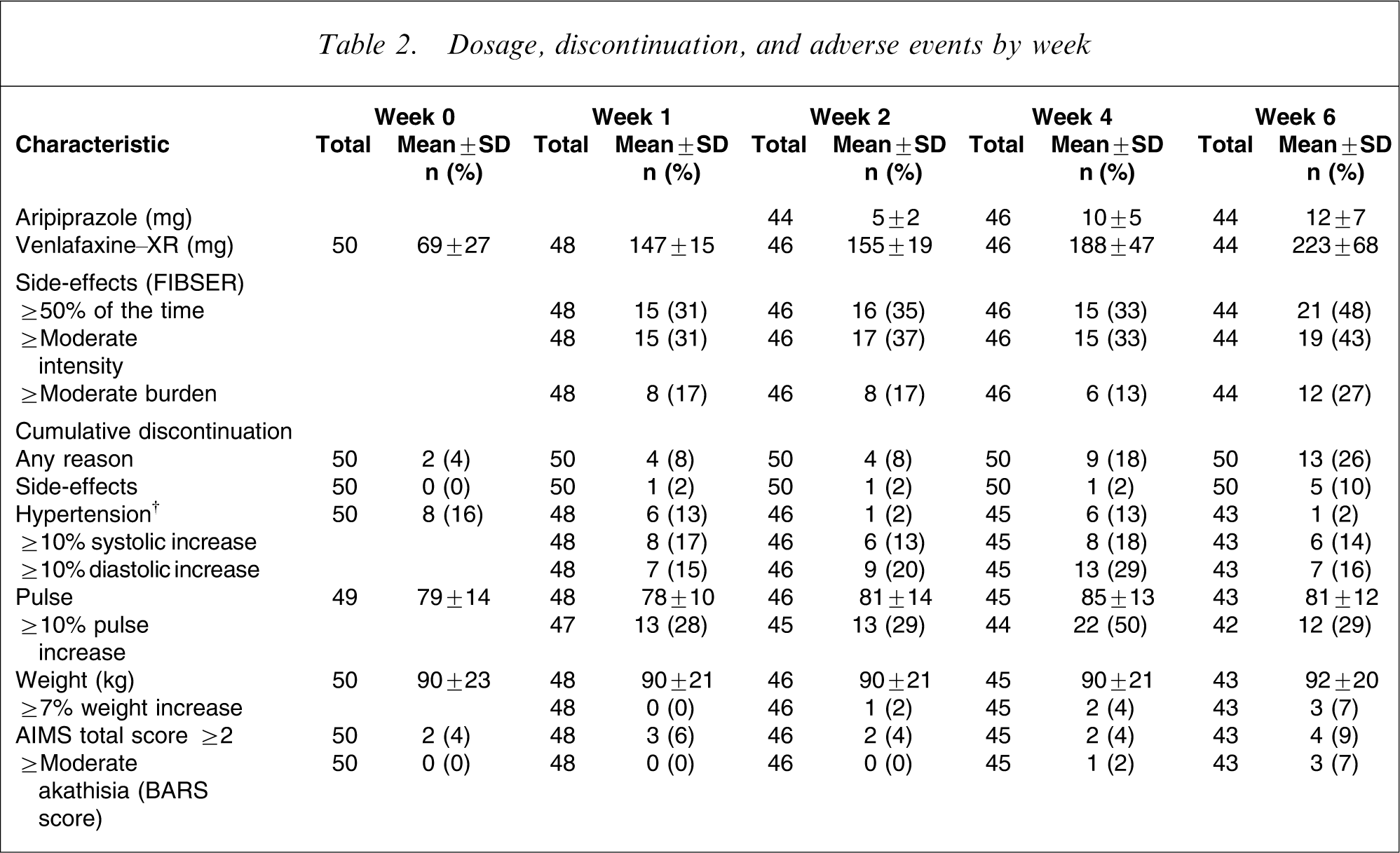
AIMS, Abnormal Involuntary Movement Scale; BARS, Barnes Akathisia Rating Scale; FIBSER, Frequency, Intensity, and Burden of Side-Effects Rating Scale. †Systolic blood pressure >140 XX and diastolic blood pressure >90 mmHg.
Treatment-emergent adverse events, remission, and response†
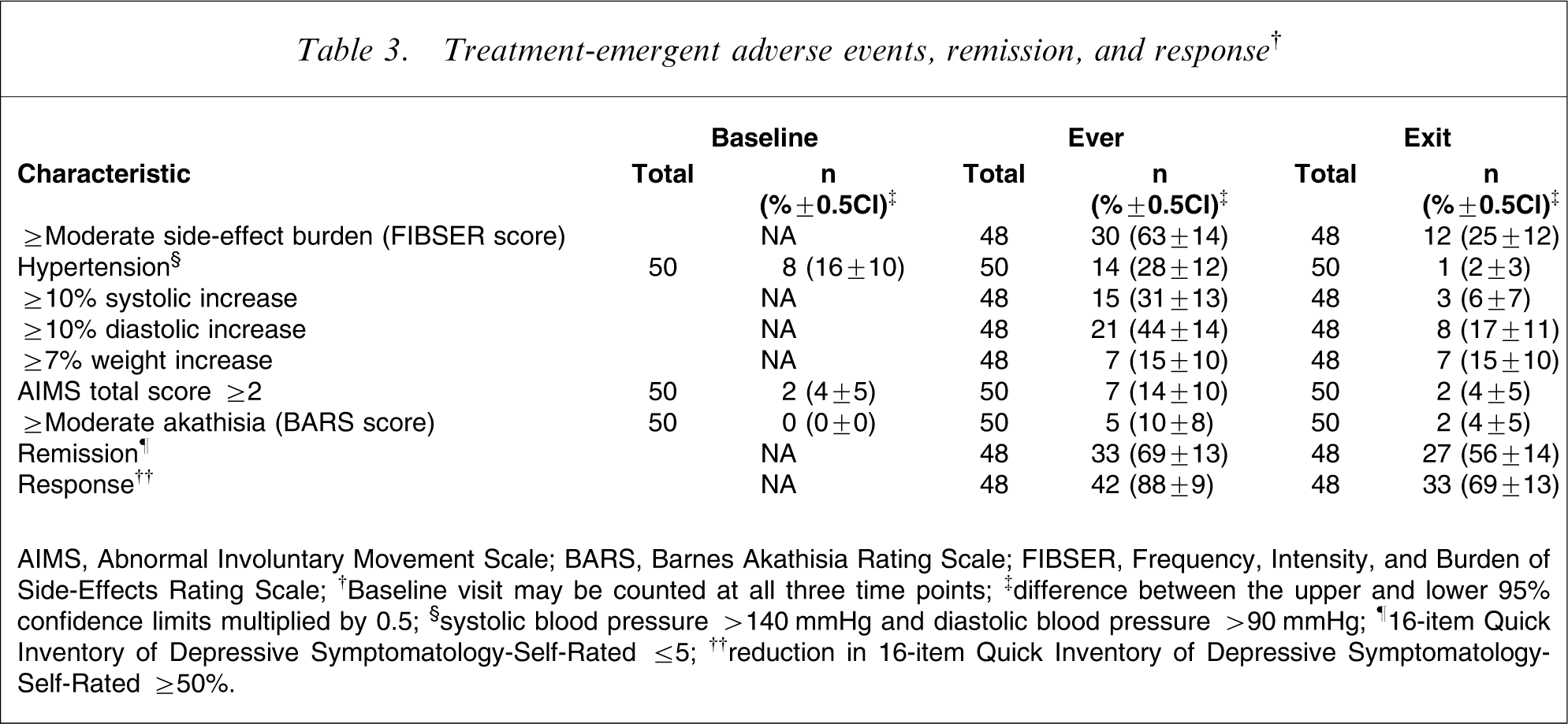
AIMS, Abnormal Involuntary Movement Scale; BARS, Barnes Akathisia Rating Scale; FIBSER, Frequency, Intensity, and Burden of Side-Effects Rating Scale; †Baseline visit may be counted at all three time points; ‡difference between the upper and lower 95% confidence limits multiplied by 0.5; §systolic blood pressure >140 XX and diastolic blood pressure >90 XX; ¶16-item Quick Inventory of Depressive Symptomatology-Self-Rated ≤5; ††reduction in 16-item Quick Inventory of Depressive Symptomatology-Self-Rated ≥50%.
Adverse events vs remission at exit
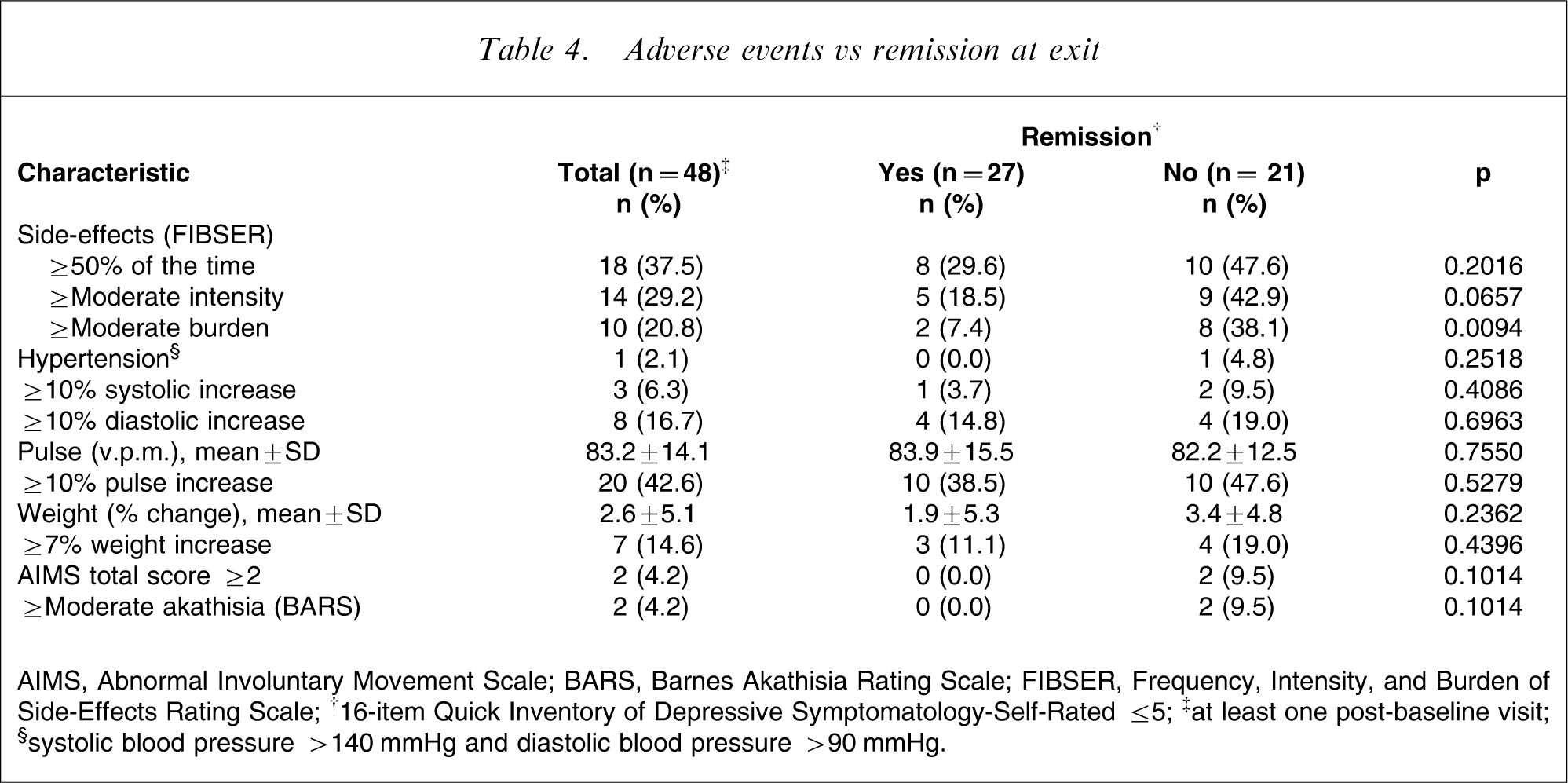
AIMS, Abnormal Involuntary Movement Scale; BARS, Barnes Akathisia Rating Scale; FIBSER, Frequency, Intensity, and Burden of Side-Effects Rating Scale; †16-item Quick Inventory of Depressive Symptomatology-Self-Rated ≤5; ‡at least one post-baseline visit; §systolic blood pressure >140 XX and diastolic blood pressure >90 XX.
Systematic assessment for treatment emergent effects:%present and%change
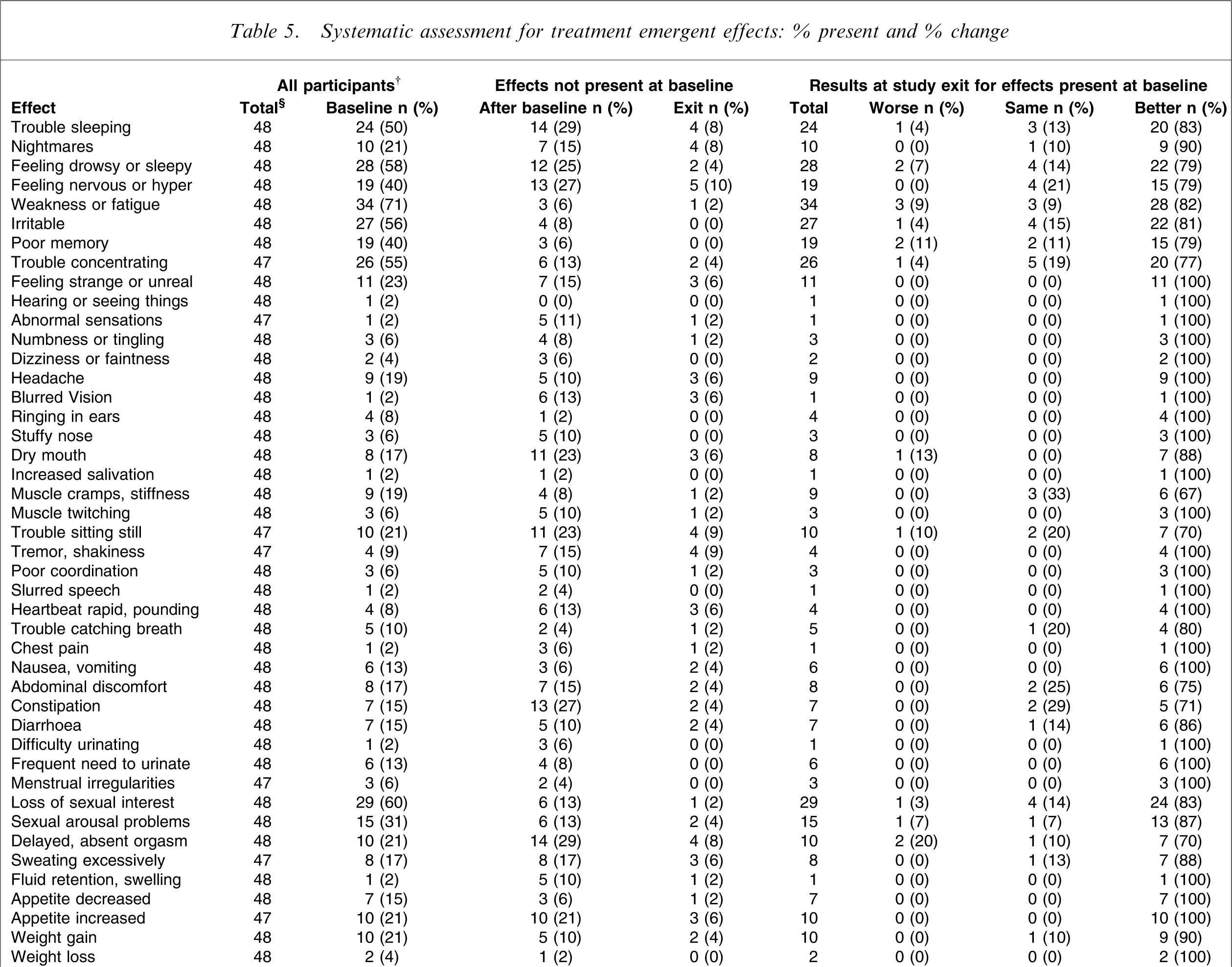
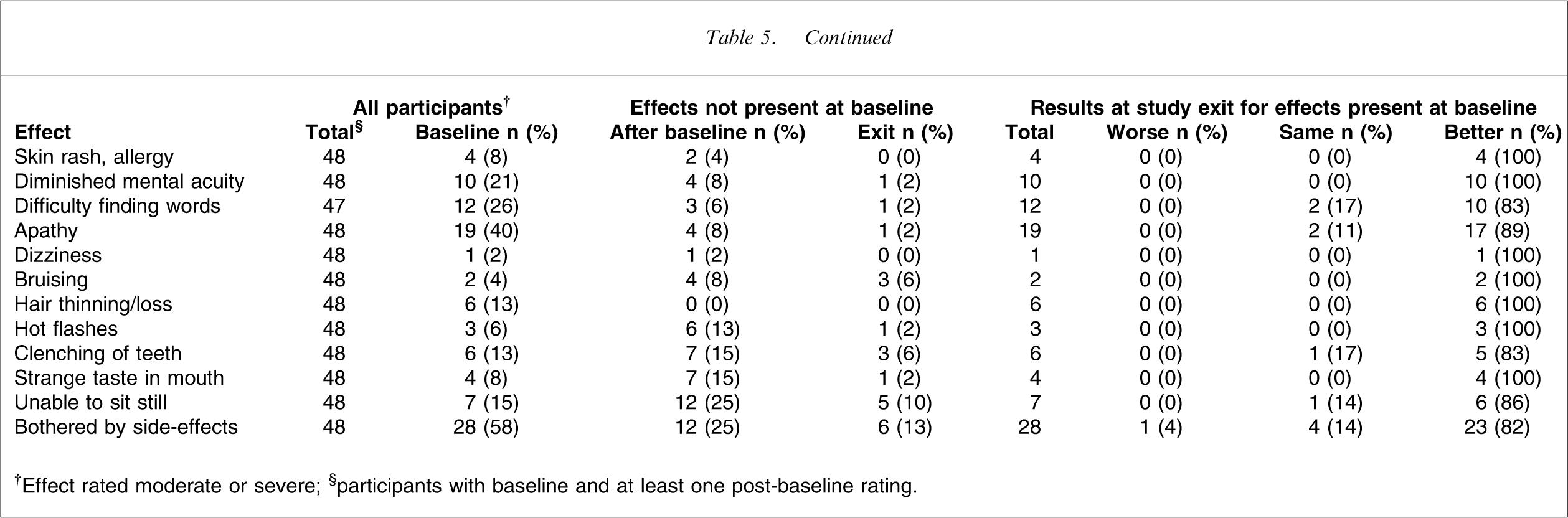
†Effect rated moderate or severe; §participants with baseline and at least one post-baseline rating; ¶not present at baseline.
Depressive symptom severity, remission and response rates are listed versus week in Table 6. The group started with a mean baseline QIDS-C16 score of approximately 15 (severe range; HRSD17 equivalent approx. 18–19), and by week 12 the group had a mean QIDS-C16 score of approximately 6 (mild range; HRSD17 equivalent approx. 9). Clinician- and self-rated QIDS scores had excellent agreement. The remission rate at exit decreased as the number of prior failed trials increased but these differences were not statistically significant (p = 0.096). Seventy-three per cent (16/22) of those with no prior failed treatments were in remission at exit, compared to 44% (8/18) with one and 38% (3/8) with two or more prior failed treatments. Cumulative remission and response rates showed that at the end of the open trial, two-thirds met criteria for remission and >80% met criteria for response. Time to first remission, first response, and discontinuation are presented in Figure 1. For those participants who achieved remission, the percentages achieving first remission versus week are shown in Figure 2.
Cumulative probability of discontinuation, remission and response vs week. Distribution of first week of remission (16-item Quick Inventory of Depressive Symptomatology–Self-Report score <5) for participants who went into remission at exit.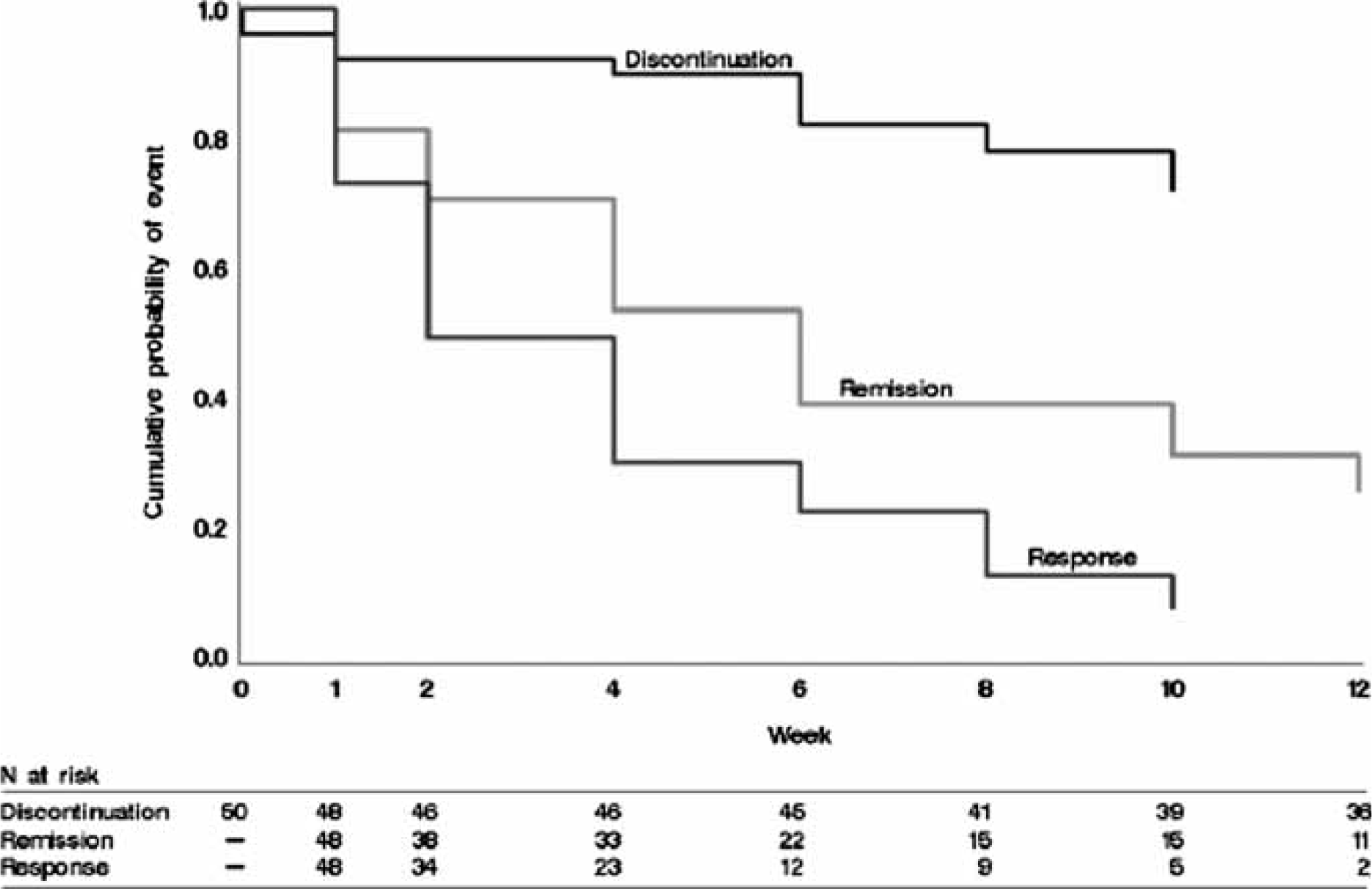
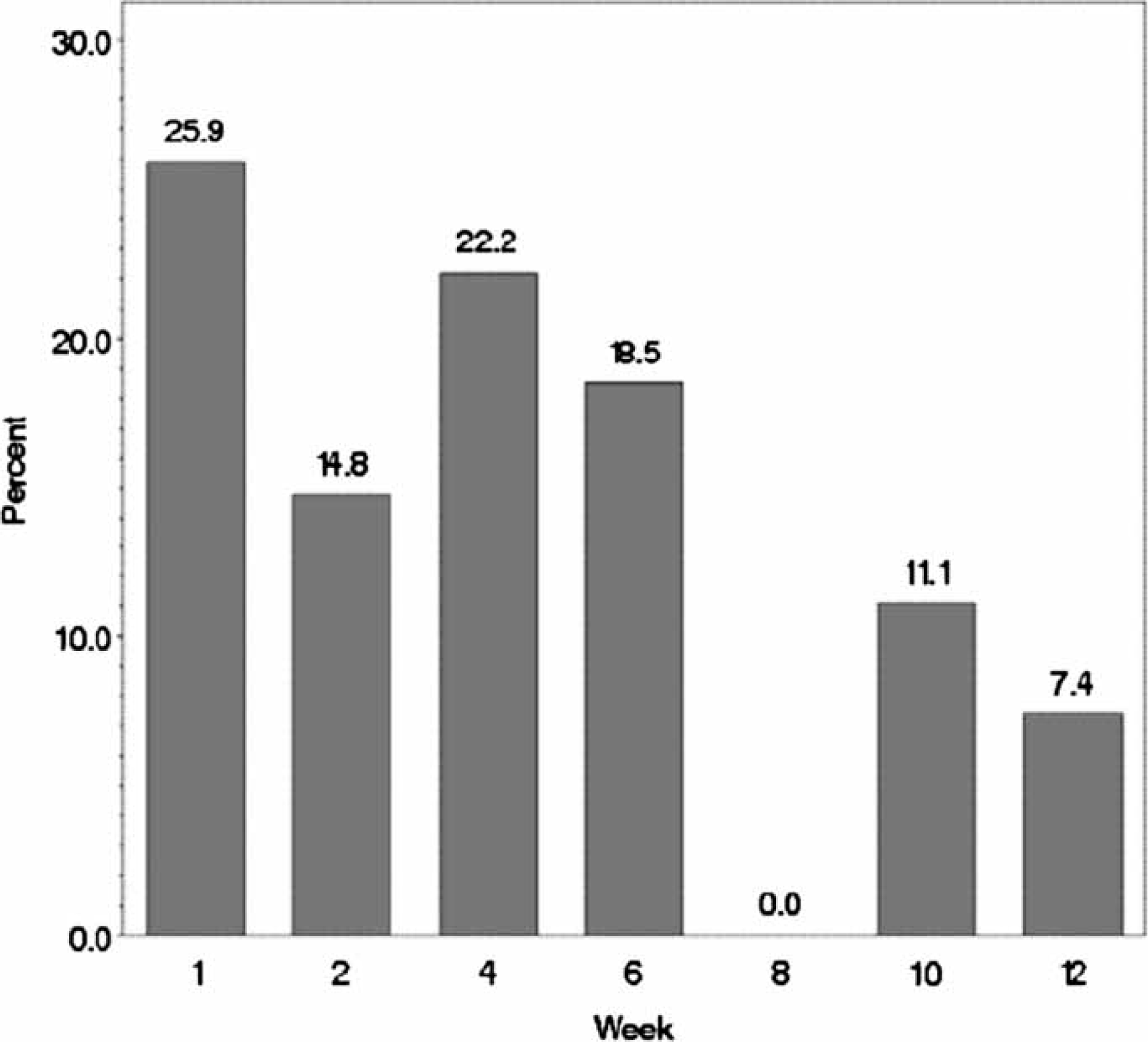
Outcomes vs week
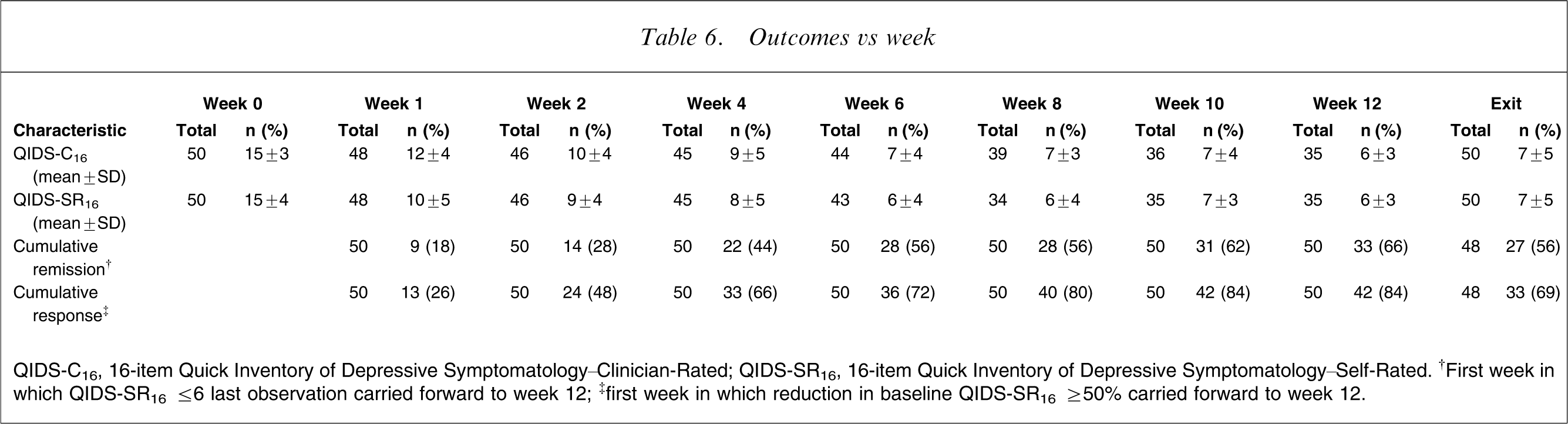
QIDS-C16, 16-item Quick Inventory of Depressive Symptomatology–Clinician-Rated; QIDS-SR16, 16-item Quick Inventory of Depressive Symptomatology–Self-Rated. †First week in which QIDS-SR16 ≤6 carried forward to week 12; ‡first week in which reduction in baseline QIDS-SR16 ≥50% carried forward to week 12.
Serious adverse events included hypertensive episodes with transient ischaemic attack (n = 2), a fall (n = 1), a hypoglycaemic episode in a patient with pre-existing diabetes mellitus, and a suicide attempt in the context of relationship problems (n = 1). There were no completed suicides.
Discussion
This is the first open effectiveness study to evaluate the safety, tolerability, and remission rates for the combination of venlafaxine-XR and aripiprazole for CRMD. The combination, starting with venlafaxine-XR and then adding aripiprazole 2 weeks later, was well-tolerated by most participants. Overall, few participants dropped out due to intolerable side-effects, and drop-out rates for side-effects were similar to those found in monotherapy studies of antidepressants. The most common adverse effects included those that would be expected for each medication separately, and these did not appear to be exacerbated by the presence of the second medication. The results of this effectiveness trial suggest that the combination is well-tolerated and can be used safely.
Participants using this combination achieved higher response and remission rates than those typically seen for initial treatment with monotherapy. Note also that this study required participants to have either CRMD while STAR∗D did not have this requirement, although approximately 80% of STAR∗D patients did have CRMD. The remission rate obtained with the venlafaxine-XR/aripiprazole combination in this non-treatment-resistant group (up to 70%) was, as expected, higher than the remission rates obtained when aripiprazole was added to antidepressants in a treatment-resistant group (26%) [15].
Initial treatment with the combination of venlafaxine-XR and aripiprazole has heuristic and theoretical importance. Venlafaxine-XR, through its simultaneous uptake inhibition of serotonin and norepinephrine, most likely has downstream effects within the neuron to promote neurogenesis via cyclic-AMP regulatory element binding protein-mediated gene expression of brain-derived neurotrophic factor and vasoepithelial growth factor [42, 43], resulting in neuroprotection and neurogenesis within the hippocampus [44]. Aripiprazole has neuroanatomically diverse effects on dopaminergic receptors, including acting like a D2 agonist as well as a functional dopaminergic blocker [44–46]. In addition, aripiprazole is a 5HT2A antagonist and 5HT1A partial agonist [47]. Thus, the uptake inhibition of serotonin and norepinephrine from venlafaxine-XR, coupled with the dopaminergic partial agonist, 5HT2A antagonist and 5HT1A partial agonist effects from aripiprazole, could be synergistic or it could benefit a wider range of patients.
The limitations of the present study include its effectiveness design with open treatment and the use of non-blinded raters. Participants were not assessed for extrapyramidal symptoms and this should be done in future studies for a complete evaluation of risks and benefits. Further, the sample size was modest and the study enrolled a mixture of participants with and without resistance to at least one treatment in the current MDD episode. Finally, given the long duration of the current episode of MDD, it is possible that some of these participants had a personality disorder that might have been responsive to the aripiprazole.
In summary, the results of the present pilot study suggest that the combination of venlafaxine-XR and aripiprazole was tolerable, safe, and resulted in a favourable acute remission rate when used as an initial treatment for non-treatment-resistant CRMD. This challenges the predominant treatment paradigm of initial antidepressant monotherapy followed by sequential switching or augmentation. Further studies will be required to assess the durability of the combination and its ability to prevent depressive relapse and recurrence, as well as the risk of aripiprazole-induced movement disorders. Further research should also focus on finding other medication combinations that are effective initial treatments for MDD.
Financial Disclosures
In the past 3 years, Dr Nierenberg consulted to and served on advisory boards of Abbott Laboratories, Appliance Computing, Brain Cells, Bristol Myers Squibb, EpiQ, Pam Labs, PGX Health, Forest Pharmaceuticals, Eli Lilly & Co, GlaxoSmithKline, Janssen, Pharmaceutica, Jazz Pharmaceuticals, Merck, Novartis Pharmaceuticals, Pfizer Pharmaceuticals, Schering-Plough, Sepracor, Shire, Somerset, Takeda, and Targacept. He has received research support from Cyberonics, Cederroth, Ortho-McNeil-Janssen, Pfizer, Pam Labs and Shire. He received honoraria from the MGH Psychiatry Academy (MGHPA are supported through Independent Medical Education (IME) grants from the following pharmaceutical companies in 2008: Astra Zeneca, Eli Lilly, and Janssen Pharmaceuticals). Dr Nierenberg owns stock options in Appliance Computing. Dr Rush has provided scientific consultation to or served on Advisory Boards for Advanced Neuromodulation Systems; AstraZeneca; Best Practice Project Management; Bristol-Myers Squibb Company; Cyberonics; Forest Pharmaceuticals; Gerson Lehman Group; GlaxoSmithKline; Jazz Pharmaceuticals; Eli Lilly & Company; Magellan Health Services; Merck & Co.; Neuronetics; Ono Pharmaceutical; Organon USA; PamLab, Personality Disorder Research Corp.; Pfizer; The Urban Institute; and Wyeth-Ayerst Laboratories. He has received royalties from Guilford Publications and Healthcare Technology Systems, and research/grant support from the Robert Wood Johnson Foundation, the National Institute of Mental Health, and the Stanley Foundation; has been on speaker bureaus for Cyberonics, Forest Pharmaceuticals, GlaxoSmithKline, and Eli Lilly & Company; and owns stock in Pfizer.
Footnotes
Acknowledgements
We would like to thank the members of the Data Safety and Monitoring Board; Andrew C. Leon PhD, Jan Fawcett MD, Pedro Delgado MD, Craig Nelson MD, and Richard Shader MD. Medications were supplied at no cost by Organon and Wyeth-Ayerst Laboratories. This project was funded by the National Institute of Mental Health under Contract N01MH90003 to UT Southwestern Medical Centre at Dallas (P.I.: A.J. Rush). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We appreciate the support of Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, King Pharmaceuticals, Organon, Pfizer, and Wyeth for providing medications at no cost for this trial. We would also like to acknowledge the editorial support of Jon Kilner, MS, MA, and the support of Fast Word Information Processing (Dallas, Texas).