
Abstract
Pfizer Inc. is a research based global pharmaceutical company committed to the discovery and development of innovative medicines that improve the quality of life of individuals throughout the world. In order to gain a competitive advantage over competitor companies in the discovery of new medicines, we must speed up drug discovery and drive down costs. Using non-combinatorial chemistry techniques increases the efficiency with which we discover new ‘leads,’ leveraging our drug discovery efforts through retaining complete control over the properties of each product made. Compound purification, managed through in-house software, augments the quality of data obtained upon biological screening of compounds.
DISCUSSION
One way of achieving the increase in productivity sought by the pharmaceutical industry, embraced over the last decade, is to use combinatorial chemistry techniques to increase the efficiency with which we discover new ‘hits’ and ‘leads’ as the source material to leverage drug discovery. 1 –4 Pfizer believes that this paradigm has benefits for drug discovery though not in the form practised by other pharmaceutical companies. Combinatorial chemistry may be defined as the rapid synthesis of chemical entities derived from all possible combinations of monomers used for synthesis; for example, 10 carboxylic acids coupled to 10 amines would lead to 100 amide products as the combinatorial set. This practice can lead to the synthesis of large numbers of compounds, but we believe that numbers alone are not sufficient for efficient drug discovery. Lipinski et. al. of Pfizer recently published a set of guidelines 5 coined the ‘rule-of-five’ which provided ranges for the physical property parameters of compounds that are likely to lead to good oral bioavailability, an important ‘drug-like’ property. The process of combinatorial chemistry leading to synthesis of all combinations will clearly lead to the production of some compounds within the library not obeying Lipinski's ‘rule-of-five,’ and hence not likely to possess good oral absorption. Thus, we do not synthesise combinatorial arrays but instead prepare non-combinatorial or ‘sparse matrix’ libraries. We only synthesise those combinations, from all possible ones, which produce ‘drug-like’ compounds meeting the ‘rule-of-five.’
Non-combinatorial chemistry as an approach allows the design, synthesis, purification and screening of any group of individual compounds from a virtual library, not just arrays of compounds. This facilitates compound synthesis in a flexible manner from virtual libraries that are as large as possible, whilst retaining complete control over the properties of each product made. Thus, our desire to include careful experimental planning and meticulous observation of results, made possible by non-combinatorial library design, provides for faster ‘traditional’ medicinal chemistry. The strategy to accomplish this requires:
High quality data
We believe that in order to minimise misleading data arising through screening of reactive intermediates and/or by-products present in crude samples, all compounds produced for biological screening should be purified by HPLC. Pfizer has been at the forefront of developing the parallel purification technology required to rapidly purify large numbers of compounds. Purification of library samples minimises misleading results caused by reactive contaminants, provides compounds of equal ‘worth’ as medicinal chemistry derived samples, expands the chemical scope of the library protocol by allowing utilisation of low-yielding reactions, and provides analytical data of all compounds entering the screening file. However, these gains come at the cost of the purification phase requiring approximately twice the time for synthesis of the compounds.
Consider development at an early stage
Non-combinatorial design and synthesis techniques allow the accumulated knowledge that medicinal chemists possess relating to such issues as why compounds may fail in development to be more easily incorporated into the up-front library design. This may be accomplished by considering pharmacokinetic predictors, such as Lipinski's ‘rule-of-five,” 5 and disallowing groups that are not desired to be present within the library, such as potential toxicophores. 6 Coping with these issues becomes more difficult if combinatorial techniques are used for library design.
Maximise chemical opportunity
Automated chemistry is 102–103 times more efficient than traditional medicinal chemistry synthesis in terms of the formers ability to synthesise larger numbers of compounds in a similar time frame, compared to traditional hand-crafted medicinal chemistry ‘wet’ synthesis. This has obvious benefits in producing drug candidates earlier if used in conjunction with an appropriate design strategy. Thus, we propose that automated chemistry be used as far as possible in the drug discovery process, aimed at producing compounds relevant to the design hypotheses. For this to be possible, validated protocols are required together with large virtual libraries to exploit in synthesis.
Exploit the virtual library with maximum flexibility
Utilise design strategies that allow one to ‘cherry pick’ compounds for synthesis and use automation that is also capable of synthesising these cherry picked selections and not just combinatorial arrays, as illustrated in Figure 1.
Cherry picked design versus a combinatorial array exemplifying maximal flexibility in exploiting a virtual library with the former design. In ‘follow-up 1,’ an active on which the library design is to be based is indicated by the red diamond. Each axis consists of a set of monomers used to construct the compound, in this case a two component library. The monomers are ordered by similarity (according to a judgement made by medicinal chemists). The design consists of keeping one monomer constant and varying exhaustively the other monomer set, and selecting only those for synthesis which meet the design criteria. This design is then repeated for the other monomer set. Additionally a selection is made based on similarity to the active, and encompassing two monomers either side of the active on each monomer axis. In reality, this selection would encompass a larger set than two monomers, but this is used for exemplification. Each blue diamond, therefore, represents a compound chosen for synthesis after filtering through the chosen design criteria. ‘Follow-up 2’ may represent a further design based on several actives now obtained from screening of the compounds from ‘follow-up 1’ against the chosen biological target. In this case, a similarity selection is made around each axis, again the axes are ordered by monomer similarity, to provide the selected compounds for synthesis after filtering against the design hypothesis.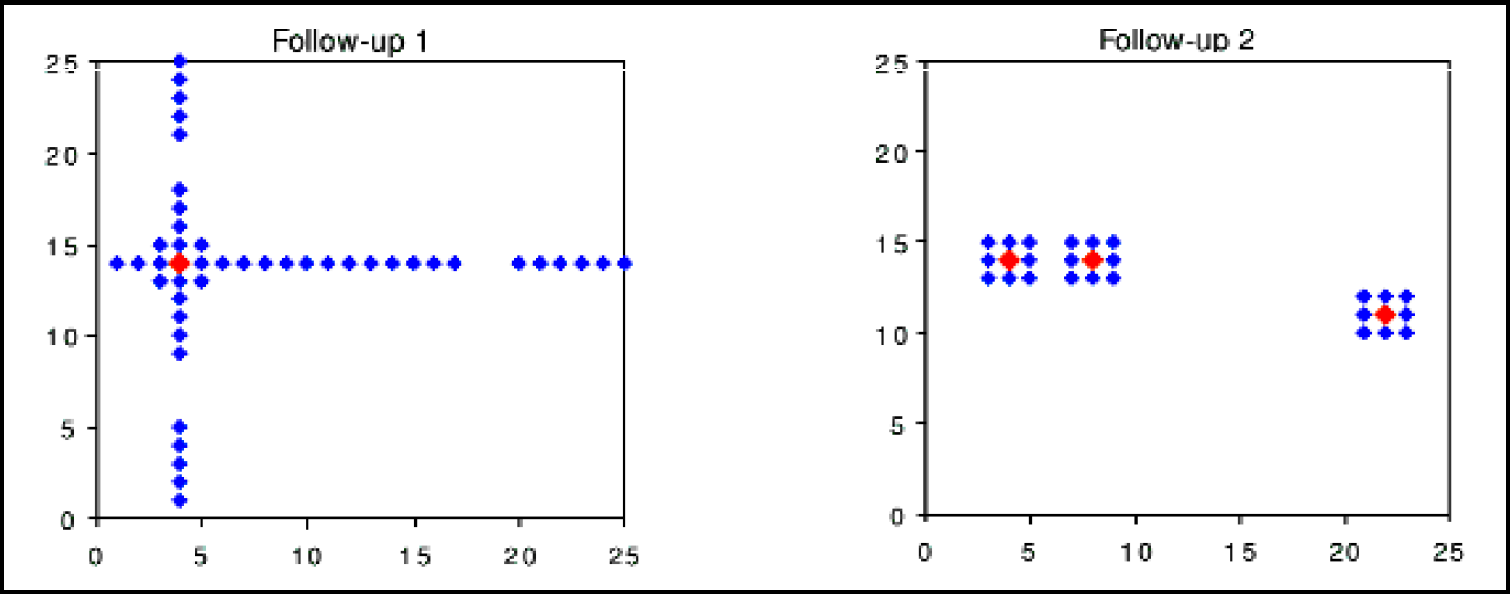
AN INTEGRATED DESIGN AND SYNTHESIS MANAGEMENT SUITE - LIBRARY CREATION, REGISTRATION AND ADMINISTRATION (LiCRA)
Pfizer set up the Library Design & Production group (LDP) in 1997 which set about the task of making the aforementioned process a reality through a number of technology developments. These innovations, vide infra, enable the production of purified single compound libraries matching Lipinski's ‘rule-of-five.’ Many of these developments have taken place in-house, primarily as a result of a dearth of relevant technology being available commercially at the time.
Development of ‘in-house’ software enabling non-combinatorial ‘sparse matrix’ library creation - ‘Selection in 1 Dimension’ (S1D)
An algorithm was developed in-house which would define only those compounds meeting Lipinski's ‘rule-of-five’ for synthesis. This allowed our stringent ‘drug-like’ criteria to be met. The library monomers are stored electronically in an ISIS database (see Figure 2), and a list containing properties such as molecular weight is imported into S1D. The algorithm sorts those compounds meeting ‘rule-of-five’ into a list ready for synthesis that is then loaded into LiCRA, vide infra. It is possible to view property profiles for the libraries that are created. In addition to the use of S1D, LDP also uses third party design software for creating library designs.
Typical design process using software developed ‘in-house:’ S1D.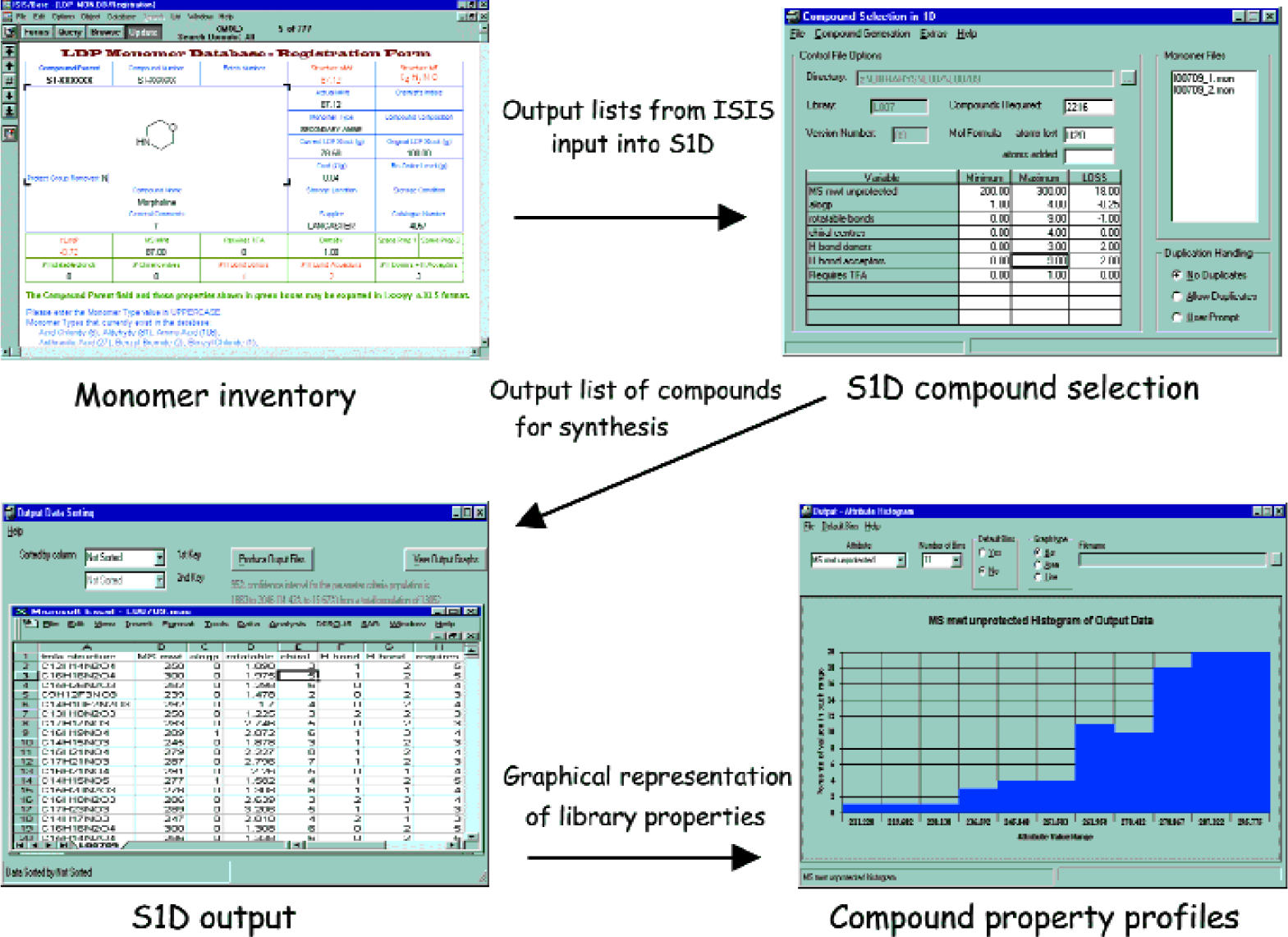
Development of ‘in-house’ software (LiCRA) enabling tracking of Library Creation, synthesis, purification, registration and administration
The output from S1D is then fed into LiCRA which manages library synthesis, purification and registration. All data is tracked from within one Oracle database. Figure 3 illustrates some of the data we are able to track from LiCRA. The initial library screen provides information relating to the point at which library creation, synthesis, purification or registration has reached. From here it is possible to command synthetic steps and manage the purification process followed by registration. The plate format is given which can provide structure, HPLC and mass spectral details on any compound from stored data.
The integrated software suite for library compound synthesis and purification.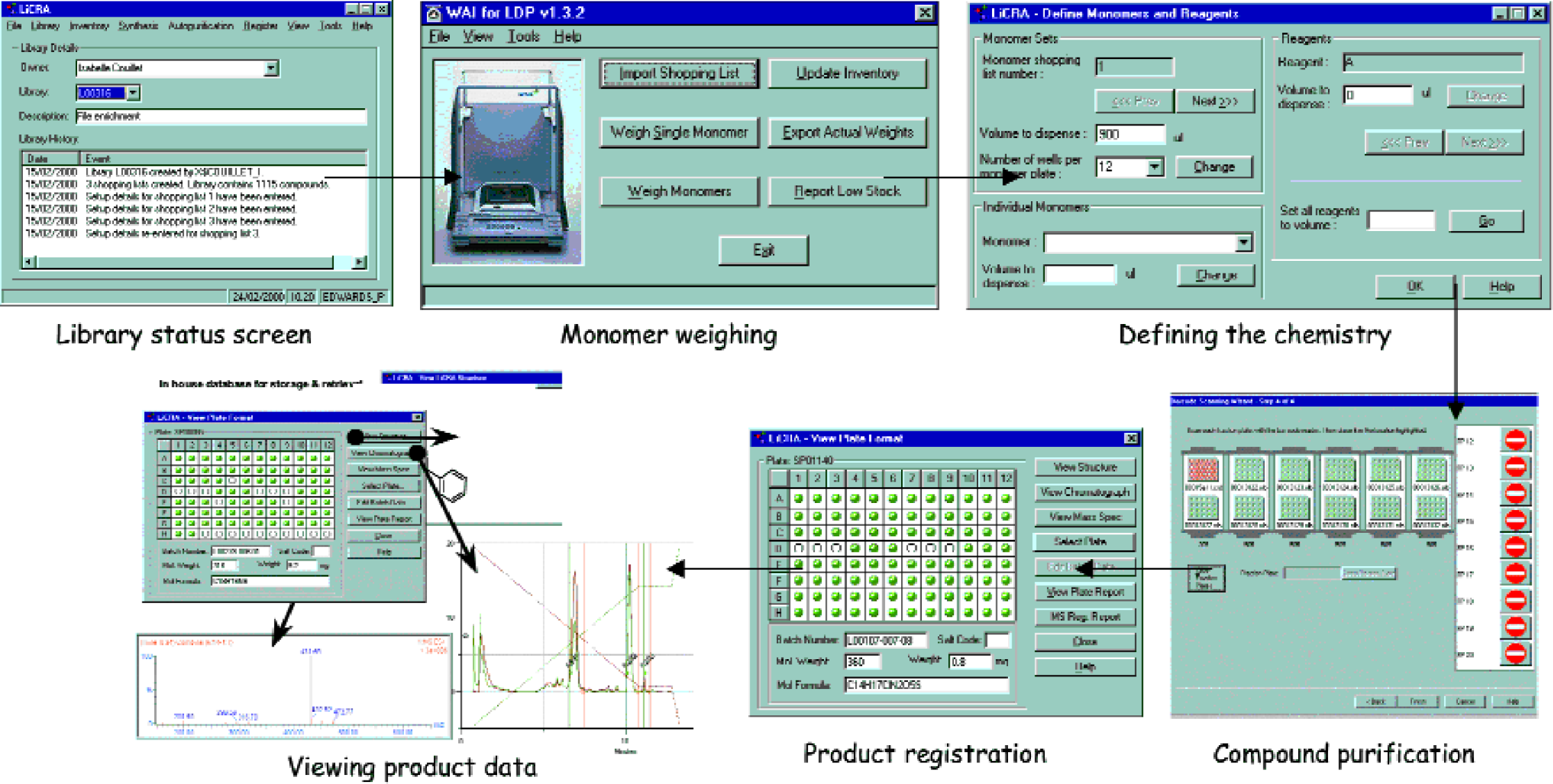
Development of software/hardware allowing efficient high throughput autopurification of library compounds
The non-combinatorial chemistry approach to library synthesis generates large numbers of single compounds. Purifying each compound will minimise the presence of impurities and the generation of misleading results caused by reactive contaminants in biological screens. There are currently no commercial purification systems on the market that can effectively process, quantify and track large numbers of compounds in an automated way. In order to purify compounds in the kind of numbers we wished to, Pfizer enabled these technological developments by entering into various collaborations with third parties. Figure 4 illustrates the main components of the autopurification system we use to purify our compounds.
Main components of the autopurification system.
96-Well plates containing a single crude synthesised sample per well are entered into the system by scanning a barcode incorporating a unique plate identifier. A liquid handling robot then takes each crude compound in turn and this is purified by reverse phase HPLC. Fractions are collected, triggered by UV detection. Once purification has taken place, the purified samples contained within fraction plates are taken, automatically by a Sagian ORCA® robot, for mass spectral analysis, with on-line Evaporative Light Scattering Detection (ELSD) to determine sample weight. All required fractions, which meet certain purity criteria, are then automatically ‘consolidated’ by the ORCA® and liquid handling robots into sample tubes based on an ‘in-house’ design which are ready for evaporation. The selection of desired fractions is made automatically based on a software analysis of the purity of each fraction. If our minimum purity criteria are exceeded the sample is selected. All of the above processes are managed from within LiCRA. ELSD is necessary to determine the amount of material present, allowing the samples to be made up to a known concentration for biological testing. Figure 5 depicts some key screen shots illustrating this process.
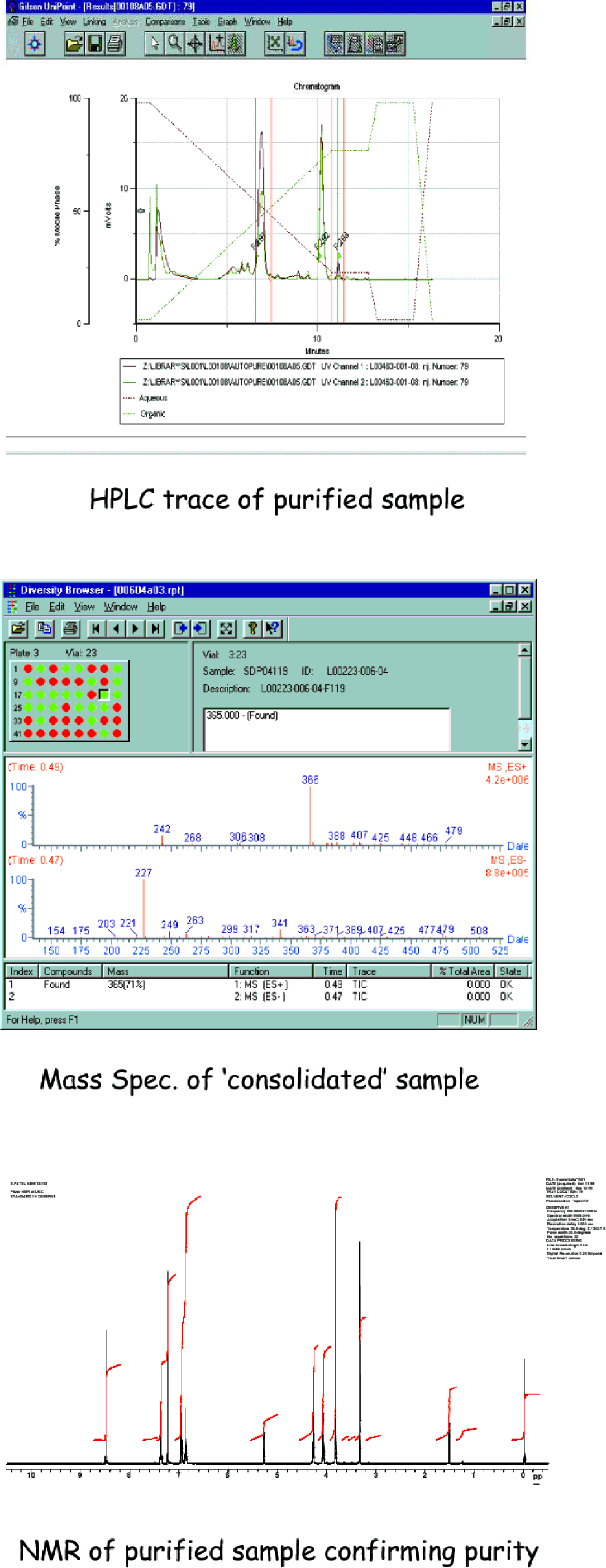
Sample screenshots from within LiCRA illustrating part of the autopurification process.
APPLICATION OF S1D AND LiCRA NON-COMBINATORIAL CHEMISTRY TO ‘HIT-TO-LEAD’ OPTIMISATION
We have applied the S1D and LiCRA systems to a number of targets. A recent example comes from an HTS campaign that was run against a GPCR target ‘X.’ A number of actives were discovered upon screening of the Pfizer corporate file and many of the active hits came from several chemical series which were amenable to parallel synthesis. Typically activity ranged from several hundred nanomolar up to 10 micromolar. One series was chosen for study based primarily on the ease of synthesis of compounds from this series. For example, a single disconnection led back to readily available monomers, and the low molecular weight (typically less than 300) preferred for pharmacokinetic reasons was seen with many of the actives. However, compounds from this series typically contained a toxicophore derived from one of the (amine) monomers used in compound construction. Thus we set for ourselves the early goal of removing this toxicophore and utilised non-combinatorial techniques to go about achieving this aim. Maintaining or improving potency was also considered an important goal, especially as we moved to different chemical series having replaced the toxicophore. From the actives isolated from the HTS, we used S1D to design a non-combinatorial library of 855 compounds where compound synthesis aimed to fill chemical space occupied by the HTS actives, and used medicinal chemical thinking to replace the toxicophore with more benign alternatives. All compounds synthesised were within Lipinski's ‘rule-of’-five’. Figure 6 illustrates the library design, depicted in two dimensions. Each monomer is ordered by a chosen ‘similarity’ metric and points represent compounds chosen for synthesis. It is evident that S1D has been used for library design as we have avoided choosing compounds for synthesis at the top right on the diagram which is occupied by high molecular weight and lipophilic compounds.
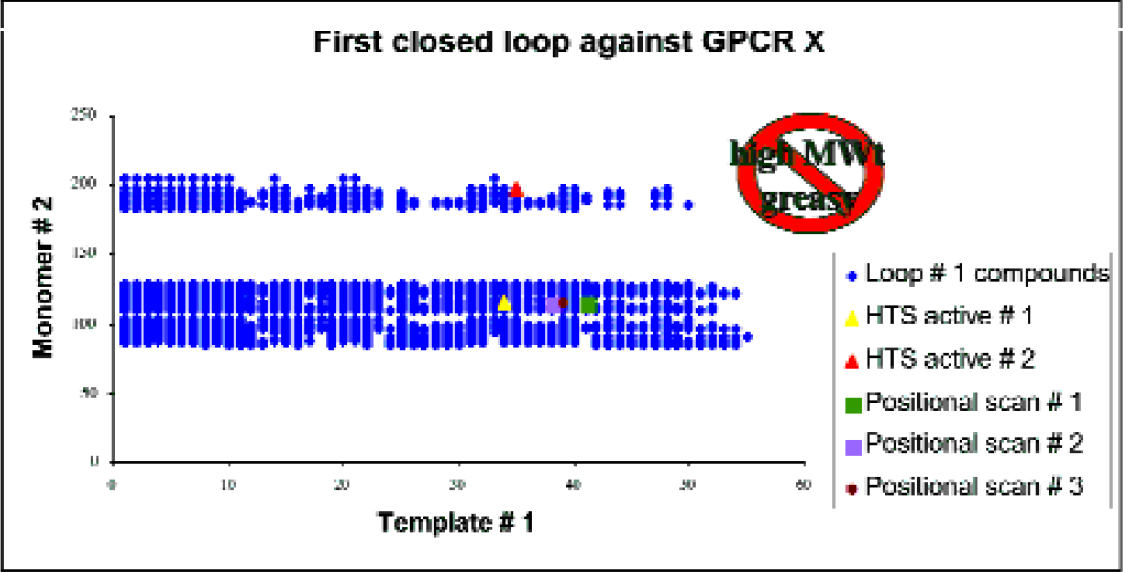
Non-combinatorial selection of compounds using S1D for library design. Non-combinatorial selection of 855 compounds. The HTS active matter, upon which this library design is based, are shown as triangles. The ‘positional scans’ represent keeping one of the monomers found in an active compound constant and exhaustively varying the other monomer set, choosing only compounds for synthesis that meet our design criteria. Each axis of the chart consists of a list of the monomers used in each position. The monomers were ordered on each axis by similarity (according to a judgement made by medicinal chemists). On each axis, as numbers increase this indicates increasing lipophilicity of the monomers used and hence of products formed from their combination. Thus the top right of the diagram indicates the most lipophilic compounds, as judged by cLogP.
The first library consisted of the synthesis of 855 compounds. Upon biological screening of this set of compounds, seven active compounds were obtained which originated from a distinct chemical series. This distinct series displayed activity in the low micromolar region, giving us slightly reduced potency compared to the HTS actives, but importantly we had successfully replaced the toxicophore and provided low molecular weight (<350) actives. These actives were then used as the basis for the design, synthesis and biological screening of a further round of compounds. 7 Encouraged by this success we ran a second synthesis loop of 990 compounds and this time we obtained a 4% activity rate (screening concentration of 10 μM). In this loop, we had made significant potency gains in the novel series, devoid of any known toxicophores, through the synthesis and screening of a large number of active compounds. Currently, design of further loops is in progress where we aim to provide potency gains and make consideration of pharmacokinetic properties of final compounds.
CONCLUSIONS
Pfizer has been able to create an integrated library synthesis group capable of efficient and robust chemical production and purification of many thousands of medicinally relevant compounds per year. Over this time we have demonstrated successful closed loop optimisations, such as with the example presented here, and have generated hits and ‘hit-to-lead’ libraries giving leads that have had a beneficial impact on Pfizer's drug discovery process. 8 We have thus shown that non-combinatorial chemistry is an effective method for lead optimisation, a method that has as its cornerstones the ‘quality’ of design and ‘quality’ of its products; 9 we have achieved this with our S1D and LiCRA systems.
ACKNOWLEDGEMENTS
The author would like to thank Pfizer Global Research & Development, members of LDT, LDP, MISD, SASS, HDG, DRi, and Therapeutic Area Chemistry and Biology who have been intimately involved with the work presented here.