
Abstract
Objective
Alveolar hypoxia as a result of high altitude leads to increased pulmonary arterial pressure. The renin-angiotensin system is involved in the regulation of pulmonary arterial pressure through angiotensin-converting enzyme 2 (ACE2). It remains unknown whether ACE2 administration alters pulmonary vascular pressure in hypoxia.
Methods
We investigated 12 anesthetized pigs instrumented with arterial, central venous, and Swan-Ganz catheters exposed to normobaric hypoxia (fraction of inspired oxygen = 0.125) for 180 minutes. After taking baseline measurements in normoxia and hypoxia, ACE2 400 μg·kg−1 was administered to 6 animals, and another 6 served as control. Ventilatory variables, arterial blood gases, ventilation/perfusion (
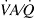
Results
We observed lower pulmonary arterial pressure (maximum: 30 vs 39 mm Hg, P < .01) and lower pulmonary vascular resistance (maximum: 4.1 vs 7.5 Wood units, P <.01) in animals treated with ACE2. There was a trend (P =.09) toward lower angiotensin II plasma concentrations among ACE2-treated animals. Cardiac variables and systemic arterial pressure in hypoxia remained unaffected by ACE2. Ventilation/perfusion relationships and Pa
Conclusions
In acute pulmonary hypertension, administration of ACE2 blunts the rise in pulmonary arterial pressure that occurs in response to hypoxia. Recombinant ACE2 may be a treatment option for high altitude pulmonary edema and hypoxia-associated pulmonary hypertension.
Keywords
Introduction
The alveolar hypobaric hypoxia associated with high altitude leads to increases in pulmonary arterial pressure and pulmonary vascular resistance (PVR). 1 Increased pulmonary pressure at altitude may predispose to (but does not necessarily lead to) high altitude pulmonary edema (HAPE). Chronic hypobaric hypoxia may lead to vascular remodeling, right ventricular hypertrophy, and death. 1 The etiology of HAPE and pulmonary hypertension is complex and various; however, the pulmonary vascular endothelium plays a pivotal role in the balance of pulmonary vascular tone. 1
It has been known for more than 10 years that the renin-angiotensin system (RAS) is involved in the regulation of pulmonary vascular tone by the endothelium. 2 Moreover, the RAS may promote vascular remodeling in the lung: renin is released from the juxtaglomerular cells of renal arterioles and cleaves the circulating precursor angiotensinogen into the inactive angiotensin I (Figure 1). 3 The latter is then hydrolyzed by angiotensin-converting enzyme (ACE) into angiotensin II (Ang II), a potent vasoconstrictive agent and promoter of vessel hypertrophy through the G-protein coupled angiotensin (AT1) receptor. 4 In a rodent model, blocking AT1 during 7 days of exposure to hypoxia led to decreased pulmonary hypertension, decreased right ventricle hypertrophy, and decreased vascular remodeling. 5
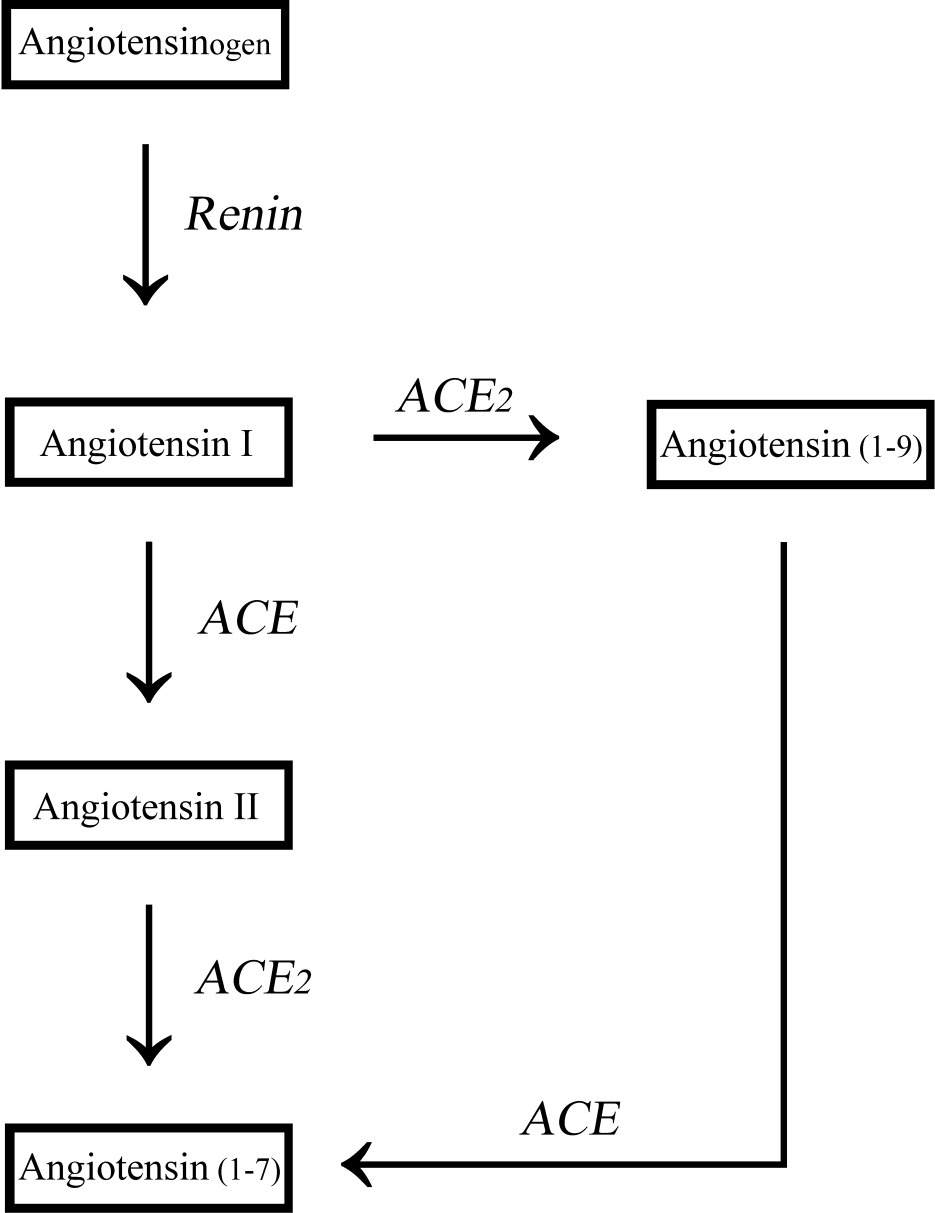
The renin-angiotensin system. Angiotensin-converting enzyme 2 (ACE2) reflects both ACE2 and ACE, the classic angiotensin-converting enzyme.
This vasoconstrictive component of the RAS is balanced with a vasodilatory component that has only recently been discovered: a homologue of ACE called ACE2. 2 ACE2 degrades vasoconstrictive Ang II to the vasodilator peptide angiotensin (1-7) (Ang [1-7]; Figure 1). 6 As a result, ACE2 compensates the vasoconstrictive axis of the RAS by lowering Ang II and producing vasodilatory Ang (1-7). Thus, ACE2 and Ang (1-7) may be considered the vasoprotective axis of the RAS. 6
Interestingly, an alternative pathway to generate Ang (1-7) is the following:
where Ang (1-9) is an inactive precursor. In this setup, the antagonistic homologues ACE and ACE2 work hand in hand to cleave the vasodilatory Ang (1-7) from angiotensin I. That is, classic ACE also plays a role in the vasodilatory component of the RAS. However, this pathway seems to be less important.
The ACE inhibitors, such as captopril, do not result in pulmonary hypertension, although Altieri et al 7 reported a transient elevation of pulmonary arterial pressure immediately after administration. ACE2 itself is insensitive to ACE inhibitors. 2 For a graphic outline of the RAS, see Figure 1. ACE2 is expressed in cardiac and renal, and also in pulmonary vascular, endothelium; and ACE2 seems to be central to cardiac and pulmonary endothelial function. 6 Experimentally observed effects by inhibition of endogenous ACE2 on circulation and lung include: 1) attenuation of the severe acute respiratory distress syndrome in mice; 8 2) upregulation of hypoxia-inducible genes in ACE2 null mice; and 3) decreased cardiac contractility in ACE2 null mice. 9
As Ang II and the AT1 receptor were found to play a role in rat hypoxia-associated pulmonary hypertension,
5
we theorized that ACE2 administration lowers pulmonary arterial pressure and resistance. These 2 variables are pivotal in the development of HAPE.
10
Furthermore, to observe the effect of a possible vasodilation on pulmonary gas exchange, we used the multiple inert gas elimination technique (MIGET) and hypothesized that ACE2 leads to a redistribution of pulmonary blood flow to lung units with subnormal ventilation/perfusion
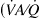
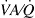
Methods
Protocol
Twelve pigs were anesthetized and instrumented. Experiments were performed at an altitude of 540 m above sea level. A baseline measurement in normoxia (fraction of inspired oxygen [FI
Animal Anesthesia and Instrumentation
All experiments conformed to the guidelines of the US National Institutes of Health and were approved by the Austrian Federal Animal Investigational Committee. Twelve healthy, 4-week-old cross-breed pigs (German Landrace × Pietrain) of either gender were selected from a local stock regularly used for experimental research. For induction of anesthesia, animals received ketamine (25 mg·kg−1 intramuscularly) and atropine (0.01 mg·kg−1 intravenously). After tracheal intubation, propofol was started at a rate of 10 to 15 mg·kg−1·h−1. All pigs were mechanically ventilated in volume-controlled mode (Evita2; Dräger, Lübeck, Germany) with a FI
Throughout the experiment, Ringer's solution (6 mL·kg−1·h−1) and a colloid (gelatin solution, 4 mL·kg−1·h−1) were infused to compensate for insensible loss (ventilation, evaporation). The colloid helps to keep infusion volumes small, as it remains longer in the circulation than a crystalloid. A standard lead II electrocardiogram was used to monitor cardiac rhythm. Body temperature was measured using the indwelling thermistor-tipped Swan-Ganz catheter and maintained between 38°C and 39°C (physiologic body temperature of the pig) by using an electric heating blanket. A 8.5F arterial catheter was advanced from the internal jugular vein into a branch of the pulmonary artery to measure central venous pressure, mean pulmonary arterial pressure (mPAP), and pulmonary capillary wedge pressure (PCWP). Cardiac output was assessed using the thermodilution technique (10 mL cooled saline in triplicates). A 6.0F arterial catheter introduced into the femoral artery was used to monitor systemic blood pressure and to take blood samples. All catheters were filled with saline and connected to pressure transducers zeroed to ambient pressure at the level of the right atrium. Measurements included hemodynamic, ventilatory, and blood gas and inert gas parameters. Arterial blood and multiple inert gas samples were drawn at normoxic baseline, hypoxic baseline, and at 30, 90, and 150 minutes. Ventilator settings were not changed during the experimental period.
Multiple Inert Gas Elimination Technique
The MIGET11,12 helps to determine pulmonary gas exchange and, particularly, diffusion limitation, which are typically altered during exercise and altitude. Six inert gases dissolved in saline were continuously infused at a rate in mL/min equal to one fourth of the respiratory minute ventilation in L/min.11,12 Samples of mixed expired gas and arterial blood samples were collected into gas-tight glass syringes. The concentrations of the 6 inert gases in expired air and arterial blood were measured using gas chromatography (Hewlett-Packard 5890, series II; Hewlett-Packard, Palo Alto, CA). The
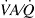
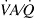
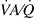
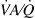
Ventilatory Measurements
Ventilatory measurements and calculations included expiratory tidal volume and ventilatory minute volume using a calibrated Wright Respirometer (Ferraris Medical Inc, Holland, NY). Dead space ventilation (alveolar or conducting areas in the lung, where no gas exchange occurs) was obtained from MIGET.
ACE2 Isolation
The ACE2 was expressed recombinantly in stable transfected CHO cells under protein-free conditions in a synthetic medium (Polymer Scientific, Ontario, NY). The expression product was purified to homogeneity (purity >98%). Three laboratory scale ACE2 batches were used in this study. All ACE2 batches were compared with the commercially available ACE2 933-ZN (R&D Systems, Minneapolis, MN) and to an in-house standard. Biochemical and immunological properties of all products were almost identical in terms of biochemical activity: specific enzymatic activities were characterized using the fluorescent-labeled triple tide adenosine-5′-phosphosulfate-kinase (APK) and Ang II as substrates.
Statistical Evaluation
A 2-way analysis of variance was used to determine intergroup and intragroup differences. Significant results were post hoc analyzed using the Newman-Keuls test. The post-hoc tests helped to eliminate false-positive observations (alpha error). Results are given as mean ± SEM in the Figures and as mean ± SD in the Tables. All P values ≤.05 were considered significant.
Results
Blood Gas Variables
Hypoxia with an FI
Arterial blood gas and inert gas measurements
Values are mean ± SD.
ACE2, angiotensin-converting enzyme 2; Pa
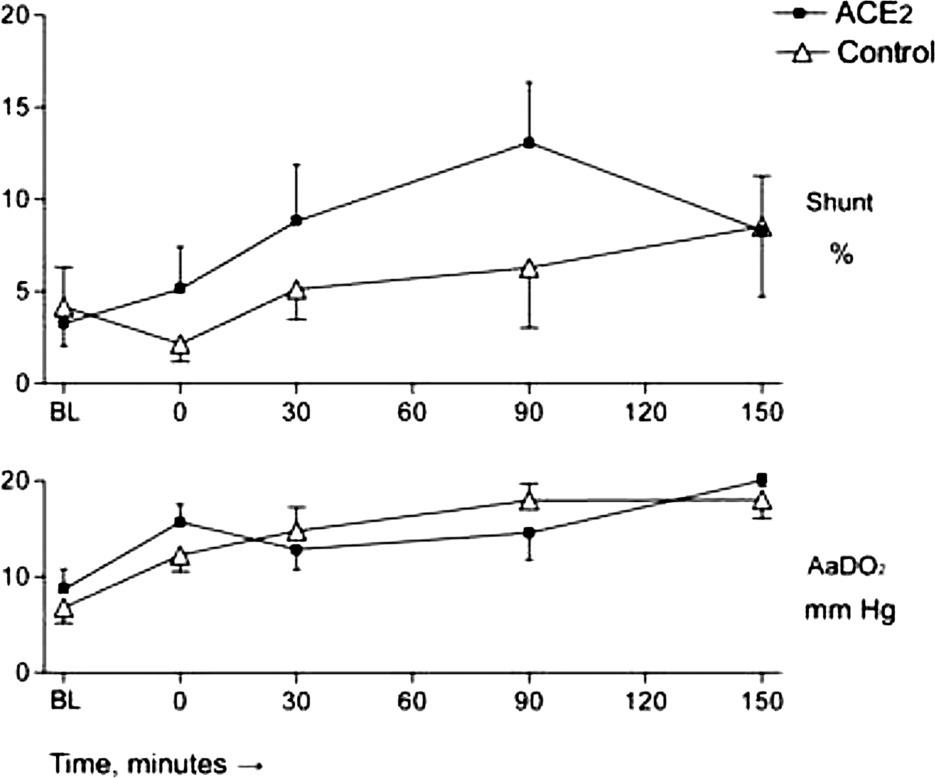
Multiple inert gas elimination technique parameters. Shunt refers to blood flow to essentially unventilated lung units; AaDO2 is the alveolo-arterial partial pressure difference in mm Hg. Hypoxia or hypoxia and angiotensin-converting enzyme 2 (ACE2) were introduced at time point zero (0). BL, baseline.
Inert Gas Variables
Hypoxia expectedly caused heterogeneity in pulmonary blood flow, as indicated by Log
Hemodynamic Variables
There were no intergroup differences in systemic blood pressures or cardiac output. Animals treated with ACE2 had lower mPAP and a lower PVR 60 to 90 minutes after administration (Figure 3). Indexed pulmonary vascular resistance (PVRI) and mPAP in percent of the normoxic baseline values were different between groups (Table 2). The PCWP was unaffected by ACE2 administration (Table 2). The PVR is no longer different between groups after 120 minutes, an observation that may be associated with the application of ACE2 as a single dose.
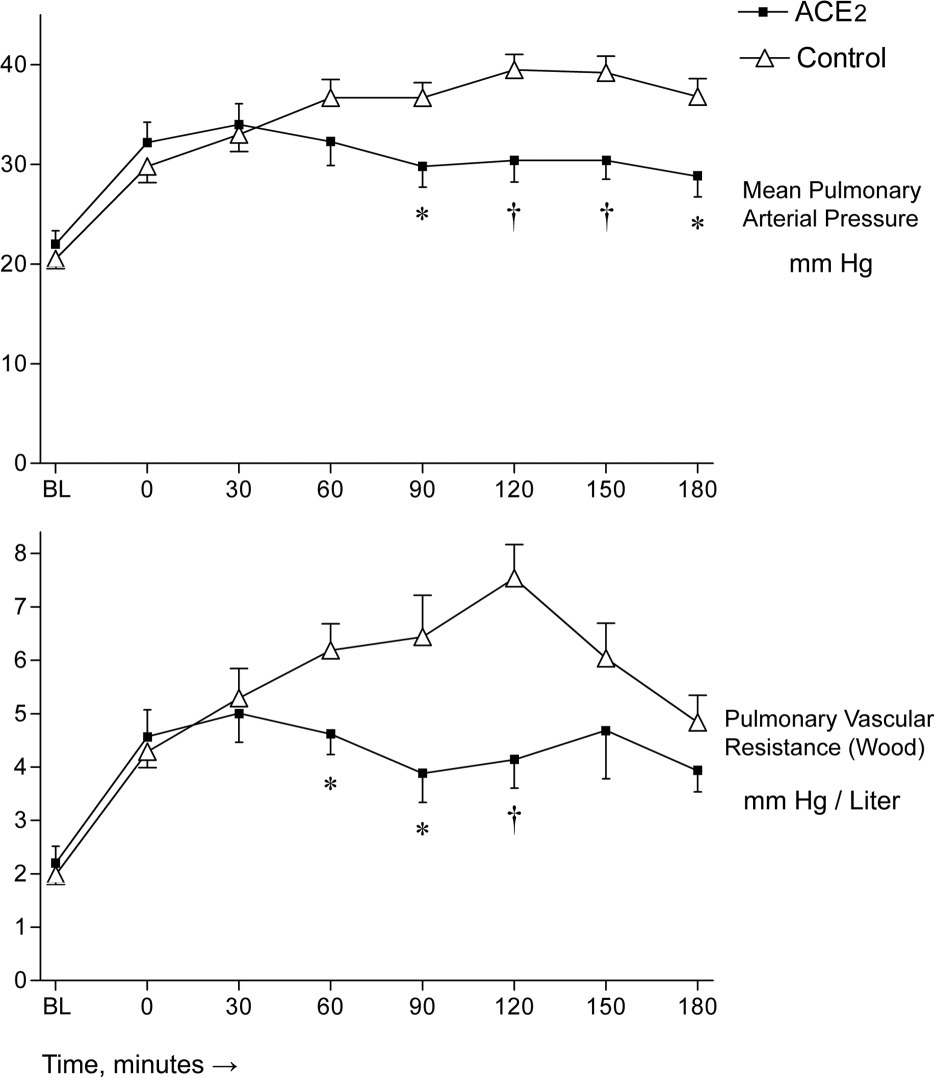
Pulmonary vascular parameters. Hypoxia or hypoxia and angiotensin-converting enzyme 2 (ACE2) were introduced at time point zero (0). *P value <.05; †P value <.01. BL, baseline.
Hemodynamic measurements and calculations
Values are mean ± SD.
ACE2, angiotensin-converting enzyme 2; PVRI, pulmonary vascular resistance index, dyn·s·cm−5·m−2; PCWP, pulmonary capillary wedge pressure, mm Hg; MAP, mean arterial pressure, mm Hg; CVP, central venous pressure, mm Hg; HR, heart rate; mPAP % BL, mean pulmonary arterial pressure in percent of baseline.
P <.05.
P <.01.
Ventilatory Variables
The respiratory minute volume needed to achieve a Pa
Angiotensin II Plasma Concentrations
There were no intergroup differences between ACE2 treatment and placebo (Figure 4).
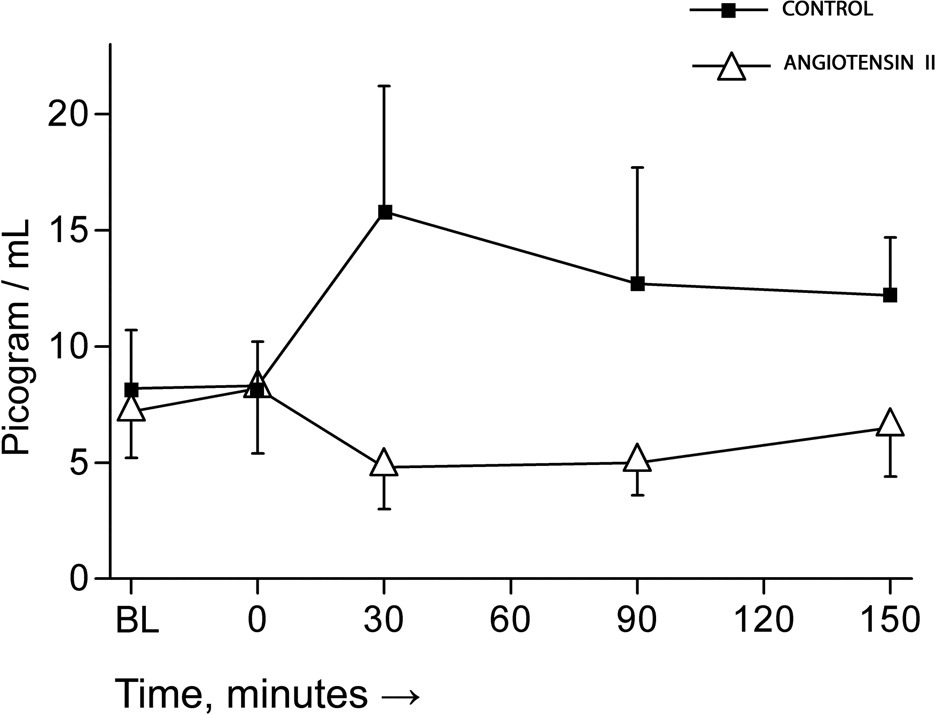
Angiotensin II plasma concentrations before and after administration of hypoxia or hypoxia and angiotensin-converting enzyme 2.
Discussion
In this experiment involving anesthetized hypoxic pigs, administration of ACE2 blunted the rise in pulmonary arterial pressure and decreased PVR. Pulmonary gas exchange, cardiac performance, and systemic arterial pressure remained unaffected by ACE2 administration, and therefore, ACE2 selectively affected the pulmonary circulation in hypoxia.
Hypoxemia at any altitude is based on increases in pulmonary blood flow heterogeneity, diffusion limitation, and shunt, and also on the hypoxia itself. Pulmonary blood flow heterogeneity and shunt are likely to be changed by a pulmonary vasodilator. In our experiment, there was a trend toward more shunting in animals treated with ACE2; however, the study was underpowered to study this hypothesis. This change was also too small to be reflected in Pa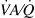
The ACE2 administration did not change the heterogeneity of pulmonary perfusion. According to current understanding, hypoxia results in irregularly distributed vasoconstriction in the lung; therefore, in these more constricted and less constricted sections, ACE2 could have led to selective dilation of the more constricted segments, resulting in a decrease in PVR and Log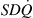
Possible Applications in Altitude Medicine
Drugs used to prevent or treat HAPE include nifedipine, sildenafil, and tadalafil—the latter two being widely used phosphodiesterase type 5 inhibitors.1,13 By its effect on mPAP, recombinant ACE2 could be useful in treating HAPE. However, intravenous infusion is certainly hard to perform at altitude, and the effects of subcutaneous or intramuscular administration are yet to be established. Oral administration is not possible with a peptide.
Other Potential Clinical Applications
The World Health Organization classifies pulmonary hypertension associated with hypoxia and pulmonary hypertension without hypoxia as two separate entities. 13 ACE2 may be a further treatment option for the pulmonary hypertension, particularly since its effect on preventing pulmonary vascular remodeling has already been reported. 5 Because ACE2 lowers mPAP and PVR but does not lower systemic pressure, it may have advantages in a critical care setting over most other drugs that lower mPAP and PVR and also lower systemic pressure.
Future Research and Directions
At present, the ACE blockers and angiotensin-receptor blockers are used to attenuate the vasoconstrictive leg of the renin-angiotensin-aldosterone system (RAAS). These drugs are central in the treatment of hypertension and its consequences. The relatively recent discovery of “the other ACE,” ACE2, indicates that the RAAS is more complex than previously thought, and that it is also involved in pulmonary hypertension and lung failure.
It would be interesting to see whether ACE2 could prevent the development of HAPE in the HAPE-susceptible population, namely, persons who have already been diagnosed with HAPE. It would also be interesting to examine ACE2 plasma concentrations in persons chronically exposed to hypoxia, for example, those living in high altitude locations such as the Andes; furthermore, HAPE may develop when such persons return to high altitude (reentry HAPE) after a sojourn to a lower altitude.
Given the effect of ACE2 on acute respiratory distress syndrome and lipopolysaccharide-induced sepsis, ACE2 may be a treatment option after tracheal aspiration, and for persons presenting to the Emergency Department with aspiration pneumonia.
Study Limitations
As always, data obtained from anesthetized animals cannot directly be applied to humans. Plasma levels of ACE2 have not been measured in this experiment, and that may be important for determining the pharmacokinetics of ACE2. Doses for humans cannot be deduced from this study, and dose-response studies warranted. The rationale for the dose used in this experiment was a dose-finding study in the pig done for our prior experiment.
14
The MIGET does not give a unique result for 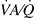
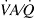
Conclusions
Administration of recombinant ACE2 to anesthetized hypoxic pigs blunted the rise in mPAP and PVR, while all other physiologic variables observed remained unaffected. ACE2 may be a treatment option for cases of pulmonary hypertension in which hypoxia is part of the etiology.