
Abstract
We discuss the use of stochastic optimization methods for biomolecular structure prediction, particularly applied to protein structure prediction and receptor ligand docking. After a brief introduction we give an overview of the dominating physical effects that are important for protein structure prediction and outline our strategy to address this problem. We discuss the strengths and weaknesses of several possible optimization methods, including the stochastic tunneling method. Finally we give examples of applications of this methodology both for protein structure prediction and receptor ligand docking.
Keywords
Introduction
Biomolecular structure prediction remains one of the main outstanding problems of theoretical biophysical chemistry. 3 One of its primary goals is the prediction of the three-dimensional, tertiary structure of proteins on the basis of their amino acid sequence (protein structure prediction — PSP). Proteins are the building blocks of the cellular machinery of all living organisms and plants. 10 Their enzymatic capabilites often surpass the efficiency of competing chemical processes by orders of magnitude. Consider the light harvesting mechanism of chlorophyll and the synthesis of the penicillin family of antibiotics as two examples. 37
Experimental methods to determine the sequence of a particular protein, either directly through protein sequencing, or indirectly through the use of genetic information, have made enormous progress in the last decade, resulting in a pool of several hundred thousand sequenced proteins for various organisms. Unfortunately, sequence information alone is often insufficient to elucidate the biological function or mechanism of a protein. To understand the mechanism of a protein, knowledge of its three-dimensional structure is very helpful. Experimental methods for protein structure determination are orders of magnitude more involved and more expensive than sequencing techniques. Although their number is steadily growing, the protein database (PDB) presently contains about 13,000 spatially resolved structures. Therefore, there is a large gap between the pool of available sequences and the set of structurally resolved proteins, which is likely to contain a wealth of important biological and biomedical information. There are entire families of important proteins, in particular transmembrane proteins, that are almost impossible to resolve structurally with present day experimental techniques. Theoretical methods for PSP may be helpful to close this gap in a number of relevant experimental contexts36,3, e.g., the elucidation of structure-function relationships in transmembrane proteins.
In addition there are many questions regarding the details of protein functions, in particular those of dynamical nature, that are difficult to address experimentally at the present time. Many of these problems, as well as questions regarding protein-protein association or protein-ligand interactions, would benefit from accurate theoretical methods for PSP. In particular simulation techniques addressing protein-ligand interactions would contribute significantly to the development of new pharmaceutical agents for a variety of diseases. 19
Closely related to the PSP challenge is the protein folding problem22,11,15, where one tries to understand the thermodynamic mechanisms of protein folding. The reason why molecules of such complexity have a unique three-dimensional structure and how they attain it is presently not well understood. Obviously an efficient solution to the protein folding problem would also provide a mechanism for PSP, but presently this seems to be far out of reach. Even though a solution to the protein folding problem may not be directly useful for PSP, investigations into this problem provide crucial information for the design of PSP methods that attempt to predict the native structure without recourse to the folding dynamics.
One approach of PSP focuses on secondary structure prediction, which remains an active area of research to date. The best methods available today, most based on neural network models, are able to assign between 60–80% of the secondary structure of a given protein with reasonable reliability. Unfortunately, from the point of tertiary structure prediction, the most successful methods available today are based on neural networks, so that little information about the underlying physical mechanisms can be derived from these predictions. Tertiary structure prediction is less well developed. Most methods competing in the CASP 26 structure prediction contest are based on homologies to structurally resolved proteins and heuristic input. As with secondary structure prediction, methods that are based on non-physical input are difficult to evolve systematically. Hybrid approaches, combining experimental data, sequence homologies and theoretical prediction, nevertheless yield important insights in present day applications. 9
One approach to PSP is to model the folding process with molecular dynamics (MD) in a classical biomolecular forcefield. Two major obstacles must be overcome in this approach: First, the protein folding process is slow compared to typical MD timescales: typical protein folding times range from 10 ms to 1 s. In order to resolve the trajectory of the system with sufficient accuracy, most presently available MD methods operate on a femtosecond timescale. An atomistic simulation of a 10 ms folding process would thus require O(1013) simulation steps. A typical protein has of order O(104) atoms, each of which interacts via long-range interactions with a significant number of neighbors, say O(1000). Folding the protein thus requires O(1020) force evaluations. Since the solvent has a significant impact on the folding process, the entire system has to be embedded in a sufficiently large bath of water, which may increase the total CPU requirement by another order of magnitude. Using 1 GFLOP processors, folding a single protein can thus be expected to consume about 30000 CPU years and great efforts are being made to make hardware resources at that scale available for this purpose. 7 Even so, it presently appears unlikely that a PSP method that simply extrapolates this strategy will be able to significantly augment experimental procedures in the foreseeable future. 16
One may therefore ask whether it is possible to devise alternate simulation strategies that circumvent the folding bottleneck described above. 24 An obvious starting point is the elimination of the explicit treatment of the solvent molecules 18 which often consumes the majority of the numerical effort associated with the simulation of the overall system. Upon closer inspection of this approximation, we find that the introduction of an implicit solvent model has far deeper implications on PSP than the obvious reduction of the computational effort resulting from the reduction of the degrees of freedom of the simulation. We note that the overwhelming majority of the entropic contribution to the folding process are solvent contributions, mediated by hydrophobic and hydrophilic effects of the different amino acid side chains. Incorporating these terms into an implicit solvent model, we obtain, in conjuction with the internal energy of the protein, a good model for the total free energy of the system. 13 As indicated above, most proteins attain a unique stable native structure. If the protein is in thermodynamic equilibrium with its environment, this structure must therefore correspond to the global minimum of its free energy surface. As is well known from the simulation of many physical systems with complex dynamics, it is possible to locate the thermodynamically stable state of the system using stochastic optimization methods without recourse to its dynamics orders of magnitude faster than in a simulation approach.4,6
In the past several years we have implemented this strategy, developing both forcefields and stochastic optimization methods suitable to this task. In the following sections we describe the components of this approach and give an overview of our results. A final section deals with the application of this methodology to a related problem in rational drug design.19,8
Biomolecular Forcefield
Over the last decades many classical forcefields17,20,23,30 have been developed to investigate numerous phenomena in physical, organic and inorganic chemistry. The difficulties encountered in PSP justify the development of specific forcefields because the molecular building blocks (i.e., the amino acids) are well defined and limited in number and the chemical complexity associated with the design of a forcefield specific to peptides and proteins is less than that of generic organic substances. By exploiting the fact that only a limited number of protein building blocks will occur, their ingredients may be specifically adapted to provide a more accurate description of the system. Secondly, we are interested only in the low-energy conformations of the model. As a result, many degrees of freedom that are associated with covalent interactions, e.g., bond stretching, may be neglected in the description of the system. The accuracy of this approximation can be checked against the available data in the PDB. The remaining degrees of freedom are thus only rotations about the dihedral angles of the backbone and of freely rotatable single bonds of the sidechains. This reduction of the number of degrees of freedom leads to a dramatic increase in the efficiency of the simulation. Generically the intramolecular forcefield should include Lennard-Jones (LJ) interactions that provide for steric repulsion and van-der-Waals attraction, electrostatic interactions and hydrogen-bonding interactions.
Our ab-initio protein forcefield (AIPF) represents all atoms except apolar CHn individually. CHn groups are approximated by a single sphere comprising both the carbon and the hydrogen atoms (united atom approach). Comparison with PDB data shows that many proteins have clashing configurations when Lennard-Jones parameters of standard organic forcefields are employed. We therefore developed a strategy to fit the LJ radii in accordance with the experimental data and fitted the LJ radii of our model to a subset of 134 proteins of the PDB database. 1 The associated LJ interaction strength was taken from the OPLS forcefield. 24 Note that the choice of LJ interaction strength should not be crucial in the PSP problem. Proteins in native or near-native configurations almost always assume relatively dense configurations which attain a packing density of up to 75% hexagonal close packing. As a result, all near-native configurations have similar attractive LJ contributions. Unfolded contributions, for which the number of LJ contacts may be significantly different are much higher in energy than folded configurations. Since we are only interested in the former, errors describing the latter can be tolerated (this is in contrast to folding studies, where temperature dependent equilibria between folded and unfolded configurations must be considered). Also note that in simulations with explicit solvent molecules there are LJ interactions between peptide and solvent atoms. This atom-dependent effect must be incorporated into the implicit solvent model.
Coulomb interactions in proteins are complicated, in particular regarding screening effects of the solvent. In the INT forcefield we have implemented an approach which models this effect with group-dependent and interaction dependent effective dielectric constants. 2 Using only the electrostatic interactions1,2 (including the dipolar interactions) we noted significant deviations in the backbone dihedral angles in the modeling of long helices. Since such hydrogen bonds are very important in the stabilization of the native structure we have parameterized the backbone-backbone hydrogen bonds as: where each component of the potential is modeled by a superposition of Gaussian functions that are fitted to reproduce the experimentally observed structures. For the implicit solvent model, the simplest conceivable choice assigns a free energy of solvation proportional to the effective contact area each atom of the protein/peptide has with the solvent. We have subdivided the atom types of the forcefield into suitable subgroups and fitted the resulting model to the available experimental Gly-X-Gly data 18 (see Figure 1).
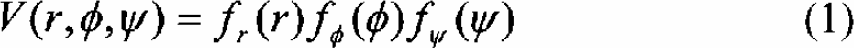
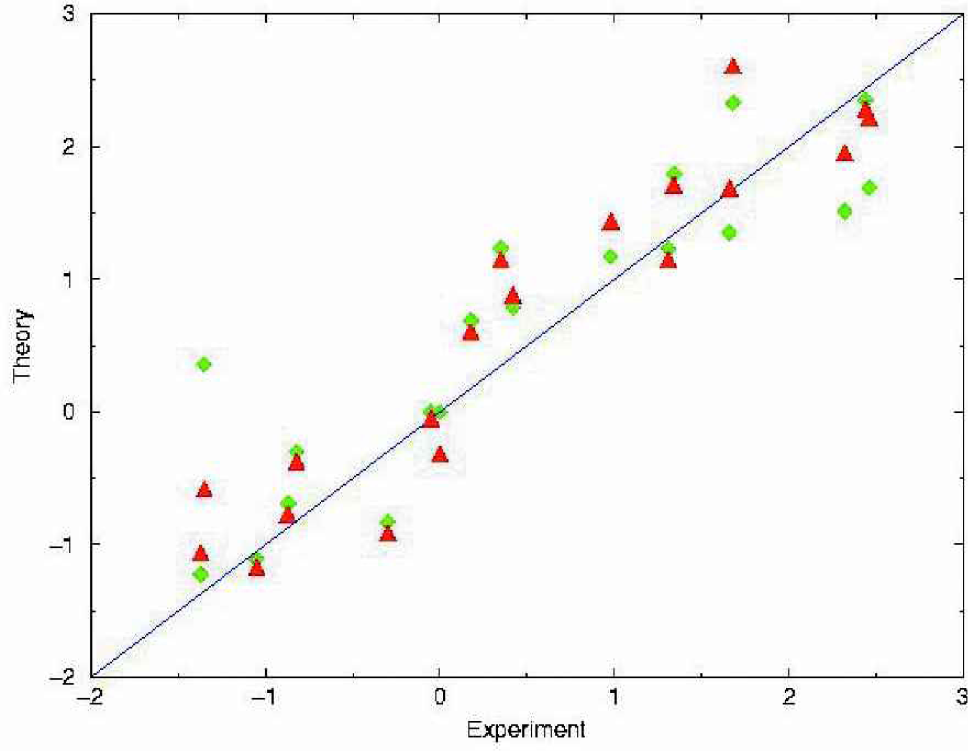
Correlation between the free energies of solvation between experimental data for Gly-X-Gly and two solvent accessible surface area based models (in units of kcal/mol) that differ in the number of atom groups used in the fit. The INT forcefield uses the fit indicated by the triangles with an RMS error of less than 0.5 kcal/mol.
Optimization Methods
Stochastic optimization methods are now being used in a multitude of applications, ranging from circuit design on silicon wafers to airline flight schedules. In these and many other applications the objective is to minimize a given cost function that depends on a large number of discrete or continuous variables.11,38 In analogy to physical problems, the cost function describes a potential energy surface (PES) in the parameter space and its global minimum optimizes the desired objective. The computational challenge in stochastic optimization methods depends strongly on the number of degrees of freedom and the complexity of the PES. The latter depends on the total number of low-lying metastable states, the ability to efficiently explore the configuration space and the average height of transition states that separate low-lying metastable states.
In simulated annealing (SA) 25 optimisation is achieved by simulating the finite temperature dynamics of the system. Starting from a configuration r with energy E(r) one generates a new configuration r' with energy E(r') which replaces the original configuration with probability
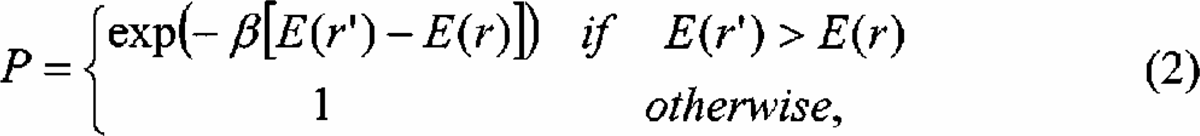
where β = 1/(kT) is the fictitious inverse temperature. At any given temperature such a (ergodic) Monte-Carlo process 31 samples the configurations r of the PES according to their thermodynamic probability. Thus, at high temperature, moves with or against the gradient are accepted with almost equal probability. At low temperature only downhill moves are accepted. In simulated annealing one thus starts with high temperature simulation and gradually cools the system to zero temperature. If ergodicity is not lost during the cooling schedule, the simulation will stop in the global minimum of the PES with a probability of one. For locally smooth PES the search is greatly improved by locally minimizing the new configuration after its generation (basin hopping technique). 38 The particle then travels only among the local minima of the PES, eliminating the costly exploration of intermediate states altogether.
In many rugged PES simulated annealing suffers from the so-called freezing problem. As illustrated schematically in Figure 2 the ruggedness of the PES depends on the ratio of the energy difference of adjacent local minima to the height of the intervening transition state. Figure 2 (b) traces the distribution of two sets of SA processes for a smooth (left) and (rugged) PES respectively. In the latter case the particles remain trapped in their respective local minima, because when the temperature is low enough to thermodynamically differentiate between adjacent minima, the probability of crossing the transition state is already exponentially suppressed. For optimisation problems, where the distance between the global optimum and its approximant is irrelevant, SA can be considered successful even for the rugged PES illustrated here. One should also note that for smooth potentials, basin hopping eliminates the freezing problem.

Schematic potential energy surfaces f(x)=Ax^{2}+cos(x/n) that differ in their ruggedness, i.e. the ratio of the energy difference of nearby local minima to the height of the intervening transition state.
For PES in which the progress of SA is slow, the freezing problem may be circumvented by allowing a trapped particle to escape from a local minimum by increasing the temperature of its simulation. Following this idea the parallel tempering method (PT) replaces the unidirectional cooling of SA by a set of concurrent simulations at different temperatures {Ti | i=1… n}, which occasionally exchange configurations with probability:
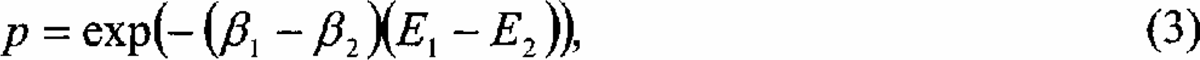
where βi and Ei (i=1,2) are the inverse temperatures and energies of the two simulations/configurations respectively. This mechanism permits each particle to alternate between low temperature simulations where only the closest local minimum is explored and high temperature simulations where it diffuses freely across potential barriers. Equation (3) allows all simulations to remain in thermal equilibrium so that thermal averages can be computed at a variety of temperatures simultaneously (detailed balance). Compared to straightforward SA, PT incurs an n-fold increase in cost for a given total simulation length. On rugged or glassy PES, however, where the escape time from a given local minium can be exponentially long, this overhead may be more than compensated for. Recently a number of methods have been proposed that provide similar mechanisms by generalizing the Monte-Carlo method21,29,28 to simulate ensembles other than the canonical. However, the efficiency of at least some of these techniques has been questioned for glassy PES. 5
The stochastic tunneling method 38 incorporates the ability to escape metastable states by letting the particle in the minimization process “tunnel” forbidden regions of the PES. As in SA we retain the idea of a biased random walk, but apply a non-linear transformation to the potential energy surface:
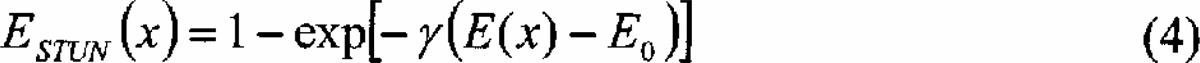
where E0 is the lowest minimum encountered by the dynamical process so far. Alternately a suitable upper bound for the global minimum can be used for E0. This effective potential preserves the locations of all minima, but maps the entire energy space from E0 to the maximum of the potential onto the interval.0, 1
While the transformation in Equation (4) is not the only possible functional, we believe that there are a number of features that constrain its construction: (i) The transformation must be strongly nonlinear in the high-energy regime, as only such a transformation will lead to a nearly constant effective PES for high energies and true “tunnelling”. (ii) There must be a parameter that modulates the degree of compression (γ), since the ratio of the energy differences of adjacent local minima to the transition state energy separating them varies from problem to problem. (iii) Requiring an essentially flat PES at high energy (for typically unbounded PES) requires a transformation that maps the interval [E0, ∞] onto some finite interval, which can be chosen as0, 1 without loss of generality. (iv) It is possible to use a fixed inverse temperature β as a second parameter and to quench the configuration whenever a configuration with an energy lower than E0 is encountered. The optimization of this additional parameter can be avoided, when one adopts the introduced self-adjusting cooling schedule. 38
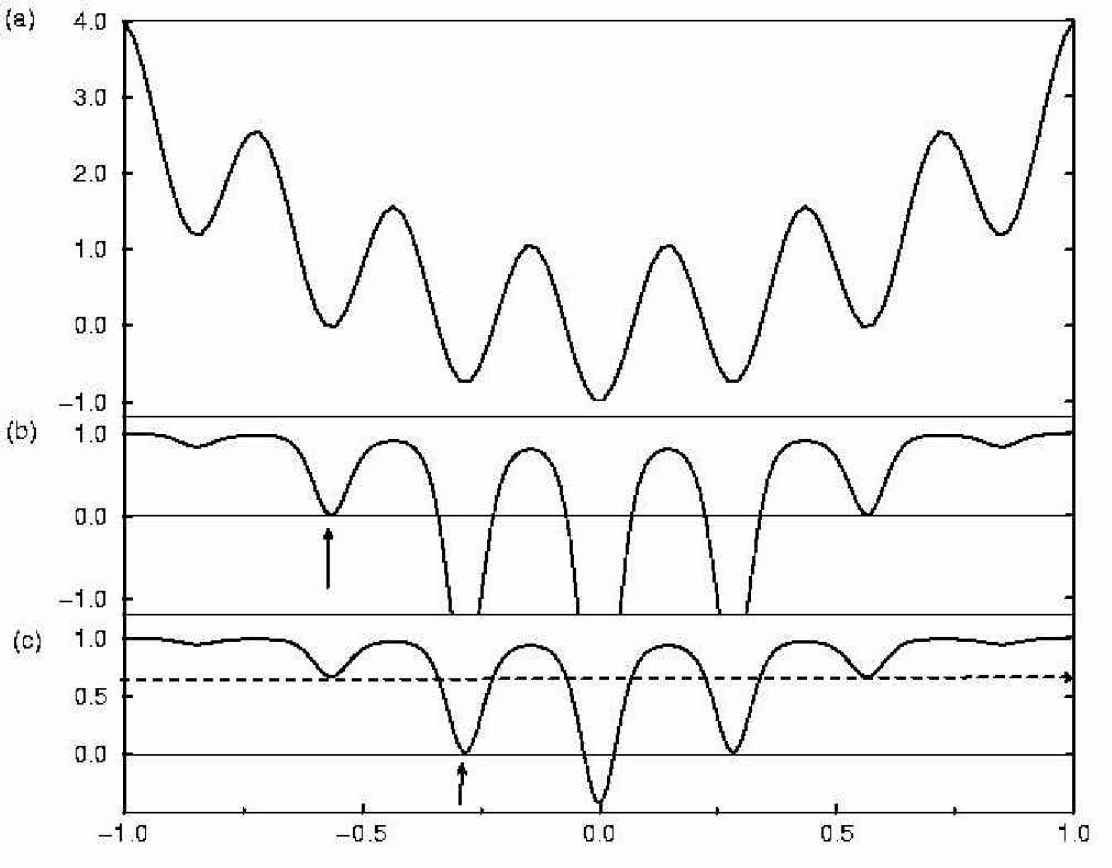
Schematic one dimensional potential energy surface and its transformations under the STUN procedure, provided that the local minima indicated by the arrows have been found. Part (a) shows the original potential energy surface as in Figure 2, parts (b) and (c) the transformed PES under the assumption that the minima indicated by the arrow are the best configurations found so far in the simulation, respectively.
Results
Protein Structure Prediction
We have first investigated the folding of small peptide fragments that are believed to assume a unique three dimensional structure even when removed from their environment in the protein. Figure 4 shows the overlay of the crystal structure of a helical 13 amino-acid residue fragment of the 1HRC protein with the structure we have obtained in ab-initio STUN simulations. Encouragingly, the backbone configurations of these two structures are identical to better than experimental resolution. Figure 5 (a) shows the evolution of the total energy of the structure from an unfolded configuration to the folded configuration as a function of the number of energy evaluations. Figure 5 (b) shows the effective energy and the effective temperature. Several heating and cooling cycles were required to fold the helix fragment and “tunnelling phases” that occur when the effective energy is relatively high and significantly aided the search process. In these phases the original energy of the system undergoes significant fluctuations that are much larger in magnitude than the difference in energy of two successive metastable states.

Overlay of the crystal structure of a 13 residue helical fragment of 1HRC (Residues 92–105) with the structure obtained in the simulation.
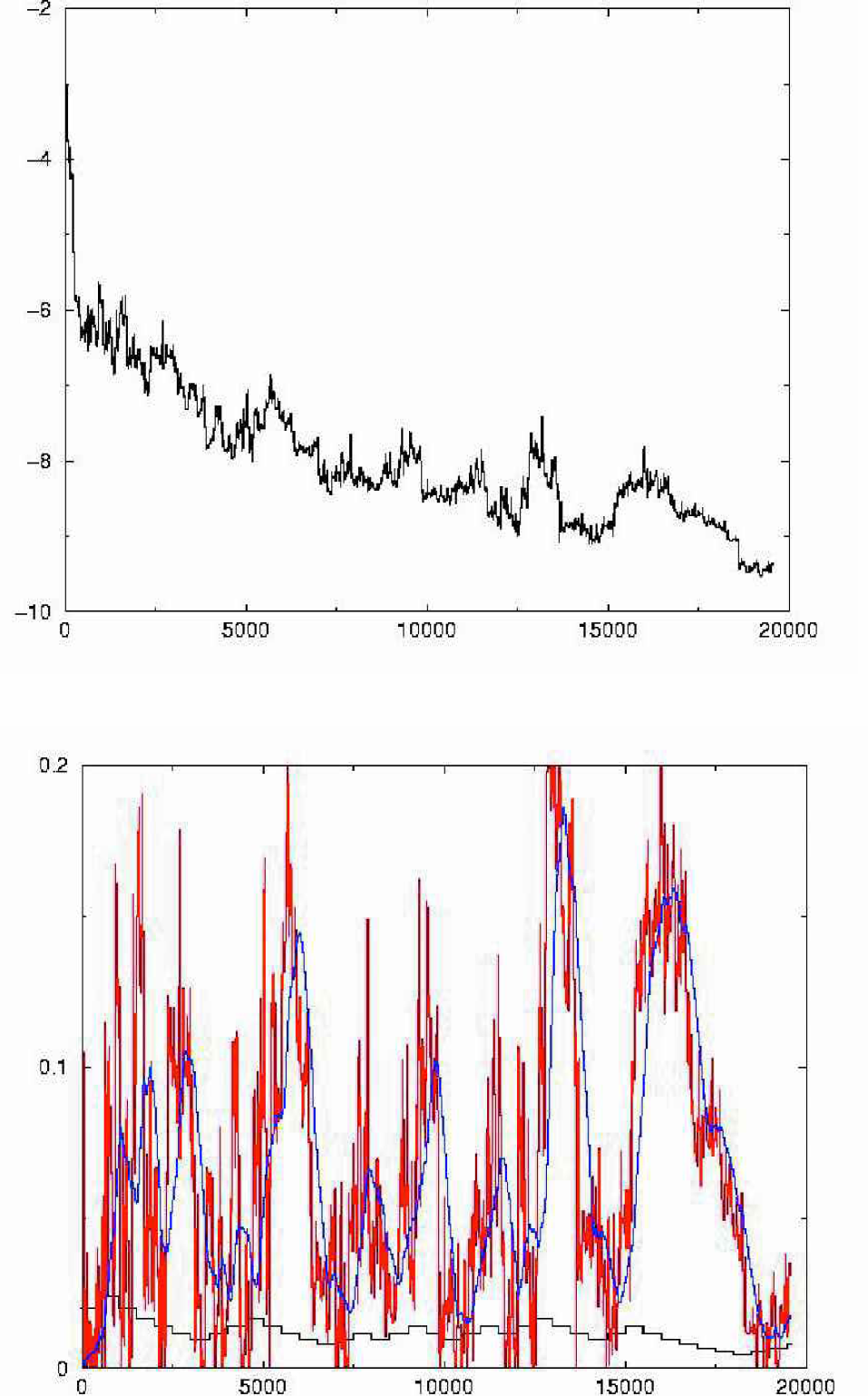
Application of the stochastic tunneling method to the folding of a 13 amino acid helix fragment of 1HRC (Residues: 92–105). The top of the figure shows the total energy of the system as a function of the number of simulation steps. The lower part shows the effective energy, its moving average (dashed) and the effective inverse temperature of the STUN procedure. Both tunneling and local search phases are relevant to determine the native structure of the peptide. Note that tunneling phases with relatively high effective energy correspond to large fluctuations of the original energy in the upper part.
Circumnavigating these energy barriers in a traditional simulation would significantly slow the optimization process. We conducted several dozen STUN runs for this, as well as for other fragments that were investigated to verify that the structure we had obtained corresponds to the global optimum of the system. For 1HRC we found no competing structures with either PT or SA. We noted that in SA the helix could not be folded even with a tenfold increase of the computational effort. Hence STUN appears to present a viable and efficient optimization strategy to optimize peptide fragments of this length.
An example for a non-helical 12 amino-acid fragment of the 1UBQ protein is shown in Figure 6. In this structure hydrogen bonding interactions that attempt to stabilize a helix compete with longer range hydrogen bonding and solvent interactions to form a structure that is part helix part bend. The figure again illustrates the good overlap (backbone RMSD 1.2 A) that was found in our STUN simulations for the simulated configuration and the corresponding crystal structure. A prerequisite for this success is a good balance between hydrogen bonding terms and solvent interactions in the forcefield.
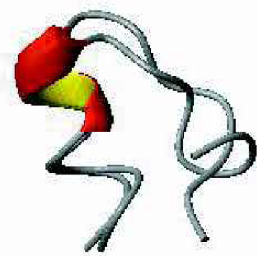
Overlay of the crystal structure of a helical bend in 1UBQ with the simulated structure.
We have also attempted to fold the 36 residue headpiece of the villin protein that was recently simulated with molecular dynamics. 16 The best configuration obtained with about a CPU week on a single PC is shown in Figure 7 (b) in comparison with the NMR structure. The fraction of native contacts was similar in both studies, although more than 85 years of CPU time were invested in the MD simulation on a 256 node CRAY-T3E supercomputer. This comparison illustrates the increase in efficiency that can be obtained through the use of stochastic optimization methods, even though both simulations failed to reach the NMR structure. We find, however that the structure obtained in our simulation has a lower energy than that of the NMR structure, indicating that this failure is not due to a failure of the optimization strategy, but is attributable to a shortcoming of the forcefield. This suggests the need for a rational decoy strategy to systematically improve the forcefield that we presently implement. We generate a large set of “good” candidates that compete with the NMR structure. As long as one of these decoys has a better energy than the native configuration, the forcefield must be modified to stabilize the native configuration in comparison to all other decoys. When this is achieved, we generate new decoys by refolding the peptide, generating either new configurations that are yet again better in energy than the NMR structure or ultimately folding the peptide. This strategy is presently implemented in ongoing work.
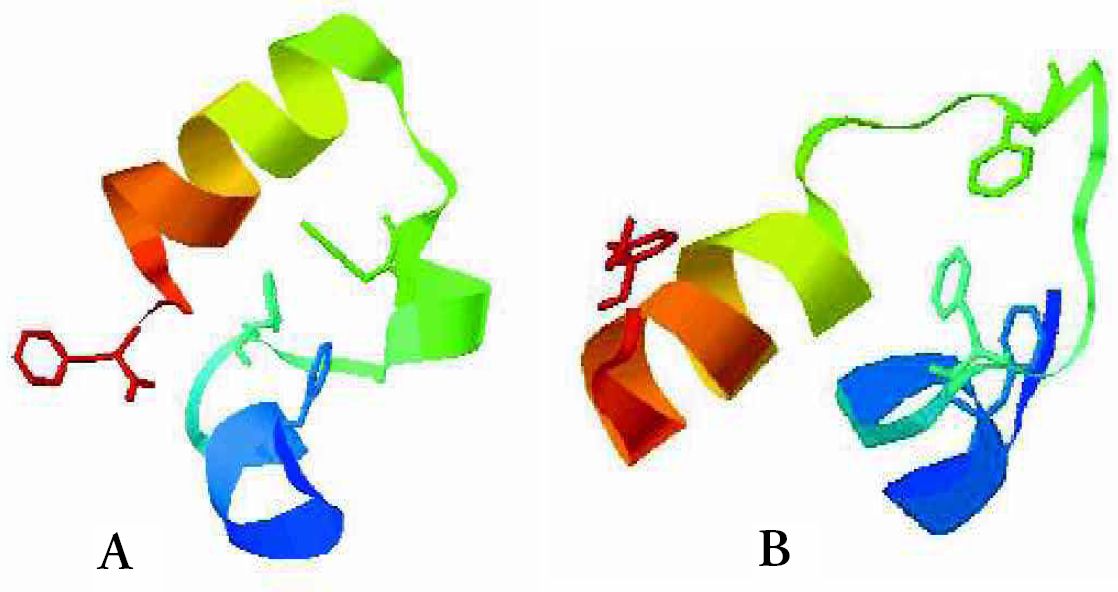
Comparison of the (a) NMR structure and the (b) simulated structure of 1VII
Receptor Ligand Docking
A related low-dimensional optimization problem of considerable practical interest is the receptor-ligand docking problem, where suitable ligands must be selected for a given, structurally characterized receptor.19,8 In order to select suitable ligands large chemical databases must be screened in-silico and for each ligand the best possible fit between ligand and receptor must be determined. Even in the most simple atomistic model, where both protein and ligand are treated as inflexible molecules, efficient numerical techniques to screen large databases in any reasonable timeframe are still lacking. The reason for this difficulty lies in the competition between two vastly different energy scales in the problem, where steric repulsion competes with attractive electrostatic forces and hydrogen bonding to determine the global minimum of the PES. The tight fit between receptor and ligand (key-lock-principle) complicates the optimization problem significantly because it is almost impossible to reorient the ligand within the receptor, while there are few specific interactions between ligand and receptor outside the receptor pocket. Here we illustrate the performance of STUN in comparison with PT and SA for two receptor-ligand pairs, dihydrofolate reductase (4dfr) with methotrexate and the retinol binding protein (1rbp) with retinol respectively. For the simulations discussed below we used a scoring function:
which contains the empirical Pauli repulsion, the Van der Waals
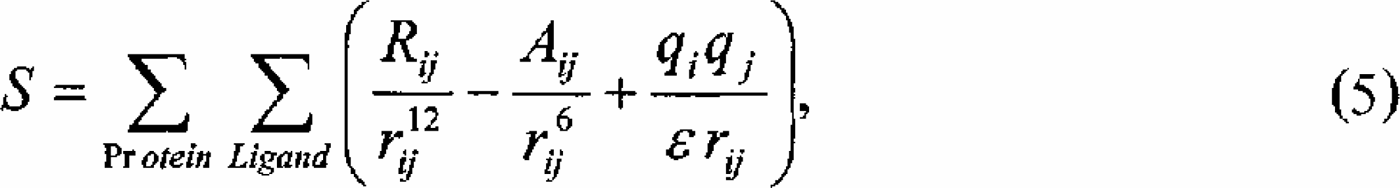
attraction and the electrostatic Coulomb potential. Neither entropic solvation effects nor dielectric screening were used in the simulations because such terms alter the specifics of the affinity of a given ligand to the receptor, but not the nature of the optimization problem. The ligands are simulated as rigid bodies, there are five degrees of freedom in the simulations. In cases where rotatable bonds exist, the x-ray crystallographic structures of the docked ligands were taken from the PDB database. The force field parameters Rij and Aij are taken from the OPLSAA force field 23 and the scoring function is pre-calculated on grids. The atomic affinity grids are interpolated using a logarithmic interpolation technique. 14 To localise the ligand in the vicinity of the receptor, we introduce a drift term that, in the absence of other forces, would lead to a Gaussian localization of the center of mass coordinates of the ligand. 12 The advantage of this approach over the introduction of a penalty function is that the structure of the potential surface remains unchanged.
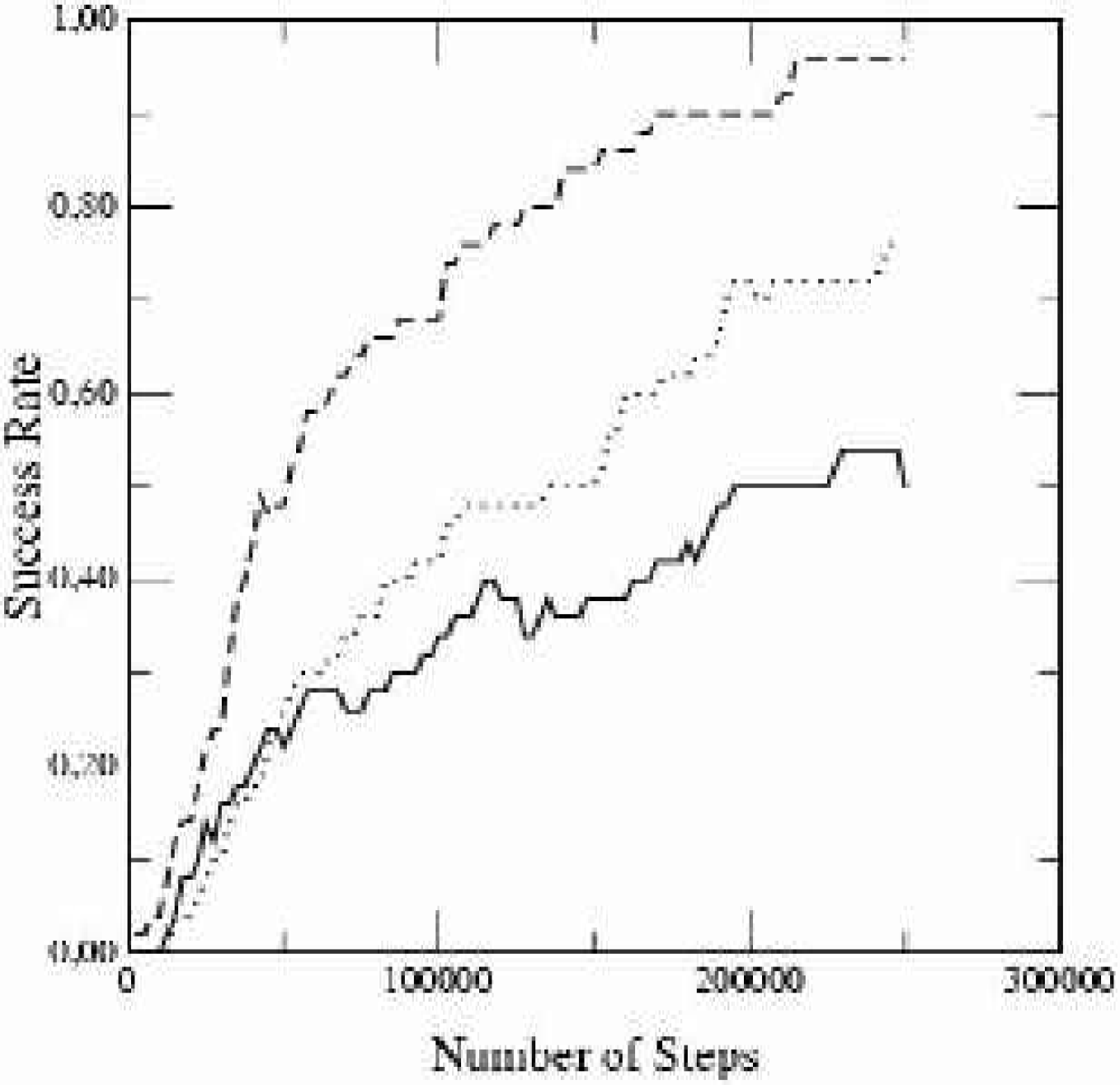
In all simulations ligands were placed in a random position outside the cavity and we averaged the results of 50 runs of pre-described step number. A ligand was defined as ‘docked’ if the average RMS deviation of the atoms from the global minimum was less than 0.1 nm. The potential values were pre-calculated on cubic grids with a grid constant of 0.04 nm and a dimension of 3×3×3nm3. Methotrexate is a prolate-shaped ligand with an axial ratio of a/b=2.5 in the ellipsoidal approximation, i.e., a fat cigar. This system presents several problems. The ligand has a strong dipole moment and tends to dock wherever there are residues with partial electric charges. This leads to a rugged potential surface with a large number of local minima. To make things worse, there exists a very deep local minimum just at the entrance of the cavity. The global minimum, the energy of which is only a few percent lower, is separated from the metastable state by a barrier of hundreds of kJ/mol. The ligand has to tunnel through the barrier, which requires a high temperature, and then to localize the minimum to an accuracy of a few percent in order to distinguish it from the local minimum in front of the barrier.
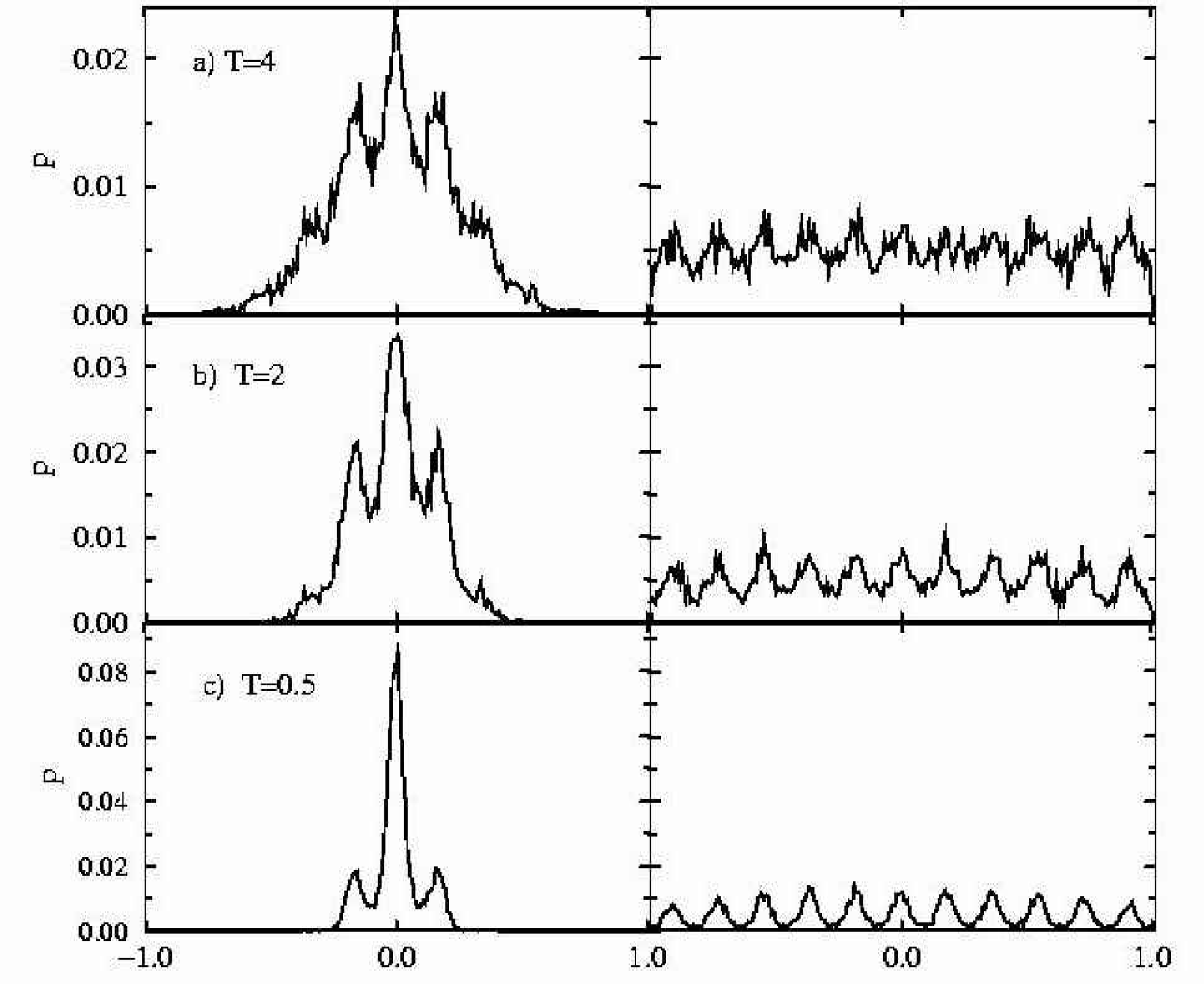
Figure 9 shows the success distribution. Where STUN reached a reliability of 0.5 after 50,000 steps, PT required 150,000 steps and SA required 200,000 steps. In previous work 35 SA was reported to fail completely for this system in the absence of a drift term. During the simulations we observed frequently that the ligand reached the inner region of the docking site in early stages of the simulation, when the temperature was still too high to probe the potential minimum, so that shortly afterwards a better score was found again in the low energy region in front of the cavity. This effect was less pronounced for STUN (tunnel parameter γ = 0.05), where the temperature is regulated by an automatic mechanism. The energy difference between the minima is resolved much earlier in the simulation.
Retinol (see Figure 8) is a prolate shaped ligand with an axial ratio of a/b = 4.5, i.e., a slim cigar. There is no significant dipole moment in retinol. In the rigid receptor approximation the binding site is almost completely enclosed and a molecule has to tunnel through a barrier of several thousands kJ/mol to reach the global minimum. On the other hand, this system presents no low-energy secondary minima. Since the cavity is quite elongated (length: 1.65 nm) only a weak drift term was applied. In agreement with previous studies 14 SA completely failed to pass the ligand through the barrier. The same held true for PT. Among 50 runs there was no successful docking. The success distribution for STUN (γ = 0.002) demonstrates that this technique is capable for a fast and reliable docking of retinol, reaching a a success rate of 0.5 after about 40000 energy evaluations.
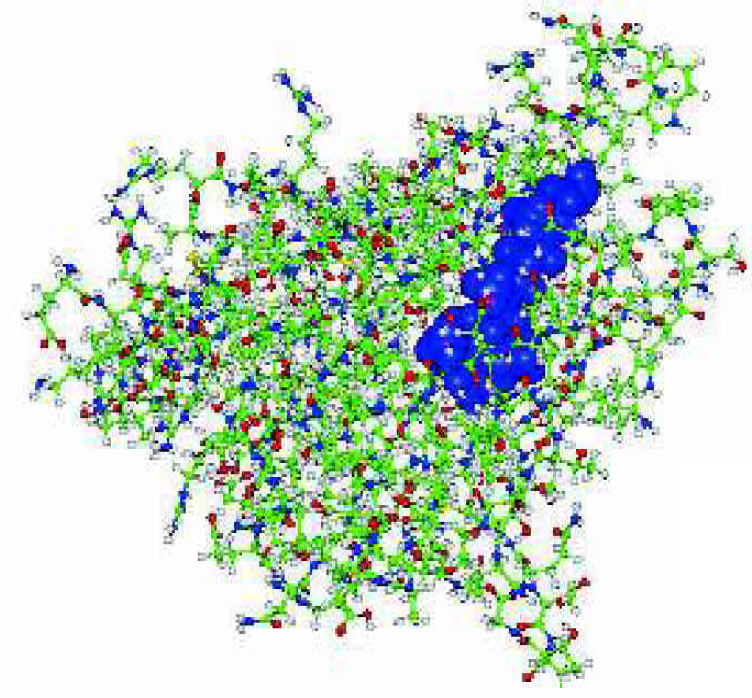
The retinol docking protein with its ligand.
Summary and Conclusions
We have discussed the use of stochastic optimization methods as a technique to predict the structure of complicated biomolecules. To implement this approach, a forcefield that parameterizes the free energy of the underlying model must be developed and such a forcefield must contain an implicit parameterization of the interactions of the biomolecule with the solvent. We have argued that there is a rational, decoy-based strategy to develop a biomolecular forcefield that can be used to predict the structure of short peptide fragments using stochastic optimization techniques such as the stochastic tunneling method. We have illustrated the success of this approach in the folding of short peptide fragments and presented an analysis of the difficulties encountered in the folding of the 36 head residues of 1VII. Stochastic optimization methods nevertheless permit an analysis of this problem and a systematic strategy for the improvement of the forcefield several orders of magnitude faster than competing simulation techniques. Finally we have illustrated the applicability of the stochastic tunneling method to a related problem of practical interest in rational drug design.
Acknowledgments
This work was funded by the Deutsche Forschungsgemeinschaft (We 1863/11–1), the BMBF and the Bode foundation.