
Abstract
In this article, two-dimensional hexamethyldisilazane (HMDS) micropatterns were generated on glass substrates using photolithographic techniques for the assembly of functional proteins. The non-HMDS patterned areas were backfilled with poly(ethylene glycol) (PEG) silane to reduce the nonspecific protein adsorption. The hydrophobic methyl-terminated HMDS monolayer was verified to be favorable for physical protein adsorption with bovine serum albumin (BSA). The PEG-silane derivatized surface significantly reduced the BSA nonspecific binding by 97% compared to the pristine glass substrate so that high patterning selectivity was achieved. A universal streptavidin template was generated using preadsorbed biotinylated BSA on HMDS surface to sequentially bind additional biotinylated antibodies. Using this patterning strategy, the biotinylated goat anti-mouse (biotin-GAM) antibodies can be specifically recognized by the fluorescently labeled mouse immunoglobulin G, which indicated that the immobilized biotin-GAM was still bioactive. Also, the immobilized alkaline phosphatase was demonstrated to retain its enzymatic functionality by the ability to convert its fluorogenic substrate fluorescein diphosphate into fluorescent products. This simple and effective protein patterning technique can also be extended to create nanoscale protein arrays. Additionally, its adaptability for the assembly of arbitrary proteins and antibodies provides great potentials for biosensor and biomicroelectromechanical systems (MEMS) applications.
Introduction
Patterning functional proteins at the micrometer and nanometer scale has drawn tremendous attention for its broad range of applications from fundamental cell biology to lab-on-a-chip biosensor. 1,2 Particularly during last decade, taking advantages of the soft lithography technique combined with the surface chemistry, various inexpensive and effective methodologies such as microcontact printing 3,4 and capillary force lithography 5 have been explored to successfully pattern functional proteins and served as important technological platforms for biological research. However, one major limitation of such soft lithography based approaches is the inability to perform precise position and dimension control on the protein patterns. This limitation hinders these techniques to be widely adapted to biosensor application, which has an essential criterion for aligning protein patterns with prefabricated structures such as microelectrodes and microcantilevers for impedance, resonance frequency, or cantilever deflection-based biosensing. 6 -11 Current methods capable of providing accurate position and dimension control on patterned proteins include self-assembly of the alkanethiol monolayer onto a patterned gold surface 12 -14 and photopattern of the alkyl silane or other organic polymers on a silicon or silicon dioxide substrate. 15,16 These methods are able to pattern proteins with precisely defined geometry and position through covalent coupling or physical adsorption. Compared to covalent coupling, physical adsorption of proteins onto a photopatternable silane surface with terminal groups favorable for protein adhesion is a relative simple and robust method and does not require special facilities for gold deposition.
In this article, we present a generic approach of using conventional photolithography techniques to create a heterogeneous surface with controllable surface hydrophilicity to physically immobilize proteins at designed sites. In our study, a commonly used photoresist promoter in micro and nano fabrication, hexamethyldisilazane (HMDS), was investigated to create the protein adhesion surface for the following reasons. Firstly, HMDS reacts with the hydroxyl-terminated silicon or silicon dioxide substrates through Si–O covalent bond even under the ambient laboratory conditions. Secondly, the methyl-terminated HMDS monolayer is hydrophobic and suitable for physical adsorption of proteins. Thirdly, HMDS can be easily patterned using photolithography techniques with high controllability on the geometries of generated patterns and their relative positions to previously fabricated microstructures on the substrate. To achieve high-protein patterning selectivity, poly(ethylene glycol) (PEG) silane passivation of non-HMDS coated areas was explored and showed to be very effective in preventing the nonspecific protein adsorption. In addition, the bioactivity of patterned proteins was tested using two strategies: antibody-antigen specific recognition and enzyme catalytic ability. Both studies verified that our patterning method had retained the functionalities of the immobilized proteins. Therefore, the ability to pattern functional proteins with high selectivity and accurate spatial control, combined with the simplicity and robustness of this proposed technique, can be explored for a wide range of applications.
Experimental
Materials and Reagents
The following materials and chemicals were used in this study: HMDS from Shin-Etsu Microsi Inc. (Phoenix, AZ), 2-[methoxy(polyethyleneoxy)propyl]trimethoxy-silane (methoxy-PEG-silane, Molecular Weight = 460–590) from Gelest (Morrisville, PA), 25 mm × 75 mm glass slides and phosphate buffered saline (PBS, pH = 7.2) from Fisher Scientific (Pittsburgh, PA), triethylamine, dimethyl sulfoxide (DMSO), acetone, isopropyl alcohol (IPA), anhydrous toluene, and ethanol from Sigma (Milwaukee, WI).
The following reagents used in this study were reconstituted to the concentrations indicated in the text with PBS unless individually specified: biotinylated bovine serum albumin (biotin-BSA) from Pierce (Rockford, IL); texas red-labeled bovine serum albumin (TR-BSA) from Invitrogen (Carlsbad, CA); fluorescein-labeled streptavidin (FITC-streptavidin); biotinylated goat anti-mouse (biotin-GAM); tetramethylrhodamine (TRITC)-labeled mouse, goat, and rabbit immunoglobulin G (IgG) from Jackson ImmunoResearch Laboratories (West Grove, PA); alkaline phosphatase (ALP) from Molecular Probe (Eugene, OR); and fluorescein diphosphate, tetraammonium salt (FDP) from AnaSpec Corporate (San Jose, CA).
Substrate Modification
Glass substrates were first cleaned with a mixture of four parts of H2SO4 and one part of H2O2 at 100 °C for 10 min, extensively rinsed with deionized (DI) water (18.3 MΩ-cm, Millipore) and passively dried with nitrogen. The surface moisture was driven out by baking the cleaned glass substrates at 150 °C on a hotplate for 15 min. After baking, the substrates were immediately placed into a flask containing 2 mL of HMDS for 15 min at room temperature to form the HMDS monolayer. Positive photoresist AZ5214E was spun onto the substrates at 3000 rpm and exposed through a chromium photomask with a Karl Suss UV exposure system. Both the exposed AZ5214E photoresist and the underlying HMDS monolayer were removed by AZ400K developer diluted with four volumes of DI water. A 2-min oxygen plasma treatment was followed to remove the residual AZ5214E photoresist and HMDS so that glass substrates were fully uncovered in the designed areas. Undeveloped photoresist was then dissolved with acetone, while the underlying HMDS monolayer was not affected by acetone. The substrates were then thoroughly rinsed with IPA and DI water and passively dried with nitrogen. To covalently couple methoxy-PEG-silane to non–HMDS-coated surface, glass substrates were then placed into 3 mM methoxy-PEG-silane dissolved in anhydrous toluene with 1% triethylamine as a catalyst for 4 h at room temperature. The physically adsorbed methoxy-PEG-silane moieties were then removed by rinsing the glass substrates in anhydrous toluene, ethanol, and DI water for 5 min, respectively, with sonication. Finally, the substrates were dried with nitrogen and used immediately for protein immobilization experiments.
Characterization Methods
Contact angles were measured by the sessile drop technique using a contact angle goniometer (First Ten Angstroms, Portsmouth, VA) under ambient conditions (∼36% humidity and 25 °C). Measurements were made within 30 s after pipetting 5 μL of DI water onto sample surface. HMDS micropatterns were characterized with a scanning electron microscope (Hitachi S4700). Protein binding effects were observed using an inverted fluorescence microscopy (Leica DMIRB) equipped with a monochrome CCD camera (Roper Scientific Photometrics, CoolSnap HQ) with a pixel size of 6.45 μm × 6.45 μm.
Results and Discussion
It has been demonstrated that HMDS ([CH3)3Si–NH–Si(CH3)3]) can be used to render a hydroxyl-terminated surface from hydrophilic to hydrophobic. 17,18 Through condensation reaction, HMDS can react with the surface hydroxyl (–OH) groups to form a monolayer of siloxane (i.e., Si–O–Si(CH3)3) through the following reaction:
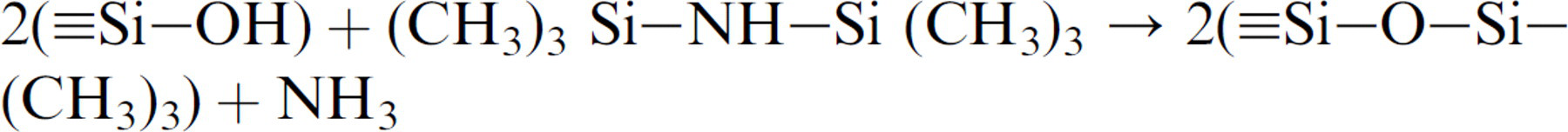
In this way, the hydroxyl groups were replaced with the hydrophobic methyl groups (–CH3) of HMDS during the surface modification. This newly formed methyl termination renders the surface more hydrophobic. The hydrophobic nature of the HMDS monolayer leads to greater wettability for the photoresist and makes HMDS a suitable photoresist adhesion promoter in micro and nano fabrication industries. In addition, HMDS can be removed by the alkaline-based positive photoresist developer (AZ400K in our study) so that it can be selectively patterned. To inhibit protein nonspecific binding and thus increase the protein patterning selectivity, low molecular weight methoxy-PEG-silane was covalently coupled to the glass substrate after the HMDS monolayer had been patterned. Figure 1 illustrates the surface modification process.
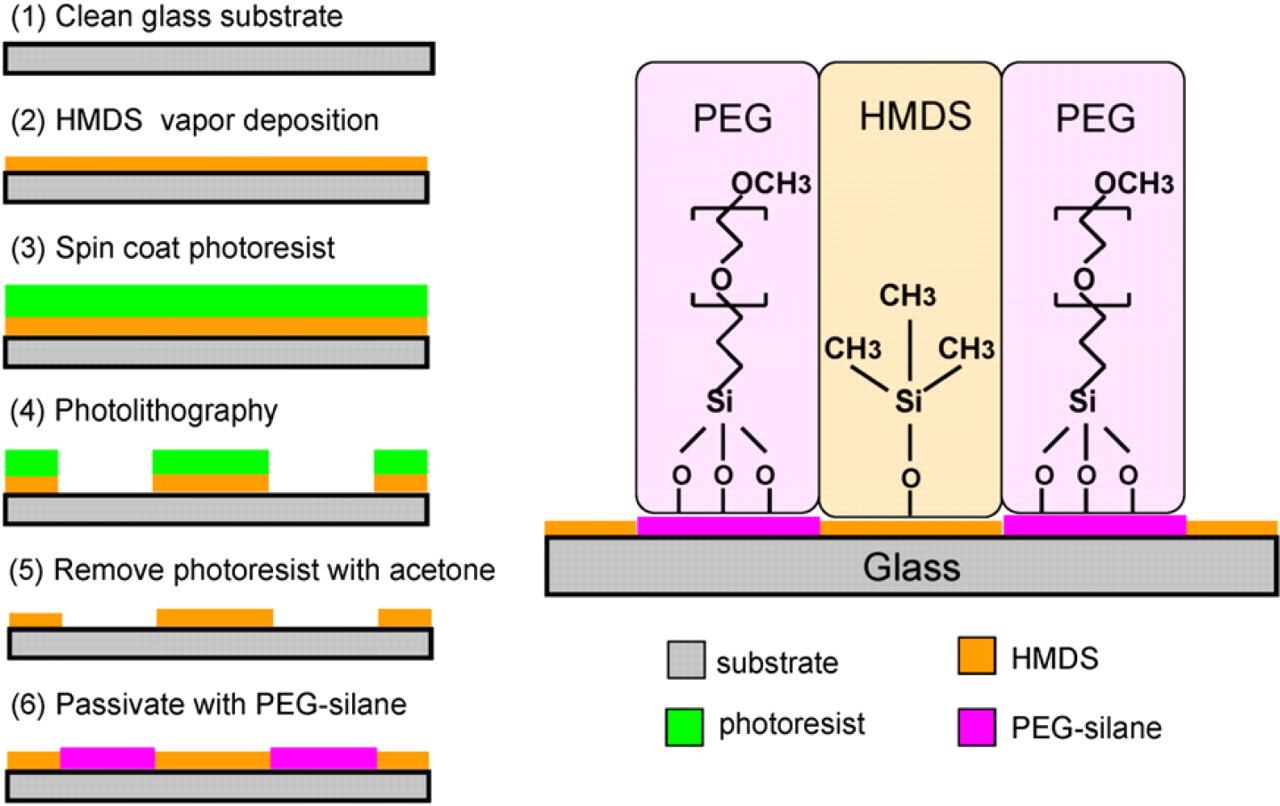
Surface modification process for generating hexamethyldisilazane protein adhesion micropatterns and poly(ethylene glycol) protein resistant surface.
The SEM image of photolithographically patterned HMDS microfeatures was shown in Figure 2. In this image, a silicon substrate was used instead of glass to enhance the second electron emission between the substrate and the HDMS derivatized substrate surface, and hence the better image contrast. 19 The contact angle of the HMDS-modified glass surface with a 15min incubation time was measured to be 72 ± 2°, which was comparable to the previously reported value. 20,21 However, the pristine glass surface is hydrophilic with a contact angle of approximate 30°. BSA, a protein well known for its propensity to adhere to surfaces, 22 was used to study the protein adsorbing ability on HMDS- and PEG-modified glass substrates. TR-BSA (10 μg/mL) was incubated on the freshly prepared HMDS monolayer for 10 min at room temperature. Unbound moieties were washed away with PBS and DI water and gently dried with nitrogen. Contact angles were measured and found to decrease to 39 ± 2°, which indicated the successful BSA adsorption on the HMDS monolayer. The adsorption of TR-BSA onto HMDS monolayer was also characterized by fluorescence microscopy and showed to follow an exponential function with a characteristic time constant of about 4 min at the concentration of 10 μg/mL (data not shown). PEG is well known for the resistance to the nonspecific protein adsorption because its highly hydrated polymer chains minimize the hydrophobic interaction between proteins and the underlying substrate. 23 Compared to the pristine glass surface, the PEG derivatized surface showed excellent protein repelling capability by significantly reducing BSA nonspecific binding up to 97%. Figure 3 shows the fluorescence image of the HMDS- and PEG-modified glass surfaces after 10 min of TR-BSA (10 μg/mL) incubation. BSA almost exclusively stayed on the HMDS surface, which demonstrated the high selective protein patterning capability of this method.

Scanning electron microscopy image of hexamethyldisilazane micropatterns on a silicon substrate.
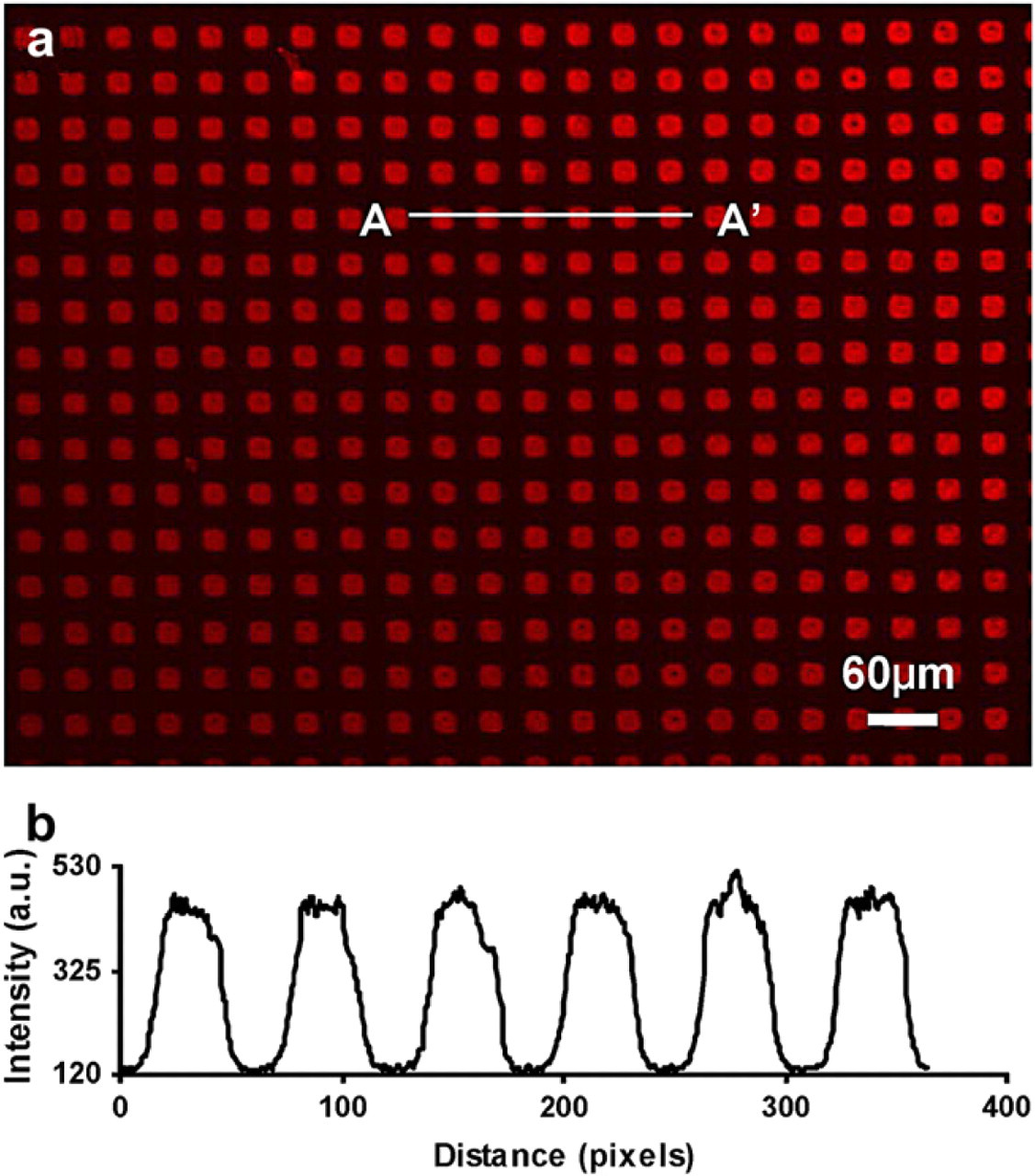
(a) Fluorescence micrograph of texas red-labeled bovine serum albumin adsorbed onto the patterned hexamethyldisilazane squares (20 μm × 20 μm) on a glass substrate. The surrounding areas were coated with methoxy poly(ethylene glycol) silane to prevent nonspecific protein adsorption. (b) Fluorescence intensity profile of A-A' cross-section in (a).
The successful patterning of BSA can be extended to other proteins and antibodies using the biotin-streptavidin specific binding. The choice of biotin-streptavidin complex as a receptor-ligand pair was due to the high binding affinity and well-studied binding mechanism. 24 Streptavidin is a globular protein with four biotin-binding sites on two opposite sites. Using biotin-BSA as an intermediate layer, streptavidin can be linked onto the surface and serves as a “universal” template for further immobilization of other biotinylated proteins. In our study, biotin-BSA (20 μg/mL) was incubated on a HMDS- and PEG-modified glass substrates using the same incubation procedure as TR-BSA mentioned above, followed by incubation of FITC-streptavidin (100 μg/mL) for 10 min and rinsed with PBS. This streptavidin layer was ready for chemisorbing other biotinylated proteins or antibodies. To test the effectiveness of this strategy, biotin-GAM IgG was incubated onto the streptavidin layer and its bioactivity was verified using a TRITC-conjugated antibody. In detail, biotin-GAM (10 μg/mL) was incubated on the top of the FITC-streptavidin layer for 10 min at room temperature and rinsed with PBS to remove any unbound moieties. TRITC-labeled IgG (10 μg/mL) from three species (mouse, goat, and rabbit) was then incubated on the biotin-GAM layer for 10 min at room temperature, rinsed with PBS and DI water, and gently dried with nitrogen. The binding effect was characterized with fluorescence microscopy. Results showed that specific recognition occurred between mouse IgG and the immobilized biotin-GAM (Fig. 4a). However, as controls, goat and rabbit IgG did not specifically interact with the immobilized biotin-GAM. Only very small amounts of goat and rabbit IgG adsorbed nonspecifically to the surface causing the fluorescence signals almost undistinguishable from the background noise (Fig. 4b, c). These results verified that the bioactivity of biotin-GAM was retained by this biotin-streptavidin template-guided protein immobilization strategy.
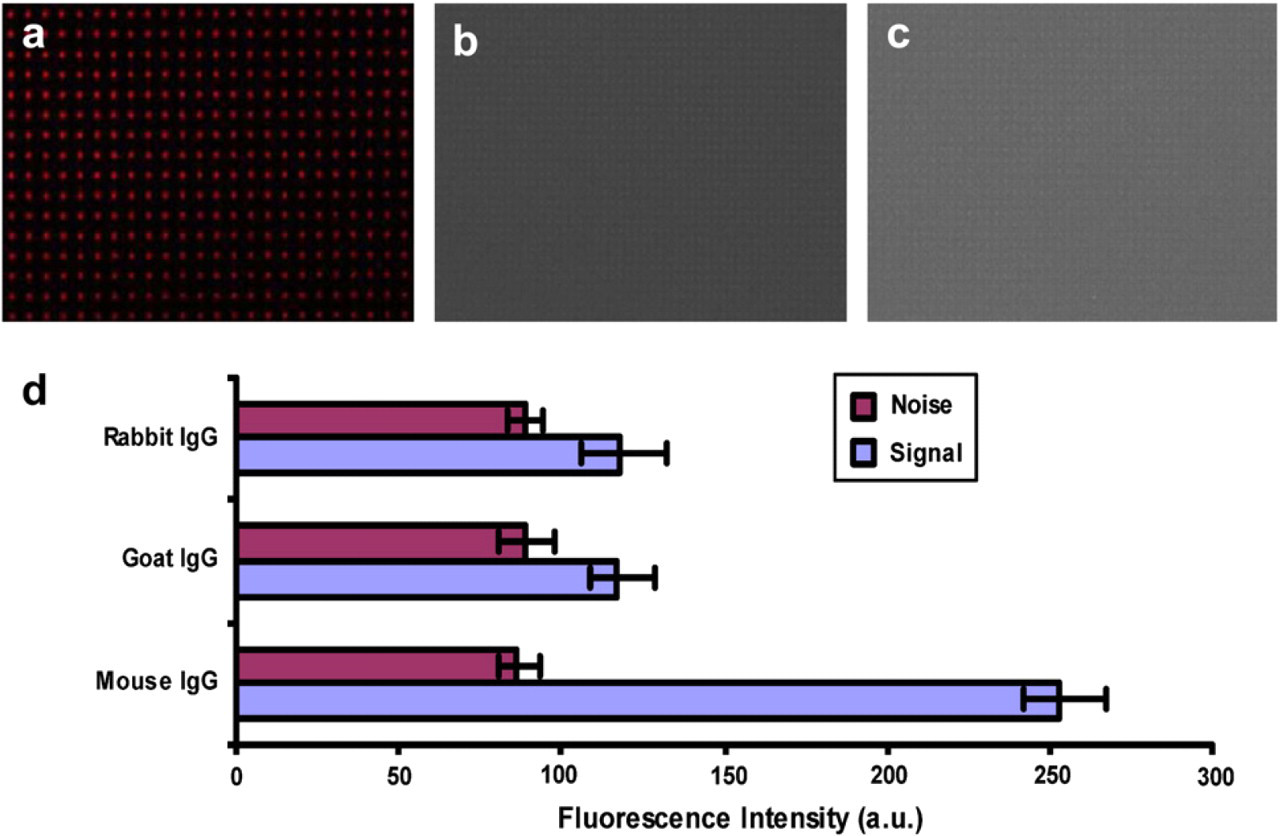
Fluorescence micrograph of tetramethylrhodamine (TRITC)-labeled immunoglobulin G (IgG) from mouse (a), goat (b), and rabbit (c) to probe the patterned biotinylated goat anti mouse (biotin-GAM). (d) Fluorescence intensity from TRITC-labeled mouse, goat, and rabbit IgG after incubation on the immobilized biotin-GAM. Signals from the mouse IgG were about two times higher compared to the goat and rabbit IgG, which were almost at the same level as the background noises.
We also tested the enzymatic functionality of ALP after being immobilized onto a HMDS-modified surface. ALP is known for being able to dephosphorylate a fluorogenic substrate, FDP, to generate highly fluorescent products fluorescein. In this experiment, we incubated ALP (200 μg/mL) dissolved in DI water onto an array of 20 μm × 20 μm HMDS squares with surrounding areas passivated by methoxy-PEG-silane. Next, FDP (100 μg/mL) dissolved in DMSO was added and the fluorescence images were taken sequentially with time. ALP was demonstrated to retain its functionality by the ability to convert FDP into insoluble products that consequently precipitated at the areas with immobilized ALP. Fluorescence signals were increasing with time and indicated the catalytic ability of ALP. After approximate 40 s with the addition of FDP, the fluorescent intensity reached the maximum level (Fig. 5). Due to the fast reaction, FDP could not be continuously washed away from the substrate. We believe that the background signal in Figure 5a was resulted from unwashed FDP due to its intrinsic fluorescence. After washing with DI water, the background signal dramatically decreased to the original level.
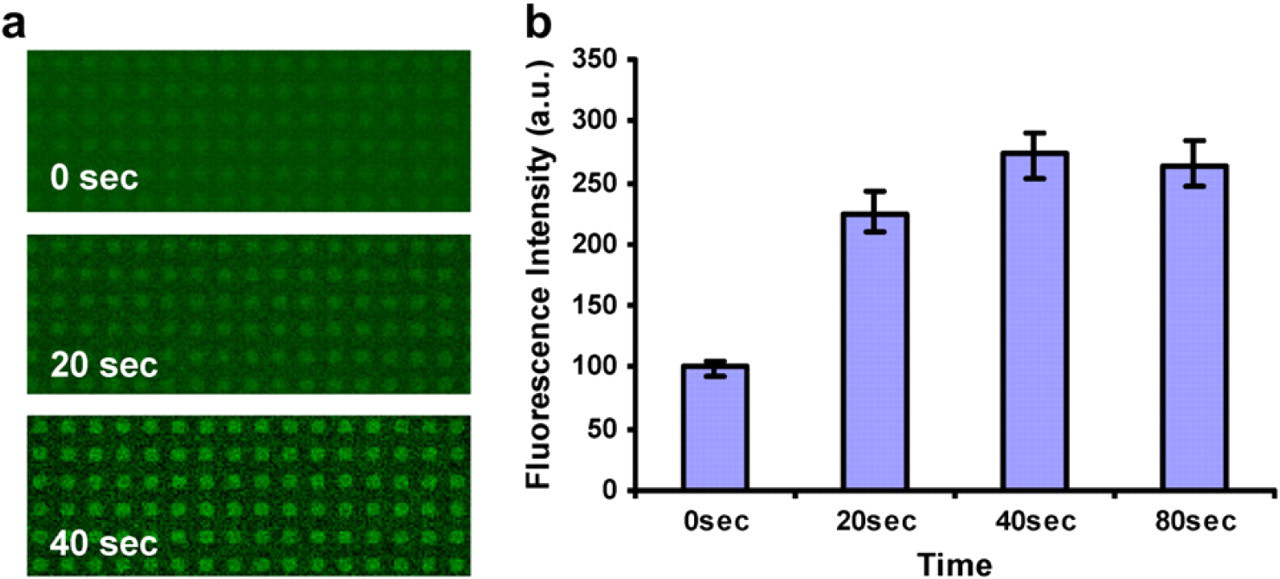
Visualization of alkaline phosphatase (ALP) functionality after being adsorbed on hexamethyldisilazane surface. (a) Time sequences of the catalytic activity of patterned ALP converting a fluorogenic substrate fluorescein diphosphate (FDP) into its fluorescent products. Each spot is 20 μm × 20 μm. (b) Fluorescence intensity of 0, 20, 40, and 80 s after the addition of FDP.
Conclusion
In this article, we demonstrated a simple and robust method to create two-dimensional HMDS micropatterns for patterning functional proteins. High selectivity was achieved through passivating the non-HMDS covered glass substrate surface with methoxy-PEG-silane. BSA was used as a model protein to characterize the effectiveness of this method. Both contact angle measurement and fluorescence microscopic observation verified that BSA was strongly adsorbed onto HMDS surface while PEG provided nearly complete resistance for BSA adsorption. Furthermore, using biotinylated BSA as an intermediate layer, streptavidin was successfully immobilized onto HMDS patterns and can be used as a template to bind a variety of biotinylated proteins or antibodies. Results also showed that the proposed patterning strategy can retain the functionality of the immobilized proteins through studies of the specific antibody-antigen recognition and the enzyme catalytic ability.
Our protein patterning method has several advantages including simplicity, high throughput, and robustness. Additionally, this method extends the capability of conventional photolithography to patterning biological species with very high spatial resolution and alignment accuracy. Finally, this technique is compatible with electron-beam lithography so that it can also be applied to generate nanoscale proteins patterns. The well-maintained bioactivity of the patterned proteins and the versatility of this technique can lead to promising applications for creating complex microscale and nanoscale protein patterns for biological studies and biosensor development.
Acknowledgment
The authors would like to thank NASA National Space Biomedical Research Institute (NCC 9–58–317) and NSF nano manufacture center SINAM (DMI-0327077) for the funding supports.