
Abstract
Most people with episodic tension-type headache (TTH) treat themselves with over-the-counter analgesics. In the absence of clear evidence of dose-related efficacy of the two most commonly used analgesics, aspirin (acetylsalicylic acid) and paracetamol (acetaminophen), this study compared two doses of each with placebo. In a double-blind, double-dummy, randomized parallel-groups comparative trial, 638 consenting subjects aged 16-65 years with episodic TTH (but not migraine) by IHS criteria were recruited from the UK general population by advertisement. They treated one episode of moderate or severe TTH with a single dose of 500 or 1000 mg aspirin, 500 or 1000 mg paracetamol or placebo. The primary objective was to compare aspirin 1000 mg with placebo, and the primary end-point was subjective pain relief (total or worthwhile) 2 h after treatment ('response'). Additionally, pain intensity on a 100-mm visual analogue scale and functional impairment were monitored regularly for 4 h and at 24 h, although rescue medication was allowed after 2 h. The analysis was of the intention-to-treat population of 542 who took treatment (all providing outcome data). Treatment groups were matched at baseline. Aspirin 1000 mg (75.7% response rate; P = 0.0009) and to a lesser extent aspirin 500 mg (70.3%; P = 0.011) and paracetamol 1000 mg (71.2%; P = 0.007), but not paracetamol 500 mg (63.8%; P = 0.104), were statistically more effective than placebo despite a high placebo-response rate (54.5%). Outcome was not affected by headache intensity at baseline. Secondary end-points including functional recovery (by median times of 4.0-13.5 h) were consistent with these findings, although a minority of subjects recorded long-duration functional impairment (37-54 h). Adverse events reported by 13.4-18.9% of subjects were mild or moderate, and transient. No safety concerns arose.
Introduction
More than one headache type goes under the name of tension-type headache (TTH) (1). Episodic TTH occurs in attack-like episodes, mostly lasting a few hours, with variable, often very low frequency. People call it ‘normal’ or ‘ordinary’ headache. Chronic TTH, much less prevalent, occurs by definition on > 15 days a month and can be daily and unremitting. In either case the pathophysiology is not known. TTH may be stress-related or associated with functional or structural cervical or cranial musculoskeletal abnormality, and these aetiological factors are not mutually exclusive. The headache, often generalized, is rarely severe but typically described as a vice or tight band around the head. It commonly spreads into or appears to arise from the neck (2).
Most people with episodic TTH treat themselves with over-the-counter analgesics, as far as is known with generally good effect. Published studies give some support for this practice (3–7). In the absence of other clear evidence of dose-related efficacy of the two most commonly used analgesics, aspirin (acetylsalicylic acid; ASA) and paracetamol (acetaminophen), this study compared single doses of 500 mg and 1000 mg of each with placebo.
The primary objective was to compare, for pain relief at 2 h after ingestion, aspirin 1000 mg with placebo. Secondary objectives were comparisons for efficacy between each dose of aspirin and paracetamol, and placebo, assessed by a range of measures. Tolerability of each treatment was observed although much evidence of this, and safety, existed already for each drug.
Methods
The study protocol and recruitment and consenting procedures were approved by the South Thames (now South-east) Multicentre Research Ethics Committee. After completion of the trial, an independent audit for compliance with European Good Clinical Practice was conducted by ClinResearch, Cologne, Germany, without adverse report.
Subjects
Episodic TTH does not, usually, provoke medical consultation. Subjects aged 16–65 years were therefore recruited from the UK general population by advertisement in GP surgeries and local newspapers by a site management organization (SMO) with 10 centres, Synexus Ltd, Chorley, UK. All gave informed consent prior to screening, which included medical history and physical examination. Those meeting the IHS diagnostic criteria for episodic TTH but not those for migraine (1), without other serious physical or mental illness or contra-indications to either treatment, were randomly allocated, in equal proportions, to five treatment groups.
Women who were or might become pregnant were excluded: a pregnancy test was carried out at entry on all women of child-bearing potential, and those who admitted to being sexually active were required, as a condition of entry, to use adequate contraception until trial completion.
Concomitant medications disallowed at entry were antidepressants and any drug known to interact with either study medication.
Study design
This was a double-blind, double-dummy, placebo-controlled randomized parallel-groups comparative trial.
Following screening and randomization, each subject received a diary card and one dose of trial medication. Instructions were to treat within 8 weeks of enrolment, at home or wherever it might occur, one episode of TTH of at least moderate intensity, not improving at the time of treatment and with onset 1–12 h earlier. (The guidance given for subjective judgement of ‘moderate’ pain was that, whilst tolerable, it could not be ignored; it interfered with concentration but not necessarily with daily activities.) Study medication was not to be used for any headache associated with a cold, influenza or other viral infection, or hangover.
Treatment
The five trial treatments were aspirin 500 and 1000 mg, paracetamol 500 and 1000 mg, and placebo. The double-dummy design required four tablets in each single dose, contained in two bottles, to be taken together with water.
No prior treatment for that headache was allowed (no analgesics or NSAIDs in the previous 24 h, or long-acting NSAIDs such as oxicams or tranquillisers, sedatives, or psychotropic drugs for 72 h). Rescue medication was permitted after 2 h, and its use recorded in the diary card.
Efficacy evaluation
Also recorded in the diary card were pain intensity on a 100-mm horizontal visual analogue scale (VAS) with left boundary annotated ‘no pain’ and right boundary ‘severe pain’, and pain relief and functional ability on categorical scales (Table 1), all at 0, 30 and 45 min and 1, 2, 3, 4 and 24 h after treatment. Time to return to normal function and global evaluation of the trial treatment on a categorical scale (Table 1) were noted at 24 h.
Categorical scales used in efficacy evaluation

Diary cards were reviewed at a follow-up visit within 14 days of treatment.
Tolerability and safety
Subjects were asked to report, in free text in the diary cards, adverse events (AEs) as they occurred and their severity and duration. These reports were reviewed, and clarified where necessary, at the follow-up visit.
Investigators were responsible for attributing causation to AEs applying their clinical expertise. The procedure was standard, and allowed the following categories of probability of being drug-related: ‘probable’; ‘possible’; ‘remote’; ‘none’; ‘not assessable’. An AE was classified as not drug-related when the investigator recorded ‘none’, and otherwise (i.e. where causation by treatment could not be excluded) as drug-related. This approach was conservative.
Analysis and statistical methods
The principal null hypothesis was that the percentages of subjects reporting worthwhile or total headache relief at 2 h (principal efficacy end-point) would be identical in the two populations treated with aspirin 1000 mg or placebo. The criterion for significance (alpha) was set at P = 0.025 for a 1-tailed Fisher's exact test of effect in the expected direction. A placebo effect of 30% was assumed and, with 100 subjects in each group, the study would be powered at 83% to detect a difference of 20% (active treatment effect of 50%), which would be reported with a 95% confidence interval of 7–33%. A drop-out rate of 15% was anticipated; recruitment aimed for 120 subjects per group.
Randomization was by computer-generated list equalizing treatment groups in block sizes of 10, this number being unknown to investigators. Complete blocks were allocated to centres which, on enrolment of each subject, were required to assign the next number within the sequence allocated.
Analysis of efficacy variables, and of adverse events and their causal attributions, was undertaken once, after the trial had ended and the database was locked and prior to code-break.
The analysis reported here was on the intention-to-treat (ITT) population, defined as all enrolled subjects randomized, taking study medication and providing any outcome data. Missing data were replaced where possible by the last observation carried forward. All comparisons other than for the principal efficacy end-point were considered hypothesis-generating in nature; P-values ≤0.05 were reported as significant by convention but interpreted cautiously. Subgroup analyses were also performed in ITT subjects with baseline pain intensity on VAS > 60 mm vs. those with VAS ≤ 60 mm.
Results
Disposition of subjects
The 10 study centres made telephone contact with 2120 people responding to advertisements or identified by GPs known to the SMO. Of these, 1041 were considered potentially eligible and offered screening appointments at the nearest centre. Of the 750 who appeared, 638 were actually eligible and were enrolled and randomized to aspirin 500 mg (n = 126), aspirin 1000 mg (n = 128), paracetamol 500 mg (n = 128), paracetamol 1000 mg (n = 128) or placebo (n = 128) (Fig. 1). There were no major protocol violations at entry necessitating exclusion, but 96 randomized subjects, given study medication, did not take it. By far the most common reasons were that an episode of TTH of moderate intensity did not occur within 8 weeks of enrolment or, if one did, it was not convenient to the subject to treat and monitor that episode according to protocol requirements. Thus, 542 took treatment, all of whom provided some outcome data to be included in the ITT analysis.

Disposition of all randomized subjects (n = 638). ASA, aspirin; PAR, paracetamol; PLA, placebo; ITT, entering intention-to-treat analysis.
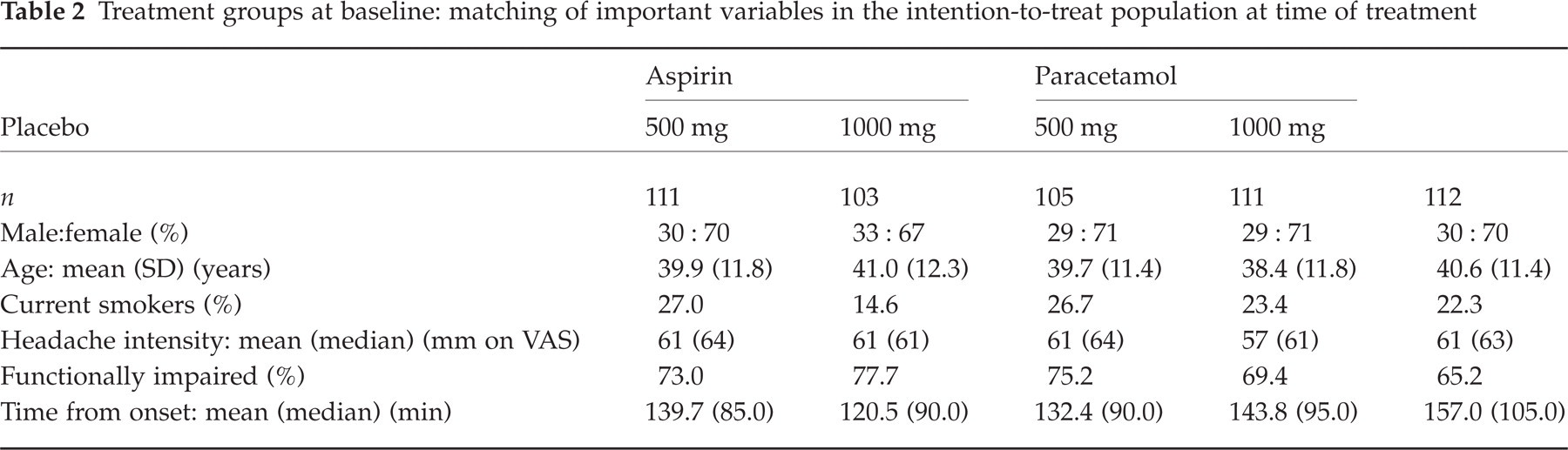
Baseline comparison of groups
Treatment groups were matched at baseline for important, potentially prognostic variables (Table 2). Of the total ITT population, 30% were male; mean age was 40 years.
Treatment groups at baseline: matching of important variables in the intention-to-treat population at time of treatment
Efficacy
Most analyses for efficacy were made over the first 4 h. Because rescue medication was allowed from 2 h onwards, no inferences were drawn for time points beyond this.
Figure 2 shows the primary efficacy analysis: percentages in each treatment group recording ‘I feel total relief’ or ‘I feel some worthwhile effect’ 2 h after treatment (‘responders’). Although placebo responder-rate (54.5%) was considerably higher than anticipated, aspirin 1000 mg (75.7%; P = 0.0009), aspirin 500 mg (70.3%; P = 0.011) and paracetamol 1000 mg (71.2%; P = 0.007), but not paracetamol 500 mg (63.8%; P = 0.104), were statistically superior to placebo. Aspirin 1000 mg was numerically but not statistically superior to paracetamol 1000 mg (P = 0.275) and aspirin 500 mg to paracetamol 500 mg (P = 0.19) (the study was not powered to show a difference between active treatments). Both drugs showed some evidence of a dose–response relationship.
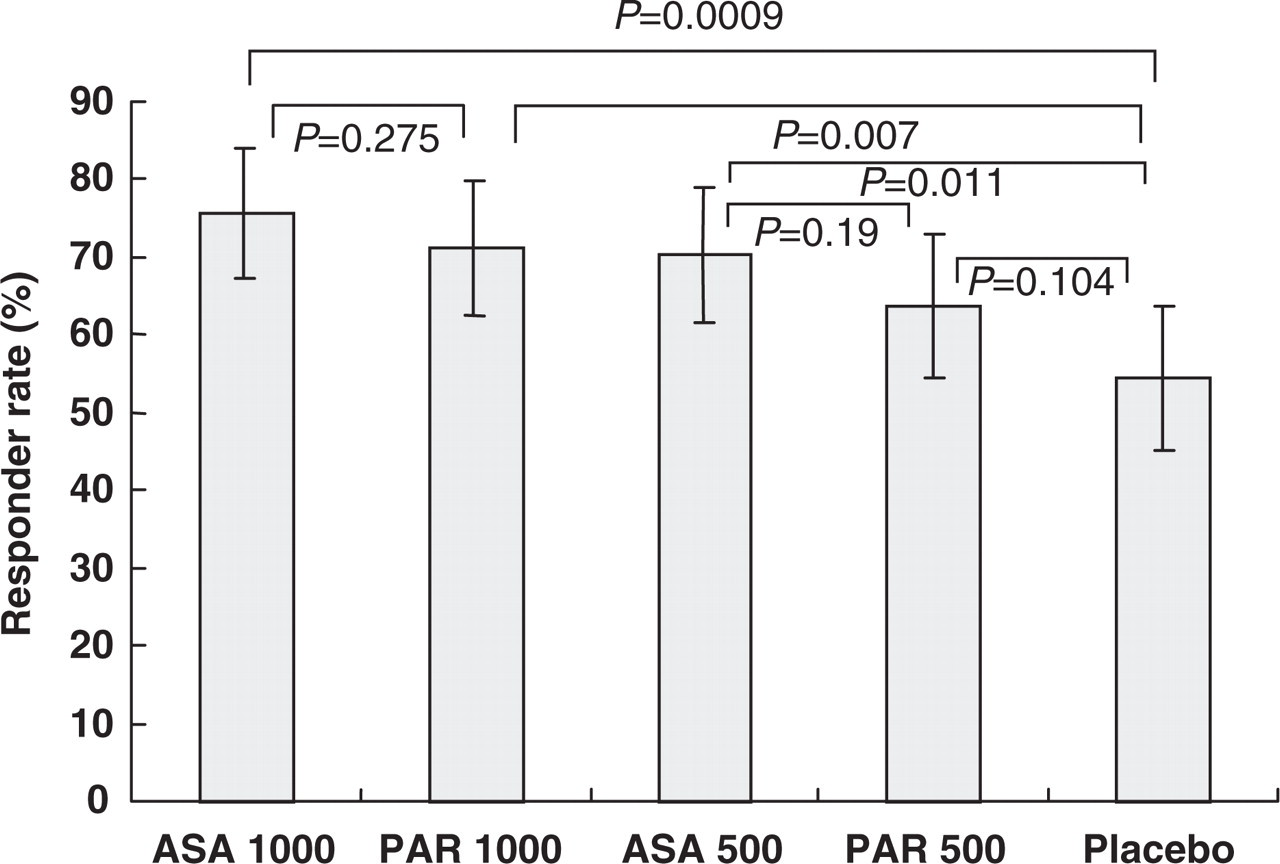
Principal efficacy analysis in the intention-to-treat population. Responders per treatment group were those recording ‘I feel total relief’ or ‘I feel some worthwhile effect’ 2 hours post-treatment (see Methods). ASA, aspirin; PAR, paracetamol. Horizontal lines indicate significance of differences and whiskers show 95% confidence intervals.
Responder rates and differences between them were similar in the subgroup with baseline pain intensity VAS > 60 mm (n = 287): aspirin 1000 mg (78.8%; n = 52; P = 0.0073), aspirin 500 mg (75.0%; n = 64; P = 0.027) and paracetamol 1000 mg (71.4%; n = 56; P = 0.035), but not paracetamol 500 mg (57.9%; n= 57; P= 0.458), were statistically superior to placebo (56.9%; n= 58). In subjects with baseline pain intensity VAS ≤ 60 mm (n = 253), only aspirin 1000 mg was statistically significantly different from placebo (P = 0.034). Two subjects who failed to record pain intensity at baseline were excluded from these analyses.
As pain relief was measured repeatedly over 4 h, its time course could be assessed in terms of responder rates (Fig. 3). The differences between aspirin 1000 mg and placebo were significant (P ≤ 0.022) at each time point from 30 min, whereas paracetamol 1000 mg and aspirin 500 mg were not significantly better than placebo until 2 h.

Time course of pain relief. Responders per treatment group were those recording ‘I feel total relief’ or ‘I feel some worthwhile effect’ (see Methods) at each time point post-treatment. • aspirin 1000; ▴ aspirin 500; □ paracetamol 1000; ○ paracetamol 500; ▪ Placebo.
Pain intensity vs. time analysis on 100-mm VAS (Fig. 4) showed all active treatments but paracetamol 500 mg clearly distinguished from placebo. The curves for aspirin 1000 mg and paracetamol 1000 mg were nearly superimposed except that the latter originated from a slightly favourable baseline. Analysis of change in headache intensity (Fig. 5) is subject to baseline-dependent floor and ceiling effects, but makes reasonable allowance for small baseline differences. For pain intensity difference (PID), where
PIDt = P0 − Pt (VAS score at baseline − VAS score at time t),

Pain intensity analysis over time. Pain intensity was recorded on 100-mm VAS. Plotted points are means per treatment group. • aspirin 1000; ▴ aspirin 500; □ paracetamol 1000; ○ paracetamol 500; ▪ Placebo.

Pain intensity difference analysis over time. Pain intensity difference (PIDt=P0– Pt) (see text) was recorded on 100-mm VAS. Plotted points are means per treatment group: higher positive values signify greater pain relief. • aspirin 1000; ▴ aspirin 500; □ paracetamol 1000; ○ paracetamol 500; ▪ Placebo.
positive scores indicate pain reduction and negative scores increased pain. The differences between aspirin 1000 mg and placebo were significant (P ≤ 0.016) at each time point from 30 min to 2 h (P = 0.0001), whereas paracetamol 1000 mg and aspirin 500 mg were not significantly better than placebo until 2 h (P = 0.0058 and P= 0.0018, respectively). This analysis visually separated aspirin 1000 mg from other active treatments in the first hour (Fig. 5), although only the differences from paracetamol 500 mg were statistically significant (P ≤ 0.017 at each point from 45 to 120 min).
Summed PID scores weighted for the time between observations (SPID) where
SPID = Σ(PIDt × Te) and Te= time in hours since previous observation
are estimates of the area under the pain intensity vs. time curve. Analysis of SPID produced results numerically and statistically comparable with those for PID; therefore, they are not separately reported.
Functional impairment analysis was of percentages of subjects in each treatment group recording, at each time point, ‘My everyday activities can be carried out as usual’ (see Methods). Two-thirds to three-quarters of subjects per group had some functional impairment at baseline, with placebo and paracetamol 1000 mg groups slightly favoured (Table 2). Visually in Fig. 6, all active treatments separated from placebo by 2 h, and statistical analysis confirmed this (P < 0.05). For aspirin 1000 mg, earlier onset of effect indicated in PID analysis was reflected in clearly evident better functional recovery by 1 h. This was confirmed statistically in an analysis, limited to those in each group recording at least some baseline functional impairment, of return rate to normal function by 1 h: placebo 19.6% (n = 73); paracetamol 1000 mg 26.1% (n = 77; P = 0.16); aspirin 1000 mg 41.7%; (n = 80; P = 0.0003 vs. placebo; P = 0.012 vs. paracetamol 1000 mg).

Functional impairment analysis over time. Subjects functionally unimpaired per treatment group were those recording ‘My everyday activities can be carried out as usual’ (see Methods) at each time point. • aspirin 1000; ▴ aspirin 500; □ paracetamol 1000; ○ paracetamol 500; ▪ Placebo.
Whilst time to return to normal function was of potential interest, only 311 subjects (79.7%) of the 390 with functional impairment at baseline made a diary entry to record this, and distributions were highly skewed by a few subjects in each group reporting times of 37–54 h. The data (n; mean [median] in hours) were: aspirin 1000 mg (63; 9.9 [4.0]); aspirin 500 mg (58, 11.8 [9,9]); paracetamol 1000 mg (55; 11.2 [5,7]); paracetamol 500 mg (61; 13.6 [13.5]); placebo (74; 13.8 [10.1]).
Use of rescue medication after 2 h, a marker of treatment failure, varied between the treatment groups. Aspirin 1000 mg (15.5%; P = 0.011) and 500 mg (16.2%; P = 0.014), but not paracetamol 1000 mg (19.8%; P = 0.065) or 500 mg (25.7%; P = 0.32), were statistically better on this measure than placebo (29.5%).
Global evaluation analysis was based on percentages of subjects in each group recording ‘The medication is very good’ or ‘The medication is good’ (see Methods). These responses would have been based on expectation derived from past experience of treatment, and presumably took into account any need for rescue and any adverse events (see below). Aspirin 1000 mg (52.4%; P = 0.0003) and 500 mg (42.3%; P = 0.022), and paracetamol 1000 mg (45.0%; P = 0.008) but not 500 mg (38.1%; P = 0.089), were better rated than placebo (28.6%).
Adverse events were not infrequent, reported by 13.4–18.9% of subjects in the five treatment groups (Fig. 7). All were mild or moderate, and transient, as expected with these agents. Those considered by investigators to be drug-related (3.9–7.2% on aspirin; 4.5–5.7% on paracetamol) were not dose-related for either active treatment although more frequent than for placebo (1.8%). A small excess of all-causality gastrointestinal events on aspirin (5.8–6.3%; paracetamol 3.6–3.8%; placebo 2.7%) was also not dose-related. No safety concerns arose in this study.

Adverse-event analysis (intention-to-treat population, n = 542). Rates were of subjects per group reporting one or more events, not of events. Therefore, rates for all events are not the sums in each treatment group of drug-related and not drug-related event rates. ‘Drug-related’ was investigator-attributed (see text). GI, gastrointestinal.
Discussion
In view of the very widespread use of aspirin and paracetamol for episodic TTH, formal confirmation of the efficacy of these inexpensive drugs is helpful for physicians, pharmacists and others who advise people with this common condition.
The strongest conclusion from this study is that moderate-to-severe headache in episodic TTH responds better within 2 h to aspirin 1000 mg than to placebo, disproving the principal null hypothesis. With high internal consistency, every secondary end-point reflected this outcome of the principal analysis, which was therefore unambiguous. Subgroup analysis of subjects with baseline pain intensity>60 mm on VAS showed the same. The study found that response to placebo by 2 h was marked (in part, no doubt, because of the self-limiting nature of episodic TTH), but aspirin 1000 mg produced a greater and earlier group response. It found also that both active treatments were superior for efficacy to placebo, with consistent though in no case statistically significant findings of a dose–response relationship for each drug.
Interestingly, the subgroup analysis of those with baseline VAS > 60 mm did not indicate that greater headache intensity at time of treatment is predictive of treatment failure for aspirin 1000 mg or 500 mg or paracetamol 1000 mg, nor did it find a reduced response to placebo. As these findings were unexpected, regression analysis was conducted for each treatment including placebo of baseline VAS against response at 2 h (categorical yes/no) and against time to worthwhile effect. Values of R2 were maximally 0.085, confirming that baseline headache intensity had very little effect on outcome.
There are some observations to be made, cautiously, from comparing outcomes on aspirin with those on paracetamol. Numerically and statistically, results for paracetamol 1000 mg were generally similar to those for aspirin 500 mg. Figure 3 demonstrates this well. Paracetamol 500 mg was not statistically different from placebo on the principal efficacy criterion or any other, although numerical differences occurred on most. It is doubtful that this dose is generally effective in the treatment of episodic TTH, although some individuals may nonetheless find it so.
Whilst years of widespread use attest to the safety in adults of both aspirin and paracetamol (8), the issue of side-effects is not unimportant. Adverse events causally attributed to treatment were not rare in this study (reported by 3.9–7.2% of subjects on aspirin, 4.5–5.7% on paracetamol and only 1.8% on placebo), but they were not dose-related for either drug. There is widespread awareness of the potential in some people for gastric intolerance to aspirin, and gastrointestinal adverse events were likely to be considered treatment-related. They were more common on aspirin than on paracetamol or placebo, and probably the differences were real, but again they were not dose-related. There are a few people who would probably better avoid aspirin for this reason, and, of course, there are well-known contra-indications to aspirin that rule out its use in a few others. Although it was not an objective, this study provides support for the use of paracetamol 1000 mg in episodic TTH by these people.
Aspirin tablets are marketed in standard strengths of 500 mg in most European countries, with a recommended dose of 1–2 tablets, and 300 mg in the UK, Ireland and Australia, with 2 tablets recommended (3 for migraine) (9, 10). Paracetamol is marketed mostly in tablets of 500 mg, again with 1–2 tablets recommended for headache treatment. Therefore, doses investigated in this study covered those that are standard, whilst paracetamol 500 mg was confirmed to be generally subtherapeutic.
A surprising incidental finding was the duration of functional impairment attributed to episodic TTH by the 311 subjects (57.4% of the total) who both reported functional impairment at baseline and recorded its duration. Whilst data were highly skewed, with durations of up to 54 h recorded, nonetheless at least 28% of all subjects apparently remained functionally impaired to a recognizable degree in their own minds for 9.4 h (median in the 311; mean 11.9 h) after treatment, even with rescue medication allowed. Undoubtedly the nonresponders (33% overall on the principal efficacy analysis) essentially account for this, and it must be remembered that entry criteria to the trial required at least moderate headache (subjectively judged) at the time of taking study medication. An obvious question, however, is whether the sample was inadvertently polluted by cases of migraine. This possibility cannot be excluded, especially since the IHS diagnostic criteria for migraine (which recruited patients must fail) are intentionally specific rather than sensitive (1); but we believe it to be extremely unlikely that large numbers of patients were misdiagnosed and certainly not 28%. The conclusion is that, in a substantial subset of people with episodic TTH, the condition is disabling.
One implication of the outcome of this study is that future trials of drugs in moderate-to-severe episodic TTH, whether placebo-controlled or not, should use aspirin as an active comparator.
Conclusions
Aspirin 1000 mg, and perhaps to a lesser extent aspirin 500 mg and paracetamol 1000 mg, are effective in treating moderate and severe episodic TTH. The placebo-response rate is high in this condition, which is self-limiting. Nonetheless, functional impairment appears to persist for many hours in a substantial minority of people.