
Abstract
A total of 729 migraine sufferers with moderate to severe baseline pain evaluated a single 200, 400 or 600 mg dose of a new liquigel formulation of ibuprofen over 8 h. Ibuprofen liquigels were significantly superior to placebo for cumulative headache response (pain reduced to mild or none) from 0.5 (600 mg) or 1 h (200 and 400 mg) to 8 h. At 2 h, respective headache response rates for ibuprofen 200, 400 and 600 mg and placebo were 64%, 72%, 72% and 50%. All three doses were also significantly superior to placebo for 2-h pain-free (25%, 28%, 29% and 13%, respectively) and for proportions with mild or no limitation of activity (2-8 h). Ibuprofen liquigels were generally superior to placebo for reducing photophobia, phonophobia, or nausea (1-4 h) and for global evaluation. All doses were well tolerated. These data demonstrate that ibuprofen liquigels relieve the pain, ancillary symptoms, and limitation of activity, of migraine.
Introduction
Approximately 18% of women and 6% of men have at least one migraine headache annually, which projects to 23–25 million migraine sufferers in the United States of America (USA) (1). Migraine attacks are relatively common, with a median attack frequency of one to two attacks per month (2). The severity of attacks varies widely both between and within individuals, ranging from mild pain and minimal disability to incapacitating pain and total disability (3). However, the majority of attacks (60–70%) are characterized by both moderate to severe pain and disability (4).
Population-based studies have suggested that less than half of migraine sufferers have ever been diagnosed by a physician (5, 6). Most (> 90%) treated their headaches with over-the-counter (OTC) medications, and the majority (> 60%) used OTC drugs to the exclusion of prescription drugs (5, 7). A poll published in 1995 indicated that 45% of migraineurs who used OTC medications (e.g. non-steroidal anti-inflammatory drugs (NSAIDs), analgesics) found them to be at least somewhat effective and 20% reported that they were very effective (8). A recent survey indicated that the percentage of migraineurs who have ever consulted a physician for headache has increased to 69%, but the majority (72.5%) still use OTC medications and 50% use OTC agents exclusively (9).
Despite the widespread use of OTC agents for the acute management of migraine as of this writing, only one OTC medication in the USA (acetaminophen + aspirin + caffeine) is approved for acute migraine treatment. This combination has been reported to significantly reduce the pain and functional disability associated with a migraine attack compared to placebo (10). Since OTC medications are less expensive, more accessible, and have favourable safety profiles relative to prescription treatments, additional OTC analgesics should be evaluated for the treatment of migraine.
The non-prescription NSAID ibuprofen is an attractive candidate for an OTC migraine treatment. Several clinical studies have reported that NSAIDs are effective for migraine; however, only a few of these studies were placebo-controlled, and most used either prescription NSAIDs or OTC NSAIDs at prescription doses (11). In more recent studies, ibuprofen demonstrated significant anti-migraine efficacy compared to placebo with single doses of 200–1200 mg (12–15), but none of these trials explored a dose–response relationship. One of the objectives of the present study was to determine if ibuprofen exhibits a positive dose–response relationship for the relief of migraine headache.
When prescription ibuprofen is used to treat migraine, doses up to 800 mg are commonly used. For this reason and since migraine headache is generally more severe than tension-type headache (16), we wanted to explore doses above 400 mg, the maximum single dose currently approved for OTC use. We reasoned that 600 mg may be particularly advantageous for subjects with more severe pain and greater disability. A single-dose of ibuprofen 600 mg has not yet been studied for its effect on migraine.
Migraine sufferers value rapid pain relief (17). A liquid formulation of ibuprofen, ibuprofen liquigel (a soft gelatin capsule filled with solubilized ibuprofen) has been shown to reach a higher peak plasma level (Cmax) in a shorter time (Tmax) than a solid tablet, and this kinetic profile has been shown to correlate with a greater and faster peak analgesic response (18, 19). Further, ibuprofen liquigels (400 mg) were recently shown to provide faster and more complete relief than acetaminophen caplets (1000 mg) in the treatment of episodic tension-type headache (20). The gastric stasis which typically accompanies migraine headache could potentially retard the disintegration, dissolution and absorption of a solid dosage form, possibly delaying the time to pain relief and to peak relief. Therefore, the liquigel formulation is particularly promising for migraine, because it provides the potential for rapid pain relief without requiring stomach agitation for disintegration and dissolution.
The present study was designed to evaluate the efficacy and safety of ibuprofen liquigels (at single 200, 400 and 600 mg doses) compared to placebo in the acute treatment of migraine headache of at least moderate severity in subjects representative of the OTC population of migraineurs.
Methods
Subjects
This was a single-dose, double-blind, randomized, placebo-controlled, parallel group, multicentre study. Potential subjects were recruited by using population-based screening (i.e. community screening by random digit dialing using a validated computer assisted telephone interview), advertisements, or an existing database. To be eligible, males and females (≥ 12 years old) must have had at least a 1-year history of migraine with or without aura as defined by International Headache Society (IHS) criteria (21). Subjects estimated that their average migraine headaches, if left untreated, were at least moderate in pain intensity, and occurred at an average frequency ranging from one every 2 months to eight per month during the previous year. Onset of these headaches had to be before age 50, and subjects must have previously experienced some relief from OTC analgesics.
Subjects were ineligible if they had: a head or neck injury or a change in headache pattern during the previous 6 months; a history during the last year of chronic daily use (> 30 days) of analgesics, NSAIDs, sedatives/hypnotics, tranquillisers, or a history of drug or alcohol abuse; hypersensitivity or other contraindications to aspirin, ibuprofen or any other NSAID; migraine attacks which were accompanied by vomiting 20% of the time or more; migraines that were usually severely disabling or incapacitating; or atypical migraine with aura subtypes, cluster headache, or serious causes of headache other than migraine. All subjects provided written informed consent (for subjects less than 18 years of age, a parent or legal guardian provided consent). The protocol and informed consent form were approved by an Institutional Review Board (Western Institutional Review Board,® Olympia, Washington, USA).
Study procedures
Eligible subjects were stratified by gender and caffeine consumption (low: ≤ 3 servings of caffeinated beverages daily; high: > 3 servings daily) and randomly assigned to receive ibuprofen liquigels 200 mg, 400 mg, 600 mg, or matching placebo, under double-blind conditions. Subjects were discharged from the study sites with a diary, study medication, and a timer (to help ensure assessments were performed at the proper time), and were instructed to treat one eligible migraine attack within approximately 2 months. To determine if a particular headache was eligible, subjects first completed a checklist based on the IHS criteria for migraine and other study-specific requirements. Pain severity was required to be at least moderate for the migraine headache episode to be eligible for evaluation.
Pain intensity was assessed by the subjects using a standard 4-point categorical scale (0 = none; 3 = severe) at baseline, and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8 and 24 h after ingestion of study medication. Pain relief was evaluated at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7 and 8 h using a 5-point categorical scale (0 = complete relief; 4 = no relief). Four-point categorical scales were also used to record ratings of nausea, photophobia and phonophobia at baseline, 1, 2 and 4 h after dosing. Limitation of activity was assessed (4-point categorical scale) at baseline, 2, 4, 6 and 8 h. Subjects also provided a global evaluation (0–10) of study medication at the end of the 8-h assessment period, where 0 was ‘poor’ and 10 was ‘excellent.’ If required, subjects were permitted to use rescue medication of their choice after the 2-h time point. Pain intensity, relief and global evaluation were assessed at the time of rescue medication use, if applicable.
Subjects returned to the study sites after treating a headache, at which time their headache checklist and diary were reviewed to confirm that an eligible migraine headache was treated and the assessments were completed properly. All adverse experiences (AEs) recorded in the diary were reviewed and their relationships to study medication were evaluated by a study physician.
Formulation of study medication
The liquigel formulation used in this trial is a unique patented dosage form developed to enhance drug absorption. The liquigel consists of a soft gelatin capsule containing solubilized ibuprofen. The soft gelatin capsule rapidly dissolves in the acidic environment of the stomach, releasing the solubilized ibuprofen which is readily absorbed.
Statistical methods
All analyses were performed using SAS Version 6.12 (SAS Institute, Cary, North Carolina, USA). Differences were considered statistically significant if P < 0.05 (two-sided). It was estimated that a sample size of 200 subjects in each active treatment group would provide 84% power to detect a minimum of a 15% difference in cumulative headache response rates at 2 h (primary efficacy variable) between any two active treatments. With 150 subjects in the placebo group, there was an 87% power to detect a minimum of a 17% difference between any active treatment and placebo (assuming a 33% response rate for placebo).
The four treatment groups were compared with respect to demographic, headache
history and baseline characteristics. Treatment group comparability for
continuous variables (e.g. age, weight) was assessed using analysis of variance
(
The primary efficacy variable was the headache response by 2 h (i.e. the cumulative proportion of subjects whose pain intensity was reduced from moderate or severe at baseline to none or mild by 2 h). Other key variables included: cumulative percentage of responders at time points other than 2 h; cumulative percentage of subjects with no pain at each time point; pain intensity difference score (PID; = baseline score minus score at post-dosing time point) and actual pain relief rating (PRR) at each time point; times to response and to no pain (measures of times to pain relief); global evaluation at 8 h; cumulative percentage of subjects with no or mild limitation of activity at 2, 4, 6, and 8 h; cumulative percentages of subjects with no nausea, photophobia or phonophobia at 1, 2 and 4 h; time to use of rescue medication (duration of relief); and percentage of subjects with migraine recurrence over 24 h (i.e. of those subjects who were responders at 2 h, the proportion whose headache returned to moderate or severe by 24 h).
The cumulative percentages of subjects who were responders, who achieved no pain,
who had no or mild limitation of activity, no nausea, no photophobia, no
phonophobia, and who took rescue medication by each analysis time point, were
analysed by the CMH test controlling for site, the corresponding baseline score,
gender, and caffeine consumption stratum. The actual percentage of subjects who
experienced headache recurrence over 24 h was analysed similarly. The median
times to response, no pain, and to rescue medication, and their 95% confidence
intervals, were derived by the method of Simon and Lee (22). Treatment and covariate
effects were assessed using the Cox proportional hazards model (23). Global
evaluation, PIDs and PRRs were analysed using
The primary efficacy analysis was conducted on the intent-to-treat (ITT) population, which included all randomized subjects who evaluated baseline pain intensity, took study medication, and performed at least one post-baseline pain intensity assessment. A secondary analysis was performed on the evaluable population, which excluded subjects who were discontinued, had significant protocol violations, or took rescue medication prior to 2 h post-dosing. The ITT analysis is the primary focus of this report.
All subjects who received study medication and had follow-up safety data were included in the safety analysis. All reported AEs were categorized according to COSTART terms, and analysed using Fisher's exact test.
Results
Subject population
This study was conducted at three investigative sites. Subject disposition is summarized in Table 1. A total of 972 subjects were enrolled and randomized into the study. Of these, 237 subjects did not take study medication or were lost to follow-up (the majority did not treat an eligible migraine headache within 8 weeks of enrolement). The remaining 735 subjects took study medication and were included in the safety analysis. Six of these did not complete either baseline or a single post-baseline pain assessment and therefore were excluded from the primary analysis of the ITT population (n = 729). Before the treatment blind was broken, a total of 57 subjects were discontinued or deemed to be unevaluable: 19 assessed an ineligible headache, 17 for administrative reasons, 10 for protocol violations, four for taking rescue prior to 2 h, and one was determined to be ineligible (inappropriate migraine diagnosis). The remaining 672 subjects were included in the secondary evaluable analysis.
Summary of subject disposition

The demographic characteristics and migraine headache history data of the ITT population are summarized in Table 2. The four treatment groups were comparable for all demographic variables. Similar to the general migraine population, female subjects outnumbered males by a ratio of approximately 3:1 (75.4% vs. 24.6%). Most subjects were Caucasian (72.8%), and most (85.7%) were ‘low’ caffeine consumers (i.e. 0–3 caffeinated beverages consumed daily). Overall, 4.8% (35) of the subjects were adolescents (12–19 years of age). The treatment groups were also comparable for all headache history variables, except for nausea associated with headache, which was significantly lower in the ibuprofen 400 mg group. However, this was not considered clinically relevant. The majority (91.9%) of subjects reported a history of migraine without aura; 11.9% reported migraine with aura. The intensity of a typical untreated migraine attack was nearly evenly divided between moderate (51.6%) and severe (48.4%), and the majority of subjects indicated that they usually did not suffer from severely disabling/incapacitating migraines. Most subjects (89.4%) treated their migraines with non-prescription medicines, although more than 10% used prescription medication either alone or in addition to non-prescription drugs (Table 2).
Demographics and migraine headache history – intent-to-treat subjects

The baseline symptom profiles of treated migraine headaches were comparable for all treatment groups (Table 3). Most subjects treated a migraine without aura (89.5%). Subjects treated attacks with moderate (69.4%) or severe (29.6%) baseline pain. Contrary to instructions, seven subjects (1%) treated a migraine with mild pain, and were excluded from the evaluable analysis. Approximately 71% had moderate or severe limitation of activity at baseline, and most reported at least some photophobia (97.4%), phonophobia (95.0%) and nausea (51.7%).
Baseline characteristics of treated headaches – intent-to-treat subjects

Efficacy results
Pain intensity and pain relief
The cumulative percentage of responders (i.e. pain improving from moderate or severe at baseline to none or mild, or from mild at baseline to none) over time are shown in Fig. 1 (left panel). Ibuprofen 600 mg was significantly better than placebo beginning at 30 min after dosing, while 200 and 400 mg reached significance at 60 min. At 2 h post-dosing (primary efficacy parameter), each ibuprofen dose was significantly superior to the placebo group; ibuprofen 400 mg (72.3%; P < 0.001) and 600 mg (71.7%; P < 0.001) had the highest cumulative percentages of responders at 2 h, followed by 200 mg (64.1%; P < 0.01) and placebo (50%). All three doses were comparable throughout the 8-h duration, except at 60 min when 600 mg (51.0% cumulative responders) was significantly (P < 0.05) better than 200 mg (38.4%). By the end of the study (i.e. 8 h), respective values for placebo, 200 mg, 400 mg and 600 mg ibuprofen were 71.1%, 86.4%, 89.5% and 86.9%. When the seven subjects in the ITT population with mild baseline pain were excluded from these analyses, the results were essentially identical to those obtained using all ITT subjects.

Cumulative percentages of subjects with a headache ‘response’ (i.e. pain reduced to mild or none) and with ‘no pain’ after treatment with placebo, 200 mg, 400 mg or 600 mg of ibuprofen (IBU) liquigels. ∗P ≤ 0.001, ‡P ≤ 0.01, †P ≤ 0.05 compared to placebo; #P ≤ 0.05 compared to IBU 400 mg; §P ≤ 0.05 compared to IBU 200 mg.
For cumulative percentage of subjects with no pain (pain-free endpoint), statistical significance relative to placebo emerged at 1 h for ibuprofen 600 mg, 1.5 h for 400 mg, and 2 h for 200 mg (Fig. 1, right panel). At 2 h post-dosing, the respective pain-free values for placebo, 200 mg, 400 mg and 600 mg ibuprofen were 13.4%, 25.3% (P < 0.01), 27.7% (P < 0.001) and 29.3% (P < 0.001). All three doses were comparable throughout the time-course of the study except at 1 h, when 600 mg (9.6%) was significantly (P < 0.05) better than 400 mg (4.2%). By the end of the study, 43.7%, 63.1%, 66.5% and 65.7% of subjects in the placebo, 200 mg, 400 mg and 600 mg ibuprofen groups, respectively, were pain-free.
The results for PID and PRR at 2 h (Table 4) were similar to those obtained for cumulative percentage of responders and for subjects without pain: all three doses of ibuprofen were superior (P < 0.001) to placebo and comparable to each other.
Summary of results – intent-to-treat subjects

Headache response = cumulative percentage of subjects with moderate or severe pain at baseline reduced to none or mild at thespecified post-dosing time interval.
PID = pain intensity difference from baseline (pain intensity score at baseline – pain intensity score at specified time point).
PRR = pain relief rating (actual rating on a 5-point scale).
P ≤ 0.001 compared to placebo.
P ≤ 0.01 compared to placebo.
P < 0.05 compared to placebo.
P < 0.05 compared to IBU 200 mg.
Associated symptoms
The results for cumulative percentages of subjects without the ancillary symptoms of nausea, photophobia and phonophobia are shown in Fig. 2 and in Table 4 (for 2 and 4 h). For nausea, ibuprofen 200 and 400 mg were superior to placebo at most time points, whereas 600 mg did not statistically separate from placebo at any time. For photophobia, 200 mg was not different from placebo, while 400 and 600 mg were superior to placebo at 2 and 4 h. Ibuprofen 600 mg was superior to 400 mg at 1 h, and better than 200 mg at 4 h. For phonophobia, 600 mg was superior to placebo at all time points, 400 mg was better at 2 and 4 h, and 200 mg was superior only at 4 h. All three doses were comparable at all time points.
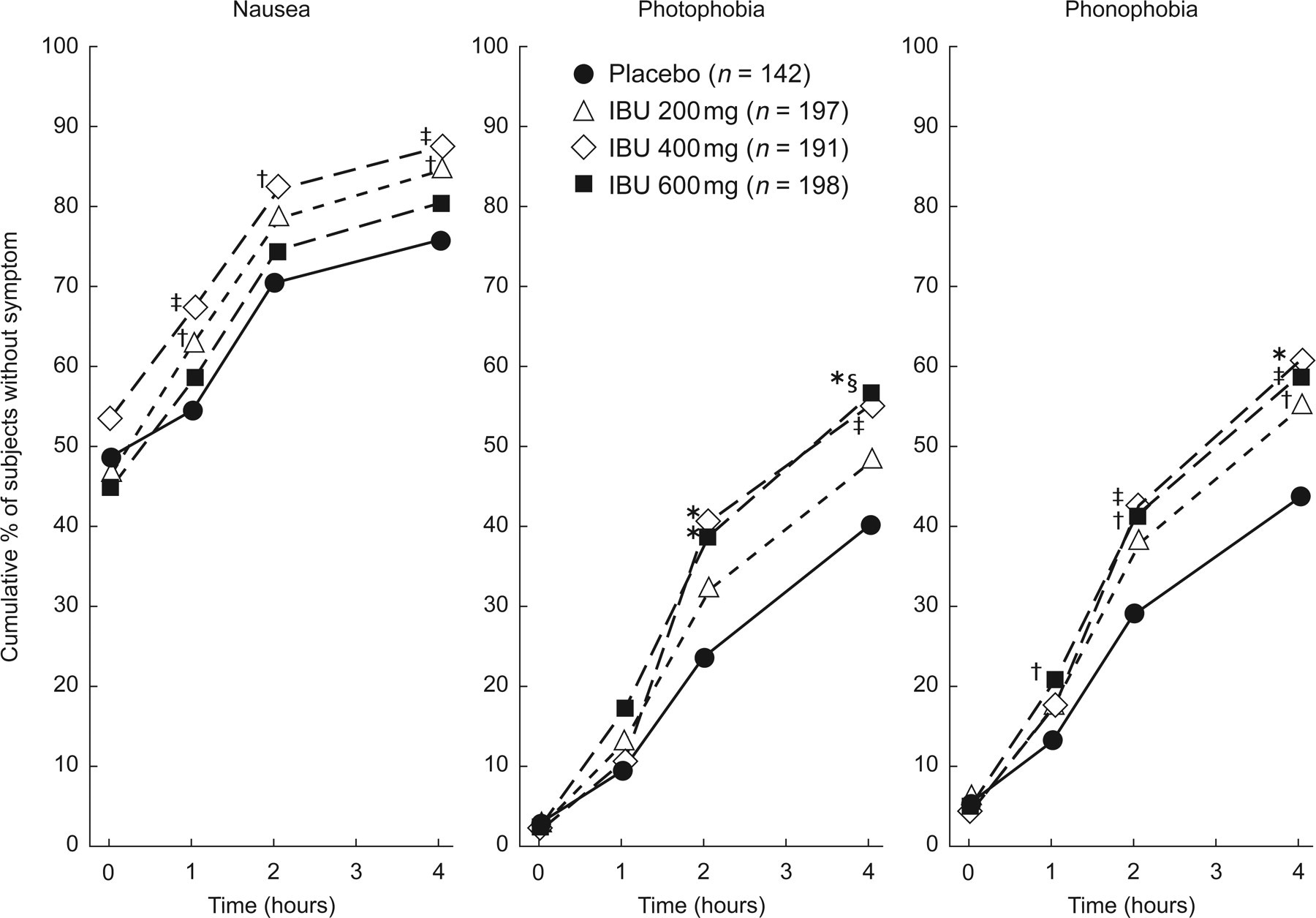
Cumulative percentages of subjects without nausea, photophobia or phonophobia after treatment with placebo, 200 mg, 400 mg, or 600 mg of ibuprofen (IBU) liquigels. ∗P ≤ 0.001, ‡P ≤ 0.01, †P ≤ 0.05 compared to placebo; #P ≤ 0.05 compared to IBU 400 mg; §P ≤ 0.05 compared to IBU 200 mg.
Limitation of activity
The results for cumulative percentages of subjects with mild or no limitation of activity at 2 and 4 h are shown in Table 4. All three doses of ibuprofen were superior (P < 0.001) to placebo and comparable to each other at both time points (and also at 6 and 8 h). By the end of the study (8 h), 72.5%, 88.4%, 91.1%, and 89.3% of subjects in the placebo and 200, 400 and 600 mg groups, respectively, reported mild or no limitation of activity.
Times to response and no pain
The results for times to response and to no pain, measures of the ‘onset’ of pain relief, are shown in Table 4. All three doses of ibuprofen provided significantly (P < 0.001) faster times to response and to no pain than placebo. There was a trend suggesting that the median times to headache response and pain-free response became generally shorter as the dose of ibuprofen increased, but differences did not reach statistical significance. Two standard measures of time to ‘onset’ of analgesia, median time to first perceptible relief (the first time point at which the sum of pain relief and pain intensity difference (PRID) is at least 1, confirmed by a PRID of at least 1 at the next time point), and the first time point statistically separating from placebo for cumulative percentage with first perceptible relief, were also determined. Both 400 and 600 mg ibuprofen had median times to first perceptible relief of 30 min; median times for 200 mg and placebo were 36 and 52 min, respectively. Thirty minutes was the first time point at which all three doses of ibuprofen liquigel statistically separated from placebo for cumulative percentage with first perceptible relief.
Rescue medication
The proportion of subjects taking rescue medication during the 8-h evaluation period was numerically lowest with ibuprofen 400 mg (13.1%), followed by 600 mg (13.6%), 200 mg (16.7%) and placebo (30.3%). All three doses had significantly lower cumulative percentages of rescuing than placebo from 3 h until the end of the study, and 600 mg was significantly better than 200 mg at 2 h. Since less than half of the subjects in each group took rescue medication, the median times to rescue were > 480 min for all treatments. However, statistical comparison of the distribution of times to rescue indicated that all three doses of ibuprofen had significantly (P < 0.005) longer durations of relief (i.e. times to rescue) than placebo and were comparable to each other.
Global evaluations
At 8 h (or at the time of rescue), subjects were asked to provide a global evaluation in response to the question, ‘How would you rate your study medication for relief of your migraine headache pain?’ on a scale of 0 (‘poor’) to 10 (‘excellent’). Mean global evaluations for placebo, ibuprofen 200 mg, 400 mg and 600 mg were 4.4, 5.6, 6.2 and 6.0, respectively. Both 400 mg and 600 mg were superior (P < 0.001) to placebo, and all doses of ibuprofen were comparable.
Migraine recurrence
The proportions of subjects experiencing recurrence over 24 h (i.e. of those subjects with pain intensity of mild or none at 2 h, the percentage whose score was moderate or severe by 24 h) for placebo, 200 mg, 400 mg and 600 mg ibuprofen were 29.7%, 16.9%, 19.8% and 14.6%, respectively. Both 400 mg (P < 0.05) and 600 mg (P < 0.005), but not 200 mg, were significantly better than placebo.
Adolescent subjects
A subgroup analysis of the data for cumulative proportion of responders in adolescent subjects (age 12–19) indicated that all three doses of ibuprofen liquigel were numerically better than placebo from 60 min through to 180 min (data not shown). Due to the small number of subjects in this subgroup (total n = 35), the results were not statistically significant. However, borderline significance (0.05 < P < 0.10) was attained for the comparison of 600 mg (n = 16) vs. placebo (n = 5) at 120 and 180 min. Pooling together the results for all three ibuprofen doses, ibuprofen was significantly better than placebo at 120 and 180 min. At 240 min and beyond, the placebo response was 80% to 100%.
Evaluable subjects analysis
The results of the secondary analysis of the 672 evaluable subjects were substantially similar to those based on the ITT subjects. The only difference was that ibuprofen 200 mg was also significantly better than placebo for the global evaluation in the evaluable group.
Safety results
Adverse experience (AE) data from the 735 subjects included in the safety analysis are summarized in Table 5. There were no significant differences between the four treatment groups with respect to overall AEs, AEs assessed as possibly, probably or definitely related to treatment, or severe AEs. There were no serious AEs reported, and no subjects withdrew from the study due to an AE. All four treatment groups were comparable with respect to the incidence of AEs in any COSTART body system (e.g. digestive system). The only individual AEs reported at an incidence greater than 2% in any treatment group were dyspepsia, dizziness (2.6% and 2.1%, respectively; ibuprofen 400 mg), and somnolence (2.5%; ibuprofen 600 mg). However, there were no significant differences between the treatment groups for the occurrence of any individual AE except for tremor, which had a higher incidence (1.4%) in the placebo group than in the ibuprofen groups (0 for all three doses).
Summary of adverse experience (AE) data – safety subjects

Significant at P ≤ 0.05.
Discussion
The results of this study indicate that a solubilized formulation of ibuprofen (liquigels) significantly relieves the pain and associated symptoms of migraine headache at single doses of 200, 400 or 600 mg compared to placebo in subjects representative of a typical OTC migraine population. These findings are consistent with previous studies demonstrating the efficacy of solid formulations of ibuprofen for migraine at single doses of 200, 400, 800 or 1200 mg (12–15). In addition, the current study demonstrated the efficacy of 600 mg.
To our knowledge, this is the first dose-ranging study performed on ibuprofen for the treatment of migraine. For the primary study endpoint, all three doses were comparable in efficacy and a significant dose–response relationship was not demonstrated. Nevertheless, there was a consistent numerical trend towards greater efficacy at the two higher doses for most efficacy parameters. Although there was a ‘plateau’ effect at 400 mg (i.e. 600 mg generally did not provide any additional efficacy than 400 mg), 600 mg was significantly superior to 200 mg for several parameters (proportion of responders at 1 h, proportion rescuing by 2 h, and proportion without photophobia at 4 h).
In addition, higher doses of ibuprofen separated from placebo at earlier time points for both the cumulative proportion of responders and proportion of subjects pain-free. As shown in Fig. 1, 400 and 200 mg were significantly better than placebo beginning at 1 h for headache response, and at 1.5 and 2 h, respectively, for ‘pain-free’ response. The 600 mg dose separated from placebo for headache response at 0.5 h, and for ‘pain-free’ response at 1 h. These findings are reflected in Table 4, which demonstrates a trend towards faster median times to headache ‘response’ and to ‘pain-free’ response with increasing dose. In summary, although the individual time point data and maximal values for ‘responders’ and ‘pain-free’ do not indicate a significant dose–response, the data suggest that increasing doses may provide faster migraine pain relief.
The ‘upside sensitivity’ (i.e. ability to separate higher doses) in this study may have been limited due to the relatively high placebo response rate. This may have been due, at least in part, to the large proportion (69.4%) of subjects treating attacks with moderate baseline intensity, as opposed to 29.6% with severe migraines. Supporting this, the placebo group had a 55.6% headache response at 2 h for subjects with moderate pain, compared to 32.4% for those with severe baseline pain. It is noteworthy that all doses of ibuprofen were also effective in the severe subgroup, with 2-h response rates of 32%, 56%, 67% and 60% for placebo, 200, 400 and 600 mg, respectively (all P < 0.01 compared to placebo).
Although it is difficult to make cross-study comparisons, ibuprofen liquigels 400 mg and 600 mg had efficacy similar to acetaminophen + aspirin + caffeine (500 mg + 500 mg + 130 mg). For example, for headache response at 2 h, the overall ‘therapeutic gain’ (i.e. difference between active treatment and placebo) for ibuprofen 400–600 mg was about 22%, while for acetaminophen + aspirin + caffeine, the therapeutic gain pooling across three studies was 26% (10). Using the headache response values for subjects with severe baseline pain in the previous paragraph, the ‘therapeutic gain’ for ibuprofen 400–600 mg in the severe subgroup was even greater, approximately 28–35%.
In addition to a beneficial effect on migraine pain, ibuprofen liquigels significantly improved the limitation of activity associated with migraine at all three doses, as assessed by the proportion with ‘no or mild’ limitation of activity at 2–8 h. These results suggest that ibuprofen helps restore ability to function. These findings are similar to those reported for acetaminophen + aspirin + caffeine using a ‘functional disability’ scale (10).
The present results also indicate that ibuprofen liquigels are effective in relieving the ancillary symptoms which accompany migraine headache. Specifically with headache-associated nausea, 200 and 400 mg of ibuprofen significantly increased the proportion of subjects without nausea in a dose-related manner. With respect to photophobia and phonophobia, the results consistently indicate that ibuprofen liquigels, especially the two higher doses, significantly improved these ancillary symptoms. Taken together, these findings indicate that ibuprofen liquigels relieve the entire symptom complex of a migraine attack.
The duration of analgesia (i.e. time to taking rescue medication) was significantly longer for all three doses of ibuprofen in comparison to placebo. Furthermore, 200 mg numerically reduced, whereas 400 mg and 600 mg significantly reduced, 24-h migraine recurrence rates compared to placebo.
Although only a small number of adolescents (n = 35) were included in this study, the results suggest that ibuprofen liquigels would also be efficacious in the treatment of migraine in this age group. The very high placebo response (> 80%) at 240 min and beyond in adolescent subjects suggests that the migraine episode had ended.
The safety data indicate that all three doses of ibuprofen liquigel were safe and well-tolerated. No dose increased the incidence or severity of overall or individual AEs compared to placebo, and all doses were comparable. Further, there were no serious AEs and no discontinuations due to an AE. Of particular interest, there was no increase in the incidence of digestive system AEs. These results are consistent with recent favourable publications on the safety of OTC ibuprofen (24–26), and also indicate that ibuprofen retains its favourable safety profile in the treatment of migraine. In contrast, the incidences of overall and certain individual AEs for the pooled acetaminophen + aspirin + caffeine trials were significantly greater than placebo (10). Specifically, the incidences of nausea (4.9% vs. 1.7%) and nervousness (4.4% vs. 0.8%) were significantly greater for acetaminophen + aspirin + caffeine than for placebo (10), whereas the incidences of nausea and nervousness for ibuprofen in the present study (Table 5) were very low (maximal incidences of 1.0% (200 mg) and 0.5% (600 mg), respectively) and similar to placebo (0.7% and 0%, respectively).
In conclusion, the results of this study indicate that ibuprofen liquigels reduce the pain, ancillary symptoms and limitation of activity associated with migraine headache over the dose range of 200 mg to 600 mg. All doses were safe and well-tolerated. These data indicate that ibuprofen liquigels may provide a safe and effective alternative to caffeine-containing medications for the acute self-treatment of migraine headache.