
Abstract
A synthetic Candida antarctica lipase B (CALB) gene open reading frame (ORF) for expression in yeast was constructed, and the lycotoxin-1 (Lyt-1) C3 variant gene ORF, potentially to improve the availability of the active enzyme at the surface of the yeast cell, was added in frame with the CALB ORF using an automated PCR assembly and DNA purification protocol on an integrated robotic workcell. Saccharomyces cerevisiae strains expressing CALB protein or CALB Lyt-1 fusion protein were first grown on 2% (w/v) glucose, producing 9.3 g/L ethanol during fermentation. The carbon source was switched to galactose for GAL1-driven expression, and the CALB and CALB Lyt-1 enzymes expressed were tested for fatty acid ethyl ester (biodiesel) production. The synthetic enzymes catalyzed the formation of fatty acid ethyl esters from ethanol and either corn or soybean oil. It was further demonstrated that a one-step-charging resin, specifically selected for binding to lipase, was capable of covalent attachment of the CALB Lyt-1 enzyme, and that the resin-bound enzyme catalyzed the production of biodiesel. High-level expression of lipase in an ethanologenic yeast strain has the potential to increase the profitability of an integrated biorefinery by combining bioethanol production with coproduction of a low-cost biocatalyst that converts corn oil to biodiesel.
Introduction
Most of the fuel ethanol produced in the United States is made from corn starch in either wet-mill or dry-grind ethanol facilities. Projections indicate that corn supplies will not be able to meet the increasing demand for biofuels. Lignocellulosic biomass, an abundant and renewable carbon source, has the potential to supplement or replace starch feedstocks for the production of fuel ethanol, 1 but current technology is constrained by production costs. The profitability of ethanol production from lignocellulosic biomass will be improved if high-value coproducts are also generated. Current processes for fuel ethanol production from starch yield substantial amounts of corn oil as a byproduct. The corn oil is used for the manufacture of biodiesel, thereby removing the oil from the dried distiller grain solubles to give more digestible defatted animal feed. Corn oil triacylglycerides (TAGs) are converted to fatty acid ethyl esters (biodiesel) and glycerol by transesterification with ethanol. One method of catalyzing this transesterification reaction is using lipase enzymes. 2 An integrated biorefinery combining starch ethanol and cellulosic ethanol facilities may become more cost effective if biodiesel is produced as a coproduct using lipase-catalyzed single-step column trans-esterification, 3 with low-cost lipases expressed in large quantities in a recombinant yeast strain capable of cellulosic ethanol production. Such a strain is the recently developed yeast using the major sugars, glucose, mannose, arabinose, and xylose (GMAX), in woody or agricultural biomass hydrolysate anaerobically to produce ethanol. 4,5 The GMAX recombinant yeast strain has been engineered to use cellulosic biomass as well as corn starch and could also be engineered to express lipases for biodiesel production from the corn oil byproduct of the starch ethanol processes in an integrated biorefinery.
Biodiesel is defined as the monoalkyl esters of long-chain fatty acids derived from vegetable oil or animal fats (TAGs). 6 Today, most biodiesel is produced by transesterification of these TAGs with a short-chain alcohol (usually methanol but sometimes ethanol) based on the homogeneous catalyst method using strong bases, such as sodium, potassium hydroxide, or methoxide. 7 –9 When methanol is used, the products of the reaction are fatty acid methyl esters (FAME) along with glycerol (glycerin). The most common feedstock for FAME biodiesel in the United States is soybean oil, but other oils also may be used. 2,8 In fact, corn oil biodiesel achieves superior cold flow properties compared with soybean biodiesel. 10 The catalyst is dissolved in the alcohol and charged into a closed reaction vessel. The oil or fat is added, and the mixture is heated just above the boiling point of the alcohol, normally for 1 or 2 h. 9 Excess alcohol is used to ensure total conversion to the fatty acid esters. If the levels of free fatty acids or water in the oil or fat are too high, problems, such as saponification (production of fatty acid sodium salts [soaps] rather than fatty acid esters) and difficulty in separation of the glycerol byproduct downstream, will occur. At the end of the reaction, the denser glycerol phase is eliminated by gravity separation, and the alcohol is removed by flash evaporation or distillation for reuse. Care must be taken to ensure that no water accumulates in the alcohol stream, which would drive the reaction in the reverse direction. The biodiesel is washed with water to remove residual catalyst or soaps.
However, this method has a number of drawbacks: (1) acid pretreatment is required of feedstocks high in free fatty acids; (2) residual water deactivates the alkali catalyst; (3) unwanted production of water as byproduct occurs if free fatty acids are present in the case of hydroxide catalysts; (4) excessive water is consumed during washing and large amounts of wastewater are generated; and (5) fatty acids, alkali metals, water, and glycerin must be removed from the final biodiesel product. 8 –13 These issues make it difficult to meet the American Society for Testing and Materials (ASTM) D6751 biodiesel fuel standard, which is becoming increasingly stringent. 6 Additionally, the high cost of commodity vegetable oils, such as soybean oil in the United States, represents a serious threat to the economic viability of the biodiesel industry. Currently, feedstock acquisition accounts for up to 85% of the costs associated with biodiesel production. 14,15 Producing biodiesel from corn oil extracted at an ethanol production site would be one option to reduce biodiesel production expenses.
Use of lipases as biocatalysts to accomplish transesterification would also help to decrease the costs associated with the traditional method of biodiesel production and to overcome some of the technical drawbacks, 2,16 especially if large amounts of the lipases are expressed inexpensively in the yeast used in ethanol production processes in the biorefinery. The biocatalytic process not only represents a simplification over the traditional biodiesel production process, reducing production costs, but also produces a high-quality fuel. Other advantages of using lipases include the following: (1) ease of product recovery; (2) use of mild reaction conditions while minimizing energy consumption; (3) regeneration and reuse of the enzyme over several cycles; (4) use in conjunction with a column containing ionic solid-phase catalyst; (5) use with FAME process to esterify free fatty acids with lipase at the exit port; (6) high selectivity; (7) flexibility of accepting various substrates and alcohols; and (8) ability to react in both solvent and solvent-free systems. 2 Typical biodiesel production using homogeneous alkaline catalysts requires more expensive refined fats or oils as feedstocks to avoid problematic saponification. 8,17 The lipase-catalyzed process is capable of simultaneous esterification and transesterification, thus, eliminating the need for cost-prohibitive refined oils or fats. In addition, purification of biodiesel can be accomplished on a resin to produce a high-quality product that satisfies ASTM D6751 specifications. 18,19 Although the cost of the enzymatic catalyst remains a hurdle compared with that of the less-expensive chemical catalysts, the use of recombinant DNA technology to produce large quantities of lipases and the use of immobilized lipases may lower the cost of biodiesel production, while reducing downstream processing problems. 2 20 –22
Differences have been observed in the catalytic activity of a lipase enzyme immobilized on different supports having different functional groups on their surface, allowing either physical or chemical adsorption. 2,19,23 Surface structure of the support can affect enzyme activity through direct effects (type, strength, and orientation of enzyme-support interactions) or indirect effects (substrate-product interaction with the support affecting substrate-product partitioning between medium and binding site). In one study, the lowest loading of enzyme on a support showed the highest catalytic efficiency (activity of loaded protein). Efficiency was inversely related to loading, possibly because high loading means high enzyme-support affinity as a result of very strong interactions. 19 This suggests that strong enzyme-surface interactions might distort the dynamics of the bound enzyme molecules required for catalytic activity. Weaker interactions appear more likely to preserve the enzyme in its active conformation. 19
Optimization of lipases and development of strains to express large quantities of these biocatalysts are necessary for inexpensive production of biodiesel. The scripting of automated protocols and scheduling of PCR assembly steps on the robotic workcell developed in our laboratory have the potential to be used in an iterative fashion for production of any gene open reading frame (ORF) and are demonstrated in this work for the production of a lipase gene ORF. Rapid production of gene ORFs is essential for large-scale production of libraries of ORFs from full genome sequences 24 or from systematically mutagenized optimized sequences based on a single gene ORF. 25,26
It is also possible to immobilize lipases on specialty resins to establish a continuous single-step column process for bio-diesel production, even with a high content of free fatty acids in the oil. Sepabeads EC-EP (Resindion S.r.l., Milan, Italy) are polymethacrylate beads containing epoxy functional groups on their surfaces. These beads are able to anchor lipase catalysts to their surfaces through covalent linkages with the epoxy functional groups. 27 A polyhistidine tag on the lipase facilitates a nucleophilic substitution reaction, which causes immobilization to occur. The beads are durable and may be used in stirred batch reactors or high-flow-rate column processes.
This report describes the automated PCR assembly and amplicon purification techniques used in the production of Candida antarctica lipase B (CALB) gene ORFs. The assembled CALB enzymes are expressed in yeast and assayed for the production of fatty acid ethyl esters (biodiesel). The expressed CALB enzyme is also immobilized on Sepabeads, and the activity of the immobilized enzyme in the production of biodiesel is compared with that of nonimmobilized expressed CALB enzyme.
Materials and Methods
Polymerase Chain Reaction Assembly of Oligonucleotides to Produce Candida antarctica Lipase B Gene Open Reading Frame
The assembled ORF was based on the CALB sequence registered in GenBank as Z30645. The GenBank sequence consists of 1029 nucleotides. A CACC sequence for directional cloning into pENTR D TOPO was added at the 5′ end so the final assembled CALB ORF consisted of 1030 nucleotides. In addition, the TGA stop codon at the 3′ end was modified to a TAA stop codon. An overall stepwise assembly method was implemented, in which nine increasingly longer PCR amplicons, 6 at the 5′ end of the CALB sequence and 3 at the 3′ end, were created sequentially from 38 oligonucleotides (Sigma Aldrich, St. Louis, MO), which included 36 50mer oligonucleotides, one 40mer oligonucleotide, and one 15mer oligonucleotide to construct the complete CALB ORF (Fig. 1; forward oligonucleotides are shown in yellow and the overlapping reverse oligonucleotides in tan). Each oligonucleotide overlapped at the 5′ end with 25 nucleotides of the previous oligonucleotide and overlapped at the 3′ end with 25 nucleotides of the next oligonucleotide (Table 1). The first six oligonucleotides were used to initiate the formation of the CALB ORF. The amplicon from the PCR assembly of these six oligonucleotides was added to the next PCR mixture together with the two oligonucleotides at the 3' end of the amplicon plus the next four oligonucleotides in the sequence that overlapped each other by 25 nucleotides at either end. The two-oligonucleotide overlap at the 3′ end of the amplicon formed a strongly annealed link to that template and ensured sequential assembly. Twenty-six of the 38 oligomers were added in this fashion. However, when the same strategy was attempted to add the next four oligonucleotides, no band corresponding to the expected sequence (oligomers 1–30) was obtained on the agarose gel. It is possible that one or more of the individual oligonucleotides, intended for addition at the 3′ end of the 1–26 template, by annealing to each other, and to the 3′ end of the template, instead preferentially annealed to regions within the 1–26 sequence. To conserve the oligonucleotides that were available, the strategy was modified so that the last section (oligonucleotides 25–38) of the CALB ORF was constructed separately, beginning with the oligonucleotide set Y-Z-AA-BB-CC-DD. The two sections (1–26 and 25–38) were then joined by means of PCR to give the complete CALB ORF.
Diagram of stepwise assembly strategy used to construct the CALB expression plasmids. Nine increasingly longer PCR amplicons, six at the 5ℙ end of the CALB sequence and three at the 3ℙ end, were created sequentially from 38 oligonucleotides (forward shown in yellow and reverse shown in tan) that included 36 50mer oligonucleotides, one 40mer oligonucleotide, and one 15mer oligonucleotide for CALB 1–38. Template 1–26 and template 25–38 were combined using PCR to give the CALB 1–38 construct (top). PCR assembly and addition of five oligonucleotides containing the Lyt-1 sequence to the 3ℙ end of CALB 1–38 to give CALB Lyt-1 1–43 were performed on the robotic workcell. Purification of CALB 1–38 and CALB Lyt-1 1–43 was followed by TOPO ligation of each directionally into pENTR D TOPO and LR clonase recombinational cloning into pYES2 DEST 52 yeast expression vector (bottom).
Sequences of the 43 oligonucleotides used in the PCR assembly of CALB 1–38 and CALB Lyt-1 1–43 ORFs

The PCR mixture for the step producing CALB template 1–6 (Fig. 1) contained 38 μL H2O, 10 μL 5x Phusion HF Buffer, 1 μL 10-mM deoxynucleoside triphosphate (dNTPs), 0.5 μL oligonucleotide mixture, and 0.5 μL Phusion Enzyme (Finnzymes Phusion High-Fidelity PCR kit; New England Biolabs, Ipswich, MA). The oligonucleotide mixture consisted of 10 μL (1 μg/μL) each of the six oligonucleotides, A, B, C, D, E, and F (Table 1). The reaction was prepared in a Bio-Rad hard-shell 96-well PCR plate (Bio-Rad Laboratories, Hercules, CA) on ice, placed into a PTC-225 Tetrad (Bio-Rad Laboratories) at 98 °C for 2 min and run using the following cycle: 98 °C for 40 s, 50 °C for 30 s, 12 °C for 45 s, repeated 30 times, followed by 12 °C for 7 min and 4 °C for 5 min. The amplified DNA was separated using agarose gel electrophoresis and observed with an AlphaImager 3400 (Alpha Innotech Corporation, San Leandro, CA) using a trans-ultraviolet light. A 180-base-pair fragment, corresponding to amplicon 1–6, was isolated and purified by the GENECLEAN II method (GENECLEAN II kit; MP Biomedicals, Irvine, CA). The purified DNA was used as the template for the next PCR.
The next five steps to obtain CALB sequence 1–26 (Fig. 1) were performed using similar PCR conditions, except the PCR mixture to produce CALB template 1–10 contained 2 μL (0.1 μg/μL) amplicon 1–6 and 0.5 μL of a mixture consisting of 10 μL(1 μg/μL) of forward primer A and 10 μL (1 μg/μL) each of oligonucleotides, E, F, G, H, I, and J (Table 1), yielding a 290-base-pair fragment; the PCR mixture to produce CALB template 1–14 contained 2 μL (1 μg/μL) amplicon 1–10 and 0.5 μL of a mixture consisting of 10 μL(1 μg/μL) of forward primer A and 10 μL(1 μg/μL) each of oligonucleotides, I, J, K, L, M, and N, yielding a 400-base-pair fragment; the PCR mixture to produce CALB template 1–18 contained 2 μL (0.1 μg/μL) amplicon 1–14 and 0.5 μL of a mixture consisting of 10 μL(1 μg/μL) of forward primer A and 10 μL(1μg/μL) each of oligonucleotides, M, N, O, P, Q, and R, yielding a 510-base-pair fragment; the PCR mixture to produce CALB template 1–22 contained 2 μL (0.1 μg/μL) amplicon 1–18 and 0.5 μL of a mixture consisting of 10 μL(1μg/μL) of forward primer A and 10 μL (1 μg/μL) each of oligonucleotides, Q, R, S, T, U, and V, yielding a 620-base-pair fragment; the PCR mixture to produce CALB template 1–26 contained 2 μL (0.1 μg/μL) amplicon 1–22 and 0.5 μL of a mixture consisting of 10 μL (1 μg/μL) of forward primer A and 10 μL (1 μg/μL) each of oligonucleotides, U, V, W, X, Y, and Z, yielding a 730-base-pair fragment corresponding to amplicon 1–26.
A similar approach was used to obtain amplicon 25–36 at the 3′ region. The PCR mixture to produce CALB template 25–30 (Fig. 1) contained 38 μL H2O, 10 μL 5x Phusion HF Buffer, 1 μL 10-mM dNTPs, 0.5 μL oligonucleotide mixture, and 0.5 μL Phusion enzyme. The oligonucleotide mixture consisted of 10 μL (1 μg/μL) each of oligonucleotides, Y, Z, AA, BB, CC, and DD (Table 1). A 180-base-pair fragment, corresponding to amplicon 25–30, was isolated. The PCR mixture to produce CALB template 25–34 contained the same reagents as described previously except that it had 2 μL (0.1 μg/μL) amplicon 25–30 and 0.5 μL of a mixture consisting of 10 μL (1 μg/μL) of forward primer Y and 10 μL (1 μg/μL) each of oligonucleotides, CC, DD, EE, FF, GG, and HH, yielding a 290-base-pair fragment. The PCR mixture to produce CALB template 25–38 contained 2 μL (0.1 μg/μL) amplicon 25–34 and 0.5 μL of a mixture consisting of 10 μL(1μg/μL) of forward primer Y and 10 μL(1 μg/μL) each of oligonucleotides, GG, HH, II, JJ, KK, and LL, yielding a 400-base-pair fragment.
The final sequence, CALB 1–38 (Fig. 1), was obtained using a PCR mixture containing 2 μL of a template mixture consisting of 10 μL (0.1 μg/μL) of the 730-base-pair 1–26 amplicon and 10 μL (0.1 μg/μL) of the 400-base-pair 25–38 amplicon and 0.5 μL of an oligonucleotide mixture consisting of 10 μL(1 μg/μL) of forward primer A and 10 μL (1 μg/μL) of reverse primer LL (Table 1). A 1030-base-pair fragment, corresponding to amplicon 1 —38, was isolated and sequenced.
Agarose Gel Electrophoresis and Purification of DNA from Gel
A 1% (w/v) agarose gel was prepared by combining 300 mL 1 x tris-acetate ethylenediaminetetraacetic acid (TAE) buffer (Sigma Aldrich) with 3 g agarose (Thermo Fisher Scientific Inc., Waltham, MA), microwaving for 4 min and adding 30 μL ethidium bromide (Sigma Aldrich). The first and last wells of the gel were loaded with 10 μL Bionexus DNA marker (Bionexus, Inc., Oakland, CA). Five microliters of 10 x glycerol-loading buffer (Teknova, Inc., Hollister, CA) was added to the PCR products, and 15 μL of the mixture was loaded onto the gel and run at 80 V on a Bio-Rad Power Pac 3000 (Bio-Rad Laboratories) for 1.5 h. The desired band was cut out, 900 μL NaI solution was added to the gel slice, and the mixture was incubated at 55 °C for 30 min to melt the gel. The PCR products were purified using a GENECLEAN II kit (MP Biomedicals) according to the manufacturer's directions. The supernatant with eluted DNA was used for subsequent PCR or cloning into pENTR D TOPO (Gateway entry vector for directional cloning [5′ to 3′] with DNA topo-isomerase I as restriction enzyme and ligase).
Automated Polymerase Chain Reaction Assembly of Candida antarctica Lipase B Lyt-1 Sequence on Robotic Workcell
The sequence of Lyt-1 variant C3, 26,28,29 an amphipathic peptide with pore-forming properties that previous results 30 suggest might facilitate the movement of the expressed enzyme to the surface of the yeast cell, was assembled and placed in frame with the CALB 1–38 ORF sequence using the integrated robotic workcell (Fig. 2). Amphipathic peptides form α-helical structures, in which most hydrophobic amino acid residues occur on one side of the helix while most of the hydrophilic residues are on the other side. The hydrophobic side of the helix interacts with cell membranes and with the same region in other peptide molecules to form a “pore” or channel through the membrane, whereas the hydrophilic side projects into the center of the pore, permitting passage of ions and polar molecules through the membrane. 31 Lyt-1 C3 is a mutant with optimized hydrophilic and hydrophobic regions compared with wild-type Lyt-1. 30

Diagram depicting automated routines scheduled on the integrated robotic workcell used for PCR assembly, amplification, and purification of CALB Lyt-1 1–43 ORF.
The photographs and diagrams in Figure 2 show the SoftLink-scheduled routines (Hudson Robotics, Inc., Springfield, NJ) performed on the workcell to carry out the automated protocols for PCR assembly of Lyt-1 ORF and addition to CALB 1–38 ORF, thermal cycling, and purification of the CALB Lyt-1 1–43 amplicon.
The Lyt-1 variant sequence, ATCTGGCTGACCGGGCTGAAATTTCTGGGCAAACATGCGGCGAAACATCT GGCGAAACAGCAGTTGTCGCCATGG, was present in oligonucleotides XX, YY, and ZZ (Table 1; underlined). The PCR mixture for the step to add Lyt-1 contained 21.5 μL H2O, 10 μL 5× Phusion HF Buffer, 1 μL 10-mM dNTPs, 11 μL oligonucleotide template mixture, and 0.5 μL Phusion enzyme. The oligonucleotide template mixture consisted of 0.5-μL forward primer A, 0.5 μL of a solution containing 10 μL each of the five oligonucleotides VV, WW, XX, YY, and ZZ (Table 1), 1.0 μL of pENTR D TOPO CALB 1–38 ligation mixture as template, and 9 μLH2O.
The oligonucleotide template mixture in a capped tube in a 96-tube 2D Matrix plate (Thermo Fisher Scientific) was brought by the four-axis robot (PlateCrane EX, Hudson Robotics, Inc., Springfield, NJ; Fig. 2, R) with solenoid plate gripper and telescoping arm (Hudson Robotics, Inc.) onto the track (Fig. 2, Q) on the deck of the liquid handler (Fig. 2, P). The plate was moved by the servo motor gripper of the liquid handler into position on the vacuum block (Fig. 2, D) where the tube jig and pipettes were used to pierce the Matrix tube cap and pipette the oligonucleotide template mixture into a Bio-Rad hard-shell 0.2-mL 96-well plate (Bio-Rad Laboratories) that was also brought from the clean stack (Fig. 2, M) by the PlateCrane to the track on the deck of the liquid handler and moved by the liquid handler gripper into position on the vacuum block (Fig. 2, E). After the oligonucleotide template mixture was pipetted into the hardshell plate, the liquid handler gripper moved the plate to the plate position on the cold block (Fig. 2, B), and the PCR reagent mixture was pipetted from a Matrix tube in the reagent position (Fig. 2, A) on the cold block into the hard-shell plate with the cleaned pipettes.
The hard-shell PCR plate was placed on the liquid handler StopLink (Hudson Robotics, Inc., Springfield, NJ) (Fig. 2, Q) for export to the aspirator StopLink (Fig. 2, O) and moved by the PlateCrane into the ABgene ABS 300 heat sealer (ABgene, Rochester, NJ) (Fig. 2, L) and hermetically sealed with foil. The sealed plate was moved by the Plate-Crane into the MJ PTC200 thermal cycler with autolid (MJ Research, Waltham, MA) (Fig. 2, N) and held at 98 °C for 5 min. PCR was run using the following cycle: 98 °C for 1 min, 60 °C for 1 min, 16 °C for 1 min, repeated 30 times, then held at 12 °C for 7 min and 4 °C for 2 min.
Automated Purification of Polymerase Chain Reaction Amplicon on Robotic Workcell
After the cycling program was completed, the autolid opened, the plate was moved back to the aspirator StopLink (Fig. 2, O) and was then returned to the liquid handler Stop-Link (Fig. 2, Q) for purification of the PCR amplicon. The liquid handler gripper moved the hard-shell plate with the PCR mixture to the top position in the vacuum block (Fig.2, E) where it was clamped in place. A modified GENE-CLEAN II process on the liquid handler 26 was used to purify the PCR amplicon for subsequent manual directional cloning into pENTR D TOPO and transformation of bacteria with the CALB Lyt-1 insert for LR clonase recombinational cloning into pYES2 DEST 52. The GLASSMILK/NaI reagent (MP Biomedicals) in a preassembled sealed ABgene 2.2-mL square-well 96-well pyramid bottom deep-well plate (ABgene) was shaken on the Variomag shaker on the work-cell (Fig. 2, I) and was moved by the liquid handler gripper (Fig. 2, K) to the vacuum block (Fig. 2, D). The sterile stainless steel pipette tips of the pipette arm pierced the sealed ABgene plate. The pipettes removed the PCR samples from the hard-shell plate and added them to the GLASSMILK/NaI reagent, and the mixture was aspirated to mix. The hard-shell plate was parked at the liquid handler StopLink (Fig. 2, Q) in preparation for the next PCR assembly step. A foil-sealed Qiagen Turbo Filter Plate (Qiagen, Valencia, CA) was moved to the vacuum block (Fig. 2,E) by the liquid handler gripper (Fig. 2, K) and clamped in place. The mixture in the ABgene plate was aspirated several times to mix, pulled through the pipette-pierced Turbo Filter Plate using a vacuum of 85 kPa, washed three times, 5 min each time, with 900-μL NEW Wash (MP Biomedicals) from a stainless steel trough, moved to the drying position (Fig. 2, F) by the liquid handler gripper, and dried with fan-driven heated air. The filter plate was moved to the vacuum block (Fig. 2, D top) and clamped into place. Bound purified PCR products were eluted after a 1-min delay with 90 μL of Qiagen EB (Fig. 2, S) into a clean Matrix 2D barcode 0.75-mL collection tube in a 96-tube rack brought into the vacuum block (Fig. 2, D bottom). The eluted sample was brought to the liquid handler deck for TOPO cloning or iterative PCR assembly.
Directional Cloning into pENTR D TOPO and Plasmid Preparation
The full-length purified assembled CALB or CALB Lyt-1, with a CACC sequence at the 3′ end to facilitate cloning into pENTR, was TOPO cloned into pENTR D TOPO vector (Life Technologies Corporation, formerly Invitrogen, Carlsbad, CA), and the resulting reaction was transformed into TOP10 chemically competent Escherichia coli cells (Life Technologies Corporation), as described previously. 25 Volumes of 20 and 100 μL of the bacterial culture were spread onto two lysogeny broth kanamycin (LB Kan; Teknova, Inc.) plates and incubated overnight at 37 °C. Bacterial colonies were picked from the plates into each well of an ABgene-0932 96-well deep-well plate filled with 1600-μL LB Kan medium and incubated overnight at 37 °C with shaking. The resulting colonies were subjected to a plasmid preparation procedure using QIAprep 96 Turbo Kit protocol (Qiagen, Valencia, CA) as described previously. 26 The presence of insert was verified by sequencing.
Restriction Enzyme Digestion and DNA Sequencing
Forty-four microliters of pENTR D TOPO containing CACC CALB or CACC CALB Lyt-1 (no histidine [His] codons) were combined with 6 μL of the restriction enzyme mixture containing 0.5 μL BsrGI, 0.5 μL bovine serum albumin (BSA), and 5.0 μL buffer 2 (New England Biolabs, Ipswich, MA) in a Bio-Rad 96-well hard-shell plate and placed in an incubator at 37 °C overnight. The digestion reaction mixture was subjected to agarose gel electrophoresis to verify the presence of the inserts. The inserts were sequenced as described previously. 25
Gateway Cloning into pYES2 DEST 52 Expression Vector
Four microliters of pENTR D TOPO, containing CALB or CALB Lyt-1 inserts, 1 μL of the destination vector pYES2 DEST 52, and 3 μL 1× tris-EDTA pH 8 buffer, were combined in a 1.5-mL microfuge tube at room temperature. Gateway recombinational cloning into pYES2 DEST 52 using LR clonase II enzyme mixture (Invitrogen) was performed according to manufacturer's instruction. Volumes of 20 and 100 μL of the transformation reaction were spread onto two separate terrific broth ampicillin (TB Amp) plates (Teknova, Inc.) and incubated overnight at 37 °C with shaking. Bacterial colonies were picked from the plates the next day into each well of an ABgene-0932 96-well deep-well plate filled with 1600-μL TB Amp medium. The picked colonies were incubated overnight at 37 °C with shaking. The plasmid (pYES2 DEST 52 containing CALB or CALB Lyt-1 and 6 x His) was extracted using the QIAprep 96 Turbo Kit protocol (Qiagen) as described previously. 26
Yeast Transformation
Four isolated yeast colonies of PJ69–4 (S. Fields, Washington University, Seattle, WA) or INVSc1 (Invitrogen) from frozen glycerol stock were inoculated into two different 125-mL baffled shake flasks containing 25 mL of autoclaved yeast extract bacto-peptone and dextrose (YPD)-rich medium (10 g yeast extract, 20 g bacto-peptone, and 20 g dextrose (Sigma Aldrich) per liter H2O) and placed in the incubator at 30 °C for 2 d while shaking at 100 rpm. A 1.5-mL aliquot was taken from each flask and placed into a 1.5-mL microfuge tube. A volume of 125 μL of EZ-transformation solution (MP Biomedicals, Solon, OH), 4 μL of plasmid (pYES2 DEST 52 containing CALB or CALB Lyt-1), and 5 μL of carrier DNA (MP Biomedicals) were combined and added to the tube. The cells were resuspended by vortexing and incubated at 42 °C for 30 min. Volumes of 20 and 100 μL were then spread onto two separate complete minimal (CM) 2% (w/v) glucose minus uracil (URA) medium plates (Teknova, Inc.) and incubated at 30 °C for 2 d.
Preparation of Plates with Galactose or Glucose Medium and Corn Oil
CM 2% (w/v) galactose medium plus all amino acids, 5% (v/v) corn oil (Lincolnland Agri-Energy, Palestine, IL), and 0.1% (v/v) Triton X-100 (Roche Applied Science, Indianapolis, IN) was prepared, consisting of 1.4 g yeast synthetic drop-out medium supplement (minus histidine, leucine, tryptophan, and uracil); 0.06 g L-leucine; 0.04 g l-tryptophan; 0.02 g l-histidine; 0.02 g uracil; 20 g D-galactose (Sigma Aldrich); 15 g Bacto Agar (Thermo Fisher Scientific); 5 g ammonium sulfate; 1 mL Triton X-100; and 50 mL corn oil per liter. CM 2% (w/v) glucose minus URA-selective medium with 5% (v/v) corn oil and 0.1% (v/v) Triton X-100 was prepared using 1.4 g yeast synthetic drop-out medium supplement (minus histidine, leucine, tryptophan, and uracil); 0.06 g l-leucine; 0.04 g l-tryptophan; 0.02 g l-histidine; 20 g dextrose (d-glucose); 15 g Bacto Agar; 5 g ammonium sulfate; 1 mL Triton X-100; 50 mL corn oil; and made up to 1 L with water. The media were autoclaved for 30 min and then plated. Ten microliters of PJ69–4 yeast control and 10 μL PJ69–4 yeast transformed with CALB His or CALB Lyt-1 His, adjusted to an absorbance of 0.1 at 660 nm, were spotted on both sets of corn oil plates and incubated at 30 °C for 5 d. A sample of corn oil containing the yeast cells was taken from the surface of the CM 2% galactose plate and used for SEM analysis. 32
Fermentation
Fermentations were performed in a Fedbatch-pro fermentation system (DASGIP BioTools, LLC, Shrewsbury, MA) maintained at 30 °C with stirring (150 rpm). CM 2% (w/v) glucose medium consisted of the following per liter: 1.4 g yeast synthetic drop-out medium supplement minus histidine, leucine, tryptophan, and uracil; 0.06 g l-leucine; 0.04 g l-tryptophan; 0.02 g l-histidine; 0.02 g uracil; 20 g dextrose; and 5 g ammonium sulfate. CM 2% (w/v) glucose minus URA-selective medium was prepared in the same manner, except that uracil was not added. Yeast Peptone Dextrose (YPD) and Yeast Peptone Galactose (YPG) media consisted of 10 g/L yeast extract, 20 g/L bacto-peptone, and either 20 g/L glucose or 20 g/L galactose, respectively. Liquid precultures were inoculated with colonies of yeast and incubated for 2d at 30 °C with shaking at 100 rpm. The density of the preculture was adjusted to an absorbance equivalent to 4.0 at 660 nm, and 25 mL was added to 150 mL of YPD medium in a 400-mL DASGIP culture vessel (DASGIP Bio-Tools, LLC, Shrewsbury, MA). The absorbance at 660 nm, glucose consumption, and ethanol production were monitored. After 48 h, the cells were harvested by ultracentrifugation, resuspended in 150 mL of YPG medium, and returned to the culture vessel to continue fermentation for another 72 h. The absorbance at 660 nm, galactose consumption, and ethanol production were monitored. The cells were again harvested by centrifugation.
Biodiesel Assay Using Gas Chromatography Analysis
Liquid cultures for the fatty acid ethyl ester (biodiesel) assay were prepared by inoculating 20 mL from a 25-mL liquid preculture of PJ69–4 yeast in YPD medium or PJ69–4 yeast transformed with recombinant CALB His or CALB Lyt-1 His in glucose minus URA medium that had been incubated at 30 °C for 2 d at 100 rpm into 1 L YPD in 2.8-L Fernbach flasks and incubating at 30 °C for 2d at 100 rpm. The cultures were centrifuged for 10 min in a Beckman Avanti J20 (Beckman Coulter, Inc., Brea, CA) at 3000 rpm in a JS 4.3 swing bucket rotor using sterile 500-mL spin bottles, and the pelleted cells were resuspended in a volume of 1 L of YPG and incubated at 30 °C for 2 d at 100 rpm. Cultures were centrifuged, and 10 mL of 15:1 (v/v) ethanol:soybean oil substrate was added to 5 mL of the supernatant and to the pellets resuspended in 5 mL of yeast-protein extraction reagent (Y-PER Plus; Thermo Fisher Scientific, Inc.) to give a 2:1 (v/v) ratio of substrate mixture to supernatant or to pellet suspension. Lysed samples were prepared by adding 1.5 mL of acid-washed 425- to 600-μm glass beads to each pellet and vortexing before substrate was added. The control was prepared by spiking the pelleted, lysed PJ69–4 yeast cells from 1 L of culture with 0.5-mg purified commercial CALB (Novozym 435; Sigma Aldrich). The reaction mixtures were placed in an incubator at 50 °C and mixed at 20 rpm for 24 h.
To stop the reaction and prepare for GC analysis, 15 mL of hexane was added to each sample, and the samples were mixed for 30 s. The samples were placed in the Beckman Avanti J20 (Beckman Coulter, Inc., Brea, CA) at 3000 rpm for 10 min in a JS 4.3 swing bucket rotor using sterile 50-mL conical tubes. Twenty milliliters of the hexane upper layer was removed from each sample and separated into several 1.5-mL microfuge tubes, and the samples were concentrated in the SPD2010 SpeedVac Savant (Thermo Fisher Scientific) at 8 mtorr and 200 rpm for 1 h. The concentrated samples were recombined in 1 mL hexane and used for GC analysis.
Conversion to ethyl esters and ethanol production were monitored by an Agilent model 6890 GC-FID equipped with a model 6890 series injector and an Agilent D8–5HT column (Agilent, Santa Clara, CA). The carrier gas was helium at a flow rate of 3 mL/min. The oven temperature was initially held at 50 °C for 1 min, increased to 180 °C at 15 °C/min, to 230 °C at 7 °C/min, and to 380 °C at 30 °C/min, and held for 10 min at 380 °C. One microgram of TAG, diacylglyceride, monoacylglyceride, and ethyl ester standards was used to evaluate the retention times of the samples.
Immobilization on Sepabeads Resin
The immobilization procedure was adapted from Resin-dion (Binasco, Italy). 21,33 Liquid cultures were prepared in Fernbach flasks as described for the biodiesel assay. After incubation in YPG and centrifugation in the Beckman Avanti J20 at 3000 rpm for 10 min in a JS 4.3 swing bucket rotor using 500-mL spin bottles, the pellets were resuspended in a volume of 15 mL Y-PER Plus (Thermo Fisher Scientific, Inc., Rockford, IL). The resuspended pellet solution was passed through a 60-cc syringe with an 18-guage needle filled with 1.5 mL of acid-washed 425- to 600-μm (US sieve size, 30–40) glass beads. The syringe plunger was pushed in to drive the solution through and lyse the yeast cells. The beads are too large to pass through the needle, and because of their hardness and round shape, do not clog the needle. The plunger was pulled out, and the solution that was driven through was poured back into the syringe. This process was repeated twice. One milliliter of Qiagen Superflow nickel beads, capable of binding 20 mg of His-tagged protein per mL slurry, was added to the lysed yeast cells, and the mixture was incubated for 4 h at 4 °C with shaking. The beads were washed three times by adding 20 mL of 1 x phosphate buffered saline (PBS). Twenty milliliters of 300 mM EDTA was added to elute the lipase from the Ni beads, and the mixture was centrifuged and decanted. Two milliliters was added to 1.5 mL Sepabeads resin and made up to 5 mL with equilibrium buffer (necessary to charge the Sepabeads resins; consists of 20 mM PBS and 2 M (NH4)2SO4), and the mixture was reacted for 1 h at room temperature with shaking. After 1 h, 10 mL of 15:1 ethanol:oil substrate was added to give a 2:1 ratio, and the mixture was incubated for 12 h at 50 °C with shaking.
Coomassie Analysis
Liquid cultures were prepared in Fernbach flasks as described for the biodiesel assay. One-microliter samples of culture were taken after incubation in YPD medium and after incubation in YPG medium and placed in a 1.5-mL microfuge tube, centrifuged for 2 min at 13,000 rpm, and the supernatant was transferred to a new tube. Twenty milliliters of 2 × tris-tricine loading buffer (pH = 8.3; Invitrogen) plus 2 mL β-mercaptoethanol (Bio-Rad Laboratories) were prepared, and 40 μL was added to the pellet and supernatant samples. The samples were vortexed thoroughly and heated at 95 °C for 10 min; then, 10 μL of each sample was loaded onto a 4–20% tris-tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Invitrogen) in an Novex XCell mini-cell (Invitrogen) and run in 1 × tris-tricine running buffer (Invitrogen) at 125 V for 100 min on the Bio-Rad Power Pac Basic. Five microliters (1 μg/μL) of BSA standard and 10 μL SeeBlue Plus2 marker (Invitrogen) were loaded in the first and last lanes, respectively. After electrophoresis, the gel was stained for 1 h with Coomassie brilliant blue R-250 staining solution (Bio-Rad Laboratories) and destained for 20 h with a solution of 15% methanol and 10% acetic acid. The gel was analyzed using the AlphaImager System 3400 (Alpha Innotech Corporation) to determine the band density, and the concentration of the expressed lipase was calculated by comparison with the BSA standard.
Western Blot
One-milliliter culture samples in YPD and in YPG were taken, and pellet and supernatant samples were prepared in loading buffer as described for Coomassie analysis. The samples were loaded on a 4–20% tris-tricine SDS-PAGE gel placed into a Novex box and run in 1 x tris-tricine running buffer for 120 min at 125 V on the Bio-Rad Power Pac Basic. Ten microliters of SeeBlue Plus2 marker were loaded into the first lane. The polyvinylidene (PVDF) membrane (Invitrogen) was charged for 30 s with 20 mL methanol and 20 mL of 1 × transfer buffer (Invitrogen). The charged PVDF membrane was placed between two layers of filter papers in the X-Cell II Blot Module (Invitrogen), positioned in the electrophoresis apparatus, and run in 1 × tris-tricine running buffer for 10 h at constant 200 mAmps. The PVDF membrane was removed and processed according to the manufacturer's directions in the Western Breeze kit (Invitrogen) using 30 μL of a solution prepared by reconstituting a 1-μg vial of Qiagen Penta-His antibody (Qiagen) with 1 mL 1 × PBS (Mediatech, Inc., Manassas, VA) as the primary antibody. The membrane was air dried overnight and analyzed by the AlphaImager System 3400 (Alpha Innotech Corporation) to determine the band density.
Results and Discussion
Polymerase Chain Reaction Assembly of Candida antarctica Lipase B Open Reading Frame
The CALB gene ORF was produced using a stepwise oligonucleotide PCR assembly strategy followed by TOPO ligation directionally into pENTR D TOPO and LR clonase recombinational cloning into pYES2 DEST 52 vector for expression and evaluation of the lipase enzyme (Fig. 1). The strategy previously described for PCR assembly of the xylose isomerase gene ORF 30 involved a cloning step after each PCR step. The strategy described here eliminates the sub-cloning step and assembles the entire ORF in sequential PCR steps, so that the process is more rapid and readily adapted for the integrated robotic workcell. The method has the potential to be used iteratively to build large libraries or single clones from sets of oligonucleotides. The fusion of the C3 variant of the Lyt-1 amphipathic peptide to the lipase may facilitate secretion and isolation of the expressed lipase outside the yeast cell for ready availability, in this case, for chemical attachment to a column resin for lipase-catalyzed biodiesel production.
The sequence of the CALB ORF, which consists of 1029 nucleotides, was based on the registered sequence in Gen-Bank (Z30645). The overall stepwise assembly method is described in Materials and Methods. In this method, nine increasingly longer PCR amplicons, six at the 5′ end of the CALB sequence and three at the 3′ end, were created sequentially from 36 50mer oligonucleotides, one 40mer oligonucleotide, and one 15mer oligonucleotide to generate the complete CALB gene ORF (Fig. 1; forward oligonucleotides shown in yellow and reverse shown in tan). The 50mer oligonucleotides in this strategy were designed so that each one overlapped at the 5' end with 25 nucleotides of the previous oligonucleotide and overlapped at the 3' end with 25 nucleotides of the next oligonucleotide, alternating forward and reverse oligonucleotides. No gaps were present between the overlapping sequences. The first six 50mers were added to initiate the formation of the CALB ORF. The amplicon from the PCR assembly of these six 50mers was added to the next PCR mixture together with the two 50mer oligonucleotides at the 3ℙ end of the amplicon plus the next four 50mer oligonucleotides in the sequence that overlapped each other by 25 nucleotides at either end. The two-50mer overlap at the 3ℙ end of the amplicon formed a strongly annealed link to that template and ensured sequential assembly. Again, there were no gaps between the overlapping sections. Twenty-six of the 38 oligomers were added in this fashion. The next set of oligonucleotides did not give the desired PCR product, possibly because they preferred to anneal to the forward section(s) of the template to form shorter amplicon(s). Therefore, the last part of the CALB ORF was assembled separately, and the front and rear sections were joined using PCR to obtain the complete CALB gene ORF.
Two differences from the GenBank sequence were found in the assembled ORF. The first nucleotide difference was at position 245 in the CALB ORF sequence, with ACG, coding for a threonine amino acid residue at position 57 in the amino acid sequence, changed to ATG, coding for methionine. Both have aliphatic side chains, but methionine has a CH3SCH2CH2- group, whereas threonine has a secondary alcohol group (CH3CHOH-). The second nucleotide difference was at position 986, with TTT, coding for phenylalanine at position 304 in the amino acid sequence, changed to TAT, coding for tyrosine. Both phenylalanine and tyrosine amino acid residues have uncharged aromatic side chains; the only difference is the presence of a hydroxy group on the benzyl group in tyrosine. These two nucleotide differences did not appear to affect the catalytic activity of the assembled lipase, because the expressed enzyme was able to catalyze the production of ethyl esters from corn or soybean oil.
Candida antarctica Lipase B Lyt-1 C3 Open Reading Frame Produced Using Automated Protocols for Polymerase Chain Reaction Assembly and Amplicon Purification
Previous work 26,30 suggested that the C3 variant of the amphipathic protein Lyt-1 with a 6× His tag migrated to the surface of the yeast cell in which this variant was expressed. The Lyt-1 C3 ORF was inserted in frame at the 3ℙ end of the CALB ORF to code for the fusion protein CALB Lyt-1 C3 to determine if the Lyt-1 C3 form, which is expected to be found outside the cell, is more active or easier to purify than the CALB without the C3 Lyt-1 sequence that would be found predominantly inside the yeast cell. A schedule was scripted to include an automated protocol for PCR assembly and purification of the PCR amplicon written for the liquid handler using SoftLink software from Hudson Robotics, Inc., to control the Hudson ProLinks Workcell. The protocol contained steps to program the movement of the PCR plate first to the sealer, then to the PCR thermal cycler, and finally back to the liquid handler to purify the resulting assembled amplicon. The addition of the Lyt-1 C3 ORF to the CALB ORF was performed using the automated PCR assembly protocol written for the workcell to test the scripting of this protocol on the liquid handler and its incorporation into the SoftLinks scheduler for the workcell (Fig. 2).
Using this protocol, a 96-well Bio-Rad hard-shell plate was brought by the PlateCrane EX from the incoming clean stacks onto the deck of the liquid handler to set up the PCR assembly (Fig. 2, step 1). The oligonucleotide template mixture in a capped tube in a 96-tube Matrix plate (Thermo Fisher Scientific) was brought by the four-axis PlateCrane telescoping arm onto the track on the deck of the liquid handler. The plate was moved by the liquid handler gripper into position on the vacuum block where the tube jig and pipettes were used to pierce the Matrix tube cap and pipette the oligonucleotide template mixture into the hard-shell plate on the track on the liquid handler. The plate was moved by the liquid handler gripper into position on the vacuum block. After the oligonucleotide template mixture was pipetted into the hard-shell plate, the liquid handler gripper moved the plate to the plate position on the cold block, and the PCR reagent mixture was pipetted from a Matrix tube in the reagent position on the cold block into the hard-shell plate.
After the plate containing the complete PCR mixture was placed on the track, it moved to the deck of the liquid handler at the liquid handler StopLink for removal by the four-axis PlateCrane to the sealer and then to the thermal cycler. The plate then moved on the track from the liquid handler Sto-pLink to the aspirator StopLink for pick up by the crane. The crane moved the plate from the aspirator StopLink to the ABgene heat sealer nest for sealing with foil, followed by crane movement of the sealed plate to the thermal cycler with open autolid (Fig. 2, step 2). The crane placed the plate in the thermal cycler, the lid closed, and the cycling program proceeded. After the cycling program was complete, the autolid opened, the plate was moved back to the aspirator StopLink and moved on the track to the liquid handler StopLink for purification of the PCR amplicon on the liquid handler (Fig. 2, step 3) for subsequent manual directional cloning into pENTR D TOPO and transformation of bacteria with the CALB Lyt-1 C3 insert for LR clonase into the pYES2 DEST 52 yeast expression vector. The Lyt-1 C3 sequence was successfully assembled in frame with the CALB ORF using the automated PCR assembly and purification protocol incorporated into the SoftLink scheduler for the workcell.
Ligation into pENTR D TOPO and Plasmid Preparation
The assembled CALB and CALB Lyt-1 C3 ORFs were directionally cloned into pENTR D TOPO and transformed into TOP10 competent cells for plasmid preparation. The pENTR D TOPO CALB and pENTR D TOPO CALB Lyt-1 C3 plasmid preparations were used for LR clonase recombinational cloning into pYES2 DEST 52.
LR Clonase II Recombinational Cloning into pYES2 DEST 52 and Plasmid Preparation
The CALB and CALB-Lyt-1 C3 inserts were moved into respective pYES2 DEST 52 expression vectors using LR clonase II recombinational cloning. The LR clonase reactions were designed to use small volumes to enable future automation of this process in addition to the automated PCR assembly and purification programs described here for the production of CALB with Lyt-1. The pYES2 DEST 52 plasmids were transformed into TOP10 competent bacteria, and plasmid preparations were performed. The resulting plasmid preparations were used for transformation into PJ69–4 diploid cells.
Transformation of PJ69–4 Yeast, Growth on Glucose-Selective Medium and Galactose Medium with Corn Oil, and Scanning Electron Microscopy Analysis
The pYES2 DEST 52 CALB and pYES2 DEST 52 CALB Lyt-1 C3 plasmid preparations were used to transform the diploid strain PJ69–4 to determine if the enzymes were heterologously expressed as CALB 6× His and CALB Lyt-1 C3 6 x His and if they possessed lipase activity. The transformed strains were spotted on plates containing CM 2% glucose minus URA medium and corn oil with Triton X-100 emulsifier. On glucose minus URA medium, the control strain PJ69–4 did not grow, because it lacks the pYES2 DEST 52 plasmid with the ura3 gene. PJ69–4 with pYES2 DEST 52 CALB and PJ69–4 with pYES2 DEST 52 CALB Lyt-1 C3 showed similar growth (Fig. 3A, right) on the selective medium, indicating the presence of the pYES2 DEST 52 plasmids. The three yeast strains were also spotted on plates containing CM 2% galactose medium, corn oil, and Triton X-100. In galactose, the GAL1 promoter drives the expression of CALB His and CALB Lyt-1 His. The region corresponding to CALB His was only slightly larger than the control, and the enzyme did not appear to be available to interact with the corn oil (Fig. 3A, left). The region corresponding to CALB Lyt-1 His was significantly larger than the region for CALB His and had spread out on the corn oil (Fig. 3A, left), suggesting that the expressed enzyme with the amphipathic Lyt-I sequence was available at the surface of the yeast cell and was capable of interacting with the corn oil. In the SEMs (Fig. 3B) of cells taken from the galactose plate, it also appeared that the PJ69–4 yeast cells expressing CALB Lyt-1 His were on top of the oil mixture (Fig. 3B, left, top), again suggesting that the lipase was interacting with the oil. The PJ69–4 yeast cells expressing CALB His were immersed in the corn oil similar to the control PJ69–4 yeast cells and appeared not to interact with the oil.
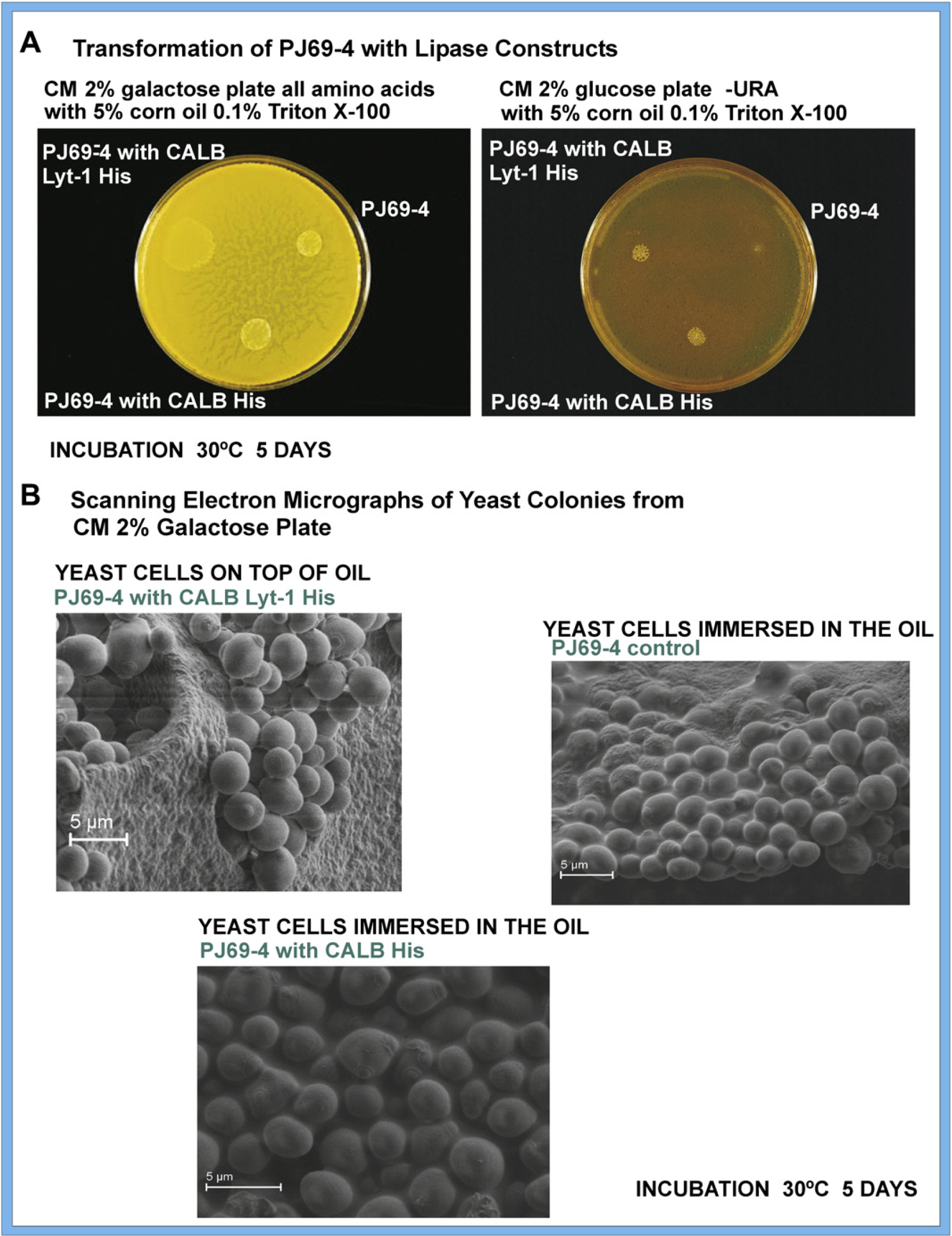
(A) Growth of PJ69–4 yeast transformed with CALB His or CALB Lyt-1 His expression plasmids on CM 2% (w/v) galactose medium and CM 2% (w/v) glucose minus URA medium. On glucose-selective medium, PJ69–4 with pYES2 DEST 52 CALB and PJ69–4 with pYES2 DEST 52 CALB Lyt-1 C3 showed similar growth, indicating the presence of the pYES2 DEST 52 plasmids; the control strain PJ69–4 showed no growth, because it lacks the pYES2 DEST 52 plasmid with the ura3 gene. In the presence of galactose, the GALI promoter drives the expression of CALB His and CALB Lyt-1 His. The region corresponding to CALB His was only slightly larger than the control; the region corresponding to CALB Lyt-1 His was significantly larger, suggesting greater availability for interaction with the corn oil. (B) Scanning electron micrographs of PJ69–4 yeast cells and PJ69–4 yeast cells transformed with CALB His or CALB Lyt-1 His taken from the surface of the galactose plates in A.
Fermentation (Run 1)
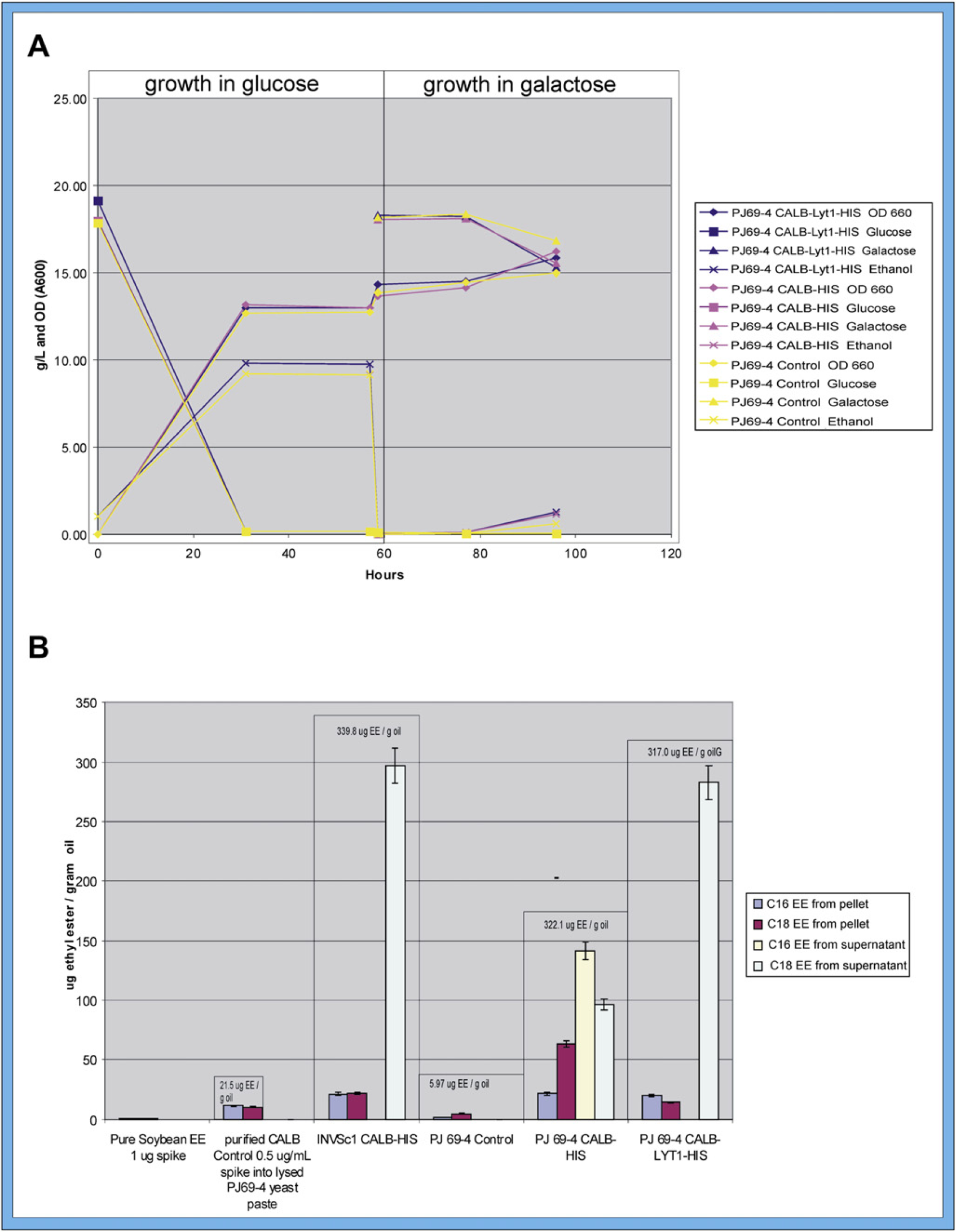
Yeast cells were incubated in YPD for 2 d to achieve a high-density culture for initiating fermentation reactions in the DASGIP reactor. Fermentation in YPD with glucose as the carbon source was incubated at 30 °C for 48 h, and ethanol production, cell growth, and glucose use were measured over that time. The results for all yeast strains through 48 h in glucose are shown in Fig. 4A (left). After 48-h fermentation, the culture was shifted to YPG. Fermentation with galactose as the carbon source was incubated at 30 °C for 72 h, and ethanol production, cell growth, and use of galactose were measured over that time. The results for all yeast strains in galactose are shown in Fig. 4A (right). With glucose as the carbon source, the maximum production of ethanol for PJ69–4 CALB His and PJ69–4 CALB Lyt-1 His strains was 9.3 g/L (Fig. 4A, 48 h). This level was as high as control PJ69–4 strain, indicating that the presence of plasmid did not affect ethanol production. All strains completely consumed the glucose substrate. With galactose as the carbon source, cell growth was greater for the PJ69–4 CALB His and PJ69–4 CALB Lyt-1 His strains than for control, possibly related to the expression of lipase. Ethanol production with galactose for both control and transformed strains was lower than that with glucose as the carbon source.
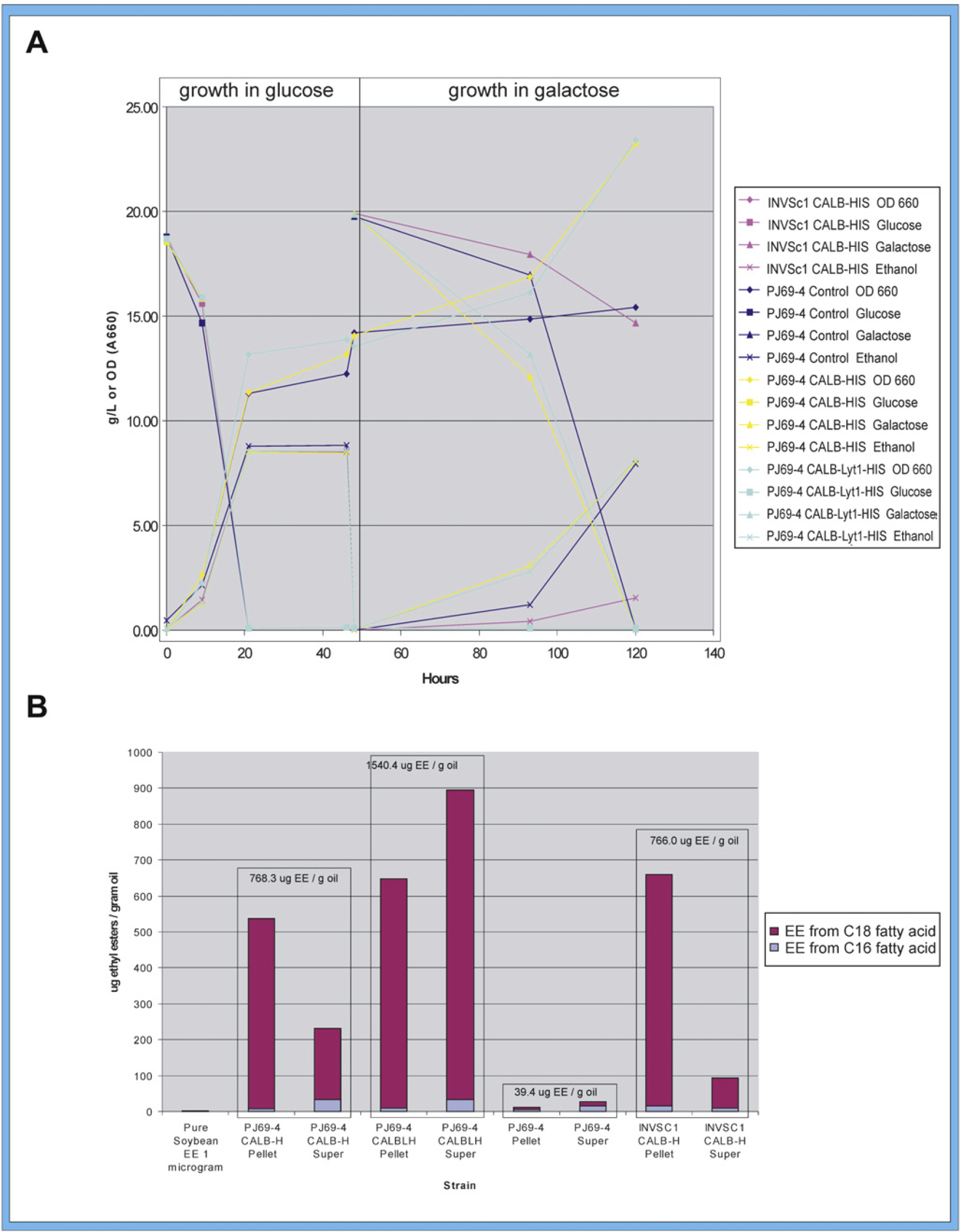
(A) Fermentation in DASGIP reactor first with glucose as carbon source for 48 h showing yeast cell growth, ethanol production, and glucose consumption, followed by galactose as the carbon source for 72 h showing cell growth, ethanol production, and galactose consumption for control strain and all transformed strains. Ethanol was isolated at 48 hours and lipase was harvested at 120 hours. (B) C16 and C18 fatty acid ethyl ester (EE) production by intact yeast cells using 15:1 ratio (v/v) of ethanol to soybean oil as substrate mixed 2:1 with resuspended pellet or supernatant. CALBH, CALB His; CALBLH, CALB Lyt-1 His.
It is possible that a single-step fermentation strategy, in which the lipase enzyme is expressed simultaneously with the production of ethanol, would save time and material costs by eliminating the need for an additional step with another carbon source and medium. The protein could be isolated from the fermentation supernatant before distillation of the ethanol product. Further research will be undertaken to determine the effect of simultaneous ethanol production and enzyme expression on the yield of ethanol and on costs for separation of the two products. The sequential strategy proposed here makes use of the same fermentation reactor for the second stage (lipase expression) after the ethanol has been removed as in the basic dry-grind process. The enzyme would be isolated for biodiesel production in the same way using either fermentation strategy.
Biodiesel Production by Intact Yeast Cells
The cells taken from the galactose culture were pelleted, and the supernatant and resuspended intact cell pellets were assayed for biodiesel production to determine if higher cell growth in galactose medium corresponded to lipase production. The results of the biodiesel assay are provided in Figure 4B. The amount of ethyl ester production was, by far, the highest for the supernatant from intact PJ69–4 yeast cells transformed with CALB Lyt-1 His (890 μg/g oil), followed in decreasing order by the pelleted intact cells of INVSc1 with CALB His (660 μg/g oil), of PJ69–4 with CALB Lyt-1 His (650 μg/g oil), and of PJ69–4 with CALB His (540 μg/g oil). Much lower levels of ethyl esters were produced by the supernatant from intact cells of PJ69–4 with CALB His (230 μg/g oil) and INVSc1 with CALB His (98 μg/g oil). The presence of the amphipathic Lyt-1 sequence appears to significantly favor the production of ethyl esters from the supernatant, suggesting that the enzyme was present at the surface of the cells and, hence, was found in the supernatant to a much greater extent than CALB without Lyt-1. In all cases, the ethyl esters produced with intact cells or supernatant contained predominantly C16 fatty acid chains (87–89%) rather than C18 chains. The percent of C16 (89%) versus C18 (11%) is the exact reverse of the ratio found in the soybean oil used as substrate (89% C18 vs. 11% C16). The 89% C18 fatty acids found in soybean oil are 85% unsaturated (23% 18:1; 54% 18:2; and 8% 18:3) and 4% saturated. The 11% C16 in soybean oil is from a saturated fatty acid (palmitic). This soybean oil fatty acid profile is very similar to that of corn oil, which is also 89% C18 and 11% C16 (palmitic), but the 89% C18 is 87% unsaturated (28% 18:1; 58% 18:2; and 1% 18:3) and 2% saturated. The properties of biodiesel are dependent on its fatty acid ester composition, and modifying the fatty acid profile has been suggested as a way to improve the low-temperature properties or oxidative stability of biodiesel. 34 An important property specified in biodiesel standards is the cetane number (CN), which is related to ignition delay time. The higher the CN, the shorter the ignition time. The CN of fatty acid esters increases with increasing saturation and increasing chain length. Favoring C16 fatty acid ester production from soybean oil increases the saturation of the fatty acid esters produced and increases the CN. Increasing the saturation also increases the oxidative stability of the biodiesel. 34 The formation of predominantly short-chain fatty acid ethyl esters is a characteristic of the native CALB enzyme, 35 but chain-length selectivity is dependent on the solvent environment 36,37 as well. Mutations also affect the chain length used. 35
Fermentation (Run 2)
Yeast cells were incubated in YPD for 2 d as for run 1 to achieve a high-density culture for initiating fermentation reactions in the reactor. Fermentation in YPD in run 2 was incubated at 30 °C for 55 h, and ethanol production, cell growth, and glucose consumption were monitored over that time. The results for all yeast strains used are presented in Figure. 5A (left). After 55 h, the culture was shifted to YPG and incubated at 30 °C for 41 h, and ethanol production, cell growth, and galactose consumption were measured. Similar to the results in run 1, with glucose medium, the maximum production of ethanol by the PJ69–4 CALB Lyt-1 His and CALB His strains was 9.9 g/L (Fig. 5, 55 h), slightly higher than the PJ69–4 control strain, indicating that the presence of the plasmid did not affect ethanol production. Cell growt for all strains was similar, and all strains completely consumed the glucose substrate. With galactose medium, as in run 1, cell growth was greater for the PJ69–4 CALB His and PJ69–4 CALB Lyt-1 His strains than for control, possibly related to the expression of lipase. Production of ethanol with galactose medium was lower than that with glucose medium and comparable with run 1 over the period of 41 h on galactose. A 12-h galactose point was not obtained for run 2.
(A) Fermentation in DASGIP reactor first with glucose as carbon source for 55 h, showing yeast cell growth, ethanol production, and glucose consumption, followed by galactose as the carbon source for 41 h, showing cell growth, ethanol production, and galactose consumption for control strain and all transformed strains. Ethanol was isolated at 55 hours and lipase was harvested at 96 hours. (B) C16 and C18 fatty acid ethyl ester (EE) production by lysed yeast cells using 15:1 ratio (v/v) of ethanol to soybean oil as substrate mixed 2:1 with resuspended pellet or supernatant. CALBH = CALB His; CALBLH = CALB Lyt-1 His.
Biodiesel Production by Lysed Yeast Cells
The results of the biodiesel assay for lysed cells are shown in Figure 5B. The supernatant of the lysed PJ69–4 cells with CALB Lyt-1 His (285 μg ethyl esters/g oil), and the lysed INVSc1 cells with CALB His (295 μg ethyl esters/g oil) produced only about one-third the amount of ethyl esters produced by the supernatant of the intact PJ69–4 cells with CALB Lyt-1 His (890 μg ethyl esters/g oil). In addition, the supernatant of these lysed cells produced exclusively (100%) C18 fatty acid ethyl esters, whereas the supernatant of the intact cells gave predominantly (81–89%) C16 fatty acid ethyl esters. On the other hand, the lysed PJ69–4 cells with CALB His, both the supernatant (243 μg ethyl esters/g oil; 145 μg [60%] C16 and 98 μg [40%] C18) and the pellet (19 μg ethyl esters/g oil; 20 μg [25%] C16 and 59 μg [15%] C18), produced ethyl esters with both C16 and C18 fatty acid chains. Commercial CALB (acrylic resin with Novozym 435; Sigma Aldrich) spiked into PJ69–4 yeast pellet lysate gave a total of 21.5 μg ethyl esters/g oil (10.0 μg [41%] C18 and 11.5 μg [53%] C16), similar to the pellets of the lysed cells. The explanation for the predominance of C18 in the supernatant for two of the strains is not clear, but it might be that there is some inhibition of the activity of the lipase by the lysed material from the yeast, whereas with the intact cells, there is less free material to inhibit the enzyme. It also suggests that some attenuation of the enzyme may occur when the cells are lysed, allowing the active site to be more “open” and to accommodate larger acyl chain lengths, so that the enzyme can more easily use the C18 chains as substrate.
Proteins Produced by the Expression of Candida antarctica Lipase B Constructs
The activity of all lipases relies on an active site catalytic triad, usually comprised of serine, histidine, and aspartate or glutamate amino acid residues. 38 The CALB enzyme is composed of a prepro region (a signal peptide of 18 amino acid residues and a propeptide of seven amino acid residues) and a mature region of 317 amino acid residues with a serine-histidine-aspartate catalytic triad at its active site. 37 Serine is at position 105, aspartate is at position 187, and histidine is at position 224 in the amino acid sequence of CALB. These positions correspond to codons TCC (nucleotides 388–390), GAC (nucleotides 634–636), and CAT (nucleotides 745–747) in the CALB ORF (highlighted in yellow in the sequence in Fig. 6, A). The ORFs that potentially can be expressed by the nucleotide sequence of the construct for CALB Lyt-1 His using each of the six in-frame ATG translation start codons (shown in color in Fig. 6A) are presented in Figure 6B. All six possible enzyme ORFs were seen in the Coomassie and Western blot analyses (Fig. 7). Four of the six contained all three amino acid residues, serine, aspartate, and histidine, which form the active site catalytic triad of CALB. The molecular weights for five of the CALB ORFs expressed from pYES2 DEST 52 CALB Lyt-1 in PJ69–4 yeast cells, four of which contain the complete catalytic triad, are indicated in Fig. 7. They are seen on the Western blot at 42.3, 34.3, 32.6, 31.3, and 26.3 kD (Fig. 7B). It is possible that some of the truncated forms containing the catalytic triad might be the enzymes giving rise to C18 versus C16 fatty acid ethyl ester production in the supernatant of the lysed cells. These truncated CALB variants are being investigated to determine if it might be possible to control the types of fatty acid esters being produced and select for products with the optimal biodiesel properties.

(A) Assembled CALB Lyt-1 His ORF sequence. Codons TCC, GAC, and CAT for amino acids in the active catalytic triad, serine, aspartate, and histidine, respectively, are highlighted in yellow. Six in-frame ATG start codons are shown in color and underlined. The two unintended differences from the GenBank sequence are indicated as capital letters, and the codons containing them are italicized. The Lyt-1 sequence is boxed in green. The 6 x His sequence is boxed in blue. The dotted underline indicates the attB2 sequence. (B) Sequence of proteins that would be expressed if all six in-frame ATG start codons were used. The residues of the catalytic triad (S, D, H) are shown in bold. The last two proteins do not contain the complete catalytic triad.
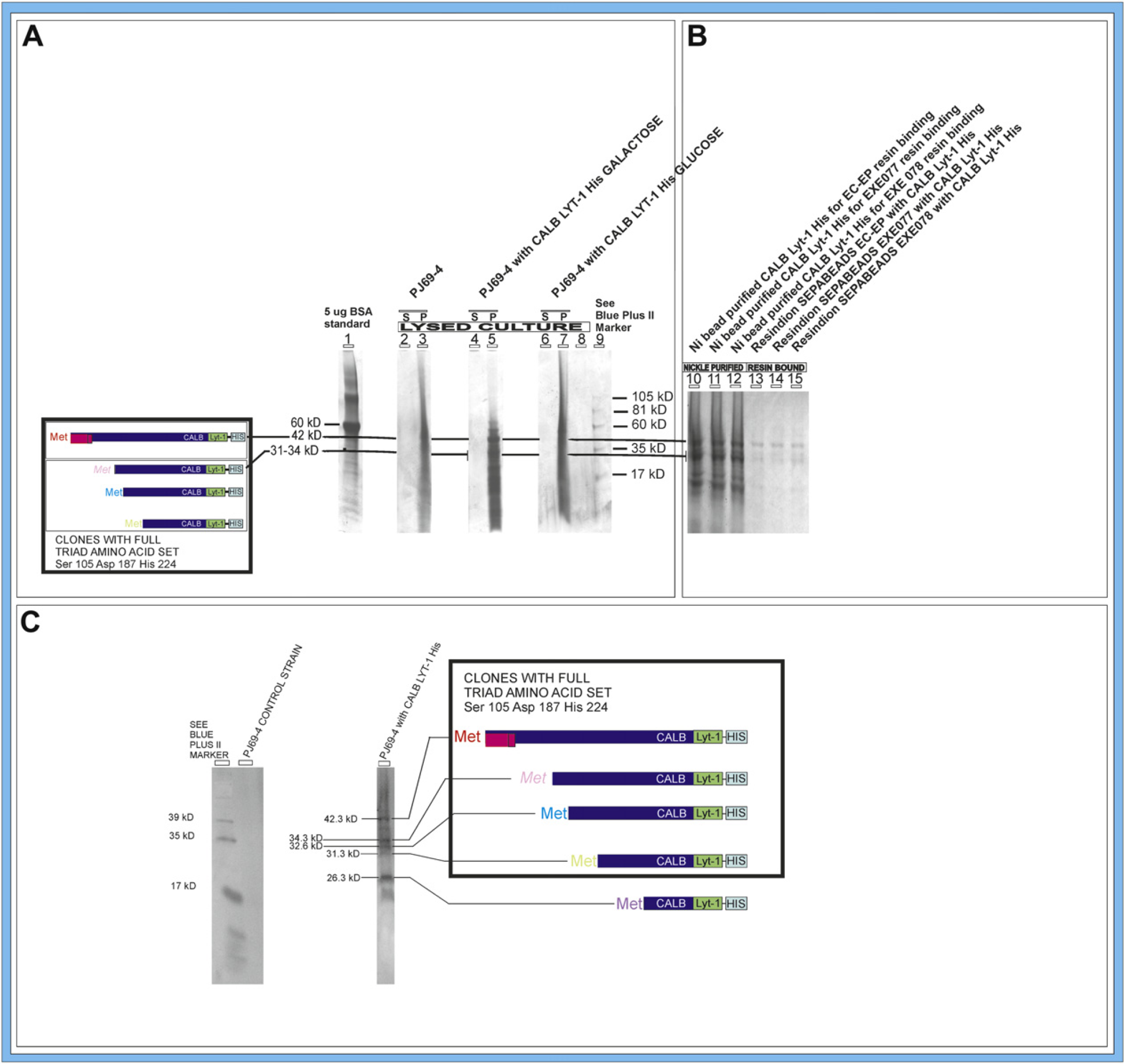
Expression of CALB constructs. (A) 4–20% gradient SDS-PAGE gel (Coomassie stain) confirming the expression of CALB constructs from four different in-frame ATG start codons in PJ69–4 yeast cells grown in galactose (lane 5) but not in glucose (lane 7). Red bar in sequence schematic represents the CALB prepro region. S, supernatant; P, resuspended pellet. BSA (5 μg) is standard for densitometer measurements of amount lipase. (B) 4–20% gradient SDS-PAGE gel (Coomassie stain) Ni bead-purified CALB Lyt-1 His and corresponding CALB Lyt-1 His bound to Resindion Sepabeads. (C) Western blot analysis of CALB Lyt-1 His constructs expressed in PJ69–4 yeast using the four different in-frame ATG start codons of sequences containing the catalytic triad. The bands corresponding to these constructs are seen at 42.3, 34.3, 32.6, and 31.3 kD. The band at 28.3 kD corresponds to an expressed construct not containing the serine residue that would form a part of the catalytic triad.
The Coomassie gel analysis (Fig. 7A) indicated that most the CALB protein was present in the yeast pellets and that sufficient CALB Lyt-1 His was available for purification and immobilization on Sepabeads resin to evaluate activity in the production of biodiesel compared with that of nonimmobilized CALB Lyt-1 His and commercially available CALB. Figure 7B shows the nickel bead-purified CALB Lyt-1 His in lanes 10, 11, and 12, which was used in the immobilization step. Lanes 13, 14, and 15 represent the CALB Lyt-1 His bound to the three types of resins after removal from the nickel beads, showing good transfer of the His-tagged CALB Lyt-1 preferentially to the Sepabeads. The original pellet yielded 9.2 mg CALB Lyt-1 His per liter culture. This was divided into three parts for binding to Ni beads for addition to each of the three resins (EC-EP, EXE077, and EXE078, Resindion S.r.l., Milan, Italy). Densitometry measurements of the gel showed that an average of 1.1 mg was bound to the Ni beads and an average of 0.8 mg of that amount (approximately 70%) was bound to the Sepabeads resin (Table 2). Western blot analysis (Fig. 7C) showed the constructs expressed in PJ69–4 yeast using the six different in-frame ATG (methionine) start codons (one from a mutation) in the CALB Lyt-1 His sequence (shown in color in Fig. 6B). The bands corresponding to the constructs containing the catalytic triad were seen at 42.3, 34.3, 32.6, and 31.3 kD. The band at 28.3 kD corresponds to an expressed construct not containing the serine residue that would form a part of the catalytic triad.
Fatty acid ethyl ester (EE) production and specific activity of nonimmobilized recombinant CALB Lyt-1 His enzyme compared with Ni bead-bound and resin-immobilized CALB Lyt-1 His enzyme
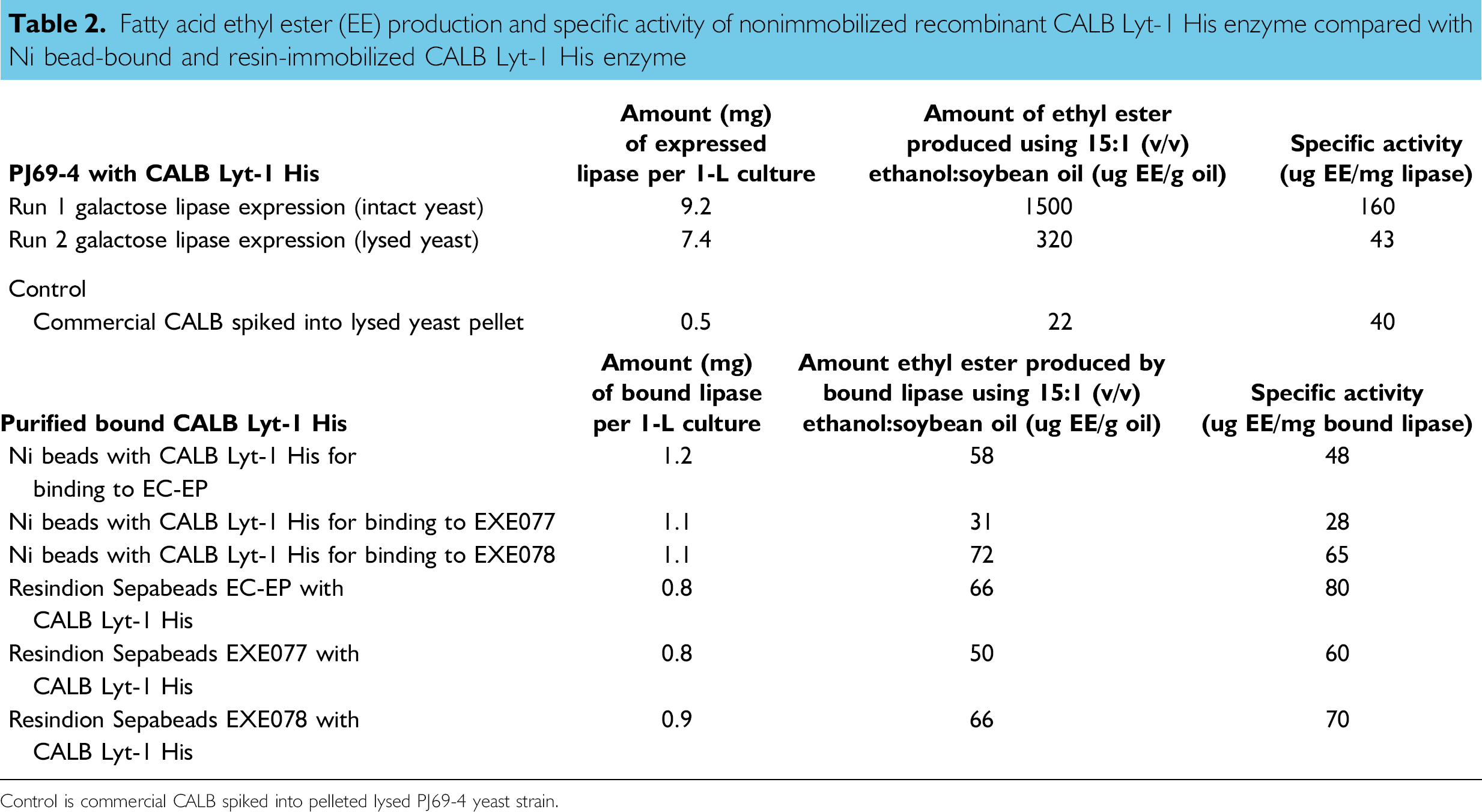
Control is commercial CALB spiked into pelleted lysed PJ69–4 yeast strain.
Specific Activity and Ethyl Ester Production from Resin-Bound Candida antarctica Lipase B Lyt-1 6 × His
CALB Lyt-1 His enzyme from cultures of PJ69–4 yeast cells was prepared, Ni bead purified, and immobilized on Sepabeads resin to evaluate its activity in the production of ethyl esters (biodiesel) compared with that of nonimmobilized recombinant CALB Lyt-1 His and of commercially available CALB. The process for isolation of the CALB Lyt-1 His enzyme involved passing the yeast cells at very high pressure through a glass bead matrix in the Y-PER plus buffer. Examination of the lysed material under a light microscope showed that this procedure caused the most complete lysis and release of material from the cells. The enzymes were purified by binding to Ni beads by means of the His tag, washing, and then covalent binding to the Resin-dion Sepabeads resins.
The specific activity of the nonimmobilized CALB Lyt-1 His enzyme was compared with the specific activities of the purified resin-immobilized recombinant CALB Lyt-1 His enzyme (Table 2). Specific activity was calculated by dividing mg ethyl esters/L by mg lipase/L. The highest specific activity (160 mg ethyl esters/mg lipase) was obtained using the secreted nonimmobilized CALB Lyt-1 His enzyme from intact cells, indicating that a large quantity of active enzyme is being expressed and secreted from the intact cells. The specific activity of CALB Lyt-1 His enzyme from lysed cells was almost fourfold lower (43 μg ethyl esters/mg lipase), indicating possible inhibition by the lipid cell debris. The commercially available resin-bound CALB enzyme (acrylic resin with Novozym 435; Sigma Aldrich) had a specific activity of 40 μg ethyl esters/mg lipase. The Ni bead-purified Sepabeads-immobilized CALB Lyt-1 His-expressed enzyme gave higher specific activities than the commercially available CALB (Table 2). Of the resins tested, the purified CALB Lyt-1 His enzyme immobilized on EC-EP resin gave the highest specific activity, 80 μg ethyl esters/mg lipase. CALB Lyt-1 His enzyme immobilized on EXE078 resin gave 70 μg ethyl esters/mg lipase and on EXE077 resin gave 60 μg ethyl esters/mg lipase.
For commercial feasibility, the immobilized enzyme must be stable through numerous cycles and must be capable of being regenerated or replaced readily. Immobilized C. antarctica lipase was shown to maintain conversion of soybean oil to methyl esters without loss of activity for 25 cycles. 23 The Sepabeads resin tested in the study reported here offers ease of attachment, and the immobilized enzyme has been shown to be highly stable as a result of multipoint covalent attachment. 27 Regeneration of the activity of deactivated immobilized C. antarctica lipase has been demonstrated. 2 The production and stability of immobilized C. antarctica lipase for routine cost-effective application in an industrial biorefinery are under investigation in our laboratory.
Conclusion
A PCR assembly strategy for producing gene ORFs that is capable of being used in an iterative fashion on an integrated robotic workcell was developed. An automated protocol was scripted to perform PCR assembly and subsequent DNA purification and was used in the final step in the production of CALB gene ORF with an in-frame gene ORF for the C3 variant of the amphipathic Lyt-1 peptide that experimental results in this and previous work suggest facilitates the movement of the enzyme out of the yeast cell. The synthetic CALB ORFs, with and without the Lyt-1 C3 sequence, were transformed into model yeast strains and evaluated in a fermentation reaction with glucose as the carbon source to evaluate ethanol production, followed by galactose as the carbon source for GAL1-driven CALB His or CALB Lyt-1 His expression. With glucose as a carbon source, ethanol production levels by the transformed strains were similar to those by the untransformed control strain.
Evaluation of the expressed CALB enzymes in a biodiesel assay showed that the lipases were active in catalyzing the formation of ethyl esters (biodiesel) from ethanol and soybean oil. The amount of ethyl ester production was, by far, the highest for the supernatant from intact PJ69–4 yeast cells transformed with CALB Lyt-1 His, followed in decreasing order by the pelleted intact cells of INVSc1 with CALB His, of PJ69–4 with CALB Lyt-1 His and of PJ69–4 with CALB His. Much lower levels of ethyl esters were produced by the supernatant from intact cells of PJ69–4 yeast with CALB His and of INVSc1 yeast with CALB His. The much greater production of ethyl esters by the CALB Lyt-1 His supernatant may be the result of Lyt-1 facilitating the movement of the enzyme to the outside of the yeast cell. Scanning electron micrographs of the yeast cells mixed with corn oil showed that yeast cells expressing CALB Lyt-1 His remained on the surface of the oil, whereas the yeast cells expressing CALB His were immersed in the oil, indicating a different interaction with the substrate. The enzymes from intact cells and intact cell supernatants produced almost exclusively C16 fatty acid ethyl esters.
About one-third the amount of ethyl esters was produced by the supernatant from lysed PJ96–4 cells with CALB Lyt-1 His or from lysed INVSc1 cells with CALB His as from the supernatant of intact PJ69–4 cells with CALB Lyt-1 His. In addition, unlike the intact cell supernatant, the lysed supernatant yielded almost exclusively C18 fatty acid ethyl esters rather than C16 fatty acid ethyl esters. Lysed pelleted cells and supernatant of PJ69–4 with CALB His gave moderate production of ethyl esters using both C16 and C18 fatty acid chains.
The predominance of C16 fatty acid ethyl esters in the supernatant from intact PJ69–4 cells with CALB Lyt-1 His may be related to the possibility that the movement of the enzyme to the supernatant is aided by the signal peptide secretion leader and Lyt-1. The signal peptide is only on the nontruncated form of the expressed CALB enzymes; hence, this is the predominant form in the supernatant of the intact cells. This form may favor the use of the shorter-chain fatty acids. Even though similar amounts of the various expressed forms of CALB (nontruncated and truncated) may be present in the intact and lysed cells, the environments are different. The lysed cells are exposed to cell lipid debris and extracellular proteases and are fully exposed to solvent. It is also possible that lysing the cells may affect the conformation of the active site. This may change the specificity of any or all forms of the recombinant CALB enzyme containing the active triad to favor the production of the C18 fatty acid ethyl esters.
The specific activities of Ni bead-purified CALB Lyt-1 His enzymes expressed from PJ69–4 yeast cells and immobilized on Sepabeads resin in the production of ethyl esters were compared with those of nonimmobilized recombinant CALB Lyt-1 His and commercially available CALB. It was demonstrated that one-step-charging resins specifically selected for binding to lipase (Sepabeads EC-EP, EXE077, and EXE078) were capable of covalent attachment of the expressed CALB Lyt-1 His and that the resin-bound enzyme was capable of catalyzing the production of biodiesel. The specific activities of the bound recombinant enzymes ranged from 60 to 80 μg ethyl esters/mg lipase for the immobilized lipases compared with 160 μg ethyl esters/mg lipase (from intact cells) for the nonimmobilized enzyme.
The recombinant lipases are designed to be bound to a resin for use in single-column production of biodiesel from ethanol and corn oil in a combined cellulosic and starch ethanol biorefinery. Lipases expressed in a cellulosic ethanol yeast strain would provide low-cost biocatalysts for the production of both bioethanol and biodiesel from cellulosic bio-mass. Use of this yeast strain could increase the profitability of an integrated biorefinery that combines starch ethanol and cellulosic ethanol facilities by catalyzing the conversion of corn oil (TAGs) byproduct to fatty acid ethyl esters (biodiesel) and glycerol in a single-step column transesterification with ethanol produced in the facility.
Acknowledgments
We thank Karen Hughes for critical reading and formatting of the manuscript. We also thank Nathane Orwig for performing the sequencing work. We acknowledge the support of USDA ARSdMitsubishi Chemical Corporation Nonfunded Cooperative Agreement, Project Number 3620-41000121-07. We thank Drs. Moreno Daminati and Paolo Caimi of Resindion S.r.l., Milan, Italy, for providing Sepabeads EC-EP, EXE077, and EXE078, and for technical assistance.
Competing Interests Statement: The authors certify that they have no relevant financial interests in this manuscript.