
Abstract

This presentation was given at the 1999 International Symposium for Laboratory Automation and Robotics held in Boston, MA, October 17-20, 1999. The full manuscript is available on CD-Rom and can be acquired by contacting Christine O'Neil, 508-497-2224; email
With the development of high throughput screening (HTS) during the last two decades new technologies have gained access to chemistry and biology laboratories. The use of both, laboratory robotics and automated workstations has greatly increased the number of chemical entities that are synthesized for and tested against new targets. While a decade ago a daily throughput of about 1,000 compounds was considered sufficient, nowadays screening laboratories aim to achieve 100 times as many samples in the same period of time. Combinatorial chemistry vice versa has increased comparably the daily output and HTS has become an important success factor during early lead finding. Nearly all drug discovery research projects in pharmaceutical industry employ HTS screening assays as initial steps to discover the chemical leads. These compounds provide the structural basis for further medicinal chemistry activities that focuses on optimization of the lead with respect to the activity and selectivity profile in order to identify the development candidate.
The pace of this technological development not only has created more benefits such as shortening the time required for lead identification but also has generated considerable issues. Among those the costs extensively increased with the number of samples investigated. Assay miniaturization and related development helped to reduce the impact. The number of chemical entities that enter the primary screening and the resultant number of hits that require further evaluation have questioned the hit selection and profiling. HTS, with a hit rate between 0.01 and 1%, depending on the target and test concentration used, produces several hundred of primary hits while in former times only a hand full of suitable candidates were available. The decision which candidate to be followed has a direct impact on the success rate during the further development phase. The generation of primary hits and the selection of leads just based on their structural properties does not predict the probability to convert the lead into a drug. The drop-out rate during development is still considerable and is associated with high financial losses in the pharmaceutical industry. Only one out of approximately ten potential drug candidates entering phase I of development will finally reach the market. Insufficient pharmacological and pharmaceutical properties represent the majority of reasons for failure (1). Information of compound properties relevant for development are often incomplete with respect to data on selectivity, solubility, pharmacokinetics and toxicology at the time when needed for decision making, since they are gathered sequentially.
HIGH THROUGHPUT PROFILING
With these specific issues in mind CEREP has built over the recent years an integrated drug discovery platform in order to serve the changing and challenging needs in lead finding. The activities are based on the well-known company's strengths such as combinatorial chemistry, molecular modeling and compound profiling. The starting point of discovery programs is the utilization of a diverse combinatorial libraries, such as Odyssey 5000, to initiate the lead finding process. This library, constructed from more than 1,500 unique monomeric building blocks, was designed to have an efficient way for the identification of initial leads in a new research program. The leads identified are passed off to the key component of CEREP's platform, the high throughput profiling (Figure 1).
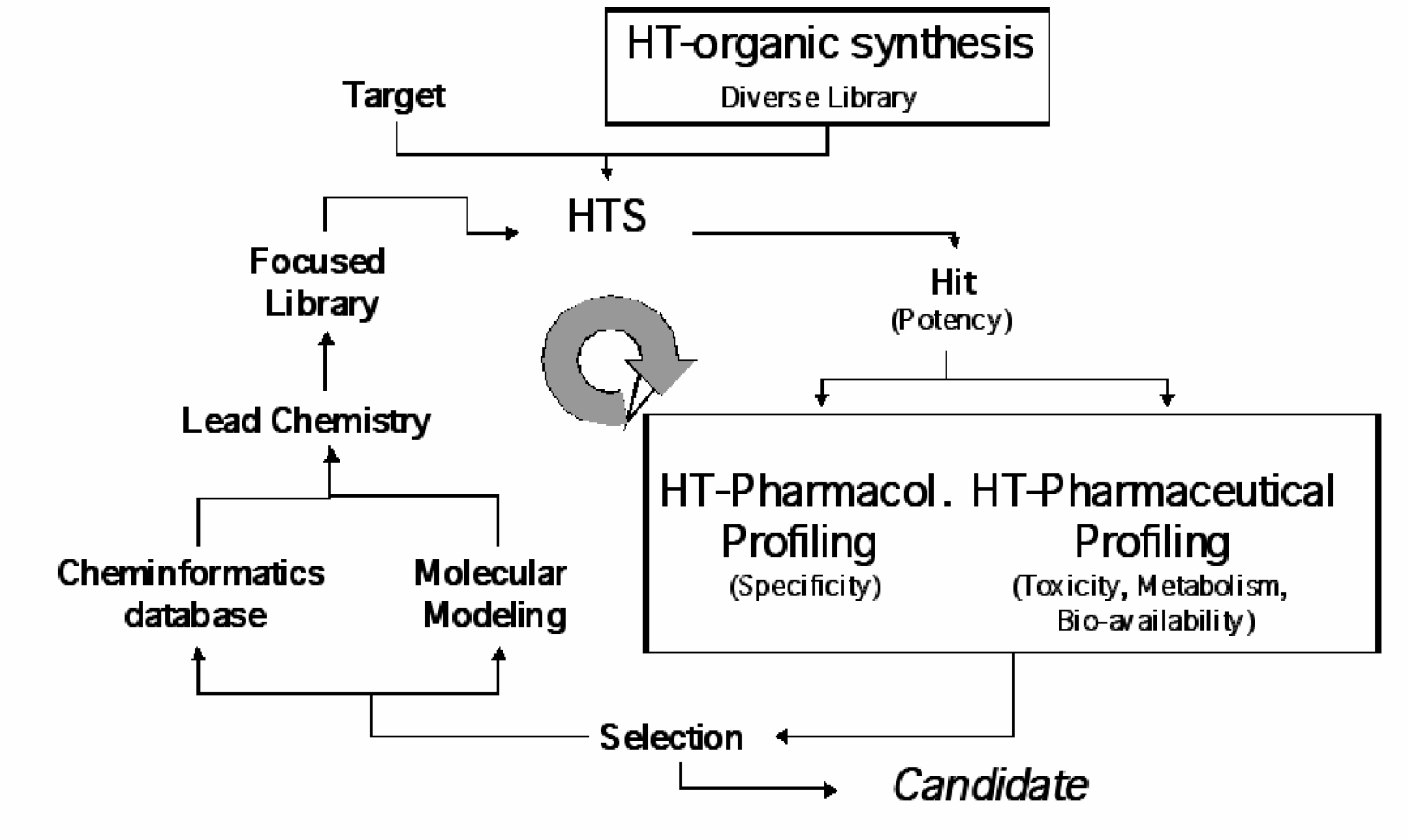
Within the discovery platform at CEREP high throughput organic synthesis and high throughput profiling are main features that enable the scientist to identify the respective candidate based on a broad resource of biological results
The high throughout profiling consists of two major components, one of which is the pharmacological profiling that addresses the issue of compound selectivity and specificity. Selectivity is determined by testing in closely related targets such as different subtypes of the target receptor (or ion channel), different isozymes of the target enzyme, or other mechanistically related targets. In most cases it is desirable to have greater than 10-fold selectivity for the desired target.
The primary purpose of specificity testing is to identify “promiscuous” compounds that interact with several unrelated targets. Such interactions can be indicative of in vivo side effects or safety problems. For these reasons selection of assays in a specificity panel are often tailored to check for known undesirable interactions.
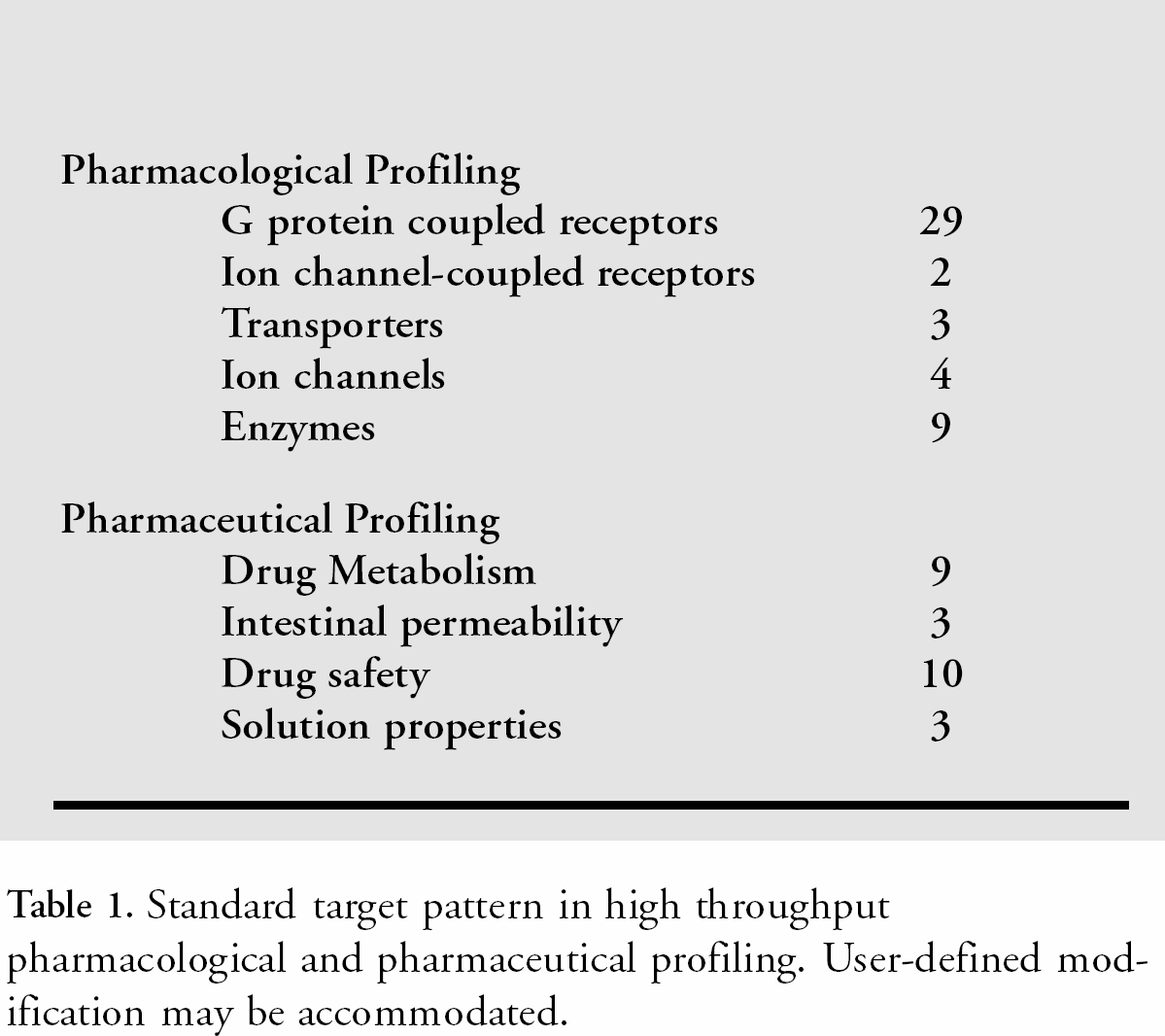
The current standard test panel consists of a variety of receptors, channels, transporter systems and enzymes but may be easily extended according to the scientists needs. The activity pattern being obtained allows the scientist to distinguish favorable and adverse drug properties with respect to its biological activity. Table 1 gives an overview on the different assay classes selected and the number of targets in each of them.
Standard target pattern in high throughput pharmacological and pharmaceutical profiling. User-defined modification may be accommodated.
Traditionally the pharmacological and pharmaceutical criteria have been assessed in a sequential manner with major focus on the compound potency and selectivity. Often there is a round of compound optimization through medicinal chemistry between each step. Each step has been used to rank a group of hits from primary screening. As it has not been practical to advance all compounds, the lowest ranked are generally dropped at each step. As a consequence, after significant time and expense many compounds are found to have high potency but display only poor specificity or pharmaceutical properties. The focus of the pharmaceutical profiling, as performed presently, addresses issues of solution properties, metabolism, intestinal permeability, and safety.
At this time it is becoming practical through application of HTS technologies to secondary assays to quickly and cost effectively generate a full data set encompassing the criteria above on each hit compound from primary screening. The advantage of this parallel (as opposed to sequential) approach is that key decisions are made with full awareness of the positive and negative attributes of each compound or compound class.
An example of compound profiling is given in Figure 2. The comparison of the activity pattern of the drugs investigated allows to chose the preferred candidate out of a list of other competitors. The broad spectrum of information obtained is the basis for and represents the first step towards a knowledge-based decision in the drug discovery. The risk of taking a compound into development can be calculated and costly failures of loosing a drug on its long way to IND maybe reduced or even completely avoided.

Activity profiles of compounds against more than 100 different targets. The grayscale coding represents their activity obtained in each test, typically expressed as % activation or inhibition depending on the target.
During the recent months we have profiled several hundreds of publicly available drugs and more than 100,000 data points have been collected using this approach. This has been made possible by a proprietary assay technology implemented on Zymark robot systems. In addition to the biological data sets structural fingerprints were established using software packages that represent the spatial orientation and distance of pharmacophoric groups within the molecules. Together with the biological results they are stored in a database. The data acquired accordingly represent a unique basis for data mining with respect to both, the clustering of compounds and biological tests. With the increase of the information content it will be possible to group development candidates according to their structure with compounds of known profile in the database. With the increasing amount of information acquired, certain predictions not only with respect to the compound's activity and physicochemical profile but also with respect to its in-vivo properties may be possible. Based on the knowledge obtained the selection of a drug candidate with an increased probability to pass through development and to reach the IND filing may be achieved.
Another approach to use the indicative potential of the data collection is the development of focussed libraries for screening against new targets. Analysis of the biological targets and the activity pattern of the drugs obtained in profiling allows the design of a test set of compounds. This may either be based on the selection of chemical entities that show activity to the respective target group, e.g. G protein-coupled receptors, kinases etc., or on lead explosion by the selection of drugs with a comparable profile obtained in profiling. The feedback of the results to molecular modeling enables the scientist to follow a more rational way in drug design that will help to reduce efforts and time normally spend.
CONCLUSION
During the last decade high throughput screening and ultra high throughput screening have significantly contributed to the advances in drug discovery. The increase in research costs associated and the bottlenecks further down the development have increased the demand for alternative approaches. Among those high throughput profiling combines both, the conventional screening strategy combined with data mining technologies that will foster drug discovery in the next millenium.
ACKNOWLEDGEMENTS
The outstanding scientific work of Dragos Horvath, John Cargil and Xianqun Wang to design the database environment and modeling algorithms is greatly acknowledged.