
Abstract
The migratory or proliferative responses mounted by wounded cell monolayers are important to drug discovery and drug safety testing, as well as to basic research across a number of disciplines, including stem-cell biology, cell biology, ophthalmology, endocrinology, microbiology, oncology, and developmental biology. Scratch wounding by mechanical means is the golden standard to achieve an appropriate model system in which to study these cellular reactions. The scratch wounding technique is plagued by wound size irregularity, release of cytosolic contents along the wound edge, and difficulty in scaling up to higher throughput screening. To address these issues, we developed a microfluidic device coupled to a well plate in which wounds were produced enzymatically using highly controlled laminar flow streams. Within the device, epithelial cells were cultivated and exposed to different compounds. Proliferation and migration were characterized by bright-field microscopy. Resulting wound size was highly uniform compared with reported variability of manual scratch wounding. The protocol for wounding was fully automated using customized software, and response to wounding was followed in real time by microscopy.
Introduction
Scratch wound assays are commonly performed to assess cellular migration and cell proliferation in response to exposure to potentially therapeutic compounds in development. Further, wounding assays are used to elucidate the molecular phenomena related to wound healing, including cell signaling, immune and healing response to bacterial and fungal infections, and tissue remodeling. Most wounding assays are performed using a mechanical means to generate a break in a confluent monolayer of cells, such as drawing a pipette tip or a pin over the mono-layer. This method is applied from very low-density cell-culture dishes to very high-throughput 384-well plates using pin tools. 1 High-throughput pin-tool wounding assays have been described to screen the effects of small molecules on wound healing. 1,2 The pin-tool—generated wounds are easy to produce and create the requisite perturbation to the mono-layer. A drawback to this method is that the scraping process causes physical damage to the cells which can cause the intracellular contents to leak into the experimental zone. This potentially complicates the interpretation of data. 3 Detritus are also left behind after the mechanical wounding, as the wound is produced in situ without further fluid disturbance of the cell culture well. A further drawback of this method is that wound size and shape are highly variable; in the literature, scratch wound assays reported wound size variability up to 33%. 4 These irregular starting conditions complicate the data analysis and make it difficult to directly compare experimental conditions.
The use of microfluidic devices has become commonplace in biology. Many devices enable very low reagent use or a novel way to perform cellular analyses. Fluidic arrangements of microfluidic devices can be designed to achieve very tight control of cellular microenvironments, including flow conditions, application of mechanical stresses, and introduction of compounds. Several devices have been reported in the literature to deliver flow to portions of channels, and thus to deliver compounds to cellular microdomains. 5 7 Nie et al. (2007) 8 describe such a three-inlet single microchannel device fabricated in polydimethylsiloxane to wound cells using trypsin, selectively perfused over the monolayer by aspiration. Although such devices offer an alternative solution to mechanical wounding, they do not transcend the limitations often seen with academic laboratory fabricated microfluidic devices, which include cumbersome fluid handling, device fabrication variability, throughput, and systems integration challenges.
Here, we adapted the BioFlux system to provide a microfluidic solution to wound-healing assays in the context of a fully integrated microfluidic system with software-controlled flow and self-contained liquid handling. The lab-on-a-chip device consisted of a high-density array of microfluidic flow cells coupled to the wells of a single SBS-standard 24-well plate with a two-inlet well to one-outlet well per channel design (Fig. 1A). There were 8 identical, independent channels on one 24-well plate providing higher throughput compared with single channel per chip laboratory made devices. The wells on the plate served as discrete fluid reservoirs for each channel, allowing simultaneous experimental preparation for all channels. Perfusion into the channel was controlled by a constant displacement pressure-driven flow system. The wounding process was initiated enzymatically using trypsin. The wounding protocol leveraged the laminar flow profile found in the microfluidic devices to only wound a fixed portion of the cell monolayer. The microfluidic channels in the BioFlux plate are lithographically reproduced with all channels within ±1 μm, and thus capable of producing highly consistent flow profiles (within ≤2% interchannel variation) and wound sizes when compared with the devices fabricated in Nie et al. 8 (data not shown). The wounds were made without mechanical damage to cells at the leading edge. Flow was used to remove unwanted cells from the monolayer. We studied both migration and proliferation after wounding in the presence or absence of epidermal growth factor (EGF), a promoter of cell migration, 9 and cytochlasin D, a motility and cell-cycle inhibitor. 10
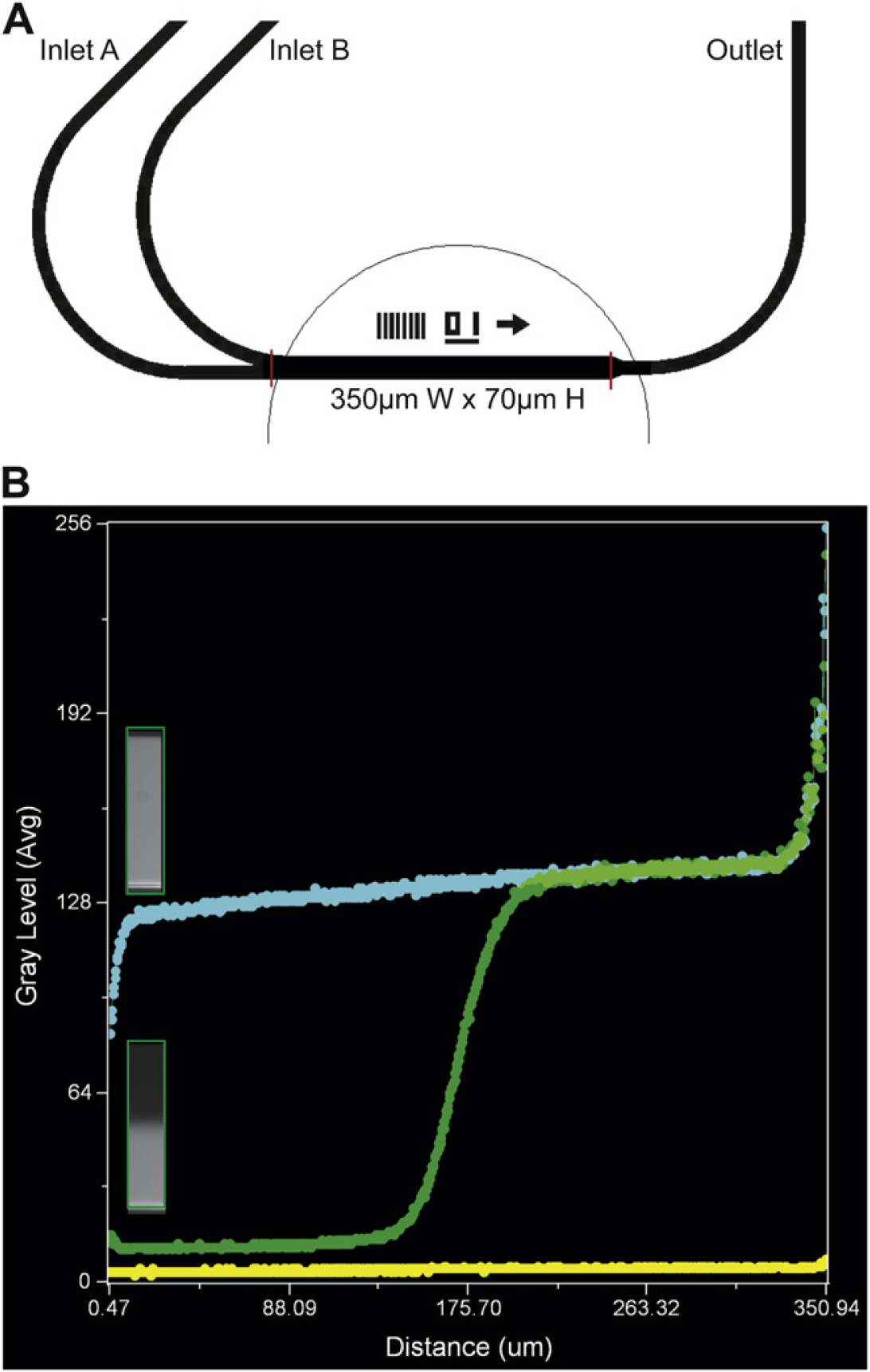
A representative two-inlet channel design for parallel flow assays. (A) Shown is an excerpt from the CAD drawing of channel 1 out of 8. Inlet A, inlet B, and outlet are connected to the wells on a 24-well plate, which serve as independently controlled fluid reservoirs. This device contains eight-independent, but identical channels. (B) Diffusion of small molecules during parallel flow. Fluorescent calcein mixed with trypsin was flowed in parallel with William's complete. Linescan measurements of fluorescence intensity were collected from channels filled with the calcein-trypsin (blue line) (fluorescent), William's complete (yellow line) (nonfluorescent), or both (green line). Insets are slices of the channel where the linescan measurements were captured. The mixing zone for calcein was approximately 67 μm; however, when trypsin mixes with serum containing media, the trypsin is inactivated confining the treatment zone to a smaller area.
Materials and Methods
Cell Culture
Rat lung epithelial cells (RLE) were cultivated under standard cell-culture conditions in William's E medium (Sigma-Aldrich, St. Louis, MO) supplemented with 2 mM Glutamax (Invitrogen, Carlsbad, CA), 10% fetal calf serum, 100 U/mL penicillin G, 100 μg/mL streptomycin, and 10 mM hepes (UCSF, CCF, San Francisco, CA) known as William's Complete. EGF was obtained from Invitrogen; cytochalasin D (CytoD) was obtained from Calbiochem (San Diego, CA).
Cell propagation for wound-healing experiments was carried out as follows: all liquid-handling steps were facilitated using the BioFlux pneumatic controller 11 ; the channels of BioFlux 200 24-well plates (Fluxion Biosciences, South San Francisco, CA) were coated with Matrigel (BD Biosciences, Franklin Lakes, NJ) at a nongelling concentration (1/50 v/v in Hank's buffered salt solution), followed by an incubation at 37 °C for 1 h. Channels were washed with prewarmed William's complete. RLE were seeded at 5 × 106 cells/mL (4 × 104 cells per channel) in the channels. Cells were allowed to attach to the channel surface for 1 h under cell-culture conditions. William's complete was added to the inlet wells of the plate to feed the cells by gravity flow overnight. Wounding experiments were performed the next day.
Parallel Flow Testing
The BioFlux controller was originally designed to provide intermittent flow from one well to another. For this experiment, the BioFlux was modified to produce parallel flow streams by rearrangement of the pneumatic lines to allow inlet A and inlet B to flow simultaneously and to be controlled independently. To define the zone of diffusion during parallel flow in the well-plate microfludic device, the channels were primed with William's complete from the outlet well. Downstream flow was initiated simultaneously from inlet B filled with trypsin mixed with calcein to a concentration of 10 μM (molecular weight is 622 Da) (Invitrogen, Carlsbad, CA) and from inlet A with William's complete (Fig. 2A). Images were captured during continuous flow at 5 dyn/cm2 using a TS100 Eclipse microscope (Nikon US, Melville, NY) with a QICAM (QIMaging, Surrey, BC, Canada) (Fig. 1B). All postexperiment data analyses were performed using BioFlux Montage software.
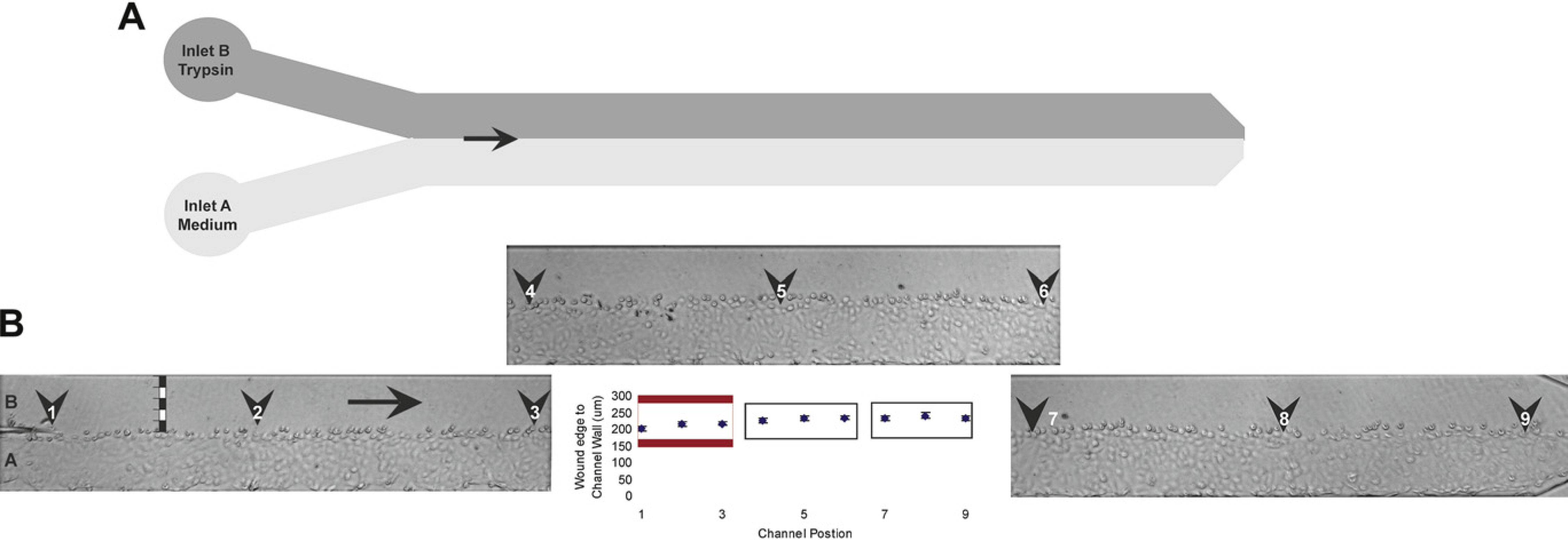
Creating wounded monolayers in the BioFlux system. (A) Parallel flow patterns in channel during wound formation. (B) Measurements of wound size in microns were made throughout the length of the viewing window at nine positions in total. The same regions were measured for each channel (black numbered arrowheads) from the leading edge of the wound to the outer wall of the channel (black and white bar). The size of the wound increased slightly as the distance from the inlet increased, regions 7–9. Error bars indicate standard error of the mean for 11 measurements.
Wounding Method, Migration, and Proliferation
For the enzymatic wounding assay, inlet B wells were washed out with PBS without divalent cations and Trypsin EDTA (0.25%) was added. Inlet A wells were filled with William's complete. Flow was initiated from both inlets concurrently at 1 dyn/cm2 for 10 min followed by a short pulse of high shear (10 dyn/cm2) to remove trypsinized cells. Perfusion from inlet B was stopped. Perfusion from inlet A was continued for 10 min to neutralize residual trypsin. Media and trypsin were removed from wells and replaced with William's complete plus or minus serum. At the same time in separate channels, CytoD, an inhibitor of migration and proliferation, or an EGF, a mediator of cellular migration, were added at the following concentrations to either serum plus or serum minus media: 1 and 5 μg/mL or 10 and 100 ng/mL for respective compounds. Plates were placed in the incubator, and data were collected at 2, 4, 5.5, 20, and 24 h post wounding using a TS100 Eclipse microscope (Nikon USA, Melville, NY) with a QICAM (QIMaging, Surrey, B.C.). All postexperimental data analyses were performed using BioFlux Montage software.
Results
Wound-Healing Reproducibility
After trypsinization, wound sizes were measured from the leading edge spanning the cell free area to the channel wall. The average wound size across the channel from the inlet to the outlet as determined by measurements at 9 points along the entire length of the viewing window was 224.5 μm ± 5.4%. The wound size did increase toward the outlet by 32 μm, which could represent diffusion and subsequent inactivation of trypsin as the length of the laminar flow streams increased from the inlet sources. The average wound size for the first three positions in the channel was 210 μm ± 3.8% (Fig. 2B, red box). This area was used for the remainder of the migration and proliferation studies, as it represents the largest gap to close with the smallest amount of variation (∼160-μm gap of a total of 350-μm channel width).
Cell Migration
To study migration after wounding, cells were allowed to recover in media without serum to separate wound-induced migration from proliferation. Measurements were made from the leading edge, the wound over the length of the monolayer to the channel wall, tracking the increase in cellular migration as movement of the cellular leading edge. The peak distance in migration was observed at 5.5 h post wounding. The highest concentration of EGF led to the furthest migration under starvation conditions at 47 μm, predictably both the media-alone control and the lower concentration of EGF led to migration as well (Fig. 3A). The unexpectedly long migration distance for the 5-μg/mL CytoD-treated monolayer occurred as clumps of rounded, presumably dead cells, massed in the channel and was erroneously measured as an increase in the distance to the wall. For future studies, the cellular morphology should be taken into consideration while performing this measurement. The initial cellular morphology observed for EGF and media-alone—treated cells after wounding was typical epithelial-like shapes and spacing; CytoD-treated cells in comparison adopted a rounded morphology by the first time point and never recovered subsequently. By 20 h post wounding, however, all cells began to round up and were lost from the mono-layer, most likely because of the starvation media conditions, which are nonpermissive for cell survival.
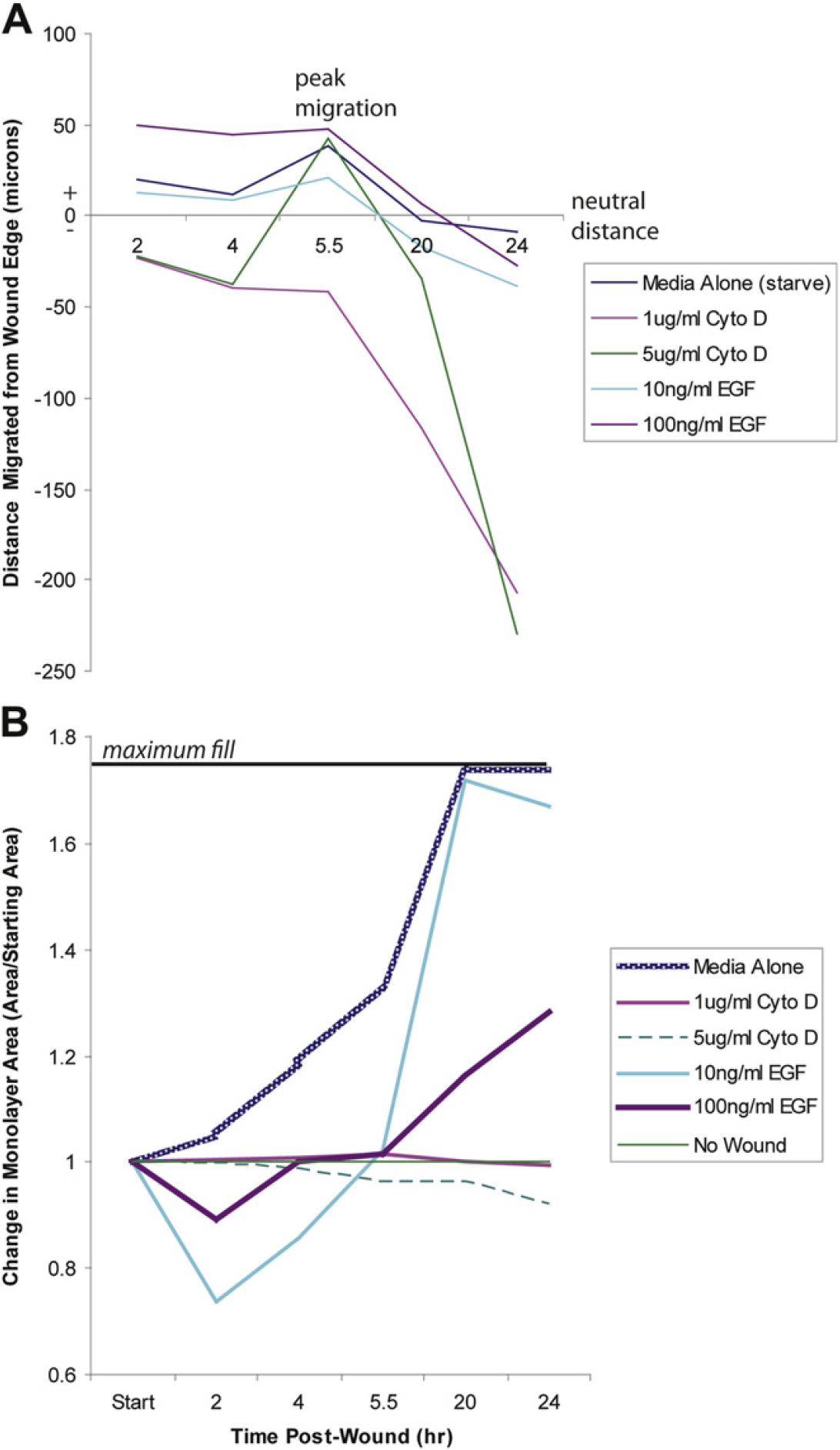
Cell migration and proliferation after wounding. Measurements were made at 2, 4, 5.5, 20, and 24 h post wounding. (A) Cell migration was measured as the distance from the mono-layer wall to the leading edge of the wound in three positions for each channel, only the first field of view was used (Fig. 2B), as the wound was the greatest size in this region. Distance migrated was calculated by subtracting the leading edge distance from the starting position of the leading edge in microns. Positive values express growth; negative values indicate contraction of or loss of cells from the monolayer. (B) Cell proliferation was measured as the total area covered by cells in μm2. The change in area shown is expressed as the area measured for each time point divided by the original area. Complete confluence of the monolayer is indicated by the bar at the top of the chart. Values below 1 indicate loss of monolayer are compared with starting size.
Wound Closure
To study cell proliferation leading to wound closure, all treatments were delivered in complete media containing fetal calf serum, which is permissive for growth. The cells in the media-alone control were tightly packed together reminiscent of the non-wounded control and closed the wound without spreading out. By 20 h, the gap was completely closed (Fig. 3B). Cells treated with EGF behaved differently from the control. EGF treatment caused cells to migrate away from the wall and spread out (Fig. 4) before closing the wound by proliferating toward the walls of the microfluidic channels. Cells with the highest EGF concentration remained disordered and never closed the gap. Cells exposed to the lowest concentration, however, came close to complete wound closure within 24 h.
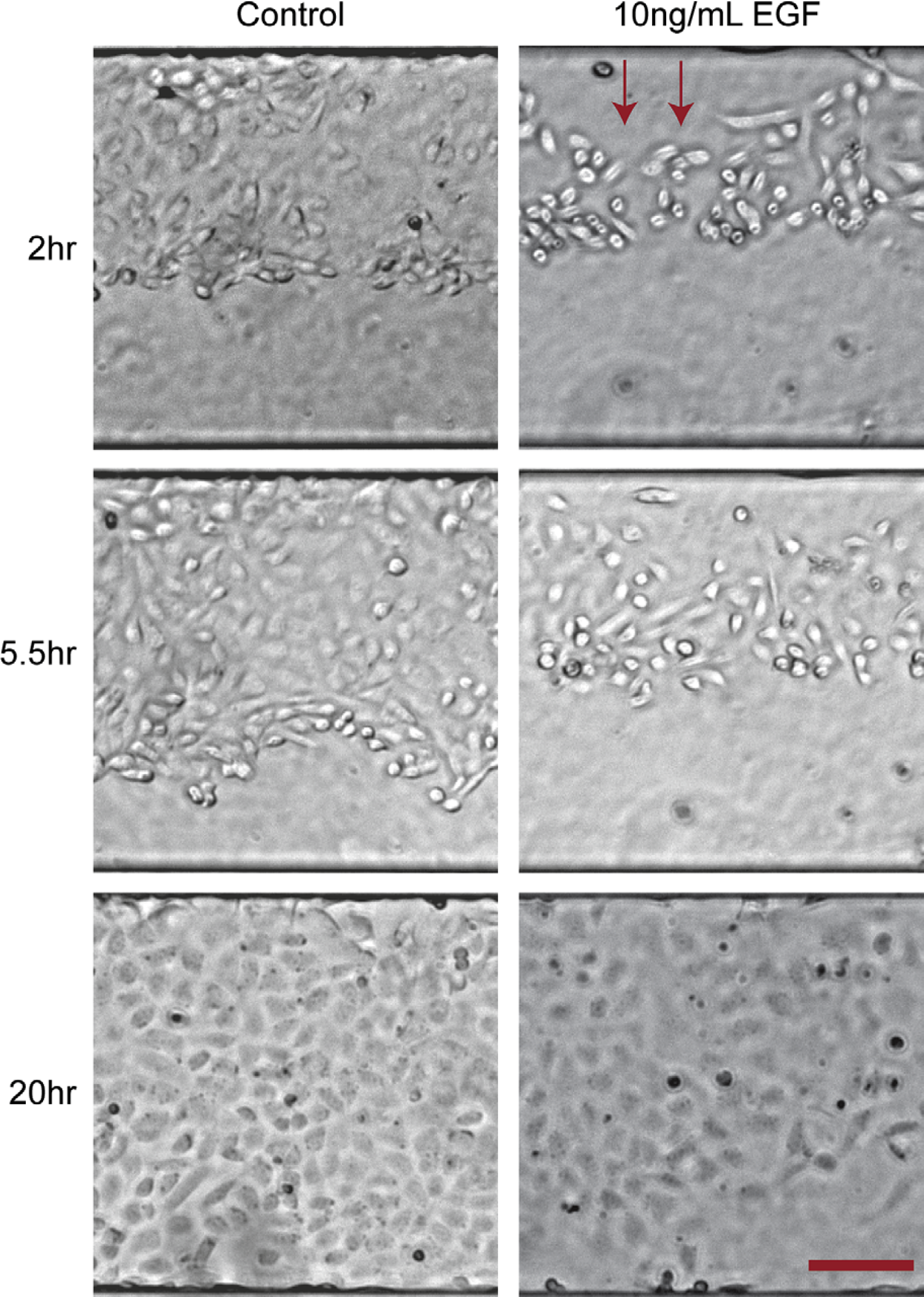
Cell morphology during proliferation. Cells treated with epidermal growth factor (EGF) displayed increased spreading and disordered morphology before proliferation under permissive conditions in contrast to the control monolayers that maintained a tightly packed cobblestone appearance and proliferated in an orderly manner from leading edge to far channel wall. Scale bar is 100 μm.
Conclusions
We adapted the commercially available BioFlux system to perform a novel wound-healing assay using parallel flow with improved reliability and reproducibility over traditional scratch wound assays. Tightly controlled wounds were generated with RLE that showed reproducibility of 5.4%. Because the wounds were generated by enzymatic means combined with fluid flow, the cells at the leading edge of the wound remained intact; thus cytosolic leakage or debris were not a factor in migration, proliferation, or the interpretation of the data. The flow capabilities of the microfluidic system were also effective in clearing away the trypsinized cells postwounding and providing a more uniform starting point for the assay. We demonstrated that cell migration can be followed using media starvation conditions with different compounds. We also showed that cell proliferation can be assessed in the same manner. For future studies, exploitation of parallel flow streams may also be applicable to live-cell assays, such as chemotaxis, invasion, compound mixing, fluid delivery to microdomains, and other assays relying on simultaneous use of different fluids.